

Abstract
In chronic myeloid leukemia, resistance against BCR-ABL1 tyrosine kinase inhibitors can develop because of BCR-ABL1 mutations, activation of additional pro-oncogenic pathways, and stem cell resistance. Drug combinations covering a broad range of targets may overcome resistance. CDDO-Me (bardoxolone methyl) is a drug that inhibits the survival of leukemic cells by targeting different pro-survival molecules, including STAT3. We found that CDDO-Me inhibits proliferation and survival of tyrosine kinase inhibitor-resistant BCR-ABL1+ cell lines and primary leukemic cells, including cells harboring BCR-ABL1T315I or T315I+ compound mutations. Furthermore, CDDO-Me was found to block growth and survival of CD34+/CD38− leukemic stem cells (LSC). Moreover, CDDO-Me was found to produce synergistic growth-inhibitory effects when combined with BCR-ABL1 tyrosine kinase inhibitors. These drug-combinations were found to block multiple signaling cascades and molecules, including STAT3 and STAT5. Furthermore, combined targeting of STAT3 and STAT5 by shRNA and STAT5-targeting drugs also resulted in synergistic growth-inhibition, pointing to a new efficient concept of combinatorial STAT3 and STAT5 inhibition. However, CDDO-Me was also found to increase the expression of heme-oxygenase-1, a heat-shock-protein that triggers drug resistance and cell survival. We therefore combined CDDO-Me with the heme-oxygenase-1 inhibitor SMA-ZnPP, which also resulted in synergistic growth-inhibitory effects. Moreover, SMA-ZnPP was found to sensitize BCR-ABL1+ cells against the combination ‘CDDO-Me+ tyrosine kinase inhibitor’. Together, combined targeting of STAT3, STAT5, and heme-oxygenase-1 overcomes resistance in BCR-ABL1+ cells, including stem cells and highly resistant sub-clones expressing BCR-ABL1T315I or T315I-compound mutations. Whether such drug-combinations are effective in tyrosine kinase inhibitor-resistant patients with chronic myeloid leukemia remains to be elucidated.
Introduction
Chronic myeloid leukemia (CML) is a stem cell disease characterized by the reciprocal translocation t(9;22) that creates the BCR-ABL1 oncoprotein, a major driver of disease evolution.1–3 Most patients with chronic phase (CP) CML achieve long-lasting cytogenetic and molecular responses when treated with the BCR-ABL1 tyrosine kinase inhibitor (TKI) imatinib.4–6 However, resistance against imatinib occurs in a substantial number of patients. Several molecular mechanisms, including BCR-ABL1 mutations, may contribute to TKI resistance in CML. Indeed, BCR-ABL1 mutations are identified in more than 50% of all resistant patients.7,8 For these patients, 2nd- and 3rd-generation TKI, including nilotinib, dasatinib, bosutinib, and ponatinib, are available and have shown beneficial effects.9–12 Using these drugs, it is now possible to cover most of the known BCR-ABL1 mutations detected in TKI-resistant CML. Ponatinib, a 3rd-generation BCR-ABL1 TKI, induces growth-inhibitory effects in TKI-resistant patients even if T315I is expressed.12 However, not all mutant forms of BCR-ABL1 are responsive to ponatinib. Moreover, it has been described that additional (multiple) mutations in BCR-ABL1, especially T315I-including compound mutations, confer resistance against ponatinib.13 Furthermore, resistance against TKI may occur independent of BCR-ABL1 mutations. In such cases, overexpression of BCR-ABL1 and/or hyper-activation of additional pro-oncogenic signaling networks and molecules, such as AKT, mTOR, MEK, STAT3, STAT5, JAK2, or SRC kinases, have been described.14–18 These molecules and pathways are often spared by the TKI used and can, therefore, contribute to drug resistance.14–20 Recently, several targeting approaches have been proposed with the aim of overcoming TKI resistance in advanced CML. One option may be to apply combinations of targeted drugs in order to cover a larger spectrum of relevant targets in TKI-resistant cells.
CDDO-Me (bardoxolone methyl) is an oleanane triterpenoid that has been described as inducing ROS-generation and to suppress a number of survival-related molecules, including AKT, mTOR, MAPK and STAT3, in malignant cells.21–26 It has also been reported that CDDO-Me promotes apoptosis in malignant cells in various neoplasms, including CML.21–26 Currently, CDDO-Me is tested in clinical trials in patients with diabetic nephropathy, a condition that may improve with CDDO-induced upregulation of the Nrf2-pathway.27,28 In addition, CDDO-Me is currently tested in clinical trials in cancer patients.29 With regard to CML, it has been reported that CDDO-Me counteracts the proliferation of BCR-ABL1+ cell lines by altering mitochondrial function and by inducing autophagy and apoptosis, regardless of the mutation status of BCR-ABL1.30 So far, the co-operative effects of BCR-ABL1 TKI and CDDO-Me on CML cells have not been analyzed. We hypothesized that, due to the large number of relevant targets blocked by CDDO-Me, this drug would be an optimal combination partner for BCR-ABL1 TKI. For example, CDDO-Me is a potent inhibitor of STAT3, a transcription factor that may play an important role in TKI-resistant CML cells, and is not blocked by BCR-ABL1 TKI.14,15,31–33 Indeed, recent data suggest that combined targeting of BCR-ABL1 and STAT3 exerts strong anti-leukemic effects in CML cells.34 Such combined targeting may be achieved by co-applying CDDO-Me and a BCR-ABL1 TKI. However, CDDO-Me is not able to suppress all pathways activated in CML cells. Notably, recent data suggest that exposure of leukemic cells to CDDO-Me results in a substantial increase in heme-oxygenase-1 (HO-1),30 a major survival factor that has been implicated in drug resistance of CML cells.35 As a result, HO-1 is considered an additional attractive target in TKI-resistant CML cells, and we hypothesized that combining CDDO-Me with an HO-1-inhibitor can enhance drug efficiency. SMA-ZnPP is an HO-1 inhibitor that counteracts growth and survival in CML cells and synergizes with imatinib in producing growth inhibition.36 In the current study, we evaluated drug interactions between CDDO-Me, 2nd- and 3rd-generation TKI, and SMA-ZnPP with the aim of enhancing anti-leukemic drug effects in TKI-resistant CML.
Methods
Reagents
Reagents used in this study are described in the Online Supplementary Appendix.
Isolation and culture of primary CML cells
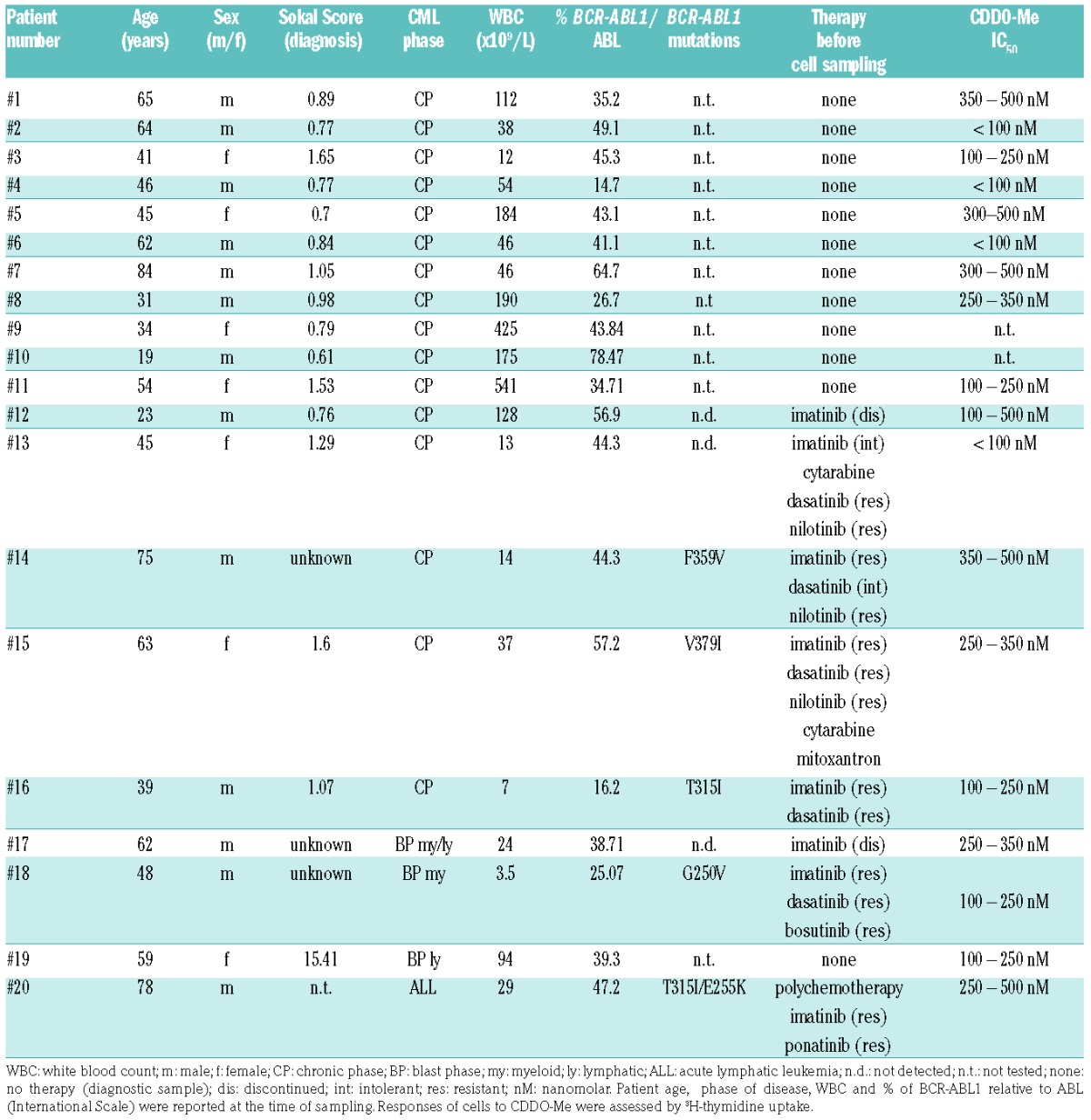
Primary leukemic cells were obtained from the peripheral blood (PB) of 16 patients with CP CML, one patient with myeloid BP, one with mixed myeloid/lymphoid BP, one with lymphoid BP, and one with relapsed BCR-ABL1+ acute lymphoblastic leukemia (ALL). Patients’ characteristics are shown in Table 1. Mononuclear cells (MNC) were isolated and kept in RPMI 1640 medium with 10% fetal calf serum (FCS) and antibiotics (without cytokines) as described.36 In all samples, cell viability was more than 90%. In 3 donors (CP, n=2; mixed BP, n=1), CD34+/CD38− leukemic stem cells (LSC) and CD34+/CD38+ progenitor cells were highly purified from MNC by high-speed sorting as reported37 using PE-conjugated monoclonal antibody (mAb) 581 against CD34, APC-conjugated mAb HIT2 against CD38, and a BD FACSAria (Becton Dickinson, San Jose, CA, USA). Normal bone marrow (BM) MNC were isolated from 7 donors (remission of acute leukemia or lymphoma patients without BM-involvement) after informed consent. Normal CD34+ BM cells were purchased from Lonza (Basel, Switzerland) and maintained in StemSpan Medium (Stemcell Technologies, Vancouver, Canada).
Table 1.
Patients’ characteristics and response of leukemic cells to CDDO-Me.
Cell lines
A characterization of cell lines used in this study is shown in Online Supplementary Table S1.
Evaluation of growth and survival of leukemic cells
Drug-exposed cells (cell lines, primary cells, LSC) were analyzed for proliferation, colony-formation, and survival. The bioassays employed are described in the Online Supplementary Methods.
Western blotting
Cell lines were incubated with CDDO-Me (0.1–1 μM) or ponatinib (1 μM) alone or in combination for 4 hours (h) or for 24 h. Thereafter, the expression of STAT5, phosphorylated (p) STAT5 (p-STAT5), STAT3, p-STAT3, CRKL, p-CRKL, p-JAK2, JAK2, ERK, p-ERK, S6, p-S6, HO-1, or Actin (loading control) was analyzed by Western blotting as described previously.36,38 Antibodies used are described in Online Supplementary Table S2.
shRNA-based knockdown experiments
For knockdown of STAT3 or STAT5, shRNA constructs were transduced in K562 and KCL22 using VSV-G pseudotyped lentiviruses, as described previously.39 The methodology and the shRNA-constructs are described in detail in the Online Supplementary Methods.
Overexpression of STAT3 in K562 cells
pMOWS retroviral vectors containing the coding sequence of GFP and wild-type (wt) STAT3 or an oncogenic mutant-form of STAT3, D661V, were kindly provided by Jürgen Scheller.40 Production of recombinant VSV-G pseudo-typed retroviruses and transduction of K562 cells were performed as described.39 After transduction, transfected cells were purified by puromycin-selection (20 ng/mL for 48 h).
Statistical analysis
To determine the significance levels in differences seen between drug-exposed and untreated cells, the Student t-test for dependent samples was applied. P<0.05 was considered statistically significant. Drug combination effects (additive vs. synergistic) were determined by calculating combination index (CI) values using Calcusyn software (Calcusyn; Biosoft, Ferguson, MO, USA).41
Approval was obtained from the Institutional Review Board (Department of Internal Medicine I, Division of Hematology and Hemostaseology, Medical University of Vienna, Austria) and from the Ethics Committee of the Medical University of Vienna for all series of experiments of this study.
Results
CDDO-Me inhibits proliferation and viability in TKI-sensitive and TKI-resistant BCR-ABL1+ cell lines
CDDO-Me was found to inhibit the proliferation of all four human CML cell lines tested, with IC50 values ranging between 0.1 and 0.5 μM (Figure 1A). A summary of growth-inhibitory effects of CDDO-Me on CML cells lines and a comparison with the effects elicited by BCR-ABL1 TKI are shown in Online Supplementary Table S3. CDDO-Me was also found to counteract growth of Ba/F3 cells expressing various imatinib-resistant forms of BCR-ABL1 (Figure 1B). In addition, CDDO-Me was found to suppress proliferation of Ba/F3 cells harboring T315I-including BCR-ABL1 compound mutations mediating resistance against all currently available TKI, including ponatinib, with IC50 values ranging between 0.1 and 0.35 μM (Figure 1C and Online Supplementary Table S4). Interestingly, in the human and Ba/F3 cell lines examined, no differences in IC50 values were seen when comparing TKI-sensitive and TKI-resistant clones, suggesting that BCR-ABL1 mutations did not influence responsiveness against CDDO-Me (Figure 1A–C). Growth-inhibition was accompanied by induction of apoptosis in all cell lines examined as demonstrated by flow cytometry (Figure 1D). In non-transfected, IL-3-dependent Ba/F3 cells, CDDO-Me also exerted growth-inhibitory effects, but the concentrations required to fully block cell growth were higher compared to that producing growth inhibition in BCR-ABL1+ Ba/F3 cells (Online Supplementary Figure S1).
Figure 1.

CDDO-Me inhibits growth and viability of Philadelphia-positive (Ph+) cell lines. (A–C) Human chronic myeloid leukemia (CML) cell lines (A) and BCR-ABL1-expressing Ba/F3 cells (B and C) were exposed to control medium (Co) or various concentrations of CDDO-Me for 48 hours (h). In case of K562-R, imatinib was removed prior to CDDO-Me exposure. Then, proliferation was measured by assessing 3H-thymidine uptake. Results are expressed in % of control and represent the mean±Standard Deviation (S.D.) of 3 independent experiments. (D) CML cell lines (K562, KU812, KCL22) were incubated in control medium or in CDDO-Me (0.5 μM) for 48 h. Thereafter, the percentage of apoptotic cells was determined by combined Annexin V/PI staining. The figure shows dot blots from one representative experiment. Almost identical results were obtained in two other experiments.
CDDO-Me counteracts proliferation in primary CML cells
Primary PB MNC of 10 patients with newly diagnosed CML (CP: n=9; BP: n=1), 5 CML patients who had developed resistance against one or more TKI (CP: n=4; BP: n=1), 2 CML patients who had discontinued imatinib and relapsed (CP: n=1; BP: n=1), and one patient suffering from ponatinib-resistant BCR-ABL1+ ALL were examined. CDDO-Me was found to suppress the proliferation of primary BCR-ABL1+ cells in all samples tested, with IC50-values ranging between 0.1 and 0.5 μM (Table 1 and Figure 2A). Drug effects were seen in samples isolated from heavily pre-treated patients with CML, in blast cells obtained from patients with BP (including one with mixed lymphoid/myeloid BP), and in primary ALL blasts harboring BCR-ABL1T315I/E255K (Table 1 and Figure 2A). No differences in IC50 values were observed when comparing newly diagnosed and relapsed patients or cells expressing or lacking BCR-ABL1 mutations (including BCR-ABL1T315I/E255K), suggesting that acquired resistance against TKI does not lead to resistance against CDDO-Me (Table 1 and Figure 2A). In control BM cells, CDDO-Me also exerted growth-inhibitory effects, but the concentrations required to fully block cell growth were higher compared to that producing growth inhibition in CML cells (Online Supplementary Figure S1).
Figure 2.

CDDO-Me counteracts the proliferation of primary chronic myeloid leukemia (CML) cells. (A) Primary CML cells were isolated from the peripheral blood (PB) of 6 patients [chronic phase (CP) n=5; primary blast phase (BP) n=1]. In 3 patients, cells were obtained at diagnosis (“new”). In all 3 TKI-resistant patients, BCR-ABL1 mutations were detected as indicated. Isolated cells were incubated in control medium (Co) or various concentrations of CDDO-Me as indicated at 37°C for 48 hours (h). Then, proliferation was measured by assessing 3H-thymidine incorporation. Results are expressed in % of control and represent the mean±Standard Deviation (S.D.) of triplicates. Patients’ numbers refer to Table 1. (B) Highly purified CD34+/CD38− stem cells (black bars) and CD34+/CD38+ precursor cells (gray bars) were sorted from peripheral blood (PB) leukocytes of 3 patients (#9, #11 and #17) and were kept in control medium (Co) or various concentrations of CDDO-Me as indicated at 37°C for 48 h. Then, proliferation was measured by assessing 3H-thymidine incorporation. Results are expressed as % of control and represent the mean±S.D. of 3 patients. *P<0.05 compared to control (Co). (C) Primary PB mononuclear cells (MNC) were isolated from 3 patients (#9, #11, #17) and kept in control medium (Co) or various concentrations of CDDO-Me at 37°C for 48 h. Thereafter, cells were subjected to flow cytometry to determine the % of apoptotic (Annexin V+) CD34+/CD38− (stem) cells (black bars) and CD34+/CD38+ (progenitor) cells (gray bars). Results represent the mean±S.D. of 3 patients. (D) PB MNC from 3 patients (#9, #10, #11) were cultured in methylcellulose with cytokines in the absence (Co) or presence of various concentrations of CDDO-Me as indicated for 14 days. Then, the numbers of granulocyte/macrophage (GM) colonies (white bars) and red cell-containing (burst-forming plus erythroid) colonies (gray bars) were counted under an inverted microscope. Results are expressed in % of control (100% = red colonies + GM colonies in the absence of CDDO-Me) and represent the mean±S.D. of 3 patients.
CDDO-Me inhibits the proliferation of primary LSC and progenitor cells obtained from patients with CML
Leukemic stem cells exhibit multiple forms of drug resistance.42,43 Therefore, we were interested to learn whether CDDO-Me would also block the proliferation of CML LSC. Indeed, we found that CDDO-Me dose-dependently inhibits the proliferation of highly enriched CD34+/CD38− stem cells and CD34+/CD38+ progenitor cells obtained from patients with CML (Figure 2B). In normal CD34+ stem- and progenitor cells, CDDO-Me produced only weak effects on proliferation (Online Supplementary Figure S1). CDDO-Me was also found to induce apoptosis in CML stem- and progenitor cells (Figure 2C). Stromal components may protect CML LSC against the effects of CDDO-Me.20,44 To address this point, the pro-apoptotic effects of CDDO-Me were analyzed in co-cultures prepared with primary CML cells and murine feeder cells. In these experiments, CDDO-Me was again found to induce apoptosis in the total CML cell compartment as well as in the CD34+/CD38− and CD34+/CD38+ cell fractions (Online Supplementary Figure S2). Finally, CDDO-Me was found to inhibit colony-formation of primary CML cells in vitro (Figure 2D).
CDDO-Me synergizes with BCR-ABL1-targeting TKI in inhibiting the proliferation of CML cells
In relapsed or TKI-resistant CML, combinations of two or more substances may be required to block both BCR-ABL1-dependent and BCR-ABL1-independent pathways and to achieve long-lasting and stable complete responses in all patients. In this study, we combined CDDO-Me with imatinib, nilotinib, dasatinib, or ponatinib at suboptimal concentrations. These combinations induced synergistic growth-inhibitory effects in all human CML cell lines tested (Figure 3A and Online Supplementary Figure S3). Synergistic effects of these drug combinations were also observed in Ba/F3 cells expressing various BCR-ABL1 mutations, including T315I and T315I-including compound mutations (Figure 3B), whereas no significant effects were seen in untransfected Ba/F3 cells when the same drug concentrations were applied (Online Supplementary Figure S3). Synergistic drug effects were confirmed by calculating CI values using calcusyn software (Online Supplementary Figure S4). Synergistic effects of CDDO-Me and ponatinib were also found in primary BCR-ABL1+ cells, including cells isolated from a patient with CML BP and one with BCR-ABL1T315I/E255K-positive relapsed ALL (Figure 3C). Synergistic drug interactions were found in all donors and cell samples, independent of the presence of BCR-ABL1 mutations, and (in case of primary cells) independent of previous treatment. By contrast, only weak effects of this drug combination (CDDO-Me+ponatinib) were observed in normal BM cells (Figure 3C). In CD34+/CD38− stem cells and CD34+/CD38+ progenitor cells obtained from 3 patients, the combination “CDDO-Me+ponatinib” was found to produce clear cooperative apoptosis-inducing effects (Figure 3D). Co-operative inhibitory effects of CDDO-Me and ponatinib were also seen in a clonogenic assay performed with primary samples obtained from 2 patients with CP-CML (Online Supplementary Figure S3). Together, these results suggest that CDDO-Me augments the anti-neoplastic effects of BCR-ABL1 TKI in CML cells, including LSC.
Figure 3.

CDDO-Me synergizes with BCR-ABL1 TKI in producing growth-inhibition in Philadelphia-positive (Ph+) cells. (A and B) Human chronic myeloid leukemia (CML) cell lines (A) or Ba/F3 cells harboring various BCR-ABL1 mutants (B) were incubated in control medium (0) or in various concentrations of CDDO-Me (●–●), BCR-ABL1-targeting TKI as indicated (■–■), or combinations of drugs at a fixed ratio of drug-concentrations as indicated (▲−▲) for 48 hours (h). Thereafter, 3H-thymidine incorporation was measured. Results are expressed in % of control and represent the mean± Standard Deviation (S.D.) of triplicates. (C) Primary neoplastic cells isolated from patient #1, #11, #17 and #20 as well as normal bone marrow (BM) mononuclear cells (MNC) obtained from 3 donors were incubated in various concentrations of CDDO-Me or ponatinib (as single drugs or in combination) as indicated. Results in the upper panels and the lower left panel are expressed in % of control and represent the mean±S.D. of triplicates. Results in the lower right panel show the mean±S.D. of 3 normal donors (gray bars) or 3 CML patients (#1, #11 and #17) (black bars). (D) Primary peripheral blood (PB) MNC were isolated from 3 patients (#9, #11, #17) and kept in control medium (0) or various concentrations of CDDO-Me or ponatinib as indicated at 37°C for 48 h. Thereafter, cells were subjected to flow cytometry to determine the % of apoptotic (Annexin V+) CD34+/CD38− (stem) cells (black bars) and CD34+/CD38+ (progenitor) cells (gray bars). Results represent the mean±S.D. of 3 patients.
CDDO-Me plus BCR-ABL1-targeting TKI induce simultaneous inhibition of STAT3 and STAT5 in CML cells
We next asked for mechanisms underlying the synergistic effect of the drug combinations (CDDO-Me plus BCR-ABL1-targeting TKI) applied. In particular, we examined potentially involved BCR-ABL1-dependent targets, including STAT5, ERK, the S6- ribosomal protein and JAK2, and the BCR-ABL1-independent target STAT3. As expected, ponatinib was found to modulate p-CRKL, p-ERK, p-JAK2, p-S6 and p-STAT5 expression in CML cells, but did only partially inhibit expression of p-STAT3 (Figure 4A and Online Supplementary Figure S5). By contrast, CDDO-Me was found to inhibit p-STAT3, p-JAK2 and p-S6, but did not block phosphorylation of CRKL, and produced only a slight effect on p-STAT5 expression (Figure 4A and Online Supplementary Figure S5). Only the combination of both compounds resulted in complete de-phosphorylation of both STAT3 and STAT5. This may explain the synergy between CDDO-Me and BCR-ABL1-targeting TKI. To further validate this hypothesis, shRNAs against STAT3 and against STAT5 were employed. shRNA induced-knockdown of STAT3- and STAT5-protein expression was confirmed by Western blotting (Figure 4B). shRNA directed against STAT3 was found to sensitize CML cell lines against nilotinib, dasatinib, and ponatinib, as well as against the more specific STAT5-inhibitor AC-3-019 (Figure 4C). Moreover, we were able to show that shRNA-mediated STAT5 knockdown enhances the effects of CDDO-Me, although the combination effect was additive rather than synergistic (Figure 4D). Together, these data suggest that simultaneous inhibition of STAT3 and STAT5 is a potent approach to block growth and survival of (TKI-resistant) CML cells. In order to confirm the role of STAT3 as a target of CDDO-Me, the effects of STAT3 overexpression in K562 cells was examined. We found that overexpression of STAT3 or STAT3 D661V results in relative resistance against CDDO-Me, confirming that STAT3 serves as a functionally relevant target in leukemic cells (Online Supplementary Figure S5).
Figure 4.

Combined inhibition of STAT3 and STAT5 with shRNA and targeted drugs results in synergistic growth inhibition in chronic myeloid leukemia (CML) cells. (A) KCL22 and K562 cells were incubated with CDDO-Me (1 μM), ponatinib (1 μM) or a combination of both drugs for 4 hours (h). Thereafter, cells were subjected to Western blot analysis using antibodies against p-STAT5, STAT5, p-STAT3, STAT3, p-CRKL, or CRKL, as indicated. (B–D) K562 and KCL22 cells were transfected with control shRNA, with an shRNA-construct directed against STAT5, or shRNA-constructs directed against STAT3 (#1 = #V3LHS_376016; #2 = #V3LHS_641818) as indicated. Protein knockdown was confirmed by western blotting using antibodies against STAT3 or STAT5. β-Actin served as loading control (B). (C) K562 cells (upper panels) and KCL22 cells (lower panels) were treated with control-shRNA (black bars) or with shRNA directed against STAT3 (construct #2, gray bars) and were then incubated with control medium (control), with BCR-ABL1 TKI (as indicated), or with the STAT5 inhibitor AC-3-019 for 48 h. Results are expressed in % of control and represent the mean±Standard Deviation (S.D.) of triplicates. *P<0.05. (D) K562 cells (left panel) and KCL22 cells (right panel) were treated with control-shRNA (black bars) or shRNA against STAT5 (gray bars) and were then incubated in control medium or in CDDO-Me for 48 h. Results are expressed in % of control and represent the mean±S.D. of triplicates. *P<0.05.
The HO-1 inhibitor SMA-ZnPP sensitizes CML cells against CDDO-Me and TKI
Recent data suggest that CDDO-Me does not suppress all survival factors in CML cells.30 Indeed, HO-1, a survival factor with anti-apoptotic activity in CML cells, has been described to be up-regulated by CDDO-Me,30 a fact that can best be interpreted as a “tumor-escape mechanism”.36,45 CDDO-Me led to an upregulation of HO-1 protein expression in KU812, KCL22, and BCR-ABL1+ Ba/F3 cells, confirming previous observations (Figure 5A).30 We therefore combined CDDO-Me with the HO-1 inhibitor SMA-ZnPP. Indeed, SMA-ZnPP was found to sensitize CML cell lines against CDDO-Me as evidenced by 3H-thymidine-uptake experiments (Figure 5B). Synergistic effects were also observed in Ba/F3 cells expressing BCR-ABL1 mutants and in primary CML cells (Figure 5C and D). No growth inhibitory effects of the drug combination on normal BM cells were seen (Online Supplementary Figure S6). Finally, we asked whether a triple-combination, including CDDO-Me, a BCR-ABL1 TKI (dasatinib or ponatinib), and SMA-ZnPP, would further enhance drug effects. In these experiments, we incubated KU812 and Ba/F3p210T315I/F311L cells with low doses of 3 compounds, either as single agents or in combination. The anti-proliferative effect of the triple-combination was superior to single drug- or 2-drug combinations (Figure 5E). These observations suggest that the triple combination “CDDO-Me+TKI+SMA-ZnPP” exerts strong anti-neoplastic effects in CML cells.
Figure 5.

SMA-ZnPP sensitizes BCR-ABL1+ cells against CDDO-Me and against the combination “CDDO-Me+BCR-ABL1 TKI”. (A) KCL22, KU812 and BCR-ABL1+ Ba/F3 cells were incubated in control medium or in 0.1 or 0.3 μM CDDO-Me for 24 hours (h). Then, cells were subjected to Western blot analysis using antibodies against HO-1 or either β-Actin or Actin (loading control) as indicated. (B–D) Cell lines (B and C) and primary chronic myeloid leukemia (CML) cells (D) were incubated in control medium or in various concentrations of CDDO-Me (●–●), SMA-ZnPP (■–■), or a combination of both drugs (at fixed ratio of drug concentrations) (▲−▲) for 48 hours (h). Then, 3H-thymidine incorporation was measured. Results are expressed in % of control and represent the mean±Standard Deviation (S.D.) of triplicates. Patient numbers in (D) refer to Table 1. (E) KU812 and Ba/F3p210T315I/F311L cells were exposed to low doses of CDDO-Me (●–●), a BCR-ABL1 TKI (dasatinib or ponatinib) (■–■), and SMA-ZnPP (▲−▲), either as single agents (blue graphs), in 2-drug combinations (green graphs; CDDO-Me+TKI: ▼–▼; CDDO-Me+SMA-ZnPP: ♦–♦; TKI+SMA-ZnPP: ○–○), or in a 3-drug combination (red graph; □–□) for 48 h. Then, 3H-thymidine uptake was measured. Results are expressed in % of control and represent the mean±S.D. of triplicates.
Discussion
Despite the availability of novel potent BCR-ABL1 TKI, drug resistance develops frequently and remains a major challenge in the treatment of CML.7,8,13 Important mechanisms underlying drug resistance are intrinsic stem cell resistance,42,43 BCR-ABL1 mutations, including compound mutations,7,8,13 and additional, BCR-ABL1-independent, signaling pathways and molecules. Among these are the JAK-STAT pathway, the RAS-RAF-MEK pathway, and several different survival-related molecules.16–20 We report here that BCR-ABL1 TKI co-operate with the STAT3-inhibitor CDDO-Me in producing growth inhibition and apoptosis in drug-resistant CML cells. This combination blocked most of the relevant signaling and survival molecules in CML cells, except HO-1, a survival molecule that is even up-regulated upon exposure to CDDO-Me in leukemic cells. In line with this observation, the HO-1 blocker SMA-ZnPP further augmented the anti-leukemic effects produced by the combination CDDO-Me+TKI in CML cells.
Recent data suggest that CDDO-Me-targets play a major role in the survival and growth of neoplastic cells in solid tumors and leukemias.21–26,30,46 In the present study, CDDO-Me was found to inhibit the proliferation of TKI-sensitive and TKI-resistant CML cells and of Ba/F3 cells exhibiting various BCR-ABL1 mutations, including compound mutations involving T315I. This finding is of interest since such T315I+ compound mutations of BCR-ABL1 often confer resistance against all available TKI, including ponatinib. IC50-values were low (<0.5 μM) in all cell lines tested, without major differences between TKI-sensitive and TKI-resistant cells. Similar observations were made using primary CML cells obtained from newly diagnosed or heavily pre-treated patients with BCR-ABL1+ CML. Finally, we were able to show that CDDO-Me inhibits the proliferation of CD34+/CD38− CML stem cells and CD34+/CD38+ CML progenitor cells at pharmacologically meaningful concentrations. All these data suggest that CDDO-Me may be an interesting drug for patients in whom neoplastic cells have lost sensitivity to 2nd- or 3rd-line TKI. The fact that CDDO-Me was found to inhibit the proliferation of TKI-resistant cells in all cell lines and all patients tested, suggests that no “cross-resistance” between TKI and CDDO-Me occurs. This was not surprising since CDDO-Me does not recognize BCR-ABL1, but inhibits cell proliferation by blocking other signaling pathways and molecules, such as STAT3.
Previous studies have shown that CDDO-Me inhibits STAT3 activation in various neoplastic cells.23,24 In the present study, we were able to confirm that CDDO-Me blocks STAT3 activity in CML cells. By contrast, CDDO-Me did not suppress STAT5 activation. Recent data suggest that STAT3 plays an essential role in the survival of CML cells,32–34 an observation that is of particular interest in light of the fact that ponatinib exerted only weak effects on STAT3 activity in the CML cell lines K562 and KCL22 in this study. We therefore asked whether a combination consisting of CDDO-Me and a BCR-ABL1-targeting TKI would result in synergistic growth inhibition. Indeed, we were able to demonstrate that CDDO-Me synergizes with imatinib and with the 2nd- and 3rd-generation BCR-ABL1 TKI applied in inhibiting growth of CML cells. These effects were seen in TKI-sensitive as well as TKI-resistant CML cells, suggesting that these combinations may be an attractive therapeutic approach in advanced CML. In this regard it is worthy to note that the drug combinations applied were also found to exert strong co-operative inhibitory effects on growth of primary blast cells obtained from a patient with mixed myeloid/lymphoid BP and in one with Ph+ ALL. Since STAT3 and STAT5 may differentially contribute to the pathogenesis of myeloid and lymphoid BP in CML, it may be of importance to block both pathways to keep all PB-associated (lymphoid and myeloid determined) sub-clones under control in these patients.
Recent data have shown that CDDO-Me enhances the expression of HO-1 in various neoplastic cells, including CML cells.30 HO-1 is a stress molecule that contributes to drug-resistance and “tumor escape mechanisms” in various malignancies, including CML.35,36,45 We therefore hypothesized that pharmacological suppression of HO-1 could potentially enhance the effects of CDDO-Me and also augment the effects of the combination “CDDO-Me+TKI” on malignant proliferation in CML cells. We therefore combined the HO-1 inhibitor SMA-ZnPP with CDDO-Me or with the 2-drug combination “CDDO-Me+TKI”. Addition of SAM-ZnPP was indeed found to augment CDDO-Me effects, and the “3-drug-combination” applied was found to exert superior anti-neoplastic effects compared to single agents or the “2-drug-combination”. This finding can best be explained by the fact that up-regulated HO-1 protects CML cells against drug-induced growth inhibition and apoptosis, and that this protective effect is eliminated by exposure to SMA-ZnPP.36,45 This concept is also in line with our previous observations.36,45
Recently, CDDO-Me has been introduced in clinical trials in patients with solid tumors and lymphomas, as well as in patients with diabetes mellitus and renal failure. A limited number of trials have been completed so far, and only very few results are available.27–29,47 In patients with malignant diseases, CDDO-Me was well tolerated.29 However, other trials have shown that CDDO-Me can cause severe cardiotoxic side effects, especially in patients with diabetes mellitus.47 These observations have to be taken into account before CDDO-Me can be developed further in patients with CML, especially when combined with nilotinib or ponatinib, both of which can also produce cardiovascular side effects.48–50 An additional concern may be drug-induced cytopenia. In fact, higher concentrations of CDDO-Me did not only inhibit growth of CML cells but also growth of normal BM cells. On the other hand, we expect that the application of such drug combinations will permit significant dose reductions of individual agents, thereby decreasing or even eliminating the risk of development of severe side effects. Indeed, we found that the drug combinations CDDO-Me+ponatinib, CDDO-Me+SMA-ZnPP and CDDO-Me+SMA-ZnPP+ponatinib did not significantly inhibit the proliferation of normal BM cells.
Together, we demonstrate that drug combinations consisting of BCR-ABL1 TKI, CDDO-Me, and SMA-ZnPP, are most effective in inhibiting the proliferation and survival of TKI-resistant CML cells. The superior effects of these drug combinations are best explained by complete suppression of multiple triggers of proliferation and survival, including STAT3, STAT5, and HO-1. Whether this concept can be applied in patients with advanced CML remains to be determined in clinical trials.
Supplementary Material
Acknowledgments
We like to thank Gabriela Stefanzl for excellent technical assistance and Jisung Park and Gary Tin for assisting in synthesizing AC-3-019 for this study.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/9/1519
Funding
This study was supported by Austrian Science Fund (FWF), grants F4701-B20 and F4704-B20 (to PV), F4705-B20 (to TL) and F4707-B20 (to RM) and by a Grant-in-Aid from the Ministry of Welfare, Health and Labor of Japan, (201220042), and A-STEP for cancer grant (AS242Z01542Q) from the Japan Science and Technology Agency (to HM).
References
- 1.Nowell PC, Hungerford DA. A minute chromosome in human granulocytic leukemia. Science. 1960;132:1497. [Google Scholar]
- 2.Rowley JD. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243(5405):290–293. [DOI] [PubMed] [Google Scholar]
- 3.Bedi A, Zehnbauer BA, Barber JP, Sharkis SJ, Jones RJ. Inhibition of apoptosis by BCR-ABL in chronic myeloid leukemia. Blood. 1994;83(8):2038–2044. [PubMed] [Google Scholar]
- 4.Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344(14):1031–1037. [DOI] [PubMed] [Google Scholar]
- 5.Kantarjian H, Sawyers C, Hochhaus A, et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med. 2002;346(9):645–652. [DOI] [PubMed] [Google Scholar]
- 6.Druker BJ, Guilhot F, O’Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408–2417. [DOI] [PubMed] [Google Scholar]
- 7.Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001; 293(5531):876–880. [DOI] [PubMed] [Google Scholar]
- 8.Shah NP, Nicoll JM, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2(2):117–125. [DOI] [PubMed] [Google Scholar]
- 9.Weisberg E, Manley PW, Breitenstein W, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7(2):129–141. [DOI] [PubMed] [Google Scholar]
- 10.Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305(5682):399–401. [DOI] [PubMed] [Google Scholar]
- 11.Puttini M, Coluccia AM, Boschelli F, et al. In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl+ neoplastic cells. Cancer Res. 2006;66(23):11314–11322. [DOI] [PubMed] [Google Scholar]
- 12.O’Hare T, Shakespeare WC, Zhu X, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16(5):401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zabriskie MS, Eide CA, Tantravahi SK, et al. BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell. 2014;26(3):428–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coppo P, Flamant S, De Mas V, et al. BCR-ABL activates STAT3 via JAK and MEK pathways in human cells. Br J Haematol. 2006;134(2):171–179. [DOI] [PubMed] [Google Scholar]
- 15.Stella S, Tirrò E, Conte E, et al. Suppression of survivin induced by a BCR-ABL/JAK2/STAT3 pathway sensitizes imatinib-resistant CML cells to different cytotoxic drugs. Mol Cancer Ther. 2013;12(6):1085–1098. [DOI] [PubMed] [Google Scholar]
- 16.Traer E, MacKenzie R, Snead J, et al. Blockade of JAK2-mediated extrinsic survival signals restores sensitivity of CML cells to ABL inhibitors. Leukemia. 2012;26(5):1140–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warsch W, Walz C, Sexl V. JAK of all trades: JAK2-STAT5 as novel therapeutic targets in BCR-ABL1+ chronic myeloid leukemia. Blood. 2013;122(13):2167–2175. [DOI] [PubMed] [Google Scholar]
- 18.Zimmerman EI, Dollins CM, Crawford M, et al. Lyn kinase-dependent regulation of miR181 and myeloid cell leukemia-1 expression: implications for drug resistance in myelogenous leukemia. Mol Pharmacol. 2010;78(5):811–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hayette S, Chabane K, Michallet M, et al. Longitudinal studies of SRC family kinases in imatinib- and dasatinib-resistant chronic myelogenous leukemia patients. Leuk Res. 2011;35(1):38–43. [DOI] [PubMed] [Google Scholar]
- 20.Tabe Y, Jin L, Iwabuchi K, et al. Role of stromal microenvironment in nonpharmacological resistance of CML to imatinib through Lyn/CXCR4 interactions in lipid rafts. Leukemia. 2012;26(5):883–892. [DOI] [PubMed] [Google Scholar]
- 21.Konopleva M, Tsao T, Ruvolo P, et al. Novel triterpenoid CDDO-Me is a potent inducer of apoptosis and differentiation in acute myelogenous leukemia. Blood. 2002; 99(1):326–335. [DOI] [PubMed] [Google Scholar]
- 22.Konopleva M, Contractor R, Kurinna SM, Chen W, Andreeff M, Ruvolo PP. The novel triterpenoid CDDO-Me suppresses MAPK pathways and promotes p38 activation in acute myeloid leukemia cells. Leukemia. 2005;19(8):1350–1354. [DOI] [PubMed] [Google Scholar]
- 23.Ling X, Konopleva M, Zeng Z, et al. The novel triterpenoid C-28 methyl ester of 2-cyano-3, 12-dioxoolen-1, 9-dien-28-oic acid inhibits metastatic murine breast tumor growth through inactivation of STAT3 signaling. Cancer Res. 2007;67(9):4210–4218. [DOI] [PubMed] [Google Scholar]
- 24.Ahmad R, Raina D, Meyer C, Kufe D. Triterpenoid CDDO-methyl ester inhibits the Janus-activated kinase-1 (JAK1)–>signal transducer and activator of transcription-3 (STAT3) pathway by direct inhibition of JAK1 and STAT3. Cancer Res. 2008;68(8):2920–2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shishodia S, Sethi G, Konopleva M, Andreeff M, Aggarwal BB. A synthetic triterpenoid, CDDO-Me, inhibits IkappaBalpha kinase and enhances apoptosis induced by TNF and chemotherapeutic agents through down-regulation of expression of nuclear factor kappaB-regulated gene products in human leukemic cells. Clin Cancer Res. 2006;12(6):1828–1838. [DOI] [PubMed] [Google Scholar]
- 26.Deeb D, Gao X, Jiang H, et al. Oleanane triterpenoid CDDO-Me inhibits growth and induces apoptosis in prostate cancer cells through a ROS-dependent mechanism. Biochem Pharmacol. 2010;79(3):350–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Zeeuw D, Akizawa T, Agarwal R, et al. Rationale and trial design of Bardoxolone Methyl Evaluation in Patients with Chronic Kidney Disease and Type 2 Diabetes: the Occurrence of Renal Events (BEACON). Am J Nephrol. 2013;37(3):212–222. [DOI] [PubMed] [Google Scholar]
- 28.Warnock DG, Hebbar S, Bargman J, et al. Prospective safety study of bardoxolone methyl in patients with type 2 diabetes mellitus, end-stage renal disease and peritoneal dialysis. Contrib Nephrol. 2012;178:157–163. [DOI] [PubMed] [Google Scholar]
- 29.Hong DS, Kurzrock R, Supko JG, et al. A phase I first-in-human trial of bardoxolone methyl in patients with advanced solid tumors and lymphomas. Clin Cancer Res. 2012;18(12):3396–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Samudio I, Kurinna S, Ruvolo P, et al. Inhibition of mitochondrial metabolism by methyl-2-cyano-3,12-dioxooleana-1,9-diene-28-oate induces apoptotic or autophagic cell death in chronic myeloid leukemia cells. Mol Cancer Ther. 2008;7(5):1130–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coppo P, Dusanter-Fourt I, Millot G, et al. Constitutive and specific activation of STAT3 by BCR-ABL in embryonic stem cells. Oncogene. 2003;22(26):4102–4110. [DOI] [PubMed] [Google Scholar]
- 32.Nair RR, Tolentino JH, Hazlehurst LA. Role of STAT3 in Transformation and Drug Resistance in CML. Front Oncol. 2012;2:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eiring AM, Kraft IL, Page BD, O’Hare T, Gunning PT, Deininger MW. STAT3 as a mediator of BCR-ABL1-independent resistance in chronic myeloid leukemia. Leuk Suppl. 2014;3(Suppl 1):S5–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eiring AM, Page BD, Kraft IL, et al. Combined STAT3 and BCR-ABL1 inhibition induces synthetic lethality in therapy-resistant chronic myeloid leukemia. Leukemia. 2015;29(3):586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mayerhofer M, Florian S, Krauth MT, et al. Identification of heme oxygenase-1 as a novel BCR/ABL-dependent survival factor in chronic myeloid leukemia. Cancer Res. 2004;64(9):3148–3154. [DOI] [PubMed] [Google Scholar]
- 36.Mayerhofer M, Gleixner KV, Mayerhofer J, et al. Targeting of heat shock protein 32 (Hsp32)/heme oxygenase-1 (HO-1) in leukemic cells in chronic myeloid leukemia: a novel approach to overcome resistance against imatinib. Blood. 2008;111(4):2200–2210. [DOI] [PubMed] [Google Scholar]
- 37.Herrmann H, Sadovnik I, Cerny-Reiterer S, et al. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood. 2014; 123(25):3951–3962. [DOI] [PubMed] [Google Scholar]
- 38.Gleixner KV, Ferenc V, Peter B, et al. Polo-like kinase 1 (Plk1) as a novel drug target in chronic myeloid leukemia: overriding imatinib resistance with the Plk1 inhibitor BI 2536. Cancer Res. 2010;70(4):1513–1523. [DOI] [PubMed] [Google Scholar]
- 39.Hoermann G, Cerny-Reiterer S, Herrmann H, et al. Identification of oncostatin M as a JAK2 V617F-dependent amplifier of cytokine production and bone marrow remodeling in myeloproliferative neoplasms. FASEB J. 2012;26(2):894–906. [DOI] [PubMed] [Google Scholar]
- 40.Aparicio-Siegmund S, Sommer J, Monhasery N, et al. Inhibition of protein kinase II (CK2) prevents induced signal transducer and activator of transcription (STAT)1/3 and constitutive STAT3 activation. Oncotarget. 2014;5(8):2131–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. [DOI] [PubMed] [Google Scholar]
- 42.Jiang X, Zhao Y, Smith C, et al. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia. 2007; 21(5):926–935. [DOI] [PubMed] [Google Scholar]
- 43.Valent P. Emerging stem cell concepts for imatinib-resistant chronic myeloid leukaemia: implications for the biology, management, and therapy of the disease. Br J Haematol. 2008;142(3):361–378. [DOI] [PubMed] [Google Scholar]
- 44.Nair RR, Tolentino J, Hazlehurst LA. The bone marrow microenvironment as a sanctuary for minimal residual disease in CML. Biochem Pharmacol. 2010;80(5):602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gleixner KV, Mayerhofer M, Vales A, et al. Targeting of Hsp32 in solid tumors and leukemias: a novel approach to optimize anticancer therapy. Curr Cancer Drug Targets. 2009;9(5):675–689. [DOI] [PubMed] [Google Scholar]
- 46.Qin D, Wang W, Lei H, et al. CDDO-Me reveals USP7 as a novel target in ovarian cancer cells. Oncotarget. 2016; 7(47):77096–77109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Zeeuw D, Akizawa T, Audhya P, et al. Bardoxolone Methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med 2013;369(26):2492–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dalzell MD. Ponatinib pulled off market over safety issues. Manag Care. 2013; 22(12):42–43. [PubMed] [Google Scholar]
- 49.Le Coutre P, Rea D, Abruzzese E, et al. Severe peripheral arterial disease during nilotinib therapy. J Natl Cancer Inst. 2001;103(17):1347–1348. [DOI] [PubMed] [Google Scholar]
- 50.Jain P, Kantarjian H, Jabbour E, et al. Ponatinib as first-line treatment for patients with chronic myeloid leukaemia in chronic phase: a phase 2 study. Lancet Haematol. 2015;2(9):e376–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.