

Abstract
Anaplastic lymphoma kinase (ALK)-positive anaplastic large cell lymphoma is characterized by 2p23/ALK aberrations, including the classic t(2;5)(p23;q35)/NPM1-ALK rearrangement present in ~80% of cases and several variant t(2p23/ALK) occurring in the remaining cases. The ALK fusion partners play a key role in the constitutive activation of the chimeric protein and its subcellular localization. Using various molecular technologies, we have characterized ALK fusions in eight recently diagnosed anaplastic large cell lymphoma cases with cytoplasmic-only ALK expression. The identified partner genes included EEF1G (one case), RNF213/ALO17 (one case), ATIC (four cases) and TPM3 (two cases). Notably, all cases showed copy number gain of the rearranged ALK gene, which is never observed in NPM1-ALK-positive lymphomas. We hypothesized that this could be due to lower expression levels and/or lower oncogenic potential of the variant ALK fusions. Indeed, all partner genes, except EEF1G, showed lower expression in normal and malignant T cells, in comparison with NPM1. In addition, we investigated the transformation potential of endogenous Npm1-Alk and Atic-Alk fusions generated by clustered regularly interspaced short palindromic repeats/Cas9 genome editing in Ba/F3 cells. We found that Npm1-Alk has a stronger transformation potential than Atic-Alk, and we observed a subclonal gain of Atic-Alk after a longer culture period, which was not observed for Npm1-Alk. Taken together, our data illustrate that lymphomas driven by the variant ATIC-ALK fusion (and likely by RNF213-ALK and TPM3-ALK), but not the classic NPM1-ALK, require an increased dosage of the ALK hybrid gene to compensate for the relatively low and insufficient expression and signaling properties of the chimeric gene.
Introduction
Anaplastic large cell lymphoma (ALCL) expressing ALK (ALK+ ALCL) is a rare but well-defined subtype of peripheral T-cell lymphoma (PTCL).1 It accounts for approximately 3% of all non-Hodgkin lymphomas (NHL) in adults, 10–15% of pediatric lymphomas and 60–80% of all ALCLs. ALK+ ALCL is hallmarked by various 2p23/ALK chromosomal rearrangements leading to an aberrant expression and constitutive activation of the ALK tyrosine kinase. The most prevalent lesion occurring in more than 80% of ALK+ ALCL is t(2;5)(p23;q35) involving ALK and NPM1 (nucleophosmin) genes, respectively.2 The translocation generates a chimeric protein comprising the N-terminal oligomerization domain of NPM1 and the C-terminal region of ALK, including its intracellular tyrosine kinase domain.2 The fusion acts as an oncogene and its transforming potential was proven in a number of in vitro and in vivo studies.3–5 The remaining ALK+ ALCL cases harbor variant 2p23/ALK rearrangements affecting at least nine partner genes: TPM3 (1q21.3; previously assigned to 1q25),6 ATIC (2q35), TFG (3q12.2), TRAF1 (9q33.2), CLTC (17q23.1), ALO17/RNF213 (17q25.3), TPM4 (19p13.12), MYH9 (22q12.3) and MSN (Xq12).7 These partners play a key role in the constitutive activation of the chimeric protein by mediating its oligomerization and determining the subcellular localization of ALK fusion (cytoplasmic and nuclear in NPM1-ALK-positive cases, and cytoplasmic-only or membranous in variant fusions).8,9 In addition, they impact a range of biological activities of ALK chimeras, including proliferation, transformation and metastatic capacities.10,11 Comparative analysis of the biological properties of ALK oncoproteins, however, is hampered by the relative low-frequency of particular variant ALK fusions.
Interestingly, ALK rearrangements have also been detected in large B-cell lymphoma and various tumors of mesenchymal and epithelial origin, including inflammatory myofibroblastic tumors, lung cancer, esophageal squamous cell carcinoma and others.7 Notably, the same ALK fusions, such as TPM3-ALK, TPM4-ALK, TFG-ALK, SEC31A-ALK, ATIC-ALK, CLTC-ALK and EML4-ALK occur in ALK+ malignancies of different cells of origin, highlighting the crucial role of ALK in tumorigenesis.7,12 These findings prompted the development of new therapeutic strategies regarding ALK+ tumors. Of particular importance is the recent discovery of specific ALK tyrosine inhibitors,13,14 one of which, crizotinib, has proven to have clinical efficacy in the treatment of ALK+ non-small cell lung cancer and ALCL.15,16
Herein, we report the molecular characterization of ALK fusions in eight ALK+ ALCL cases exhibiting a cytoplasmic-only ALK staining pattern recently diagnosed in our institution. Intriguingly, all cases analyzed by fluorescence in situ hybridization (FISH) at the time of diagnosis revealed copy number gain of the rearranged ALK gene.
Methods
Case selection
Eight cases of ALK+ ALCL with a cytoplasmic-only expression of ALK and available bioarchived material were selected from the database of the Center for Human Genetics and Department of Pathology, University Hospital Leuven, Belgium. Morphology, immunophenotype and clinical records of the reported cases were reviewed. The institutional review board “Commissie Medische Ethiek” of the University Hospital approved this retrospective study and renounced the need for written informed consent from the participants (S56799, ML10896: 12/08/2014).
Cytogenetics and FISH
Conventional Giemsa (G)-banding chromosomal analysis and FISH analysis followed routine protocols. The probes applied on patient material and Ba/F3 cell lines are listed in the Online Supplementary Table S1. Non-commercial probes were directly labeled with SpectrumOrange- and SpectrumGreen-dUTP (Abbott Molecular, Ottignies, Belgium) by random primed label ing. FISH images were acquired with a fluorescence microscope equipped with an Axiophot 2 camera (Carl Zeiss Microscopy, Jena, Germany) and a MetaSystems Isis imaging system (MetaSystems, Altlussheim, Germany).
Array-based genomic hybridization (aCGH)
Total genomic DNA was isolated from frozen lymphoma samples using standard procedures. High-resolution aCGH was performed using Affymetrix Cytoscan HD, following the manufacturer protocols. Downstream data analysis of copy number alterations was conducted using the software Array Studio, version 6.2.
Cell culture and growth curves
Ba/F3 cells constitutively expressing Cas9 (Ba/F3 Cas9) were generated using a retroviral expression vector containing a Cas9 expression cassette (Online Supplementary Figure S1). Ba/F3 Cas9 cells were cultured with interleukin 3 (IL3) in Roswell Park Memorial Institute (RPMI) medium + 10% fetal bovine serum (FBS) before and after electroporation. For growth curves, viability and proliferation were measured on a Guava flow cytometer (Merck Millipore, Darmstadt, Germany) for several consecutive days.
Quantitative real time polymerase chain reaction (QRT-PCR)
QRT-PCR with the GoTaq qPCR Master Mix (Promega, Madison, WI, USA) was used to analyze relative messenger ribonucleic acid (mRNA) expression of two Alk partner genes (Npm1 and Atic) in Ba/F3 Cas9 cells. Mouse Hprt1 and Rpl4 were used as an internal control. Primers are listed in the Online Supplementary Table S2. All samples were run in triplicate and data were analyzed with the comparative Ct (DDCt) method.
CRISPR/Cas genome editing
Guide (g)RNAs were designed using the Zhang lab’s clustered regularly interspaced short palindromic repeats (CRISPR) design tool (Broad Institute of MIT and Harvard, Cambridge, MA, USA) (Online Supplementary Table S3). gRNAs were cloned into an expression plasmid (pX321, derived from pX330, Zhang lab; Online Supplementary Figure S2). Electroporations of Ba/F3 Cas9 cells were carried out using a Gene Pulser Xcell electroporation system (Bio-Rad Laboratories, Hercules, CA, USA). After electroporation, cells were kept in RPMI medium + 10% FBS + IL3 for three days before IL3 depletion was carried out.
Other applied techniques, including the 5′ Rapid Amplification of Complementary (c)DNA ends (RACE) PCR, Low Coverage Full Genome Sequencing (LCFGS), Targeted Locus Amplification (TLA) and Nested RT-PCR are briefly described in the Online Supplementary Methods.
Results
Clinical and pathological features
Relevant clinical and pathological features of the reported cases of ALK+ ALCL are shown in Table 1. There were two children (a 5-year-old boy and a 13-year-old girl) and six adults (two female and four males) ranging from 49 to 78 years of age (mean 64.8 years). All patients presented with lymph node involvement and one displayed additional skin lesions (stages I [cases 3 and 4], II [cases 5 and 8], III [cases 2 and 7] and IV [cases 1 and 6]). Five adult patients (cases 2–6) were treated with the chemotherapy regimen cyclophosphamide, adriamycin, vincristine and prednisone (CHOP), reached complete remission (CR) and are still alive. The sixth adult patient (case 7), initially diagnosed with classical Hodgkin lymphoma, was treated with doxorubicin, bleomycin, vinblastinea and dacarbazine (ABVD) and radiotherapy, and also reached CR. He relapsed very recently (after 165 months) and died due to disease-related complications after the third course of CHOP. Two other patients also experienced a more aggressive disease course: one (case 2) achieved a second CR following treatment with dexamethasone, cytarabine and cisplatin (DHAP), and the first pediatric patient (case 1) initially treated according to the ALCL99 protocol, experienced three relapses. The latter achieved CR after treatment with crizotinib and allogenic stem cell transplantation, but died 72 months after diagnosis due to severe graft-versus-host disease (GvHD) and respiratory failure. The second pediatric patient (case 8) received six cycles of polychemotherapy (ALCL99) and remains in CR.
Table 1.
Relevant clinical and pathological data.
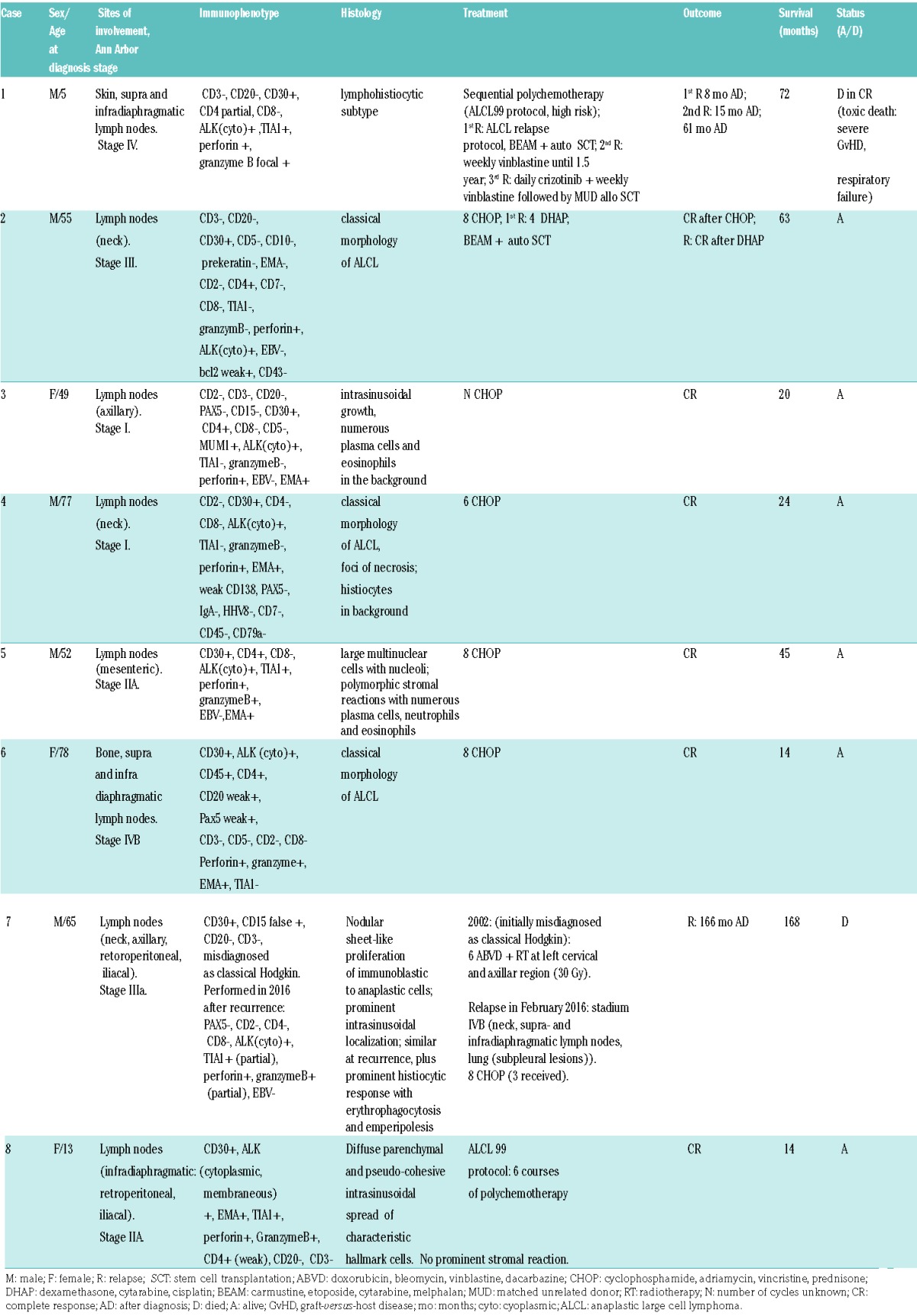
Histopathology showed proliferation of anaplastic lymphoid cells and the presence of hallmark doughnut cells in all cases. The immunophenotype of the individual tumors is shown in Table 1 and illustrated in the Online Supplementary Figure S3. All cases were CD30 positive, expressed a CD4+ or null cell phenotype in conjunction with variable cytotoxic markers, and overexpressed ALK1 in a strictly cytoplasmic pattern. The stromal infiltrate was prominent in all cases with a variable amount of histiocytes and/or neutrophils.
Cytogenetic and molecular studies
Case 1
Cytogenetic analysis showed a complex karyotype with del(2)(p23) (Table 2). FISH with LSI ALK demonstrated a break apart (BA) pattern with the green (5′ALK) signal on del(2)(p23) and two red (3′ALK) signals on a marker chromosome of postulated chromosome 11 origin (Figure 1A). Whole chromosome painting proved that der(11) indeed harbors the duplicated fragment of chromosome 2 inserted at 11q. The t(2p23/ALK) rearrangement was further investigated using the 5′ RACE PCR approach. The analysis identified an in-frame fusion transcript in which exon 8 of EEF1G, the gene located at 11q12.3, was fused to exon 20 of ALK (Figure 1B). More precisely, the fusion joined nucleotide 1161 of EEF1G (The National Center for Biotechnology Information (NCBI) ref: NM-001404), corresponding to an intronic region between exons 8 and 9, with nucleotide 4226 of ALK (NM-004304) situated in exon 20. The EEF1G-ALK fusion was subsequently confirmed by Sanger sequencing of the fragment obtained by nested RT-PCR (Figure 1B) and by FISH using probes for 5′EEF1G and 3′ALK (data not shown). Array Comparative Genomic Hybridization (CGH) analysis demonstrated that 3′ALK is involved in the gain of 2p23.2p25.3 and that the gain of 11q11q13.4 affects EEF1G (Figure 1C,D). In addition, the gain of five other regions, loss of three large regions, including that of 9p21.3p24.3/CDKN2A/B, and three microdeletions were detected (Figure 1C; Table 1). Interestingly, the focal deletion at 8q24.21 covered ~280 kbp sequences flanking the centromeric border of MYC. The loss was confirmed by FISH with LSI MYC; one of two apparently normal looking chromosomes 8 [der(8)] revealed a significantly diminished red signal (Figure 1A).
Table 2.
Results of cytogenetic and molecular studies.
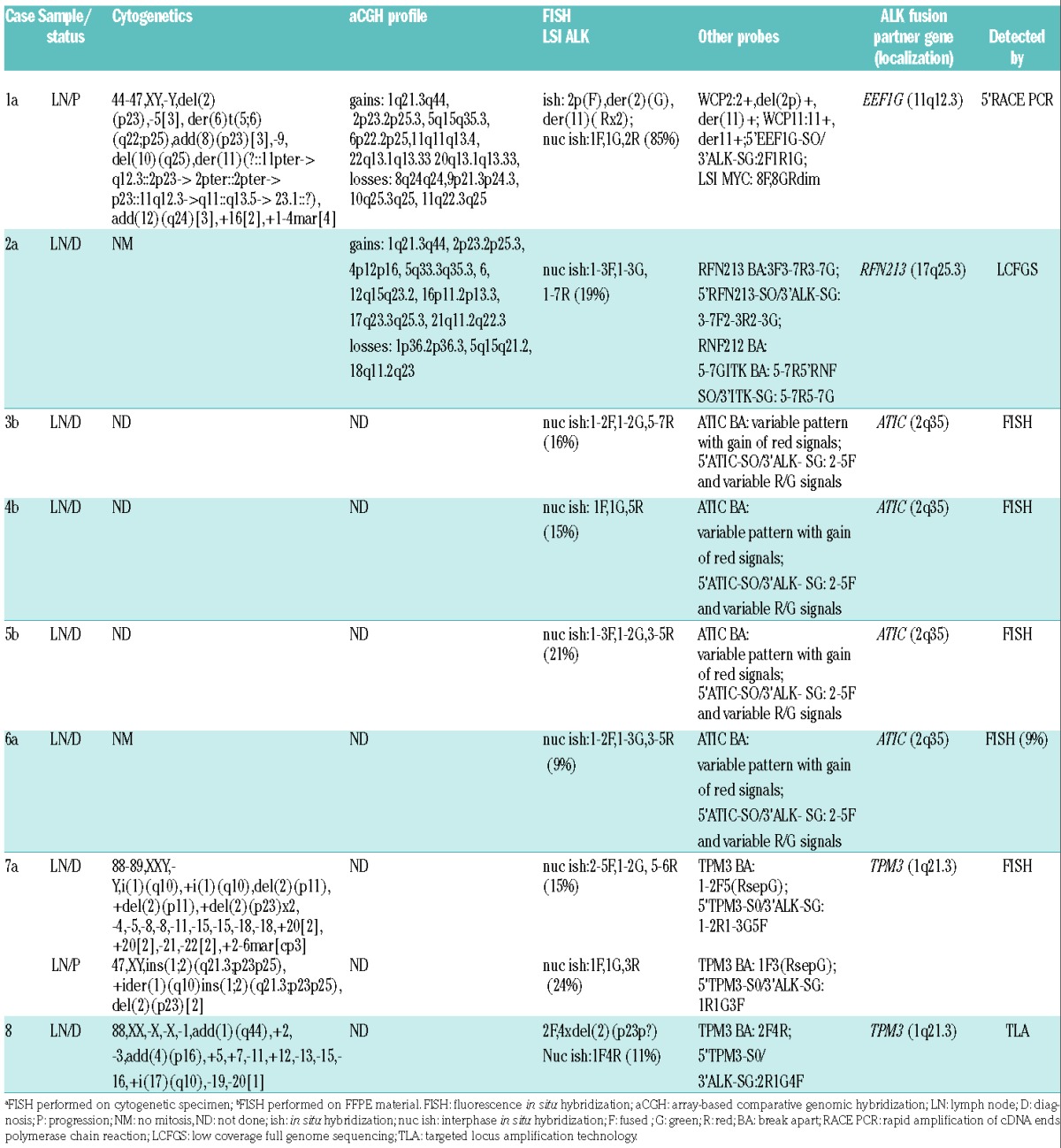
Figure 1.

Cytogenetic and molecular analysis of case 1.(A) Examples of FISH experiments with LSI ALK and LSI MYC BA probes. Metaphase FISH demonstrated BA LSI ALK pattern associated with a duplication of the red/3′ALK signal on der(11) (left image). Paints for chromosomes 2 (red) and 11 (green) confirmed insertion of 2p23p25 at 11q12 (inset). The same aberrant LSI ALK pattern (one colocalized-one green-two red signals) was observed in interphase cells (middle image). Metaphase FISH with LSI MYC showed a diminished red signal on der(8), confirming a focal deletion at 8q24.21 (right image). (B) Schematic representation of the EEF1G, ALK and EEF1G-ALK protein structures (upper panel). Sequencing of the fragment amplified by EEF1G-ALK nested RT-PCR identified an in-frame fusion between exon 8 of EEF1G (breakpoint between exon 8 and 9) and exon 20 of ALK (breakpoint in the middle of exon 20) as shown in the electropherogram (lower panel). (C) Array CGH profile of case 1 showing several unbalanced regions, including gain of 2p23pter and 11q11q13.4 (marked). (D) The selected 2pter (upper panel) and 11q (lower panel) regions. Note the 2p23pter gain-associated break within the ALK gene (gain of 3′ALK) and localization of EEF1G in the middle of gained 11q11q13.4 region.
Case 2
Cytogenetic analysis was unsuccessful. Interphase FISH with LSI ALK showed a BA pattern associated with the gain of up to seven red (3′ALK) signals (Figure 2A). The FISH pattern suggested a diploid and tetraploid status of abnormal cells. The ALK rearrangement was further investigated using LCFGS. The analysis identified the RNF213-ALK fusion (supported by at least eight read pairs) whereby exon 8 of RNF213 is fused to exon 20 of ALK, resulting in an in-frame fusion transcript equal to the previously described cases (Figure 2B).6,17,18 The fusion and its gain were confirmed by FISH (Figure 2A). The performed aCGH analysis demonstrated the ALK- and RNF213-associated gain of 2p23.2p25.3 and 17q23.3q25.3, respectively (Figure 2C,D). In addition, aCGH detected the gain of six other regions and loss of three regions, all listed in Table 2. Notably, breakpoints of the amplified 4p12p16 and 5q33.3q35.3 regions affected the RNF212/4p16 and ITK/5q33.3 genes, respectively. FISH with the respective BA probes (see Online Supplementary Table S1) confirmed the unbalanced rearrangement and partial gain of both genes. Furthermore, FISH with the probes for 5′RNF212 and 3′ITK showed multiple, but not colocalized, signals, precluding the RNF212-ITK hybrid gene (data not shown). Unfortunately, LCFGS did not identify any fusion of RNF212 and ITK.
Figure 2.

Cytogenetic and molecular analysis of case 2. (A) Examples of interphase FISH experiments. Note a BA pattern of LSI ALK and RNF213 BA probes associated with gain of six 3′ALK (red) signals (left image) and six 5′RNF213 (red) signals (middle image). Several colocalized 5′RNF213 (red) and the 3′ALK (green) signals in interphase cells confirm presence and gain of the RNF213-ALK hybrid gene in this case (left image). (B) Schematic representation of the RNF213, ALK and RNF213-ALK protein structures (upper panel). LCFGS resulted in at least eight single read pairs covering the in-frame fusion between exon 8 of RNF213 and exon 20 of ALK (lower panel). (C) Array CGH profile of case 2 showing several unbalanced regions, including gains of 2p23pter and 17q23qter (marked). (D) The selected 2p23pter (upper panel) and 17q23qter (lower panel) regions evidencing the gain-associated breaks within the ALK and RNF213 genes, respectively.
Cases 3–6
The cases were not subjected to cytogenetic analysis. FISH with LSI ALK applied on formalin-fixed paraffin-embedded (FFPE) material confirmed ALK rearrangement and displayed the presence of three to seven red (3′ALK) signals in all cases (Table 2, Figure 3A). Although a number of fused (F), red (R) and green (G) signals in analyzed cells varied, all cases showed a clear predominance of the red signals and variations of the 1F1G3R pattern. As a similar pattern was observed in ATIC-ALK-positive lymphoma,19 we initially examined the status of this gene. FISH with the ATIC BA assay revealed a BA pattern and the presence of extra 5′ATIC signals in all four cases (Figure 3B). Subsequent FISH with the 5′ATIC and 3′ALK probes demonstrated three to five fused signals per cell, confirming the ATIC-ALK rearrangement and gain of the chimeric gene in all four cases (Figure 3C).
Figure 3.

Cytogenetic and FISH analysis of cases 3, 7 and 8. (A–C) Note a BA pattern of LSI ALK and ATIC BA probes associated with a gain of four 3′ALK (red) and four 5′ATIC (red) signals, respectively, in case 3. Several colocalized 5′ATIC/3′ALK signals in interphase cells demonstrate gain of the ATIC-ALK hybrid gene in this sample. Similar FISH results were obtained in cases 4 and 5. Case 7: (D) Partial karyotype (at time of relapse) illustrating insertion of 2p23p25 at 1q21.3 (arrowhead), isochromosome 1q containing duplicated long arm of ins(1;2)(q21.3;p23q25) (two arrowheads) and del(2)(p23) (arrow). (E) Note rearrangement of ALK and gain of two extra 3′ALK (red) signals, and (F) three separated red and green TPM3 BA probes, likely marking ins(1) and ider(1). (G–I) Similar FISH patterns were observed in the diagnostic sample. Case 8: (J) Note four copies of der(2) marked by red signals of LSI ALK, (K) unbalanced rearrangement of TPM3 and (L) loss of TPM3 from one chromosome 1 and four copies of der(2)/TPM3-ALK.
Case 7
The biopsy taken at time of relapse (2016) showed a complex diploid karyotype with a presumed der (1)ins(1;¿)(q21;¿) and ider(1)(q10) containing a duplicated 1q arm of der(1)ins(1;¿)(q21;¿) and del(2)(p23) (Table 2, Figure 3D). Interphase FISH with LSI ALK revealed the 1F1G3R pattern (Figure 3E), as found in ATIC-ALK-positive cases. Considering that the 1q21 breakpoint merges with the localization of TPM3, the known partner of ALK,6 a possible involvement of this gene was investigated by FISH with the designed TPM3 BA assay (Online Supplementary Table S1). Neoplastic cells revealed the 1F3(RsepG) pattern, thus postulating the rearrangement of TPM3 due to ins(1;2)(q21.3;p23p25) (Figure 3F). The TPM3-ALK fusion was subsequently confirmed by FISH using the 5′TPM3/3′ALK probes. In the proceeding step, we revised the diagnostic sample (the case was initially misdiagnosed as classical Hodgkin lymphoma), whereby cytogenetic analysis identified rare hypertetraploid cells with i(1)(q10) and del(2)(p23) (Table 2). FISH with LSI ALK (Figure 3G), TPM3 BA (Figure 3H) and the 5′TPM3/3′ALK probes (Figure 3I) demonstrated the presence, at the time of diagnosis (2002), of the TPM3-ALK rearrangement and copy number gain of the ALK fusion.
Case 8
Cytogenetic analysis identified only one abnormal (near tetraploid) metaphase cell harboring five copies of chromosome 2 (Table 2). LSI ALK applied on this metaphase cell hybridized with all chromosomes 2: one showed a fused signal and the other four were marked by red signals (Figure 3J). The abnormal FISH pattern (1–2F4R) was detected in 11% of interphase cells. Loss of the 5′ALK sequences suggested a focal interstitial del(2)(p23p23) leading to a novel fusion of ALK with a gene located at the centromeric breakpoint of the deletion. To identify a potential partner gene, we used the TLA approach. Unexpectedly, the analysis identified the TPM3-ALK fusion with the breakpoints at the intron 7 of TPM3 and the intron 19 of ALK (Online Supplementary Figure S4). FISH confirmed the TPM3 rearrangement, which was associated with loss of the 3′TPM3 sequences, and showed that four out of five chromosomes 2 harbor one copy of TPM3-ALK (Figure 3K,L). Altogether, we identified a cryptic insertion of the 5′TPM3 into the rearranged ALK locus, loss of the 5′ALK and 3′TPM3 sequences, and gain of two copies of TPM3-ALK.
Functional studies
Gain of the EEF1G-, RFN213-, ATIC- and TPM3-ALK hybrid genes found in the present cases contrasts with a constant occurrence of a single copy of NPM1-ALK in t(2;5)-positive ALCL.20–22 We hypothesized that ALCLs harboring the variant rearrangements compensate an insufficient expression of the ALK fusions (driven by relatively weak promoters of the partner genes) via an increased dosage of the chimeric gene. To validate this concept, we analyzed expression levels of NPM1, EEF1G, RNF213, ATIC and TPM3 in various normal lymphoid cells and T-cell malignancies using previously generated RNA sequencing (Seq) data.23,24 The analysis revealed a significantly lower (P-value <0.001) expression of RNF213, ATIC and TPM3 in all samples when compared to NPM1 (Figure 4A). In contrast, expression of EEF1G was similar to that of NPM1. Based on these findings, we hypothesized that oncogenic transforming properties of the three variant ALK fusions, RFN213-ALK, ATIC-ALK and TPM3-ALK, could be lower than the strong NPM1-ALK kinase. To validate our hypothesis, we attempted to generate the Npm-Alk and Atic-Alk fusions in the genome of Ba/F3 cells using CRISPR/Cas9 genome editing. The expression level of Npm1 is around 50-fold higher than the expression level of Atic in this cell line, making it a suitable model to test our hypothesis (Figure 4B). We designed a gRNA targeting exon 20 of Alk and gRNAs targeting Npm1 and Atic in regions corresponding to the breakpoints observed in our patient samples (Figure 4C). gRNAs targeting Alk and a fusion partner were simultaneously introduced in Ba/F3 Cas9 cells by electroporation. Upon IL3 depletion, both endogenous Alk fusions were able to transform the Ba/F3 Cas9 cells, although cells harboring an endogenous Atic-Alk fusion needed more time to recover from this depletion (Figure 4D). In addition to the slower transformation rate, cells harboring an endogenous Atic-Alk fusion presented with a lower growth rate after transformation than cells with an endogenous Npm1-Alk fusion (Figure 4E). FISH demonstrated the presence of one copy of the Npm1-Alk and Atic-Alk chimeric genes per cell in the respective cell lines (Figure 4F,G). After keeping the cells in culture for three months, we observed gain of one to three copies of the Atic-Alk fusion gene in approximately 20% of these cells. In contrast, the Npm1-Alk cell line kept a single copy of the chimeric gene in all cells (Figure 4H). Altogether, these data demonstrate that the Npm1-Alk fusion is a more potent driver of proliferation than Atic-Alk in Ba/F3 cells, and that the expression level of the fusion partner is a key factor in the transformation potential of the oncogenic fusions.
Figure 4.

Functional analysis of the NPM1-, EEF1G-, RNF213-, TPM3- and ATIC-ALK fusions. (A) Expression analysis of the five ALK partner genes using previously generated RNA-Seq data.23,24 In contrast to EEF1G, expression of TPM3, RNF213, and ATIC is significantly lower (P-value <0.001) when compared to NPM1 in different malignant and non-malignant cell types (HSTL: hepatosplenic T-cell lymphoma [n=4]; T-ALL: T-cell acute lymphoblastic leukemia [n=5]; PTCL: peripheral T-cell lymphoma [n=2]; Spleen [n=1]; Thymus [n=1]; LN: lymph nodes [n=3]; Th1: T helper 1 cells [n=5]; Th2: T helper 2 cells [n=5]; Treg [n=5], and CD4 naïve T-cells [n=4]). Error bars represent the standard deviation (SD). (B) QRT-PCR on Ba/F3 Cas9 cells showing the expression levels of Npm1 and Atic. The expression of Atic is significantly lower than Npm1 (P<0.001). Error bars represent the SD. (C) Representation of the Alk, Npm1, and Atic mouse genes. Exons are indicated by vertical bars. Red arrows indicate the location of the gRNA target sites. (D) Growth curve showing the transforming capacities of Ba/F3 Cas9 cells harboring an endogenous Npm1-Alk or Atic-Alk fusion. Error bars represent the SD. (E) Growth curve showing the growth rate of Ba/F3 Cas9 cells after transformation by the endogenous Alk fusion. Error bars represent the SD. (F–H) Examples of metaphase and interphase FISH results showing the endogenous Alk fusions in Ba/F3 Cas9 cells. Arrows indicate colocalized signals/chimeric genes. Note a constant presence of a single copy of Npm1-Alk and gain of Atic-Alk in late cultures. w/o: without; IL3: interleukin 3.
Discussion
Our study provided evidence that ALCL driven by at least three variant ALK fusions, RNF213-ALK, ATIC-ALK and TPM3-ALK, is characterized by a copy number gain of the ALK hybrid gene. Gain of RNF213-ALK was already observed by us in the previously reported case of ALK+ ALCL, harboring two copies of der(17) t(2;17)(p23.2;q25.3)/RNF213(ALO17)-ALK.17 Genetic mechanisms underlying the amplification of RNF213-ALK in case 2, however, remain unknown. Notably, the four ATIC-ALK-positive cases presented herein, as well as all reported ATIC-ALK cases documented by FISH,19,25–27 showed extra copies of the chimeric gene. We previously documented that the ATIC-ALK fusion is generated by a pericentric inv(2)(p23q35) and is constantly accompanied by isochromosome 2q [derivative of inv(2)] comprising two extra copies of ATIC-ALK.19,25 Based on these data, we presume that the cases reported herein also carry inv(2)(p23q35) and one to three copies of ider(2q). Intriguingly, a similar mechanism of gain of ALK hybrid emerged to underpin the TPM3-ALK rearrangement detected in case 7. The fusion was likely created by the insertion of 3′ALK/2p23p25 at 1q21.3/TPM3 and gained by a subsequent formation of ider(1)(q10). In the second TPM3-ALK-positive case, however, the chimeric gene was produced by a cryptic insertion of 5′TPM3 into the rearranged ALK locus and gained by a duplication of the der(2), as in the case reported by Siebert et al.28 Notably, one more informative case with TPM3-ALK also presented with two to three copies of the 3′ALK.29 Lamant et al.,6 who originally described the TPM3-ALK fusion in ALK+ ALCL, linked it to t(1;2)(q25;p23). Considering, however, an opposite transcriptional orientation of both genes, generation of the in-frame TPM3-ALK fusion requires more complicated events, as illustrated in the present and previously reported cases.28
Altogether, our genetic findings, supported by the published data,17,19,26–29 highlight a strong association of RNF213-ALK, ATIC-ALK and TPM3-ALK with a copy number gain of the chimeric gene. A spectrum of such ALK fusions is probably broader, but a frequent lack of cytogenetic and/or FISH data hampers their identification. These observations contrast with ALCL driven by NPM1-ALK20–22 and at least three variant fusions, CLTC-ALK,17 TPM4-ALK30,31 and TRAF1-ALK,27,32 shown to carry a single copy of the ALK hybrid gene. We initially included the novel EEF1G-ALK variant found to be duplicated on der(11) in the former category. Given, however, that two recently published ALCL cases with EEF1G-ALK did not show copy number gain of the ALK fusion gene,33 duplication of EEF1G-ALK in our case was likely associated with progression of the disease, similar to a case of leukemic ALK+ ALCL with an extra copy of NPM1-ALK.34 This conclusion is additionally supported by our other data illustrating a strong expression of EEF1G in normal and malignant T cells, similar to that of NPM1. Thus, there is no reason why the EEF1G-ALK fusion gene would require an increased copy number.
In contrast, expression of RNF213, ATIC and TPM3 in lymphoid cells was significantly lower than NPM1 and EEF1G. These findings support the concept that ALCL driven by the RNF213-ALK, ATIC-ALK and TPM3-ALK fusions might require an increased dosage of the ALK chimeric gene to compensate an insufficient expression of ALK in neoplastic cells. To test if the best documented ATIC-ALK fusion is indeed less transforming than the NPM1-ALK fusion, we generated the Npm1-Alk and Atic-Alk fusion genes by inducing Cas9 mediated chromosomal rearrangements in Ba/F3 cells. Growth properties of these engineered cells showed that the transformation potential of Atic-Alk is significantly lower when compared to Npm1-Alk, likely due to a lower expression level of Atic. Interestingly, the tendency of the Atic-Alk-positive Ba/F3 cells to gain extra copies of the Atic-Alk chimeric gene recapitulates the events observed in the original tumors and confirms the selective pressure of the cells to acquire additional copies of the Atic-Alk fusion. We also attempted to generate an endogenous Rnf213-Alk fusion, but since Rnf213 is very lowly expressed in Ba/F3 cells, this fusion could not transform the cells. Tpm3 was not included in the functional studies, since it was only recently added to the study. Of note, Giuriato et al.35 previously showed that in conditional knock-in mice both TPM3-ALK and NPM1-ALK could induce B-cell lymphoma/leukemia with a similar disease latency. In these transgenic mice, however, the expression levels of both ALK fusions were similar, since their expression was driven by an external promoter. This could explain the observed equal tumorigenic potential of both ALK fusions.
The novel EEF1G-ALK fusion which we identified in a child with ALK+ ALCL extends the already long list of ALK fusion partners comprising approximately 20 genes.7 The very recent report of two pediatric patients with EEF1G-ALK33 indicates that the fusion is recurrent and strongly associated with pediatric ALK+ ALCL. EEF1G, located at 11q12.3, has 10 exons encoding for an eukaryotic translation elongation factor 1γ. Together with α, β and δ subunits, it forms the eukaryotic elongation factor complex, which is predominantly involved in protein biosynthesis with an elongation of the polypeptide chains.36,37 EEF1G comprises a glutathione-S-transferase (GST)-like N-terminal domain and a C-terminal (CT) domain.37 Although all four subunits of the elongation factor complex are highly expressed in most eukaryotic cells, the role of human EEF1G is poorly understood. Notably, an increased expression of EEF1G has been detected in various human carcinomas.37–40 It has been suggested that overexpression of EEF1G stimulates the overgrowth of neoplastic cells.41 In the present case of ALK+ ALCL, exons 1 to 8 of EEF1G are fused to exon 20–29 of ALK, resulting in a chimeric protein with the complete GST-like N-terminal domain and part of the CT domain of EEF1G, fused to the complete intracellular protein tyrosine kinase (PTK) domain of ALK (Figure 2B). Even though the complete PTK domain is involved, the ALK fragment contains the final 513 amino acids, which is shorter in comparison to the fragment involved in most other ALK fusions, containing the final 563 amino acids.42 Of note, both cases reported by Palacios et al.33 revealed breakpoints within intron 7 of EEF1G and intron 20 of ALK. As the fusions were identified by RNA-Seq, genetic mechanisms underlying these rearrangements are unknown.
The postulated t(2;11)(p23;q12.3), however, is unlikely due to an opposite transcriptional orientation of both genes, similar to TPM3 and ALK. Functional studies performed by the authors provided evidence of the dimerization properties of the EEF1G-ALK fusion, constitutive activation of ALK kinase and its strong oncogenic potential similar to that of NMP1-ALK.
Our patient with EEF1G-ALK experienced an unfavorable and fatal clinical course. Although ALK+ ALCL has a relatively good prognosis, chemoresistant and aggressive forms of this lymphoma, recurrently featured by MYC aberrations, have been recurrently reported.32,43–45 Notably, our case displayed a microdeletion neighboring MYC and loss of CDKN2A/B at 9p21, a known poor prognostic factor in tumors.46 We presume that a focal del(8)(q24q24) could activate MYC by loss of negative regulatory elements upstream of the gene, for example. Although clinical data of the EEF1G-ALK-positive cases reported by Palacios et al.33 are not available, we believe that an aggressive clinical course and unfavorable prognosis of certain ALK+ ALCL is not determined by the type of ALK fusion, but rather by secondary hits affecting potent oncogenes and tumor suppressor genes (e.g., MYC, TP53, PRDM1 and CDKNA2/B).32,44,45,47
In summary, we investigated eight recently diagnosed cases of ALK+ ALCL with a cytoplasmic-only expression of ALK and identified one novel EEF1G-ALK rearrangement and three already known fusions, RNF213-ALK, TPM3-ALK and ATIC-ALK. Occurrence of the latter rearrangement in four out of eight (50%) of the cases studied confirms that ATIC-ALK is the most prevalent variant fusion in ALK+ ALCL. Importantly, RNF213-ALK, TPM3-ALK and ATIC-ALK fusions were recurrently present in two or more copies, contrasting with the NPM1-ALK chimeric gene which constantly occurs in one copy. Our functional studies show that ALK+ ALCL driven by ATIC-ALK compensates the weak expression and possibly weak oncogenic properties of this variant ALK fusion by an increased gene dosage. We propose a similar explanation for the copy number gain of RNF213-ALK and TPM3-ALK. Altogether, our findings support the hypothesis that the transforming capacities of ALK fusions depend on the biological features of the partner genes.48
Supplementary Material
Acknowledgments
The authors would like to thank Vanessa Vanspauwen and Emilie Bittoun for their excellent technical assistance, and Rita Logist for the editorial help.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/9/1605
Funding
This study was supported by the concerted action grant from the KU Leuven no. 3M040406 (JAvdK, PV, TT, JC and IW), research grants from the FWO Vlaanderen (G081411N to TT) and “Stichting tegen Kanker” (PV). MVDB holds a SB Fellowship of the Research Foundation-Flanders. PV is a senior clinical investigator of the FWO-Vlaanderen. TT holds a Mandate for Fundamental and Translational Research from the “Stichting tegen Kanker” (2014-083).
References
- 1.Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: International Agency for Research on Cancer: 2008. [Google Scholar]
- 2.Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263(5151):1281–1284. [DOI] [PubMed] [Google Scholar]
- 3.Chiarle R, Gong JZ, Guasparri I, et al. NPM-ALK transgenic mice spontaneously develop T-cell lymphomas and plasma cell tumors. Blood. 2003;101(5):1919–1927. [DOI] [PubMed] [Google Scholar]
- 4.Kuefer MU, Look AT, Pulford K, et al. Retrovirus-mediated gene transfer of NPM-ALK causes lymphoid malignancy in mice. Blood. 1997;90(8):2901–2910. [PubMed] [Google Scholar]
- 5.Lange K, Uckert W, Blankenstein T, et al. Overexpression of NPM-ALK induces different types of malignant lymphomas in IL-9 transgenic mice. Oncogene. 2003; 22(4):517–527. [DOI] [PubMed] [Google Scholar]
- 6.Lamant L, Dastugue N, Pulford K, Delsol G, Mariame B. A new fusion gene TPM3-ALK in anaplastic large cell lymphoma created by a (1;2)(q25;p23) translocation. Blood. 1999;93(9):3088–3095. [PubMed] [Google Scholar]
- 7.H Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer. 2013;13(10):685–700. [DOI] [PubMed] [Google Scholar]
- 8.Falini B, Pileri S, Zinzani PL, et al. ALK+ lymphoma: clinic-pathological findings and outcome. Blood. 1999;93(8):2697–2706. [PubMed] [Google Scholar]
- 9.Boi M, Zucca E, Inghirami G, Bertoni F. Advances in understanding the pathogenesis of systemic anaplastic large cell lymphomas. Br J Haematol. 2015;168(6):771–783. [DOI] [PubMed] [Google Scholar]
- 10.Drexler HG, Gignac SM, von WR, Werner M, Dirks WG. Pathobiology of NPM-ALK and variant fusion genes in anaplastic large cell lymphoma and other lymphomas. Leukemia. 2000;14(9):1533–1559. [DOI] [PubMed] [Google Scholar]
- 11.Stein H, Foss Hd, Du H, et al. CD30+ anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood. 2000;96(12):3681–3695. [PubMed] [Google Scholar]
- 12.Sakamoto K, Nakasone H, Togashi Y, et al. ALK-positive large B-cell lymphoma: identification of EML4-ALK and a review of the literature focusing on the ALK immunohistochemical staining pattern. Int J Hematol. 2016;103(4):399–408. [DOI] [PubMed] [Google Scholar]
- 13.Mologni L. Inhibitors of the anaplastic lymphoma kinase. Expert Opin Investig Drugs. 2012;21(7):985–994. [DOI] [PubMed] [Google Scholar]
- 14.Pall G. The next-generation ALK inhibitors. Curr Opin Oncol. 2015;27(2):118–124. [DOI] [PubMed] [Google Scholar]
- 15.Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gambacorti PC, Farina F, Stasia A, et al. Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase-positive lymphoma patients. J Natl Cancer Inst. 2014; 106(2):djt378. [DOI] [PubMed] [Google Scholar]
- 17.Cools J, Wlodarska I, Somers R, et al. Identification of novel fusion partners of ALK, the anaplastic lymphoma kinase, in anaplastic large-cell lymphoma and inflammatory myofibroblastic tumor. Genes Chromosomes Cancer. 2002;34(4):354–362. [DOI] [PubMed] [Google Scholar]
- 18.Hernandez L, Bea S, Bellosillo B, et al. Diversity of genomic breakpoints in TFG-ALK translocations in anaplastic large cell lymphomas: identification of a new TFG-ALK(XL) chimeric gene with transforming activity. Am J Pathol. 2002;160(4):1487–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wlodarska I, De Wolf-Peeters C, Falini B, et al. The cryptic inv(2)(p23q35) defines a new molecular genetic subtype of ALK-positive anaplastic large-cell lymphoma. Blood. 1998;92(8):2688–2695. [PubMed] [Google Scholar]
- 20.Mathew P, Sanger WG, Weisenburger DD, et al. Detection of the t(2;5)(p23;q35) and NPM-ALK fusion in non-Hodgkin’s lymphoma by two-color fluorescence in situ hybridization. Blood. 1997;89(5):1678–1685. [PubMed] [Google Scholar]
- 21.Perkins SL, Pickering D, Lowe EJ, et al. Childhood anaplastic large cell lymphoma has a high incidence of ALK gene rearrangement as determined by immunohistochemical staining and fluorescent in situ hybridisation: a genetic and pathological correlation. Br J Haematol. 2005; 131(5):624–627. [DOI] [PubMed] [Google Scholar]
- 22.Cataldo KA, Jalal SM, Law ME, et al. Detection of t(2;5) in anaplastic large cell lymphoma: comparison of immunohistochemical studies, FISH, and RT-PCR in paraffin-embedded tissue. Am J Surg Pathol. 1999;23(11):1386–1392. [DOI] [PubMed] [Google Scholar]
- 23.Finalet Ferreiro J, Rouhigharabaei L, Urbankova H, et al. Integrative genomic and transcriptomic analysis identified candidate genes implicated in the pathogenesis of hepatosplenic T-cell lymphoma. PloS One. 2014;9(7):e102977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Atak ZK, Gianfelici V, Hulselmans G, et al. Comprehensive analysis of transcriptome variation uncovers known and novel driver events in T-cell acute lymphoblastic leukemia. PLoS Genet. 2013; 9(12):e1003997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma Z, Cools J, Marynen P, et al. Inv(2)(p23q35) in anaplastic large-cell lymphoma induces constitutive anaplastic lymphoma kinase (ALK) tyrosine kinase activation by fusion to ATIC, an enzyme involved in purine nucleotide biosynthesis. Blood. 2000;95(6):2144–2149. [PubMed] [Google Scholar]
- 26.Colleoni GW, Bridge JA, Garicochea B, Liu J, Filippa DA, Ladanyi M. ATIC-ALK: A novel variant ALK gene fusion in anaplastic large cell lymphoma resulting from the recurrent cryptic chromosomal inversion, inv(2)(p23q35). Am J Pathol. 2000; 156(3):781–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takeoka K, Okumura A, Honjo G, Ohno H. Variant translocation partners of the anaplastic lymphoma kinase (ALK) gene in two cases of anaplastic large cell lymphoma, identified by inverse cDNA polymerase chain reaction. J Clin Exp Hematop. 2014;54(3):225–235. [DOI] [PubMed] [Google Scholar]
- 28.Siebert R, Gesk S, Harder L, et al. Complex variant translocation t(1;2) with TPM3-ALK fusion due to cryptic ALK gene rearrangement in anaplastic large-cell lymphoma. Blood. 1999;94(10):3614–3617. [PubMed] [Google Scholar]
- 29.Hoshino A, Nomura K, Hamashima T, et al. Aggressive transformation of anaplastic large cell lymphoma with increased number of ALK-translocated chromosomes. Int J Hematol. 2015;101(2):198–202. [DOI] [PubMed] [Google Scholar]
- 30.Meech SJ, McGavran L, Odom LF, et al. Unusual childhood extramedullary hematologic malignancy with natural killer cell properties that contains tropomyosin 4–anaplastic lymphoma kinase gene fusion. Blood. 2001;98(4):1209–1216. [DOI] [PubMed] [Google Scholar]
- 31.Liang X, Meech SJ, Odom LF, et al. Assessment of t(2;5)(p23;q35) translocation and variants in pediatric ALK+ anaplastic large cell lymphoma. Am J Clin Pathol. 2004;121(4):496–506. [DOI] [PubMed] [Google Scholar]
- 32.Abate F, Todaro M, van der Krogt J, et al. A novel patient derived tumorgraft model with TRAF1-ALK anaplastic large cell lymphoma translocation. Leukemia. 2014;(6):1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palacios G, Shaw TI, Li Y, et al. Novel ALK fusion in anaplastic large cell lymphoma involving EEF1G, a subunit of the eukaryotic elongation factor-1 complex. Leukemia. 2017;31(3):743–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ries S, Rnjak L, Mitrovic Z, Kuvezdic KG, Nola M, Sucic M. CD13+ anaplastic large cell lymphoma with leukemic presentation and additional chromosomal abnormality. Diagn Cytopathol. 2010;38(2):141–146. [DOI] [PubMed] [Google Scholar]
- 35.Giuriato S, Foisseau M, Dejean E, et al. Conditional TPM3-ALK and NPM-ALK transgenic mice develop reversible ALK-positive early B-cell lymphoma/leukemia. Blood. 2010;115(20):4061–4070. [DOI] [PubMed] [Google Scholar]
- 36.Kumabe T, Sohma Y, Yamamoto T. Human cDNAs encoding elongation factor 1 gamma and the ribosomal protein L19. Nucleic Acids Res. 1992;20(10):2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Achilonu I, Siganunu TP, Dirr HW. Purification and characterisation of recombinant human eukaryotic elongation factor 1 gamma. Protein Expr Purif. 2014;99:70–77. [DOI] [PubMed] [Google Scholar]
- 38.Chi K, Jones DV, Frazier ML. Expression of an elongation factor 1 gamma-related sequence in adenocarcinomas of the colon. Gastroenterology. 1992;103(1):98–102. [DOI] [PubMed] [Google Scholar]
- 39.Ender B, Lynch P, Kim YH, Inamdar NV, Cleary KR, Frazier ML. Overexpression of an elongation factor-1 gamma-hybridizing RNA in colorectal adenomas. Mol Carcinog. 1993;7:18–20. [DOI] [PubMed] [Google Scholar]
- 40.Lew Y, Jones DV, Mars WM, Evans D, Byrd D, Frazier ML. Expression of elongation factor-1 gamma-related sequence in human pancreatic cancer. Pancreas. 1992;7:144–152. [DOI] [PubMed] [Google Scholar]
- 41.Mimori K, Mori M, Inoue H, et al. Elongation factor 1 gamma mRNA expression in oesophageal carcinoma. Gut. 1996;38(1):66–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Roosbroeck K, Wlodarska I. Oncogenic anaplastic lymphoma kinase rearrangements in lymphoma. European Haematology. 2009;3:50–56. [Google Scholar]
- 43.Moritake H, Shimonodan H, Marutsuka K, Kamimura S, Kojima H, Nunoi H. C-MYC rearrangement may induce an aggressive phenotype in anaplastic lymphoma kinase positive anaplastic large cell lymphoma: Identification of a novel fusion gene ALO17/C-MYC. Am J Hematol. 2011; 86(1):75–78. [DOI] [PubMed] [Google Scholar]
- 44.Liang X, Branchford B, Greffe B, et al. Dual ALK and MYC rearrangements leading to an aggressive variant of anaplastic large cell lymphoma. J Pediatr Hematol Oncol. 2013; 35(5):e209–e213. [DOI] [PubMed] [Google Scholar]
- 45.Monaco S, Tsao L, Murty VV, et al. Pediatric ALK+ anaplastic large cell lymphoma with t(3;8)(q26.2;q24) translocation and c-myc rearrangement terminating in a leukemic phase. Am J Hematol. 2007; 82(1):59–64. [DOI] [PubMed] [Google Scholar]
- 46.LaPak KM, Burd CE. The molecular balancing act of p16(INK4a) in cancer and aging. Mol Cancer Res. 2014;12(2):167–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boi M, Rinaldi A, Kwee I, et al. PRDM1/BLIMP1 is commonly inactivated in anaplastic large T-cell lymphoma. Blood. 2013;122(15):2683–2693. [DOI] [PubMed] [Google Scholar]
- 48.Armstrong F, Duplantier MM, Trempat P, et al. Differential effects of X-ALK fusion proteins on proliferation, transformation, and invasion properties of NIH3T3 cells. Oncogene. 2004;23(36):6071–6082. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.