

RUNX1/ETO, the product of the t(8;21) chromosomal translocation, is required for the onset and maintenance of one of the most common forms of acute myeloid leukemia (AML). RUNX1/ETO has a modular structure and, besides the DNA-binding domain (Runt), contains four evolutionary conserved functional domains named nervy homology regions 1–4 (NHR1 to NHR4). The NHR domains serve as docking sites for a variety of different proteins and, in addition, the NHR2 domain mediates tetramerization through hydrophobic and ionic/polar interactions. Tetramerization is essential for RUNX1/ETO oncogenic activity. Destabilization of the RUNX1/ETO high molecular weight complex abrogates RUNX1/ETO oncogenic activity.1–4 Using structure-based virtual screening, we identified several small molecule inhibitors mimicking the tetramerization hot spot within the NHR2 domain of RUNX1/ETO.5 One of these compounds, 7.44, was of particular interest as it showed biological activity in vitro and in vivo.
Compound 7.44 mimics three essential amino acids (WED) at the tetramerization hot-spot of the NHR2 dimer, thereby blocking RUNX1/ETO tetramerization (Online Supplementary Figure 1A and 1B).4 We analyzed the DNA-binding capacity of fusion-proteins containing the DNA-binding domain of RUNX1/ETO juxtaposed to the oligomerization domain of either RUNX1/ETO (RUNX1/NHR2) or the unrelated oncogenic tyrosine kinase BCR/ABL (RUNX1/BCR).6 A biotinylated oligonucleotide derived from the RUNX1/ETO binding region within the RUNX3 promoter was used as DNA-binding target. Incubation of the double-stranded RUNX3-oligonucleotide with RUNX1/NHR2 or RUNX1/BCR resulted in binding of the polypeptides to the RUNX3 target, as demonstrated by ABCD assay (Online Supplementary Figure 1C; See Online Supplementary Methods). Compound 7.44 specifically blocked binding of RUNX1/NHR2 to the oligonucleotide while binding of RUNX1/BCR was not affected (Online Supplementary Figures 1C and 1D). A control compound, 7.38, which does not interfere with NHR2 dimer-tetramer transition5, did not significantly influence DNA-binding of either polypeptide to the oligonucleotide (Online Supplementary Figures 1C and 1D), suggesting that compound 7.44 impairs DNA-binding of RUNX1/NHR2 through inhibition of NHR2-mediated tetramerization as the DNA binding capacity of the RUNX1/BCR peptide was not affected. We next analyzed the proliferation capacity of t(8;21)-positive and RUNX1/ETO-dependent SKNO-1 and Kasumi-1 cells upon treatment with compounds 7.44 and 7.38. Compound 7.44 (but not 7.38) triggered a significant reduction in the proliferation of RUNX1/ETO-expressing cells. In contrast, proliferation of K562 cells, which are independent of RUNX1/ETO expression, was unaffected by either compound (Figures 1A–C). The antiproliferative effect of 7.44 was accompanied by a significant increase in differentiation as demonstrated by elevated CD11b expression levels (P=0.0001) (Figure 1D). This effect was absent in K562 cells treated with 7.44 or in RUNX1/ETO-expressing cells treated with the control compound 7.38 (Figure 1D). Granulocytic differentiation of SKNO-1 cells in response to 7.44 was evidenced by the presence of segmented nuclei, whereas compound 7.38-treated cells remained undifferentiated similar to untreated control cells (Figure 1E). In line with this, SKNO-1 cells exhibited a significant reduction in the nucleus-to-cytoplasm ratio (P=0.003) in response to compound 7.44 treatment, while treatment with compound 7.38 did not alter cell morphology (Figure 1F). Upon treatment with compound 7.44, Kasumi-1 cells showed reduced c-KIT surface receptor expression levels in a dose-dependent manner, while c-KIT expression in the non-RUNX1/ETO dependent HEL cell line remained unaffected (Figure 1G). Moreover, the colony forming ability of compound 7.44-treated SKNO-1 and Kasumi-1 cells was significantly reduced (P=0.01), while RUNX1/ETO negative K562 cells did not show this effect (Figure 1H). In addition, treatment of SKNO-1 cells with compound 7.44 led to an accumulation of dead cells by apoptotic or necroptotic processes as demonstrated by AnnexinV/PI staining at day seven of treatment (Figure 1I).
Figure 1.

Compound 7.44 inhibits the growth of and induces myeloid differentiation in RUNX1/ETO-expressing cells. A. Proliferation of SKNO-1 and B. Kasumi-1 cells in the presence or absence of compound 7.44 or compound 7.38. C. Compound 7.44 does not affect the proliferation of the RUNX1/ETO-negative K562 cells. D. Differentiation of SKNO-1, Kasumi-1 and K562 cells after daily treatment with compound 7.44 or compound 7.38 for five days (c = 25 μM for SKNO-1 and K562 cells, 50 μM for Kasumi-1 cells). The percentage of CD11b-positive cells is depicted. E. Morphological visualization of myeloid differentiation of SKNO-1 cells after 4 days treatment with compound 7.44 or 7.38 (c = 10 μM). Arrows depict differentiated cells. F. Quantification of the nucleus/cytoplasm ratio in SKNO-1 cells shown in E. G. c-KIT expression in Kasumi-1 cells at day five after daily treatment with compound 7.44 or 7.38 and in HEL cells after daily treatment with compound 7.44 at the indicated concentrations. H. Colony formation by SKNO-1, Kasumi-1 and K562 cells before and after treatment with compounds 7.44 or 7.38. SKNO-1 and K562 were treated for 3 days (c = 10 μM). Kasumi-1 cells were treated for 4 days (c = 50 μM). The percentage of colony numbers relative to the controls is depicted. Statistical significance according to paired two-tailed t-test. I. Compound 7.44 triggers apoptotic or necroptotic processes in SKNO-1 cells. SKNO-1 cells were treated with compounds 7.44 or 7.38 for 7 days and stained with Annexin-V and propidium iodide (PI). The percentage of living (AnnexinV/PI-) and apoptotic/necroptotic (Annexin-V/PI+) cells is shown. Statistical significance determined by unpaired two-tailed t-test unless otherwise stated in the text. n=3. Bar diagrams show mean ± SD. * P < 0.05, ** P < 0.01, *** P < 0.001.
Thereafter, we analyzed the effect of compound 7.44 on RUNX1/ETO-mediated repression of gene expression. SKNO-1 cells treated with 20 μM 7.44 or 7.38 for 3 days were analyzed for the expression levels of the RUNX1/ETO target genes CD244, HCK, NFE2, OGG1, C/EBPα and PU.1 7 using real time PCR. We found a significant increase in the expression levels of all analyzed genes in cells treated with 7.44 compared to cells treated with 7.38 (Online Supplementary Figure 2A). Subsequently, we carried out chromatin immunoprecipitation (ChIP) assays to demonstrate that RUNX1/ETO binding to CD244, OGG1 and RUNX1 promoters was reduced in the presence of compound 7.44, but unchanged in the presence of 7.38 (Online Supplementary Figure 2B), which explains the up-regulation of RUNX1/ETO target genes in the presence of compound 7.44.
Furthermore, we analyzed the inhibitory activity of compound 7.44 on RUNX1/ETO-expressing primary human hematopoietic CD34+ cells and primary CD34+ AML samples. G-CSF-mobilized peripheral blood CD34+ cells were transduced with a retroviral construct containing a C-terminal truncated form of RUNX1/ETO similar to RUNX1/ETO9a,8 hereafter called RUNX1/ETOtr. In this system, RUNX1/ETOtr confers increased self-renewal capacity to CD34+ cells, which then grow continuously in a cytokine-dependent manner. After 50 days in culture, only RUNX1/ETOtr-expressing cells survive (Figure 2A). These cells are then completely dependent on sustained RUNX1/ETOtr expression and oligomerization for continuous expansion ex vivo.9 We treated the RUNX1/ETOtr-transformed CD34+ cells with compound 7.44 or compound 7.38 at a concentration of 100 μM. As expected, only compound 7.44 triggered a significant growth reduction (P = 0.0002) of RUNX1/ETOtr-expressing human primary progenitors, while non-treated cells or RUNX1/ETOtr cells treated with compound 7.38 were insensitive to treatment (Figure 2B). This antiproliferative effect of compound 7.44 was accompanied by increased cellular differentiation as measured by CD11b surface marker expression, and reduction in colony forming ability (Figures. 2C and 2D). In contrast, treatment with compound 7.38 did not have any effect on cell differentiation or colony forming ability (Figures 2C and 2D). Likewise, treatment of non-transduced CD34+ cells with compound 7.44 did not affect colony formation potential. Similar to the observations with Kasumi-1 and SKNO-1 cells, 7.44 treatment of RUNX1/ETO-dependent CD34+ cells triggered apoptotic/necroptotic processes as estimated by Annexin V staining (Figure 2E). Moreover, a reduction in cell numbers was observed upon treatment of primary CD34+AML samples with compound 7.44 in culture (c = 75 μM; Figure 2F), most likely caused by decreased proliferation as estimated from Ki67-labeling experiments (Online Supplementary Figure 3).
Figure 2.
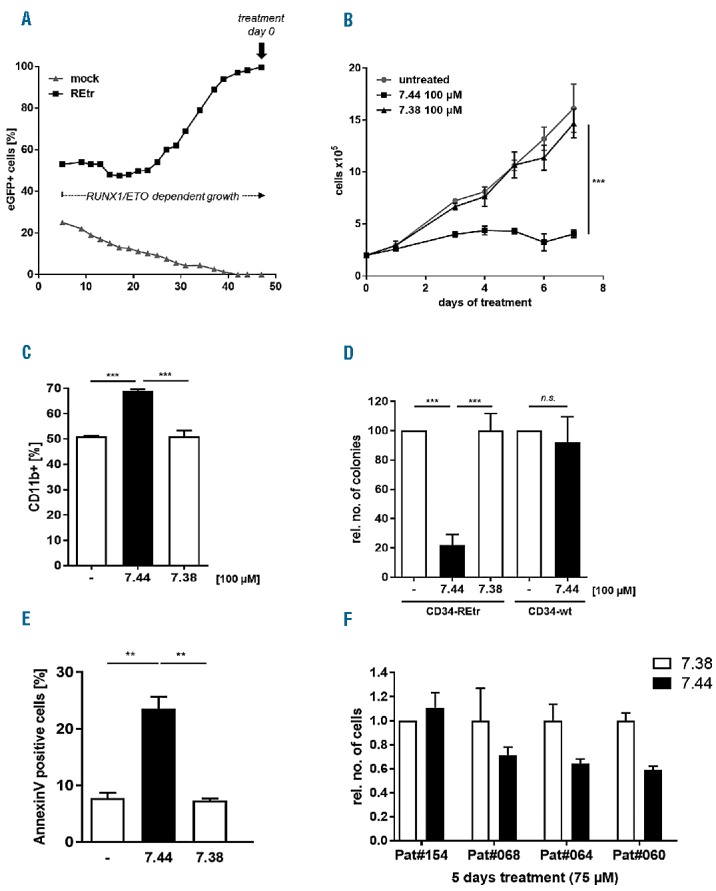
Compound 7.44 induces growth-arrest and differentiation of RUNX1/ETOtr-expressing CD34+ progenitor cells. A. G-CSF-mobilized peripheral blood CD34+ cells were transduced with a retroviral vector expressing REtr-ires-eGFP (REtr) or left untransduced (mock). Tr denotes a truncated form of RUNX1/ETO with enhanced oncogenic function. Cell growth (% eGFP positive cells) was monitored over 50 days. B. RUNX1/ETO-transformed CD34+ cells were treated with compound 7.44 or 7.38 daily for seven days (c = 100 μM). The growth kinetic of the treated cells is shown in comparison to untreated cells. C. Differentiation of RUNX1/ETOtr-expressing CD34+ progenitor cells after daily treatment with 100 μM of 7.44 or 7.38. CD11b expression was measured at day 8 of treatment. The percentage of CD11b-positive cells is depicted. D. Colony formation by RUNX1/ETOtr-expressing CD34+ cells after daily treatment with 100 μM of 7.44 or 7.38 for 7 days. Non-transduced fresh CD34+ cells were used as controls. The colony forming ability of the cells was tested at day 8 post-treatment. The percentage of colonies (treated vs. untreated) is depicted. E. Compound 7.44 triggers apoptotic or necroptotic processes in REtr-expressing CD34+ cells. Cells were treated with compounds 7.44 or 7.38 for 7 days and stained with Annexin-V and 7-AAD. The percentage of apoptotic/necroptotic (Annexin-V/7-AAD+) cells is shown. n=3. Statistical significance determined by unpaired two-tailed t-test. F. Primary AML samples were cultured with compound 7.44 or 7.38 (c = 75 μM) for five days. The relative number of cells in the cultures treated with compound 7.44 vs. 7.38 is shown. n=2. Bar diagrams show mean ± SD. ** P < 0.01, *** P < 0.001.
Lastly, we asked if treatment with compound 7.44 also reduces RUNX1/ETO driven tumor growth in vivo. We intravenously injected 2 × 106 Kasumi-1 cells expressing a luciferase reporter gene into immunocompromised NSG mice. At week 8 post-injection, animals were randomized into two groups and treated i.p. with compounds 7.44 (n = 8) or 7.38 (n = 7) at a dose between 200 and 250 μg/Kg per animal per day, five times per week. Leukemia development was monitored by bioluminescence imaging for a total of 130 days. Compound 7.44 significantly reduced the dissemination of leukemic cells over the treatment period when compared to animals treated with compound 7.38 (P=0.0047; Figure 3A–B). At day 130 post-treatment, 75% of the animals treated with compound 7.44 were still alive, whereas 85% of those treated with the control compound 7.38 had to be sacrificed due to the rapid growth and dissemination of leukemic cells.
Figure 3.
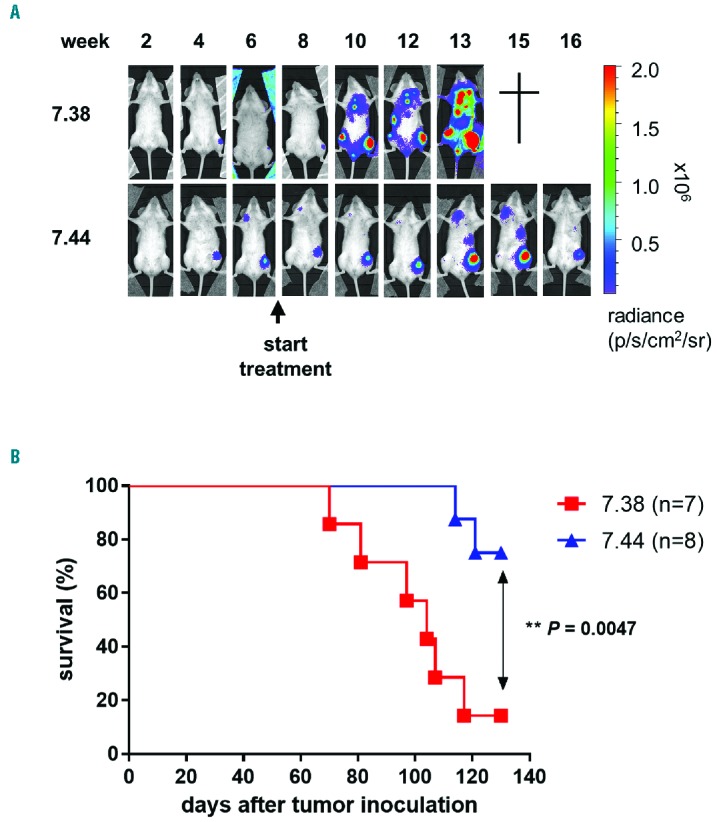
Compound 7.44 delays tumor growth of RUNX1/ETO cells in mice. A. Luciferase-expressing Kasumi-1 cells (2×106 cells) were inoculated i.v. into irradiated (D=2Gy) NSG mice. Animals were treated i.p with compound 7.44 or 7.38 at 200–250 μg/Kg per animal per day, five days per week, beginning at week 8 post inoculation of Kasumi-1 cells. Tumor proliferation was monitored weekly by in vivo bioluminescence. One representative result is shown. B. Kaplan-Meier survival curve of recipient mice treated with compound 7.44 or 7.38. Data are summarized from two independent experiments. Log-rank test was used for statistical survival analyses.
To date, several other inhibitors of RUNX1/ETO tetramerization have been described. Oridonin, a diterpenoid isolated from medicinal herbs, has been shown to mediate RUNX1/ETO cleavage at D188 in a caspase 3-dependent manner, thereby generating polypeptides that interfered with RUNX1/ETO tetramerization.10 We have used a-helical peptides mimicking the NHR2 domain for similar purposes.9 In all of these cases, RUNX1/ETO oncogenic function was abrogated, leading to a decrease in self-renewal capacity, colony-forming ability, and increased differentiation of RUNX1/ETO expressing cells, clearly demonstrating that targeting RUNX1/ETO tetramerization is a reasonable approach to inhibit its oncogenic function. Importantly, the complete disruption of RUNX1/ETO tetramers is not necessary for blocking RUNX1/ETO’s transforming capacity. A shift from tetramer to dimer is already sufficient to induce cellular differentiation in RUNX1/ETO-expressing cells.4,9 Compound 7.44 has been shown to interfere with c-Jun phosphorylation and JNK signaling.11 As RUNX1/ETO function partially depends on JNK function,12,13 the effects of 7.44 on JNK activity may also account for the effects described in our work. However, compound 7.44 has no effect on the proliferation potential of the BCR/ABL-positive K562 cells, in which the JNK pathway is essential for BCR/ABL transformation and maintenance.14,15 These observations suggest that the main mode of action of compound 7.44 is by interfering with the formation or the stability of the RUNX1/ETO tetramer complex.
Compound 7.44, the first-in-class small molecule to disrupt RUNX1/ETO tetramerization, could serve as a lead structure to guide the development of structurally related compounds with increased binding affinity, improved bioavailability, and enhanced anti-leukemic effects to inhibit RUNX1/ETO oncogenic function in t(8;21) acute myeloid leukemia.
Supplementary Material
Acknowledgments
The authors would like to thank M. Hartmann, E. Rudolf and T. Syzonenko (GSH) and K. Thomas and J. Wagner (Dept. of Pediatric Stem Cell Transplantation, University of Frankfurt) for excellent assistance during the animal, cell and molecular biology experiments. We are grateful to the NCI/DTP Open Chemical Repository (http://dtp.cancer.gov/) for compound samples and to OpenEye for an academic license.
Footnotes
Funding: this work was supported by grants from the LOEWE Center for Cell and Gene Therapy Frankfurt (funded by the Ministry of Higher Education, Research and the Arts of the State of Hessia (HMWK), ref.: III L 5–518/17.004 (2013)) to M.G. and C.W., the Jose Carreras Leukemia Foundation (DJCLS R 12/28) to C.W., the Deutsche Forschungsgemeinschaft grants (SCHE 550/6-1, ED34/4-1) and the H.W.&J. Hector Stiftung (to M.S. and M.E.). The Georg-Speyer-Haus is funded jointly by the German Federal Ministry of Health (BMG) and the HMWK.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Zhang J, Hug BA, Huang EY, et al. Oligomerization of ETO is obligatory for corepressor interaction. Mol Cell Biol. 2001;21(1):156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sun XJ, Wang Z, Wang L, et al. A stable transcription factor complex nucleated by oligomeric AML1-ETO controls leukaemogenesis. Nature. 2013;500(7460):93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Y, Cheney MD, Gaudet JJ, et al. The tetramer structure of the Nervy homology two domain, NHR2, is critical for AML1/ETO’s activity. Cancer Cell. 2006;9(4):249–260. [DOI] [PubMed] [Google Scholar]
- 4.Wichmann C, Becker Y, Chen-Wichmann L, et al. Dimer-tetramer transition controls RUNX1/ETO leukemogenic activity. Blood. 2010; 116(4):603–613. [DOI] [PubMed] [Google Scholar]
- 5.Metz A, Schanda J, Grez M, Wichmann C, Gohlke H. From determinants of RUNX1/ETO tetramerization to small-molecule protein-protein interaction inhibitors targeting acute myeloid leukemia. J Chem Inf Model. 2013;53(9):2197–2202. [DOI] [PubMed] [Google Scholar]
- 6.Zhao X, Ghaffari S, Lodish H, Malashkevich VN, Kim PS. Structure of the Bcr-Abl oncoprotein oligomerization domain. Nat Struct Biol. 2002;9(2):117–120. [DOI] [PubMed] [Google Scholar]
- 7.Gardini A, Cesaroni M, Luzi L, et al. AML1/ETO oncoprotein is directed to AML1 binding regions and co-localizes with AML1 and HEB on its targets. PLoS Genet. 2008;4(11):e1000275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan M, Kanbe E, Peterson LF, et al. A previously unidentified alternatively spliced isoform of t(8;21) transcript promotes leukemogenesis. Nat Med. 2006;12(8):945–949. [DOI] [PubMed] [Google Scholar]
- 9.Wichmann C, Chen L, Heinrich M, et al. Targeting the oligomerization domain of ETO interferes with RUNX1/ETO oncogenic activity in t(8;21)-positive leukemic cells. Cancer Res. 2007;67(5):2280–2289. [DOI] [PubMed] [Google Scholar]
- 10.Zhen T, Wu CF, Liu P, et al. Targeting of AML1-ETO in t(8;21) leukemia by oridonin generates a tumor suppressor-like protein. Sci Transl Med. 2012;4(127):127ra138. [DOI] [PubMed] [Google Scholar]
- 11.Kaoud TS, Yan C, Mitra S, et al. From in Silico Discovery to intra-Cellular Activity: Targeting JNK-Protein Interactions with Small Molecules. ACS Med Chem Lett. 2012;3(9):721–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elsasser A, Franzen M, Kohlmann A, et al. The fusion protein AML1-ETO in acute myeloid leukemia with translocation t(8;21) induces c-jun protein expression via the proximal AP-1 site of the c-jun promoter in an indirect, JNK-dependent manner. Oncogene. 2003; 22(36):5646–5657. [DOI] [PubMed] [Google Scholar]
- 13.Gao FH, Wang Q, Wu YL, Li X, Zhao KW, Chen GQ. c-Jun N-terminal kinase mediates AML1-ETO protein-induced connexin-43 expression. Biochem Biophys Res Commun. 2007;356(2):505–511. [DOI] [PubMed] [Google Scholar]
- 14.Burgess GS, Williamson EA, Cripe LD, et al. Regulation of the c-jun gene in p210 BCR-ABL transformed cells corresponds with activity of JNK, the c-jun N-terminal kinase. Blood. 1998;92(7):2450–2460. [PubMed] [Google Scholar]
- 15.Raitano AB, Halpern JR, Hambuch TM, Sawyers CL. The Bcr-Abl leukemia oncogene activates Jun kinase and requires Jun for transformation. Proc Natl Acad Sci USA. 1995;92(25):11746–11750. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.