

Acute lymphoblastic leukemia (ALL) is the most common type of cancer in childhood and its incidence has risen steadily over the past decades. Growing evidence from epidemiological studies strongly suggests that the increased leukemia rate is likely related to an abnormal immune response to infections early in life.1,2 Recent experimental support for the hypothesis of “delayed infection” proposed by Mel Greaves as a cause of childhood leukemia came from Martin-Lorenzo et al. and Swaminathan et al. who demonstrated that exposure of genetically susceptible mice to infection can cause leukemia.3,4 Martin-Lorenzo et al. showed that mice with monoallelic loss of the B-cell transcription factor PAX5 are genetically predisposed to the development of precursor B-cell ALL, but develop leukemia only if they are exposed to common pathogens.3 Swaminathan et al. demonstrated that pre-leukemic ETV6-RUNX1 translocation bearing pre-B cells respond to bacterial lipopolysaccharide (in the absence of protective IL-7) with strong induction of gene recombination activating enzymes and accelerated mutagenesis in pre-leukemic cells, resulting in leukemia in mice.4
The identity of the infectious agent in ALL remained elusive, whereas much progress has been made in understanding the contribution of infection to aggressive B-cell lymphoma where Plasmodium falciparum, Epstein-Barr virus (EBV), Helicobacter pylori, and hepatitis C virus were identified as triggers of transformation.5,6
Viruses have been suggested to play a role in the pathogenesis of ALL. Transforming viruses may integrate into the genome of precursor B cells, disturbing differentiation and proliferation control.2 Alternatively, common pathogens may act indirectly, eliciting an unusual response in genetically and immunologically susceptible children, resulting in autonomous precursor B-cell proliferation.1
Previous attempts to identify candidate viruses by biased, low-throughput techniques were unsuccessful or detected known common human pathogens (reviewed in Table 1). Representational difference analysis (RDA), the most sensitive approach so far, achieved a 95% probability of detecting viral genomes larger than 9 kb. But many genomes of oncogenic viruses are smaller (eg. 5.3 kb Merkel cell polyoma virus or 3.5 kb Rous sarcoma virus genome), and low virus copy numbers, low tumor cell content and subclonality posed additional problems to the limited detection sensitivity of the techniques used.
Table 1.
Overview on previous virus detection studies in childhood acute lymphoblastic leukemia.

In contrast, high-throughput next generation sequencing (NGS) is a highly sensitive approach that has been proven to have the potential to detect known and novel viruses.21 A recent NGS-based study that attempted to identify viral integration sites in whole genome sequencing data of 10 patients with TCF3-rearranged childhood ALL has failed to detect any true virus/host chimeras.20 Therefore, we developed a novel bioinformatics pipeline for the detection of viral sequences in data derived from whole genome sequencing that is not restricted to the detection of rare integration sites (Online Supplementary Figure S1 and Online Supplementary Methods). First, five data sets derived from the 1000 genomes database (www.1000genomes.org) were used as test sets for the pipeline. More than 25,000 viral genomes deposited in the Genome Information Broker for Viruses database were tested. Probability estimations and in silico simulations demonstrated that the pipeline has a very high probability of detecting viral genomes (≥2 kb) and viral integration sites (Figure 1A and Online Supplementary Figure S2).
Figure 1.
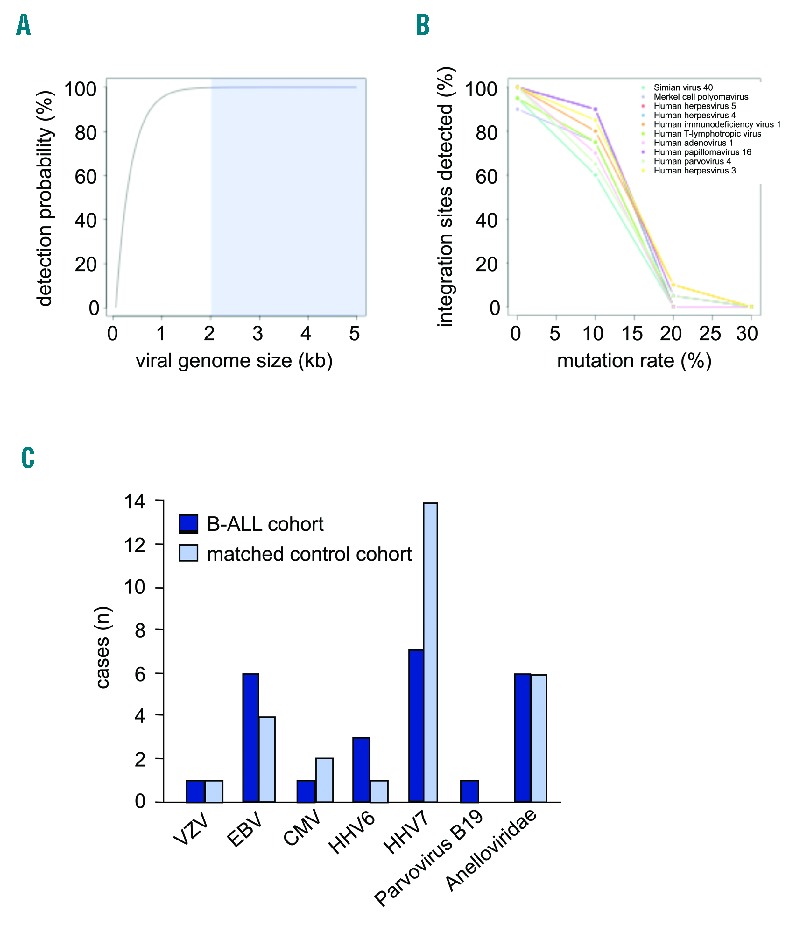
Common viruses were detected in B-precursor acute lymphoblastic leukemia (ALL) and healthy matched controls at approximately the same frequency. (A and B) Bioinformatic detection of integrated viruses and simulation of virus detectability in whole-genome sequencing data sets using the developed pipeline. (A) The probability to detect a virus increases with its genome size. The detection probability depending on genome size within 10,000,000 sequencing reads of a human genome is presented. The typical virus size of 2 kb or more is indicated (blue shaded box). (B) Viral genetic instability impairs detection only when the mutation rate exceeds 10%. Ten common viruses were selected for simulation experiments (Online Supplementary Table S1). The percentage of detected virus integration sites decreases with an increase in the mutation rate of the annotated virus sequences. For each virus, 20 integration sites were randomly distributed across the human chromosome 1. The respective wild-type virus sequences correspond to 0% mutation rate. Results for simulated paired end reads of 50 bp length and 5-fold sequencing depth are shown. (C) In ALL, viral DNA corresponding to common human pathogens (Anelloviridae, Herpesviridae, and Parvoviridae) was detected in 11 of 14 cases. A similar pattern was observed in the age- and sex-matched control cohort. Only HHV7 was more frequently detected in the control cohort (14 of 14 healthy cases compared to 7 of 14 ALL cases), but this was not the case in the adult healthy 1000 genomes test set (5 of 7 cases HHV-7 positive).
Compared to non-viral genomes, viruses have an increased mutagenesis rate allowing for greater genetic variability and rapid adaptation to changing environments that may be detrimental for sequence identification. We selected ten clinically relevant viruses: simian virus 40, Merkel cell polyoma virus, adenovirus-1, human papilloma virus-16, varicella zoster virus, EBV, cytomegalovirus, human immunodeficiency cirus, parvovirus 4, human T-lymphotropic virus-1 (Online Supplementary Table S1) ranging from 5.2 to 235.6 kb in genome size and simulated mutation rates of up to 30%. Alignment quality and number of aligned sequencing reads declined with an increasing mutation rate, but virus sequences and integration sites mutated by 10% were still well detectable (>60% for all simulated viruses) (Figure 1B and Online Supplementary Figure S3). Virus mutagenesis of 20% became detrimental for the analyses.
We used NGS combined with this pipeline to test whether viral DNA is detectable in 14 pediatric B-cell ALL cases. We chose ETV6-RUNX1-positive (n=7) and high hyperdiploid (n=7) ALL, because these two subtypes account for 50%–60% of all B-cell ALL cases. It is commonly acknowledged that both primary lesions (ETV6-RUNX1-translocation or high hyperdiploidy) are not sufficient to induce overt leukemia. Both subtypes have a long latency period after birth and infection has been discussed as a likely transforming trigger. We performed whole genome sequencing for diagnosis, remission and (if applicable) relapse samples. To this end, mononuclear cells were derived by Ficoll density centrifugation from bone marrow, DNA was isolated using standard protocols and sequencing was carried out (Online Supplementary Methods). On average, we generated 442 million sequencing reads per patient sample (Online Supplementary Table S2). Approximately 10% of these reads could not be mapped to the human reference and may potentially encode viral sequences. As a control data set, whole genome sequencing data (comparable in sequencing quality and coverage) of non-leukemic blood samples from 14 age- and sex-matched children were used [chosen from the International Cancer Genome Consortium (ICGC) PedBrain cohort; www.pedbraintumor.org] (Online Supplementary Table S3).
Viruses that are integrated into the genome of precursor B cells may directly promote leukemogenesis by acting on differentiation and proliferation. In this case, viral DNA should be persistent and detectable in the leukemic cells. Applying the developed bioinformatics pipeline to the leukemic patient samples, we detected viral DNA in 11 of 14 cases (Table 2 and Figure 1C). However, the detected virus sequences corresponded exclusively to known, common human pathogens (Anelloviridae, Herpesviridae, and Parvoviridae). A similar pattern was observed in age- and sex-matched controls. No evidence was found for the presence of other viruses.
Table 2.
Viruses detected in 14 sporadic cases of childhood B-precursor acute lymphoblastic leukemia (ALL) employing whole genome sequencing.

Integrated viruses that are truly essential for leukemic cell characteristics can be expected to be persistent at relapse. Persistence of viruses was analyzed in 6 patients for whom both diagnosis and relapse bone marrow was available. Five patients were positive for at least one virus, but none of these cases presented with the same virus at diagnosis and relapse.
Viruses may promote leukemogenesis indirectly by eliciting an abnormal immunological response resulting in autonomous B-cell precursor proliferation. In this scenario, virus DNA could persist during remission or be solely detectable in remission, in which case, widespread, low-pathogenic viruses may be seen. Similar to the results in leukemic samples, we detected only known common human pathogens in remission samples. The incidence of Anelloviridae increased from one case at diagnosis to 6 at remission; this is likely to have been due to contaminated blood transfusions. Many of the detected viruses were found only at remission and not in the leukemic samples of the same patient. In 3 patients, no viral DNA was identified at any time point.
Taken together, the analysis of 14 ALL cases, showed that only common human pathogens (Anelloviridae, Herpesviridae, and Parvoviridae) were detected in the majority of B-ALL cases and similar viruses and frequencies were observed in age- and sex-matched controls (Figure 1C and Online Supplementary Tables S4–S7). Direct alignment against the viral sequence database or alignment using a Blast-like tool did not alter this result (Online Supplementary Tables S8 and S9 and Online Supplementary Figure S3). However, non-integrating viruses and viruses working in a ‘hit-and-run’ mode would not be detected by our approach, and for low copy number viruses, detection on RNA level may be more sensitive.
Epidemiological studies suggested that the sought-after virus should persistently infect B lymphocytes, provoke minimal symptoms of primary infection, and have a prolonged viremia; for example, the transforming virus EBV detected in our study fulfills the suggested criteria. Infection can take place in utero and reactivation of maternal EBV infection has been associated with an increased risk of childhood ALL.22 Fetal progenitor and pre-B cells are susceptible to EBV transfection,23 although the EBV receptor CD21 (complement receptor 2) is more commonly known as a marker for mature B cells which can be aberrantly expressed on blast cells.24 Furthermore, early transplantation studies suggested that EBV is present in the bone marrow where quiescent pre-leukemic cells are thought to reside.25
By the age of three years, approximately 95% of children have become infected with minimal symptoms. Infection is usually latent, but reactivation can take place in immune-suppressed or genetically susceptible individuals. In keeping with this, reactivation of EBV has been identified as a major factor in post-transplant lymphoproliferative disease26 and knockdown of Ikaros (a master regulator of B-cell development similar to PAX5 and frequently deleted in B-ALL) or interference with its function as a transcription factor leads to reactivation of EBV.27
In the case of Hodgkin lymphoma, absence of viral genome sequences but strong evidence for viral pathogenesis was reconciled by a ‘hit-and-run’ mechanism.28 EBV DNA in an episomal (not integrated) form may initiate transformation, but is later lost. This could be a possible scenario in the case of human pre-B-ALL and accounts for a lower than expected rate of EBV-positive patient samples. Alternatively, cytomegalovirus, another common herpesvirus with transforming potential may play a role, as proposed in current studies.29
Our analyses of ALL cases suggest that the sought-after agent could be a common virus or other infectious agent (eg. transforming bacteria or unicellular organisms).
Supplementary Material
Acknowledgments
The authors wish to thank Monika Schmitz for data acquisition and assistance.
Footnotes
Funding: the study was funded by a grant of the Katharina-Hardt-Stiftung and the German “Kinderkrebsstiftung”. UF was supported by the “Deutsche Krebshilfe – Dr. Mildred Scheel Stiftung” (grant 108613) and two intramural grants (05/2014 and 2016–70) of the research commission of the medical faculty of the Heinrich-Heine-University Düsseldorf.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Greaves MF, Alexander FE. An infectious etiology for common acute lymphoblastic leukemia in childhood? Leukemia. 1993;7(3):349–360. [PubMed] [Google Scholar]
- 2.Kinlen LJ. Epidemiological evidence for an infective basis in childhood leukaemia. Br J Cancer. 1995;71(1):1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martin-Lorenzo A, Hauer J, Vicente-Duenas C, et al. Infection Exposure is a Causal Factor in B-cell Precursor Acute Lymphoblastic Leukemia as a Result of Pax5-Inherited Susceptibility. Cancer Disc. 2015;5(12):1328–1343. [DOI] [PubMed] [Google Scholar]
- 4.Swaminathan S, Klemm L, Park E, et al. Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nature Immunol. 2015;16(7):766–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kugelberg E. Tumour immunology: Malaria alters B cell lymphoma-genesis. Nat Rev Immunol. 2015;15(9):528. [DOI] [PubMed] [Google Scholar]
- 6.Foster LH, Portell CA. The role of infectious agents, antibiotics, and antiviral therapy in the treatment of extranodal marginal zone lymphoma and other low-grade lymphomas. Curr Treat Opt Oncol. 2015;16(6):28. [DOI] [PubMed] [Google Scholar]
- 7.Luka J, Pirruccello SJ, Kersey JH. HHV-6 genome in T-cell acute lymphoblastic leukaemia. Lancet. 1991;338:1277–1278. [DOI] [PubMed] [Google Scholar]
- 8.Bender AP, Robison LL, Kashmiri SV, et al. No involvement of bovine leukemia virus in childhood acute lymphoblastic leukemia and non-Hodgkin’s lymphoma. Cancer Res. 1988;48(10):2919–2922. [PubMed] [Google Scholar]
- 9.MacKenzie J, Perry J, Ford AM, Jarrett RF, Greaves M. JC and BK virus sequences are not detectable in leukaemic samples from children with common acute lymphoblastic leukaemia. Br J Cancer. 1999;81(5):898–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith MA, Strickler HD, Granovsky M, et al. Investigation of leukemia cells from children with common acute lymphoblastic leukemia for genomic sequences of the primate polyomaviruses JC virus, BK virus, and simian virus 40. Med Pediatr Oncol. 1999;33(5):441–443. [DOI] [PubMed] [Google Scholar]
- 11.Ma XT, Song YH, Lu DM, et al. Human herpesvirus 6 in hematologic diseases in China. Haematologica. 2000;85:458–463. [PubMed] [Google Scholar]
- 12.MacKenzie J, Gallagher A, Clayton RA, et al. Screening for herpesvirus genomes in common acute lymphoblastic leukemia. Leukemia. 2001;15(3):415–421. [DOI] [PubMed] [Google Scholar]
- 13.Shiramizu B, Yu Q, Hu N, Yanagihara R, Nerurkar VR. Investigation of TT virus in the etiology of pediatric acute lymphoblastic leukemia. Pediatr Hematol Oncol. 2002;19(8):543–551. [DOI] [PubMed] [Google Scholar]
- 14.Priftakis P, Dalianis T, Carstensen J, et al. Human polyomavirus DNA is not detected in Guthrie cards (dried blood spots) from children who developed acute lymphoblastic leukemia. Med Pediatr Oncol. 2003;40(4):219–223. [DOI] [PubMed] [Google Scholar]
- 15.Hermouet S, Sutton CA, Rose TM, et al. Qualitative and quantitative analysis of human herpesviruses in chronic and acute B cell lymphocytic leukemia and in multiple myeloma. Leukemia. 2003;17(1):185–195. [DOI] [PubMed] [Google Scholar]
- 16.Bogdanovic G, Jernberg AG, Priftakis P, Grillner L, Gustafsson B. Human herpes virus 6 or Epstein-Barr virus were not detected in Guthrie cards from children who later developed leukaemia. Br J Cancer. 2004;91(5):913–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Isa A, Priftakis P, Broliden K, Gustafsson B. Human parvovirus B19 DNA is not detected in Guthrie cards from children who have developed acute lymphoblastic leukemia. Pediatr Blood Cancer. 2004;42(4):357–360. [DOI] [PubMed] [Google Scholar]
- 18.Gustafsson B, Jernberg AG, Priftakis P, Bogdanovic G. No CMV DNA in Guthrie cards from children who later developed ALL. Pediatr Hematol Oncol. 2006;23(3):199–205. [DOI] [PubMed] [Google Scholar]
- 19.MacKenzie J, Greaves MF, Eden TO, et al. The putative role of transforming viruses in childhood acute lymphoblastic leukemia. Haematologica. 2006;91:240–243. [PubMed] [Google Scholar]
- 20.Forster M, Szymczak S, Ellinghaus D, et al. Vy-PER: eliminating false positive detection of virus integration events in next generation sequencing data. Sci Rep. 2015;5:11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang P, Chiu C. Metagenomics for the discovery of novel human viruses. Future Microbiol. 2008;5(2):177–189. [DOI] [PubMed] [Google Scholar]
- 22.Lehtinen M, Koskela P, Ogmundsdottir HM, et al. Maternal herpesvirus infections and risk of acute lymphoblastic leukemia in the offspring. Am J Epidem. 2003;158(3):207–213. [DOI] [PubMed] [Google Scholar]
- 23.Katamine S, Otsu M, Tada K, et al. Epstein-Barr virus transforms precursor B cells even before immunoglobulin gene rearrangements. Nature. 1984;309(5966):369–372. [DOI] [PubMed] [Google Scholar]
- 24.Campana D, Coustan-Smith E. Advances in the immunological monitoring of childhood acute lymphoblastic leukaemia. Best Pract & Res Clin Haematol. 2002;15(1):1–19. [DOI] [PubMed] [Google Scholar]
- 25.Gratama JW, Oosterveer MA, Zwaan FE, Lepoutre J, Klein G, Ernberg I. Eradication of Epstein-Barr virus by allogeneic bone marrow transplantation: implications for sites of viral latency. Proc Natl Acad Sci USA. 1988;85(22):8693–8696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dharnidharka VR, Webster AC, Martinez OM, Preiksaitis JK, Leblond V, Choquet S. Post-transplant lymphoproliferative disorders. Nat Rev Dis Primers. 2016;2:15088. [DOI] [PubMed] [Google Scholar]
- 27.Iempridee T, Reusch JA, Riching A, et al. Epstein-Barr virus utilizes Ikaros in regulating its latent-lytic switch in B cells. J Virol. 2014;88(9):4811–4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ambinder RF. Gammaherpesviruses and “Hit-and-Run” oncogenesis. Am J Pathol. 2000;156(1):1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Francis SS, Wallace AD, Wendt GA, et al. In utero cytomegalovirus infection and development of childhood acute lymphoblastic leukemia. Blood. 2016. DOI: 10.1182/blood-2016-07-723148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.