

Abstract
Betulinic acid (BA), a natural pentacyclic triterpene found in many medicinal plants is known to have various biological activity including tumor suppression and anti-inflammatory effects. In this study, the cell-death induction effect of BA was investigated in BV-2 microglia cells. BA was cytotoxic to BV-2 cells with IC50 of approximately 2.0 μM. Treatment of BA resulted in a dose-dependent chromosomal DNA degradation, suggesting that these cells underwent apoptosis. Flow cytometric analysis further confirmed that BA-treated BV-2 cells showed hypodiploid DNA content. BA treatment triggered apoptosis by decreasing Bcl-2 levels, activation of capase-3 protease and cleavage of PARP. In addition, BA treatment induced the accumulation of p62 and the increase in conversion of LC3-I to LC3-II, which are important autophagic flux monitoring markers. The increase in LC3-II indicates that BA treatment induced autophagosome formation, however, accumulation of p62 represents that the downstream autophagy pathway is blocked. It is demonstrated that BA induced cell death of BV-2 cells by inducing apoptosis and inhibiting autophagic flux. These data may provide important new information towards understanding the mechanisms by which BA induce cell death in microglia BV-2 cells.
Keywords: Betulinic acid, Apoptosis, Autophagy, Microglia BV-2 cell
INTRODUCTION
Microglia has multiple functions in regulating homeostasis of the central nervous systems (CNS). Microglia is a resident immunological cell in the CNS and participate in both innate and adaptive immune responses. Microglia cells have been implicated as active contributors to neuron damage in neurodegenerative disorders such as Alzheimer’s disease, Parkinson’s disease and multiple sclerosis (Block et al., 2007; Saijo and Glass, 2011). Recently, several studies have reported the involvement of apoptosis and autophagy in microglia-induced neurotoxicity (Chen et al., 2016; Su et al., 2016).
Apoptosis, also known as type I programmed cell death, is a selective physiological process that plays an important role in the balance between cell replication and cell death. A wide range of stimuli can be integrated to trigger the irreversible decision to die. Most cytotoxic and neurotoxic agents cause cell death by apoptosis (Foo et al., 2015; Liu et al., 2015b). The advantage of apoptosis-inducing agents is the elimination of potentially harmful cells without causing inflammation. Autophagy is essential for cell survival and the maintenance of homeostasis. There have been reports showing that autophagy is involved in the degradation of unnecessary or defective cellular components in the lysosome (Levine and Kroemer, 2008; Mizushima and Komatsu, 2011; Mochida et al., 2015). It is also reported that autophagy plays a critical role in the progression of certain human disorders, including neurodegenerative disease and cancer (Levine and Kroemer, 2008). Recent studies indicate that autophagy also functions in cell death, and it is called type II programmed cell death (Baehrecke, 2005). Growing evidences suggest the inter-relationship between apoptosis and autophagy in controlling cell survival and cell death (Mukhtar et al., 2012; Mukhopadhyay et al., 2014).
Betulinic acid (3-beta-hydroxy-lup-20(29)-ene-28-oic acid, BA) is a natural pentacyclic triterpene that can be isolated from various plants including white birch bark (Yogeeswari and Sriram, 2005, Sami et al., 2006). BA has been reported to have a variety of biological and pharmacological activities: tumor suppression (Periasamy et al., 2014; Zhang et al., 2016), anti-inflammatory effect (Kim et al., 2016), and anti-parasitic activity (Meira et al., 2016).
It has been reported that microglia is involved in the signaling cascade that is associated with neuronal cell death in various neurological diseases such as Alzheimer disease, Parkinson disease and traumatic brain injury (Block et al., 2007; Gao and Hong, 2008; Loane et al., 2009; Lee and Jeong, 2014; Stoica et al., 2014). It was reported that microglia is involved in apoptosis of other cells such as pheochromocytoma cells (Hornik et al., 2016). Some efforts were made to find a small molecule or an extract that modulate microglia cells thus protect microglia or other neuron cells (Lee and Jeong, 2014; Li et al., 2014; Wang et al., 2014; Liu et al., 2015a). In addition, elucidation of microglial cell death induction mechanism caused by small molecules were reported (Liu et al., 2015b; Yu et al., 2015). Hence studies in the modulation of microglia cell survival and death could be useful in formulating a strategy for the treatment of neurological diseases. In this study, we evaluated the cell death induction effect of BA in BV-2 microglia cells. BV-2 cells are common microglial cells that have been widely used in studies of neuro-protective effect and neurotoxicity (Kwon et al., 2012; Hao et al., 2013; Li et al., 2014; Liu et al., 2015b). We report that BA inhibited cell proliferation and caused cell death by inducing apoptosis and inhibiting autophagic flux in BV-2 cells.
MATERIALS AND METHODS
Materials
The following chemicals were obtained from Sigma-Aldrich, St. Louis, USA: Betulinic acid (BA) dimethyl sulfoxide (DMSO), Nondet P-40 (NP-40), phenol solution and ethidium bromide. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) was purchased from Duchefa, Haarlem, Netherland. Ethanol, isopropanol and isoamyl alcohol were obtained from Hayman Chemical Co., Witham, UK, Merck Millipore, Darmstadt, Germany and Junsei, Tokyo, Japan, respectively. Nuclear Isolation Medium-4,6-diamidino-2-phenylindole dihydrochloride (NIM-DAPI) was purchased from Beckman Coulter, Brea, USA. Bradford protein assay dye reagent was purchased from Bio-Rad, Hercules, USA. Chemicals including NaF, ethylene diamine tetra acetic acid (EDTA) and Na3VO4 were purchased form Sigma Aldrich, and protease Inhibitor was obtained from Roche diagnostics, Basel, Switzerland. Polyvinylidene difluoride (PVDF) membrane was purchased from Merck Millipore. Cell culture materials were purchased from Welgene, Gyeongsan, Korea and Gibco BRL, Waltham, USA. All other chemicals were of the highest analytical grade and purchased from common sources.
Rabbit polyclonal anti-Bcl-2, rabbit polyclonal anti-caspase-3, and rabbit polyclonal anti-human anti-Bax and anti-PARP, rabbit polyclonal anti-LC3 and rabbit polyclonal anti-p62 were purchased from Cell signaling technology, Danvers, USA. Mouse polyclonal anti-α-tubulin and mouse polyclonal anti-vinculin were obtained from Santa Cruz biotechnology, Oregon, USA and Sigma-Aldrich, respectively. Anti-mouse and anti-rabbit horseradish peroxidase (HRP)-conjugated secondary IgG antibodies were from Bethyl, Montgomery, USA. West Pico Chemiluminescent substrate solution was purchased from Thermo scientific, Waltham, USA.
Cell culture BA treatment
Mouse microglia BV-2 cells were maintained in the logarithmic phase of growth in Dulbecco’s modified Eagle’s medium (DMEM) (Welgene) supplemented with 5% fetal bovine serum (FBS, Gibco BRL), 2 mM L-glutamine, and antibiotics. Cultures were maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2. Logarithmically growing BV-2 cells were used for all experiments. Betulanic acid (BA) was dissolved in DMSO at the concentration of 20 mM and diluted in tissue culture medium before use. For glucose starvation experiment, BV-2 cells were cultured in glucose free DMEM (Welgene) supplemented with 10% FBS (Gibco BRL), 2 mM L-glutamine, and antibiotics for 16 hrs.
Cytotoxicity analysis and morphology observation
Cell viability was estimated by the MTT assay. Exponentially growing cells were seeded at 3×104 cells/well in a 96-well plate and treated with various concentrations of BA. After the cells were incubated for 20 hrs, 20 μl of MTT (5 mg/ml) was added and the cells were incubated for another 4 hrs at 37°C. The supernatant was discarded and 150 μl of dissolving solvent (4 mM HCl and 0.1% NP-40 in isopropanol) was added. The plate was gently agitated until the blue formazan crystals were fully dissolved. The absorbance was measured at 550 nm using a microplate reader (Wallac Victor 3-V, Perkinelmer, USA). The data were expressed as a mean percentage of viable cells as compared to the respective control cultures. All experiments were performed at least in triplicate.
Morphology observation
Cells used in this study were constantly observed under an inverted phase-contrast microscope (Primo Vert, Zeiss, Oberkochen, Germany). Photographs were taken after BV-2 cells were incubated with various concentrations of BA for 24 hrs as described in the text and figure legend.
DNA fragmentation analysis
Cells were grown at a density of 8×105 cells/ml and exposed to BA at different concentrations as described in the text and figure legends. Cells were rinsed with ice-cold phosphate buffered saline (PBS), centrifuged and resuspended in 0.01 vol. of TE buffer (10 mM Tris-Cl, 1 mM EDTA, pH 8.0). DNA was purified as previously described (Hyun et al., 1997). The resulting purified DNA fragments were subjected to electrophoresis on 1.5% agarose gel. DNA bands were visualized by fluorescence after ethidium bromide staining, and quantified with a densitometer (Ultra-Lum Imaging System, San Diego, USA). Results shown are an example from 3 different experiments.
Flow cytometry
The effects of BA on cell proliferation were evaluated by measuring the distribution of the cells in the different phases of the cell cycle by flow cytometry. Cells were treated with BA at various concentrations and harvested by centrifugation at 750×g for 5 min. Cell pellets were rinsed with PBS and re-suspended in the staining solution containing DAPI (NIM-DAPI, 10 μg/ml, Beckman coulter). The cell suspensions were incubated at room temperature for 10 min in the dark and analyzed on a fluorescence-activated cell sorter flow cytometer (Quanta SC, Beckman coulter). All experiments were performed at least in triplicate.
Western blot analysis
BV-2 cells were treated with BA and subjected to western blot analysis. BV-2 cells were exposed to various concentrations of BA for 24 hrs or 16 hrs to analyze apoptosis-related or autophagy-related proteins, respectively. Cells were lysed in a lysis buffer (20 mM Tris, 100 mM NaCl, 0.1% NP40, 50 mM NaF, 2 mM EDTA, 1 mM Na3VO4 and protease inhibitor, pH 7.5) and protein concentrations were determined by Bradford assay. Lysis buffer L (1% triton X-100, 50 mM NaF in phosphate buffered saline) was used for the analysis of autophagy-related proteins. The total protein (5 or 10 μg) in each lysate were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electro-transferred onto PVDF membranes. The membranes were blocked with 5% non-fat milk for 1 hour at room temperature and then probed with specific primary antibody for 16 hours at 4°C. The specific protein bands were visualized by peroxidase-conjugated secondary antibody and chemiluminescent substrate solution.
RESULTS
BA inhibited proliferation of BV-2 cells
The chemical structure of test compound, BA is shown in Fig. 1. The effect of BA on cellular proliferation was evaluated using the MTT assay. A 24 hr exposure to BA dramatically decreased the proliferation of BV-2 cells in a dose-dependent manner (Fig. 2A). The concentration required to inhibit growth by 50% (IC50) was approximately 2.0 μM. Relative cell survival was also assessed at various times after exposure to 2.0 μM of BA. Prolonged exposure to BA markedly decreased the viability of these cells (data not shown). Control cells treated with vehicle alone showed no changes in cell proliferation or viability.
Fig. 1.
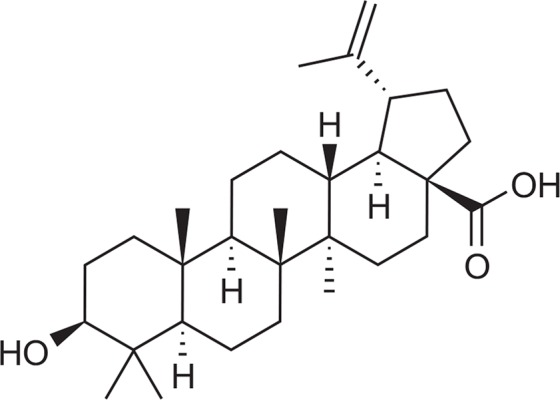
Structure of betulinic acid (BA).
Fig. 2.

Antiproliferative effect of BA in BV-2 cells. (A) Cells were incubated with indicated concentrations of BA for 24 hrs and cell survival was measured by the MTT assay. (B) Morphological changes in BV-2 cells in response to BA treatment. Cells were incubated with indicated concentrations of BA for 24 hrs. Cell morphology was observed under a phase-contrast microscope (×200 magnification) after BA treatment.
Morphological changes were observed using a phase contrast microscope. Treatment of various concentrations of BA for 24 hrs in BV-2 cells resulted in reduction of live cell numbers and morphology changes (Fig. 2B). The morphology change includes rounding, detachment and cell shrinking which are distinct morphological characteristics associated with apoptotic cells.
Induction of apoptosis by BA treatment
To determine whether BA-meditated inhibition of growth and proliferation were associated with apoptosis, the BA-induced chromosomal DNA degradation and appearance of DNA fragmentation in BV-2 cells were examined. As shown in Fig. 3, cells treated with BA showed significant degradation of chromosomal DNA and appearance of DNA fragmentation. When we measured the chromosomal DNA content after 24 hr of various concentrations of BA treatment, approximately 8% and 26% of chromosomal DNA were degraded at 2 μM and 6 μM of BA treatment, respectively (Fig. 3B).
Fig. 3.

Induction of chromosomal DNA degradation in BA-treated BV-2 cells. (A) Cells were treated with BA at indicated concentrations for 24 hrs and harvested. Chromosomal DNA was extracted and subjected to electrophoresis on 1.5% agarose gels followed by ethidium bromide staining. (B) Density of each bands were measured and plotted as relative content to the control.
The induction of apoptotic bodies in BA-treated BV-2 cells was further analyzed by flow-cytometric determination of DNA content (Fig. 4). Histograms of DNA content obtained from DAPI-stained BV-2 cells showed that the percentage of cells with reduced DNA content progressively increased as the treatment dose increased. Apoptosis was negligible up to 4 μM of BA treatment. However, the percentage of apoptotic cells (cells in sub-G1) increased to 3.7 and 7.1% at 10 and 15 μM of BA treatment, respectively. The profile for the BA-induced increase in cells with hypodiploid DNA content closely correlated with the results obtained with the chromosomal DNA degradation. In parallel to the increase in the number of cells with sub-G1 hypodiploid DNA content, there was a decrease in the number of cells with diploid DNA content.
Fig. 4.

Apoptotic bodies were induced by BA treatment. (A) BV-2 cells were treated with various concentrations BA for 24 hrs. Cells were stained with DAPI and analyzed by flow cytometry. (B) Relative number of sub-G1 apoptotic cells in BA-treated cells was plotted.
Effect of BA treatment on Bcl-2 level and caspase-3 activation
In order to investigate the mechanism by which BA induces apoptosis, we examined the expression levels of various apoptosis-related proteins. BV-2 cells were cultured in media containing 0–6 μM of BA for 24 hrs. Total proteins were isolated and Bax, caspase-3, and PARP [poly(ADP-ribosyl)polymerase] immunoreactivity levels were measured by western blotting. As shown in Fig. 5, western blot analysis revealed that BA treatment decreased the levels of Bcl-2 protein, an important regulator of apoptotic signaling pathways (Reed, 1998). No significant change in the level of pro-apoptotic protein, Bax was observed. We also found that BA induced the proteolytic processing of caspase-3 in dose-dependent manner. Activation of caspase-3 led to the cleavage of a number of proteins, one of which is poly (ADP-ribose) polymerase (PARP). Although PARP is not essential for cell death, the cleavage of PARP is another hallmark of apoptosis. BA treatment also induced a dose-dependent proteolytic cleavage of PARP, with concomitant accumulation of the 89 kDa form and the disappearance of the full-size 116 kDa molecule (Fig. 5). Taken together, these findings suggest that BA induced apoptosis through the down-regulation of Bcl-2 and the activation of caspase-3.
Fig. 5.

Changes in the expression of apoptosis-related proteins in response to BA treatment. BV-2 cells were treated with various concentrations of BA for24 hrs. Cell extracts were subjected to western blotting to determine immunoreactivity levels of Bcl-2, Bax, caspase-3, and PARP. Representative western blots are shown.
Effect of BA treatment on autophagic flux
To further confirm the cell death mechanism mediated by BA in BV-2 cells, changes in the expression levels of the protein in autophagy induction pathways were investigated. In order to examine the autophagic flux, the conversion of microtubule-associated protein light chain 3 (LC3)-I to phosphatudylethanolamine (PE) conjugate form (LC3-II), and p62 expression were observed.
BA treatment increases in the conversion of LC3-1 to LC3-II in dose-dependent manner within BV-2 cells (Fig. 6). On the other hand, the expression level of p62 was increased, suggesting that p62 was not degraded but accumulated (Fig. 6). As a positive control, cells were starved with glucose and the changes in the level of p62 and the conversion of LC3-I to LC3-II form were monitored (Fig. 6B). Under the glucose starvation condition the level of p62 was decreased and the the level of LC3-II was increased which are the well-known characteristic of autophagy induction (Kim et al., 2013). These results indicate that BA treatment induced the accumulation of LC3-II, which represents the increase in the number of autophagosome. However, there was no concomitant degradation of p62 was not occurred. Rather, p62 accumulation was observed. These data suggested that BA treatment inhibited the autophagic flux in BV-2 cells.
Fig. 6.

Change in the expression of autophagy-related protein in response to BA treatment. (A) BV-2 cells were treated with various concentrations of BA for 16 hrs in a complete medium. (B) BV-2 cells were cultured either in a complete medium or in a glucose-free medium. Cell extracts were subjected to western blotting to determine immunoreactivity levels of p62 and LC3. Representative western blots are shown.
DISCUSSION
The importance of natural products in drug discovery have been emphasized (Rosén et al., 2009; Hong, 2011). Natural products have been good source for new drug development and discovery in various diseases (Butler, 2008). Natural products and their molecular framework also have been used in medicinal chemistry for drug design for the discovery of new drugs (Rodrigues et al., 2016). Recently, many attempts have been made to find a new therapeutics of neurological diseases (Butler, 2008; Choi et al., 2011; Gu et al., 2014). BA is a natural product that can be found in many medicinal plants and contains many favorable biological activities (Periasamy et al., 2014).
In this study we evaluated the cell death inducing effect in microglial BV-2 cells. BA showed cell proliferation inhibition effect with IC50 of 2 μM. The alterations in cell morphology, the fragmentation of chromosomal DNA, and the appearance of sub-G1 hypodiploid cells in flow cytometry analysis all indicate that BA induced apoptosis in BV-2 cells. To further analyze the molecular mechanism by which BA causes cell death, we evaluated the level of proteins in apoptosis pathway.
Apoptosis is morphologically characterized by cellular shrinkage, chromatin condensation, and nuclear fragmentation. During apoptosis, double strand cleavage occurs at the linker regions between nucleosomes to produce DNA fragments which develop characteristic DNA ladder pattern on agarose gels (Wyllie, 1980; Arends et al., 1990). The appearance of chromosomal DNA fragmentation pattern seems different in various cell types. In our data the pattern of chromosomal DNA fragmentation in BV-2 cells was rather smeary degraded bands than discrete DNA ladder bands. Similar observations were made with dehydroepiandrosterone-treated BV-2 cells and primary neuronal cells (Vogel et al., 1997; Yang et al., 2000).
In our study, it was observed that caspase-3 was activated and the caspase substrate PARP was proteolytically cleaved to low molecular weight fragments in BA-treated BV-2 cells. In addition to caspase-3 activation, the level of Bcl-2, an anti-apoptotic protein, was decreased while the level of Bax, a pro-apoptotic protein, remained constant upon treatment of BA, resulting in a decrease in the ratio of Bcl-2/Bax, one of the major events that regulate apoptosis (Oltvai and Korsmeyer, 1994). Similar observation was reported that BA or BA derivative-treated human cancer cells in the induction of decrease in the level of Bcl-2 and PARP cleavage by caspase activation (Li et al., 2010; Khan et al., 2016). It was obvious that BA caused cell-death through the induction of apoptosis. However, the extend of cell death caused by apoptosis alone could not explain the cell death data observed with MTT. Therefore, we tried to find other cell death inducing mechanism that is involved in BA-induced BV-2 cell death.
Autophagy is the major lysosomal intracellular degradation system that involves the delivery of cytoplasmic cargo to the lysosome. Autophagy is essential in survival, differentiation, development and homeostasis (Levine and Kroemer, 2008; Mizushima and Komatsu, 2011). Autophagy can be activated in response to various cellular and environmental stress conditions to promote cell survival or cell death (Baehrecke, 2005). One of the best characterized proteins in autophagy pathway is p62 (also known as sequestome 1/SQSTM1) which is unbiquitously expressed. p62 interacts with LC3 and subsequently, it is incorporated into the autophagosome and degraded in autophagy pathway (Mizushima and Komatsu, 2011). During autophagy process, a cytosolic form of LC3 (LC3-I) is conjugated to PE to form LC-3-PE conjugate (LC3-II). The conversion of LC3-I to LC3-II is used as autophagosome marker for autophagy monitoring (Mizushima and Yoshimori, 2007; Tanida et al., 2008; McLeland et al., 2011). It is suggested that comparison of the amount of LC3-II between samples is more important than the comparison of the LC3-1/LC3-II ratio (Mizushima and Yoshimori, 2007). Autophagosome accumulation may indicate the induction of autophagy and, at the same time, may represent the increased generation of autophagosome, and a block in autophagosome maturation and the completion of autophagy pathway (Mizushima et al., 2010). If there is an autophagy induction, the increase in the conversion of LC3-I to LC3-II and the decrease in p62 are expected. It is known that p62 is selectively incorporated into autophagosome through binding to LC3 and degraded by autophagy (Mizushima and Komatsu, 2011). It is also reported that the accumulation of LC3-II form does not always indicate the induction of autophagy. It may be accumulated by blocking of the downstream steps (Mizushima et al., 2010).Our data showed the accumulation of p62 and the increase in LC3-II upon BA-treatment. However, under the glucose starvation condition, that is known to induce autophagy (Kim et al., 2013), the level of p62 decreased and the level of LC3-II was increased. These data indicate that the accumulation of autophagosome without fusion with lysosome and subsequent degradation of p62, hence suggesting the inhibition of autophagy flux. It seemed that the inhibition of autophagic flux contributed to cell death in BA-treated BV-2 cells.
In conclusion, we demonstrated that BA caused cell death in microglia BV-2 cells by inducing apoptosis and inhibiting autophagic flux. BA-treatment inhibited cell proliferation and, induced chromosomal DNA fragmentation and appearance of sub-G1 hypodiploid cells. Induction of apoptosis was through the decrease in anti-apoptotic Bcl-2 and the activation of caspase-3. Autophagic flux inhibition was shown through the accumulation of p62 and increase in the conversion of LC3-I to LC3-II.
Acknowledgments
This study was supported by a grant from BIO & Medical Technology Development Program of National Research Foundation of Korea grant (MEST, 2012M3A9C6049936).
REFERENCES
- Arends MJ, Morris RG, Wyllie AH. Apoptosis. The role of the endonuclease. Am J Pathol. 1990;136:593–608. [PMC free article] [PubMed] [Google Scholar]
- Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6:505–510. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Butler MS. Natural products to drugs: natural product-derived compounds in clinical trials. Nat Prod Rep. 2008;25:475–516. doi: 10.1039/b514294f. [DOI] [PubMed] [Google Scholar]
- Chen CM, Wu CT, Yang TH, Chang YA, Sheu ML, Liu SH. Green tea catechin prevents hypoxia/reperfusion-evoked oxidative stress-regulated autophagy-activated apoptosis and cell death in microglial cells. J Agric Food Chem. 2016;64:4078–4085. doi: 10.1021/acs.jafc.6b01513. [DOI] [PubMed] [Google Scholar]
- Choi DK, Koppula S, Suk K. Inhibitors of microglial neurotoxicity: focus on natural products. Molecules. 2011;16:1021–1043. doi: 10.3390/molecules16021021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo JB, Saiful Yazan L, Tor YS, Wibowo A, Ismail N, How CW, Armania N, Loh SP, Ismail IS, Cheah YK, Abdullah R. Induction of cell cycle arrest and apoptosis by betulinic acid-rich fraction from Dilleniasuffruticosa root in MCF-7 cells involved p53/p21 and mitochondrial signalling pathway. J Ethnopharmacol. 2015;166:270–278. doi: 10.1016/j.jep.2015.03.039. [DOI] [PubMed] [Google Scholar]
- Gao HM, Hong JS. Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 2008;29:357–365. doi: 10.1016/j.it.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Chen J, Shen J. Herbal medicines for ischemic stroke: combating inflammation as therapeutic targets. J Neuroimmune Pharmacol. 2014;9:313–339. doi: 10.1007/s11481-014-9525-5. [DOI] [PubMed] [Google Scholar]
- Hao F, Zhang NN, Zhang DM, Bai HY, Piao H, Yuan B, Zhu HY, Yu H, Xiao CS, Li AP. Chemokine fractalkine attenuates overactivation and apoptosis of BV-2 microglial cells induced by extracellular ATP. Neurochem Res. 2013;38:1002–1012. doi: 10.1007/s11064-013-1010-7. [DOI] [PubMed] [Google Scholar]
- Hornik TC, Vilalta A, Brown GC. Activated microglia cause reversible apoptosis of pheochromocytoma cells, inducing their cell death by phagocytosis. J Cell Sci. 2016;129:65–79. doi: 10.1242/jcs.174631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J. Natural product diversity and its role in chemical biology and drug discovery. Curr Opin Chem Biol. 2011;15:350–354. doi: 10.1016/j.cbpa.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun SJ, Yoon MY, Kim TH, Kim JH. Enhancement of mitogen-stimulated proliferation of low dose radiation-adapted mouse splenocytes. Anticancer Res. 1997;17:225–229. [PubMed] [Google Scholar]
- Khan I, Guru SK, Rath SK, Chinthakindi PK, Singh B, Koul S, Bhushan S, Sangwan PL. A novel triazole derivative of betulinic acid induces extrinsic and intrinsic apoptosis in human leukemia HL-60 cells. Eur J Med Chem. 2016;108:104–116. doi: 10.1016/j.ejmech.2015.11.018. [DOI] [PubMed] [Google Scholar]
- Kim KS, Lee DS, Kim DC, Yoon CS, Ko W, Oh H, Kim YC. Anti-inflammatory effects and mechanisms of action of coussaric and betulinic acids isolated from Diospyros kaki in lipopolysaccharide-stimulated RAW 264.7 macrophages. Molecules. 2016;21:1206. doi: 10.3390/molecules21091206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152:290–303. doi: 10.1016/j.cell.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon OW, Moon E, Chari MA, Kim TW, Kim AJ, Lee P, Ahn KH, Kim SY. A substituted 3,4-dihydropyrimidinone derivative (compound D22) prevents inflammation mediated neurotoxicity; role in microglial activation in BV-2 cells. Bioorg Med Chem Lett. 2012;22:5199–5203. doi: 10.1016/j.bmcl.2012.06.082. [DOI] [PubMed] [Google Scholar]
- Lee DS, Jeong GS. Arylbenzofuran isolated from Dalbergiaodorifera suppresses lipopolysaccharide-induced mouse BV2 microglial cell activation, which protects mouse hippocampal HT22 cells death from neuroinflammation-mediated toxicity. Eur J Pharmacol. 2014;728:1–8. doi: 10.1016/j.ejphar.2013.12.041. [DOI] [PubMed] [Google Scholar]
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, He K, Huang Y, Zheng D, Gao C, Cui L, Jin YH. Betulin induces mitochondrial cytochrome c release associated apoptosis in human cancer cells. Mol Carcinog. 2010;49:630–640. doi: 10.1002/mc.20638. [DOI] [PubMed] [Google Scholar]
- Li Z, Hu H, Lin R, Mao J, Zhu X, Hong Z, Tao J, Zhang Y, Chen L. Neuroprotective effects of Gua Lou GuiZhi decoction against glutamate-induced apoptosis in BV-2 cells. Int J Mol Med. 2014;33:597–604. doi: 10.3892/ijmm.2013.1612. [DOI] [PubMed] [Google Scholar]
- Liu J, Huang D, Xu J, Tong J, Wang Z, Huang L, Yang Y, Bai X, Wang P, Suo H, Ma Y, Yu M, Fei J, Huang F. Tiagabine protects dopaminergic neurons against neurotoxins by inhibiting microglial activation. Sci Rep. 2015a;5:15720. doi: 10.1038/srep15720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Song G, Zou C, Liu G, Wu W, Yuan T, Liu X. Acrylamide induces mitochondrial dysfunction and apoptosis in BV-2 microglial cells. Free Radic Biol Med. 2015b;84:42–53. doi: 10.1016/j.freeradbiomed.2015.03.013. [DOI] [PubMed] [Google Scholar]
- Loane DJ, Stoica BA, Pajoohesh-Ganji A, Byrnes KR, Faden AI. Activation of metabotropic glutamate receptor 5 modulates microglial reactivity and neurotoxicity by inhibiting NADPH oxidase. J Biol Chem. 2009;284:15629–15639. doi: 10.1074/jbc.M806139200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeland CB, Rodriguez J, Stern ST. Autophagy monitoring assay: qualitative analysis of MAP LC3-I to II conversion by immunoblot. Methods Mol Biol. 2011;697:199–206. doi: 10.1007/978-1-60327-198-1_21. [DOI] [PubMed] [Google Scholar]
- Meira CS, Barbosa-Filho JM, Lanfredi-Rangel A, Guimarães ET, Moreira DR, Soares MB. Antiparasitic evaluation of betulinic acid derivatives reveals effective and selective anti-Trypanosoma cruzi inhibitors. Exp Parasitol. 2016;166:108–115. doi: 10.1016/j.exppara.2016.04.007. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida K, Oikawa Y, Kimura Y, Kirisako H, Hirano H, Ohsumi Y, Nakatogawa H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature. 2015;522:359–362. doi: 10.1038/nature14506. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Panda PK, Sinha N, Das DN, Bhutia SK. Autophagy and apoptosis: where do they meet? Apoptosis. 2014;19:555–566. doi: 10.1007/s10495-014-0967-2. [DOI] [PubMed] [Google Scholar]
- Mukhtar E, Adhami VM, Khan N, Mukhtar H. Apoptosis and autophagy induction as mechanism of cancer prevention by naturally occurring dietary agents. Curr. Drug Targets. 2012;13:1831–1841. doi: 10.2174/138945012804545489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltvai ZN, Korsmeyer SJ. Checkpoints of dueling dimers foil death wishes. Cell. 1994;79:189–192. doi: 10.1016/0092-8674(94)90188-0. [DOI] [PubMed] [Google Scholar]
- Periasamy G, Teketelew G, Gebrelibanos M, Sintayehu B, Gebrehiwot M, Karim A, Geremedhin G. Betulinic acid and its derivatives as anti-cancer agent: a review. Arch Appl Sci Res. 2014;6:47–58. [Google Scholar]
- Reed JC. Bcl-2 family proteins. Oncogene. 1998;17:3225–3226. doi: 10.1038/sj.onc.1202591. [DOI] [PubMed] [Google Scholar]
- Rodrigues T, Reker D, Schneider P, Schneider G. Counting on natural products for drug design. Nat Chem. 2016;8:531–541. doi: 10.1038/nchem.2479. [DOI] [PubMed] [Google Scholar]
- Rosén J, Gottfries J, Muresan S, Backlund A, Oprea T. Novel chemical space exploration via natural products. J Med Chem. 2009;52:1953–1962. doi: 10.1021/jm801514w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol. 2011;11:775–787. doi: 10.1038/nri3086. [DOI] [PubMed] [Google Scholar]
- Sami A, Taru M, Salme K, Jari Y-K. Pharmavological properties of the ubiquitous natural prduct betulin. Eur J Pharm Sci. 2006;29:1–13. doi: 10.1016/j.ejps.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Stoica BA, Loane DJ, Zhao Z, Kabadi SV, Hanscom M, Byrnes KR, Faden AI. PARP-1 inhibition attenuates neuronal loss, microglia activation and neurological deficits after traumatic brain injury. J Neurotrauma. 2014;31:758–772. doi: 10.1089/neu.2013.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su P, Zhang J, Wang D, Zhao F, Cao Z, Aschner M, Luo W. The role of autophagy in modulation of neuroinflammation in microglia. Neuroscience. 2016;319:155–167. doi: 10.1016/j.neuroscience.2016.01.035. [DOI] [PubMed] [Google Scholar]
- Tanida I, Ueno T, Kominami E. LC3 and autophagy. Methods Mol Biol. 2008;445:77–88. doi: 10.1007/978-1-59745-157-4_4. [DOI] [PubMed] [Google Scholar]
- Vogel P, Dux E, Wiessner C. Evidence of apoptosis in primary neuronal cultures after heat shock. Brain Res. 1997;764:205–213. doi: 10.1016/S0006-8993(97)00458-7. [DOI] [PubMed] [Google Scholar]
- Wang C, Xie N, Zhang H, Li Y, Wang Y. Puerarin protects against β-amyloid-induced microglia apoptosis via a PI3K-dependent signaling pathway. Neurochem Res. 2014;39:2189–2196. doi: 10.1007/s11064-014-1420-1. [DOI] [PubMed] [Google Scholar]
- Wyllie AH. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 1980;284:555–556. doi: 10.1038/284555a0. [DOI] [PubMed] [Google Scholar]
- Yang NC, Jeng KC, Ho WM, Chou SJ, Hu ML. DHEA inhibits cell growth and induces apoptosis in BV-2 cells and the effects are inversely associated with glucose concentration in the medium. J Steroid Biochem Mol Biol. 2000;75:159–166. doi: 10.1016/S0960-0760(00)00180-1. [DOI] [PubMed] [Google Scholar]
- Yogeeswari P, Sriram D. Betulinic acid and its derivatives: a review on their biological properties. Curr Med Chem. 2005;12:657–666. doi: 10.2174/0929867053202214. [DOI] [PubMed] [Google Scholar]
- Yu DS, Lv G, Mei XF, Cao Y, Wang YF, Wang YS, Bi YL. MiR-200c regulates ROS-induced apoptosis in murine BV-2 cells by targeting FAP-1. Spinal Cord. 2015;53:182–189. doi: 10.1038/sc.2014.185. [DOI] [PubMed] [Google Scholar]
- Zhang X, Hu J, Chen Y. Betulinic acid and the pharmacological effects of tumor suppression (review). Mol Med Rep. 2016;14:4489–4495. doi: 10.3892/mmr.2016.5792. [DOI] [PubMed] [Google Scholar]