

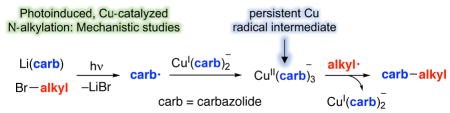
Abstract
We have recently reported that a variety of couplings of nitrogen, sulfur, oxygen, and carbon nucleophiles with organic halides can be achieved under mild conditions (−40 to 30 °C) through the use of light and a copper catalyst. Insight into the various mechanisms by which these reactions proceed may enhance our understanding of chemical reactivity and facilitate the development of new methods. In this report, we apply an array of tools (EPR, NMR, transient absorption, and UV–vis spectroscopy; ESI–MS; X-ray crystallography; DFT calculations; reactivity, stereochemical, and product studies) to investigate the photoinduced, copper-catalyzed coupling of carbazole with alkyl bromides. Our observations are consistent with pathways wherein both an excited state of the copper(I) carbazolide complex ([CuI(carb)2]− ), and an excited state of the nucleophile (Li(carb)), can serve as photoreductants of the alkyl bromide. The catalytically dominant pathway proceeds from the excited state of Li(carb), generating a carbazyl radical and an alkyl radical. The cross-coupling of these radicals is catalyzed by copper via an out-of-cage mechanism in which [CuI(carb)2]− and [CuII(carb)3]− (carb = carbazolide), both of which have been identified under coupling conditions, are key intermediates, and [CuII(carb)3]− serves as the persistent radical that is responsible for predominant cross-coupling. This study underscores the versatility of copper(II) complexes in engaging with radical intermediates that are generated by disparate pathways, en route to targeted bond constructions.
Graphical Abstract
INTRODUCTION
Since our discovery several years ago of a photoinduced, copper-catalyzed Ullmann-type N-arylation process,1 we have expanded the scope of such photoinduced, copper-catalyzed coupling reactions to a variety of nucleophiles (nitrogen, sulfur, oxygen, and carbon) and electrophiles (aryl, alkyl, alkenyl, and alkynyl halides).2,3,4 We have naturally been interested in understanding the mechanisms of these processes, recognizing that different pathways may well be operative, depending on the nucleophile, the electrophile, and the reaction conditions.
In 2016, we described a mechanistic investigation of one such process, specifically, photoinduced reactions of aryl iodides with copper(I)–thiolates (eq 1).5 Our observations were consistent with the pathway outlined in Figure 1 (A→D): irradiation of a copper(I)–nucleophile complex (A) leads to the formation of an excited-state complex (B), which donates an electron to the electrophile (R–X) to generate an organic radical and a copper(II)–nucleophile complex (C), which then combine in-cage to form a carbon–nucleophile bond. Due in part to solubility issues, this mechanistic study of photoinduced S–arylation focused on stoichiometric reactions (A→D), rather than on catalyzed processes. This pathway, wherein a copper complex serves both as a photoreductant and as a participant in the key bond-forming step, differs from that suggested for most photoredox reactions, wherein a photosensitizer (e.g., Ru(bipy)32+) generates a radical via reduction or oxidation, but is not itself engaged in the key bond construction.6
Figure 1.

Outline of a possible pathway for photoinduced, copper-catalyzed cross-couplings. For simplicity, all copper complexes are illustrated as neutral species, and all processes are depicted as irreversible; X may be an inner- or an outer-sphere group, and Ln denotes additional ligand(s) coordinated to copper.
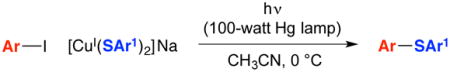 |
(1) |
The development of new methods for the construction of C–N bonds is an important objective, due to the importance of amines in a variety of disciplines, including biology, chemistry, and materials science.7 Because classical SN2 reactions of nitrogen nucleophiles with alkyl halides have rather limited scope, the discovery of catalyzed variants is of substantial interest, and in recent years the first systematic studies of transition-metal catalysis have been described.2a,d,g,8 For example, in 2013 we reported that alkylations of carbazoles by unactivated primary and secondary alkyl halides can be achieved under mild conditions in the presence of copper and light (eq 2).2a
 |
(2) |
Complementing our recent study of the photoinduced arylation of copper–thiolates,5 we have turned our attention to elucidating the mechanism of the photoinduced, copper-catalyzed alkylation of carbazole by alkyl halides. These processes differ in the nucleophile (sulfur vs. nitrogen) and in the electrophile (aryl vs. alkyl halide), while keeping the reaction conditions (light source, solvent, and temperature) constant; furthermore, in the present investigation, we examine not only stoichiometric reactions, but also catalyzed couplings.
RESULTS AND DISCUSSION
Background
In our original report of photoinduced, copper-catalyzed alkylations of carbazoles, we described preliminary mechanistic studies that employed alkyl iodides as electrophiles.2a In the present investigation, we have chosen to focus instead on alkyl bromides, in order to reduce potential complications due to undesired photoinduced side reactions. Furthermore, we have utilized discrete reaction components ([CuI(carb)2]Li (1)9 as the catalyst and Li(carb) as the nucleophile; carb = carbazolide), rather than the mixture of CuI, carbazole, and LiOt-Bu that was described in our original study. Under the new conditions, carbazole is alkylated by 2-bromo-4-phenylbutane in 64% yield at 0 °C in the presence of 5 mol% of catalyst 1 (eq 3). In the absence of light, no coupling (<1%) is observed.
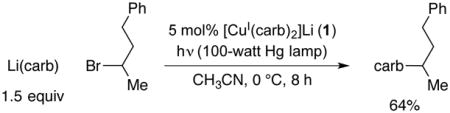 |
(3) |
Stoichiometric coupling of [CuI(carb)2]Li with an alkyl bromide (Figure 2, A’→D’)
Figure 2.

Outline of a possible pathway for the photoinduced, copper-catalyzed cross-coupling of Li(carb) with an alkyl bromide.
In an initial study, we examined the stoichiometric reaction of [CuI(carb)2]Li (1) with 2-bromo-4-phenylbutane (1:1), and we have determined that N-alkylation proceeds in 96% yield (eq 4). This is consistent with the mechanism outlined in Figures 1 and 2.
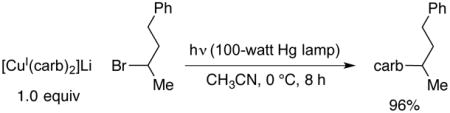 |
(4) |
Excitation of [CuI(carb)2]Li (Figure 2, A’→B’)
The UV–vis spectrum of [CuI(carb)2]Li in CH3CN exhibits an absorption at 365 nm with ε = 4300 M−1 cm−1, which overlaps with the highest-energy emission band of the medium-pressure Hg lamp used for the catalytic reactions. This absorption profile has enabled us to probe the photophysical properties of the excited-state copper complex, [CuI(carb)2]Li*, by transient absorption spectroscopy using a Nd:YAG laser source with excitation at 355 nm. For a 1.7 mM solution of [CuI(carb)2]Li (as under the catalysis conditions of eq 3), we observe a non-emissive excited state with a maximum absorption at 580 nm and a lifetime of 910 ns (Figure 3a).10
Figure 3.

(a) Difference absorption spectra of [CuI(carb)2]Li* at selected time delays (excitation of [CuI(carb)2]Li in CH3CN at 355 nm). (b) Quenching of [CuI(carb)2]Li* by 2-bromo-4-phenylbutane (RBr) in CH3CN observed by time-resolved transient absorption spectroscopy (λexc = 355 nm; λobs = 580 nm; room temperature).
Single electron transfer (SET) from [CuI(carb)2]Li* to an alkyl bromide (Figure 2, B’→C’)
To determine the viability of photoinduced SET from [CuI(carb)2]Li* to 2-bromo-4-phenylbutane, we have studied the excited-state quenching kinetics by examining the dependence of the lifetime of the transient-absorption signal at 580 nm on the concentration of the alkyl bromide. The lifetime of [CuI(carb)2]Li* in CH3CN decreases as the concentration of the alkyl bromide increases (Figure 3b), and the second-order rate constant for quenching is 4.8 × 106 M−1 s−1. In view of the lowest-energy excited state of the alkyl bromide, electron transfer, rather than energy transfer, from [CuI(carb)2]Li* to the alkyl bromide is the more likely mechanism of quenching.11
Because SET from [CuI(carb)2]Li* to an alkyl bromide should lead to the formation of a paramagnetic Cu(II) complex, we have employed electron paramagnetic resonance (EPR) spectroscopy to probe for an S = 1/2 copper photoproduct. When a freezing mixture of [CuI(carb)2]Li and 2-bromo-4-phenylbutane (5 equiv) in butyronitrile is irradiated at 350 nm, a pseudo-axial EPR signal is observed (Figure 4, black trace). A similar spectrum is obtained by treating [CuI(carb)2]Li with tris(4-bromophenyl)aminium hexachloroantimonate (Magic Blue; 0.2 equiv) in butyronitrile at −80 °C (Figure 4, red trace). Although hyperfine coupling is unresolved in these spectra, strong g anisotropy indicates the presence of a copper-containing metalloradical.
Figure 4.

EPR spectra (9.4 GHz, 77 K). Black trace: mixture of [CuI(carb)2]Li and 2-bromo-4-phenylbutane (5 equiv) in freezing butyronitrile upon irradiation at 350 nm; red trace: mixture of [CuI(carb)2]Li and Magic Blue (0.2 equiv) in butyronitrile at −80 °C. Simulated g values: g = [2.445, 2.060, 1.994]).
SET from [Li(carb)]* to an alkyl bromide
In a control reaction, Li(carb) and 2-bromo-4-phenylbutane do not couple in the absence of light (8 h at 0 °C); the alkyl bromide can be recovered quantitatively. However, when a mixture of Li(carb) and 2-bromo-4-phenylbutane is irradiated with a Hg lamp, a small amount of the C–N coupling product is generated (5% yield), although this N-alkylation is much less efficient than in the presence of a copper catalyst (Figure 5). In the copper-free reaction, side products E (32%), F (4%), and G (3%)12 are also observed (eq 5).
Figure 5.

Impact of a copper catalyst on C–N cross-coupling: Photoinduced reaction of Li(carb) (1.5 equiv) with 2-bromo-4-phenylbutane (100-watt Hg lamp, CH3CN, 0 °C) in the presence of 5 mol% [CuI(carb)2]Li and in the absence of copper.
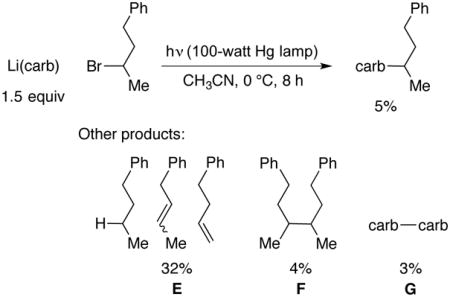 |
(5) |
Because compounds E–G likely form via bimolecular reactions of alkyl or carbazyl radicals,13 we decided to investigate a possible role for photoinduced SET from [Li(carb)]*, the excited state of Li(carb), to the alkyl bromide under our reaction conditions. The UV–vis spectra of solutions of Li(carb) in CH3CN show concentration-dependent molar absorptivity. At concentrations below 0.4 mM, Li(carb) does not absorb appreciably at 365 nm (Figure 6, black trace); however, at concentrations above 0.4 mM, absorption bands near 365 nm that feature molar absorptivity that is concentration-dependent begin to appear and are pronounced at 3.1 mM (Figure 6, blue trace). At 49 mM, which is the concentration of Li(carb) that is present under our catalysis conditions (eq 3), the molar absorptivity of Li(carb) at 365 nm is 2200 M−1 cm−1.
Figure 6.

All data were collected in CH3CN at room temperature. (a) Black trace: absorption spectrum for [Li(carb)] = 0.39 mM; blue trace: absorption spectrum for [Li(carb)] = 3.1 mM; green trace: excitation spectrum for [Li(carb)] = 1.1 mM (λem = 420 nm); red trace: emission spectrum for [Li(carb)] = 1.1 mM (λexc = 365 nm).
This dependence of the molar absorptivity on the concentration of Li(carb) is likely due to changes in aggregation.14 7Li NMR and 1H NMR spectroscopic data support the hypothesis that the aggregation of Li(carb) is concentration-dependent in CH3CN. For example, the 7Li and 1H NMR chemical shifts of Li(carb) vary with concentration. Furthermore, 1H diffusion ordered spectroscopy (1H DOSY) reveals that, at low concentrations (<2 mM) of Li(carb), a species with a hydrodynamic volume comparable to that of carbazole is observed.15 When the concentration of Li(carb) is increased to 49 mM (as under the catalysis conditions of eq 3), the volume triples, consistent with a significant change in aggregation.
Upon photoexcitation of a 49 mM solution of Li(carb) by a Nd:YAG laser at 355 nm, the excited state of Li(carb) emits at 420 nm with a lifetime of 31 ns (see SI). Stern–Volmer analysis establishes that the luminescent state of [Li(carb)]* is quenched by 2-bromo-4-phenylbutane with a second-order rate constant of 4.9 × 108 M−1 s−1 at room temperature (Figure 7). Thus, the UV–vis and Stern–Volmer data, combined with the higher concentration of Li(carb) compared with [CuI(carb)2]Li under the catalysis conditions, suggest that [Li(carb)]* may be the primary photoreductant during catalysis.
Figure 7.

Quenching of [Li(carb)]* by 2-bromo-4-phenylbutane (RBr) in CH3CN observed by time-resolved luminescence spectroscopy (λexc = 355 nm; λobs = 420 nm; room temperature).
On the basis of these observations, we propose an additional, dominant pathway for the photoinduced, copper-catalyzed coupling of carbazole with alkyl bromides (Figure 8). In this mechanism, Li(carb) is photoexcited to generate [Li(carb)]*, which transfers an electron to the alkyl bromide, resulting in the formation of a carbazyl radical and an alkyl radical. These radicals diffuse away from one another faster than they couple. The carbazyl radical reacts with [CuI(carb)2]Li (1) to generate a copper(II) complex, [CuII(carb)3]Li (2), which then couples with an alkyl radical to afford the N-alkylation product. As in the case of the original mechanism (Figure 2),16 the key C–N bond-forming step is the reaction of an alkyl radical with a Cu(II)–carbazolide complex, but the pathway for forming these intermediates is different.
Figure 8.

Outline of a new possible pathway for photoinduced, copper-catalyzed cross-coupling of Li(carb) with an alkyl bromide.
In order for this catalytic cycle to be viable and for side products (e.g., E, F, and G in the copper-free reaction illustrated in eq 5) to be minimized, the concentration of complexes 1 and 2 should be higher than the concentration of carbazyl and alkyl radicals that are being generated by photolysis. Accordingly, we sought evidence for [CuI(carb)2]Li (1) and for [CuII(carb)3]Li (2), the postulated persistent radical,17 under catalysis conditions.18
Evidence for the presence of [CuI(carb)2]Li (1) under catalysis conditions
Previously, we have crystallographically characterized [CuI(carb)2]Li and observed its presence via ESI–MS during a photoinduced, copper-catalyzed alkylation of carbazole by an alkyl iodide.2a We have now determined that, for the coupling of an alkyl bromide (eq 3), we also observe signals at m/z of 395 and 397, which correspond to the masses of [63CuI(carb)2]− and [65CuI(carb)2]−, respectively, after 60 minutes of irradiation.
Evidence for the presence of [CuII(carb)3]Li (2) under catalysis conditions
As indicated above, the efficient operation of the catalytic cycle outlined in Figure 8 requires that the concentration of [CuII(carb)3]Li (2) be higher than the concentration of the alkyl radical that is being generated through photolysis of Li(carb). This suggested that it might be possible to characterize this proposed intermediate via spectroscopy and perhaps even crystallography.
Since [CuII(carb)3]Li would have a d9 configuration, we undertook an EPR investigation of a catalyzed cross-coupling. Specifically, a reaction mixture under the model conditions (eq 3) was irradiated at 0 °C for 60 minutes and then freeze-quenched at −196 °C, leading to the observation of a strong, pseudo-axial EPR signal that shows an unpaired spin coupled to 63/65Cu (I = 3/2) (Figure 9, black trace; a 4-line coupling pattern is evident in gz). This EPR spectrum is different from that observed upon irradiation of a mixture of [CuI(carb)2]Li (1) and this alkyl bromide in the absence of added Li(carb) (Figure 4, black trace).
Figure 9.

EPR evidence for [CuII(carb)3]Li (2), generated through three independent pathways. Black trace: catalysis mixture; blue trace: mixture of Li(carb) and CuBr2; green trace: mixture of Li(carb), [CuI(carb)2]Li (1), and Magic Blue; red trace: simulated EPR spectrum (g = [2.318, 2.058, 2.050]) displaying hyperfine to Cu and three 14N atoms.
The same EPR-active compound can be generated through low temperature treatment of CuBr2 with 3 equivalents of Li(carb) (Figure 9, blue trace) and through reaction of [CuI(carb)2]Li (1) with Li(carb) and Magic Blue (Figure 9, green trace). These EPR signals are modeled well as a monomeric Cu(II) species coordinated by three equivalent nitrogen (I = 1) atoms (Figure 9, red trace), consistent with its assignment as the homoleptic [CuII(carb)3]− complex.
We have carried out corresponding studies using UV–vis spectroscopy (Figure 10). When a reaction mixture is irradiated under the standard catalysis conditions (eq 3), the solution turns deep-blue; this color diminishes when irradiation is discontinued or when the solution is allowed to warm to room temperature. Similarly, under the standard conditions but in butyronitrile at −80 °C, irradiation results in an intense blue color. The UV–vis spectrum of this solution displays an absorption maximum at 580 nm (Figure 10, black trace); this feature is not observed when the same experiment is conducted in the absence of [CuI(carb)2]Li.19 Likewise, when CuBr2 is treated with 3 equiv of Li(carb) in butyronitrile at −80 °C, a deep-blue solution with an absorption band centered around 580 nm is observed (Figure 10, blue trace). Finally, when a mixture of [CuI(carb)2]Li and Li(carb) (2 equiv) is oxidized by Magic Blue (0.3 equiv) in butyronitrile at −80 °C, the same absorption feature is evident (green trace). We hypothesize that the absorption band at 580 nm correlates with the EPR-active Cu(II) species shown in Figure 9.
Figure 10.

UV–vis spectra. Black trace: catalyzed reaction mixture irradiated in butyronitrile at −80 °C; blue trace: mixture of CuBr2 and Li(carb) (3 equiv) in butyronitrile at −80 °C; green trace: mixture of [CuI(carb)2]Li (1) and Li(carb) (2 equiv) treated with Magic Blue (0.3 equiv) in butyronitrile at −80 °C.
Although we have been unable to crystallographically characterize a lithium salt of [CuII(carb)3]−, due in part to its thermal instability, crystals of two potassium salts, [CuII(carb)3][K(THF)6] and [CuII(carb)3][K(benzo-15-crown-5)2],20 have been obtained by layering Et2O onto a solution that contained Cu(OTf)2, K(carb), and benzo-15-crown-5 in THF at −78 °C (Figure 11a). This complex represents a rare example of a structurally characterized homoleptic three-coordinate copper(II) complex.21 The powder and glass EPR spectra of [CuII(carb)3][K(benzo-15-crown-5)2] are consistent with the spectra illustrated in Figure 9.
Figure 11.

(a) X-ray crystal structure of [CuII(carb)3][K(THF)6] (thermal ellipsoids at 30% probability; hydrogen atoms and [K(THF)6]+ are omitted for clarity). (b) DFT-computed spin-density plot of [CuII(carb)3]−; 27% of the total spin resides on the three nitrogen atoms. See SI for details.
The quality of the X-ray diffraction data for [CuII(carb)3][K(THF)6] is modest, and structural metrics should therefore be viewed with caution. Nevertheless, the N–Cu–N bond angles (two large and one small: 126.7(2)°, 124.4(2)°, 108.8(2)°) indicate a significant distortion from a trigonal planar geometry, as anticipated for an 2E electronic state that is Jahn-Teller active. DFT computations show significant spin delocalization, with 0.43 e−on Cu and 0.27 e− distributed between the three nitrogen atoms of the carbazolide ligands (Figure 11b).22
Given the appreciable predicted spin density on nitrogen, [CuII(carb)3]− might display reactivity characteristic of a carbazyl radical. For example, the key bond-forming step in these photoinduced, copper-catalyzed reactions might occur through direct C–N coupling, rather than through a copper(III) intermediate. Previously, Warren has observed the reaction between trityl radical (Ph3C·) and a Cu(II)–anilido complex to furnish a C–N bond (Ph3C–NHAr) and Cu(I). 23 While the addition of trityl radical did not yield C–N coupled product, addition of TEMPO–H to a solution of [CuII(carb)3]Li generates TEMPO· as the only EPR-active species (TEMPO = 2,2,6,6-tetramethylpiperidine 1-oxyl), as well as [CuI(carb)2]Li. This reactivity is consistent with hydrogen atom abstraction from TEMPO–H by [CuII(carb)3]−.24
As outlined in Figure 8, we hypothesize that [CuII(carb)3]Li (2) is generated through the coupling of carb• with [CuI(carb)2]Li (1). DFT calculations support the viability of this elementary step, indicating that it is exergonic by ~9 kcal mol−1.25 In contrast, the potential coupling of [CuI(carb)2]Li (1) with R•, the other radical that is produced upon excitation of Li(carb) and subsequent reaction with R–Br, to form [CuIIR(carb)2]− is endergonic by ~3 kcal mol−1.
Next, using UV–vis spectroscopy, we have examined the build-up of the persistent radical, [CuII(carb)3]Li (2), under our standard conditions for a catalyzed coupling (eq 3). When a reaction mixture is irradiated in a quartz cuvette at 0 °C, an absorption band for [CuII(carb)3]Li (λmax = 580 nm) is observed within a few seconds (Figure 12a). On the basis of an extinction coefficient of ~1100 M−1 cm−1 for [CuII(carb)3]Li, a concentration of [CuII(carb)3]Li corresponding to ~50% of the original amount of copper is reached within three minutes (Figure 12b).
Figure 12.

(a) Appearance of an absorption band (λmax = 580 nm) for [CuII(carb)3]Li upon irradiation of a reaction mixture containing [CuI(carb)2]Li, Li(carb), and 2-bromo-4-phenylbutane in CH3CN in a quartz cuvette at 0 °C. (b) Concentration of [CuII(carb)3]Li as a function of time.
According to our mechanistic hypothesis, at the outset of a photoinduced, copper-catalyzed C–N coupling, when the concentration of the persistent radical, [CuII(carb)3]Li (2), is still low, a higher proportion of products derived from undesired side reactions of R• (e.g., E in eq 5) is expected; as the concentration of [CuII(carb)3]Li builds up upon irradiation of the catalysis mixture, the proportion of the C–N cross-coupling product should increase. This is indeed observed (Figure 13).
Figure 13.

Product distribution in the photoinduced, copper-catalyzed coupling of Li(carb) with 2-bromo-4-phenylbutane (eq 3).
Evidence for out-of-cage C–N bond formation via a free-radical intermediate under catalysis conditions (Figure 8, 2→1)
The mechanism outlined in Figure 8 suggests that the coupling of R• and carb• is an out-of-cage process mediated by copper. We have turned to a radical-cyclization probe to gain insight into the C–N bond-forming step of the catalytic cycle. Because the reaction of secondary bromides is the focus of the present study, we chose 6-bromo-1-heptene as the substrate. The derived secondary alkyl radical cyclizes with a rate constant of 1.0 × 105 s−1 at 25 °C,26 furnishing a primary alkyl radical with 4:1 cis:trans diastereoselectivity.27
When 6-bromo-1-heptene is subjected to our standard reaction conditions, the cyclization/coupling product (H) is formed in 62% yield with 4:1 diastereoselectivity, along with 10% of the direct-coupling product (I) (eq 6). Because the rate of diffusion (typically >108 s−1)28 is significantly higher than the rate of cyclization of the derived secondary radical, predominant generation of cyclized product H suggests that, if ring formation is occurring through radical cyclization, then C–N bond construction is proceeding primarily through an out-of-cage pathway.
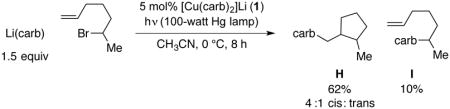 |
(6) |
The 4:1 cis:trans stereoselectivity that we observe is identical to that previously reported for the cyclization of this putative secondary alkyl radical at 0 °C.26 To provide further support for a radical, rather than an organometallic, pathway for ring formation, we have examined the photoinduced, copper-catalyzed coupling of the deuterium-labeled analogue of 6-bromo-1-heptene that is illustrated in eq 7. Analysis via NMR spectroscopy revealed a 1:1 mixture at the indicated stereocenter, which is inconsistent with a radical-free organometallic mechanism that involves uninterrupted oxidative addition, β-migratory insertion, and reductive elimination.29
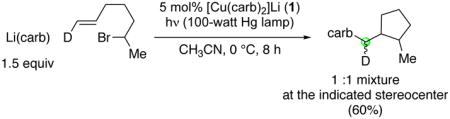 |
(7) |
To further explore the question of out-of-cage coupling, we have investigated the effect of the concentration of the reaction on the ratio of cyclized (H) to uncyclized (I) product (eq 8).30 If all of the cross-coupling is occurring in-cage, then the ratio should not depend on concentration. The trend that we observe–a greater preference for cyclization at lower concentration–is that expected if out-of-cage coupling is occurring.
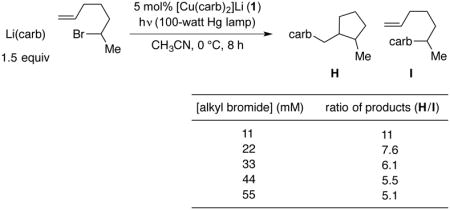 |
(8) |
Evaluation of a radical chain mechanism under catalysis conditions
A recent report by Yoon serves as a reminder of the need to consider the possibility of radical chain pathways during photoredox processes.31,32 In the present study, we have determined that the total quantum yield (ϕ) for all processes that consume 2-bromo-4-phenylbutane under the standard catalysis conditions (eq 3) is 0.10 ± 0.01,33 a value that is consistent both with a non-chain pathway and with a chain process with rapid chain termination. We have also measured the Stern–Volmer quenching fraction (Q) to be 0.33, which suggests that at most 33% of all excited states of lithium carbazolide that are generated are quenched by the electrophile. The chain length (ϕ/Q; the number of molecules of product formed per photoinduced electron-transfer event) is therefore 0.30, indicating that every quenching event of Li(carb)* leads to the consumption of 0.30 equivalents of electrophile. This low chain length, which represents a lower limit (see SI), is suggestive of a non-chain pathway.
CONCLUSIONS
Using a wide range of tools, we have investigated the mechanism of the photoinduced, copper-catalyzed coupling of carbazole with an alkyl bromide, which is one of the first examples of transition-metal catalysis of the alkylation of an amine by an alkyl halide. In addition to the originally proposed pathway, we suggest that a second mechanism is operative and dominant, wherein photoexcited Li(carb) serves as a reductant of the alkyl bromide, generating an alkyl radical and a carbazyl radical, which combine via an out-of-cage, copper-catalyzed pathway in which [CuI(carb)2]− (1) and [CuII(carb)3]− (2) are key intermediates (Figure 8).
Using ESI–MS, we have obtained evidence for the presence of [CuI(carb)2]− (1) under catalysis conditions. With the aid of UV–vis and luminescence spectroscopy, we have established that, upon irradiation, Li(carb) can undergo excitation and then electron transfer to an alkyl bromide, thereby affording a carbazyl radical and an alkyl radical. Using EPR spectroscopy, we have obtained evidence for the formation of a Cu(II) complex during catalysis; we postulated this intermediate to be [CuII(carb)3]− (2; generated by the reaction of a carbazyl radical with [CuI(carb)2]− (1)), which we then independently synthesized and structurally characterized. Through the use of UV–vis spectroscopy, we have monitored the buildup of this complex, the persistent radical that is responsible for effective cross-coupling, during a reaction. Radical-cyclization, stereochemical, and reactivity probes are consistent with the generation of an alkyl radical, which engages in out-of-cage coupling with [CuII(carb)3]− (2). The chain length for the coupling reaction is relatively low (0.3), as expected for a non-chain process.
The additional pathway illustrated in Figure 8 highlights the opportunity to achieve photoinduced, copper-catalyzed coupling reactions not only through excitation of copper–nucleophile complexes, but also of nucleophiles themselves. Regardless of the photoreductant under catalytic conditions, copper appears to play a critical role in the key bond construction step via coupling of a copper(II)–nucleophile complex with an organic radical. Ongoing studies are directed at further expanding the scope of such processes, as well as exploring their mechanisms.
Supplementary Material
Acknowledgments
Support has been provided by the National Institutes of Health (National Institute of General Medical Sciences: R01–109194), the Natural Sciences and Engineering Research Council of Canada (graduate research fellowship for J.M.A.), and the National Science Foundation (support of the Caltech EPR Facility (NSF-1531940) and a graduate research fellowship for T.S.R.). Additional support has been provided by the Arnold and Mabel Beckman Foundation through the Caltech Beckman Institute Laser Resource Center. We thank Dr. Angel J. Di Bilio, Dr. Paul Oyala, Dr. Sidney E. Creutz, Lawrence M. Henling, Dr. Marcin Kalek, Dr. Wesley Sattler, Dr. Oliver S. Shafaat, Dr. Mona Shahgholi, Dr. David VanderVelde, and Dr. Jay R. Winkler for technical assistance and helpful discussions.
Footnotes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: Procedures and characterization data (PDF)
References
- 1.Creutz SE, Lotito KJ, Fu GC, Peters JC. Science. 2012;338:647–651. doi: 10.1126/science.1226458. [DOI] [PubMed] [Google Scholar]
- 2.alkylation of nitrogen nucleophiles: Bissember AC, Lundgren RJ, Creutz SE, Peters JC, Fu GC. Angew Chem Int Ed. 2013;52:5129–5133. doi: 10.1002/anie.201301202.arylation of sulfur nucleophiles: Uyeda C, Tan Y, Fu GC, Peters JC. J Am Chem Soc. 2013;135:9548–9552. doi: 10.1021/ja404050f.arylation, alkenylation, and alkynylation of nitrogen nucleophiles: Ziegler DT, Choi J, Munoz-Molina JM, Bissember AC, Peters JC, Fu GC. J Am Chem Soc. 2013;135:13107–13112. doi: 10.1021/ja4060806.alkylation of nitrogen nucleophiles: Do H-Q, Bachman S, Bissember AC, Peters JC, Fu GC. J Am Chem Soc. 2014;136:2162–2167. doi: 10.1021/ja4126609.arylation of oxygen nucleophiles: Tan Y, Munoz-Molina JM, Fu GC, Peters JC. Chem Sci. 2014;5:2831–2835.alkylation of carbon nucleophiles: Ratani TS, Bachman S, Fu GC, Peters JC. J Am Chem Soc. 2015;137:13902–13907. doi: 10.1021/jacs.5b08452.enantioconvergent alkylation of nitrogen nucleophiles: Kainz QM, Matier CM, Bartoszewicz A, Zultanski SL, Peters JC, Fu GC. Science. 2016;351:681–684. doi: 10.1126/science.aad8313.
- 3.For independent early work, see: Sagadevan A, Hwang KC. Adv Synth Catal. 2012;354:3421–3427.
- 4.For an overview of copper in photocatalysis, see: Paria S, Reiser O. ChemCatChem. 2014;6:2477–2483.
- 5.Johnson MW, Hannoun KI, Tan Y, Fu GC, Peters JC. Chem Sci. 2016;7:4091–4100. doi: 10.1039/c5sc04709a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For leading reviews on photoredox catalysis, see: Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r.Skubi KL, Blum TR, Yoon TP. Chem Rev. 2016;116:10035–10074. doi: 10.1021/acs.chemrev.6b00018.
- 7.For example, see: Lawrence SA. Amines: Synthesis, Properties, and Applications. Cambridge University Press; Cambridge, U.K: 2004.
- 8.Peacock DM, Roos CB, Hartwig JF. ACS Cent Sci. 2016;2:647–652. doi: 10.1021/acscentsci.6b00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For simplicity, we write “[CuI(carb)2]Li”, although the lithium is in fact solvated by CH3CN. For example, see Reference 2a.
- 10.A luminescent state of [CuI(carb)2]Li* with a lifetime of 590 ps was also observed.
- 11.A concentrated sample of the alkyl bromide in CH3CN shows no absorption beyond 300 nm, whereas the Nd:YAG laser is pumping the sample at 355 nm.
- 12.Compound G is the N–N coupled product.
- 13.Alfassi ZB. In: General Aspects of the Chemistry of Radicals. Alfassi ZB, editor. Wiley; Chichester, U.K: 1999. pp. 139–173. [Google Scholar]
- 14.For examples of aggregation of carbazolide anions, see: Dinnebier R, Esbak H, Olbrich F, Behrens U. Organometallics. 2007;26:2604–2608.Bock H, Arad C, Näther C, Havlas Z. J Organomet Chem. 1997;548:115–120.
- 15.The expected value for the diffusion coefficient for carbazole is 24.2 × 1010 m2/s, which leads to a hydrodynamic radius of 2.6 Å; the measured value is 22.7 × 1010 m2/s, which gives a hydrodynamic radius of 2.8 Å.
- 16.Correspondingly, with respect to the C–N coupling pathway that proceeds via excitation of [CuI(carb)2]Li (Figure 2), R• may react with [CuII(carb)3]Li (2), rather than with CuII(carb)2.
- 17.For leading references, see: Studer A. Chem Eur J. 2001;7:1159–1164. doi: 10.1002/1521-3765(20010316)7:6<1159::aid-chem1159>3.0.co;2-i.Fischer H. Chem Rev. 2001;101:3581–3610. doi: 10.1021/cr990124y.
- 18.We use this term loosely to include reactions conducted according to eq 3, but where irradiation has been stopped (e.g., to allow spectroscopic studies).
- 19.The absorption profiles shown in Figure 3a (the transient absorption spectrum of [CuI(carb)2]Li in the absence of added electrophile) and Figure 10 (the absorption spectrum of [CuII(carb)3]–) are similar. The [CuI(carb)2]Li* excited state detected in Figure 3a presumably has MLCT character with some degree of admixed ligand-to-ligand charge transfer (i.e., MLLCT), reflecting a degree of copper(II)-carbazolide character. MLCT character in [CuI(carb)2]Li* is consistent with a molar extinction coefficient of 4300 M−1 cm−1 for this transition of [CuI(carb)2]– at 365 nm.
- 20.The X-ray diffraction data for [CuII(carb)3][K(benzo-15-crown-5)2] were of poor quality and hence were only sufficient for establishing overall atomic connectivity.
- 21.Homoleptic monomeric three-coordinate Cu(II) anions have only been crystallographically characterized in the case of alkoxide ligands: Purdy AP, George CF. Inorg Chem. 1991;30:1969–1970.Hannigan SF, Lum JS, Bacon JW, Moore C, Golen JA, Rheingold AL, Doerrer LH. Organometallics. 2013;32:3429–3436.
- 22.For examples of other copper(II)–amido complexes wherein significant spin density is believed to reside on nitrogen, see: Mankad NP, Antholine WE, Szilagyi RK, Peters JC. J Am Chem Soc. 2009;131:3878–3880. doi: 10.1021/ja809834k.Wiese S, Badiei YM, Gephart RT, Mossin S, Varonka MS, Melzer MM, Meyer K, Cundari TR, Warren TH. Angew Chem Int Ed. 2010;49:8850–8855. doi: 10.1002/anie.201003676.Wagner CL, Tao L, Thompson EJ, Stich TA, Guo J, Fettinger JC, Berben LA, Britt RD, Nagase S, Power PP. Angew Chem Int Ed. 2016;55:10444–10447. doi: 10.1002/anie.201605061.
- 23.Jang ES, McMullin CL, Käß M, Meyer K, Cundari TR, Warren TH. J Am Chem Soc. 2014;136:10930–10940. doi: 10.1021/ja5026547. [DOI] [PubMed] [Google Scholar]
- 24.For examples of other copper(II)–amido complexes that abstract hydrogen atoms, see: (a) Reference 22a. (b) Reference 23.
- 25.The computed activation barrier is less than 8 kcal/mol.
- 26.Lusztyk J, Maillard B, Deycard S, Lindsay DA, Ingold KU. J Org Chem. 1987;52:3509–3514. [Google Scholar]
- 27.This diastereoselectivity (4:1 cis:trans) has previously been observed for the cyclization of this radical at 0 °C: Reference 26.
- 28.Anslyn EV, Dougherty DA. Modern Physical Organic Chemistry. University Science Books; Sausalito, CA: 2006. p. 156. [Google Scholar]
- 29.When a photoinduced, copper-catalyzed cross-coupling is run in the presence of TEMPO, a considerable amount of the TEMPO adducts (derived from trapping of the uncyclized secondary radical or the cyclized primary radical) is observed.
- 30.For leading references on radical clocks, see: Newcomb M. In: Encyclopedia of Radicals in Chemistry, Biology and Materials. Chatgilialoglu C, Studer A, editors. Vol. 1. John Wiley & Sons; Chichester, U.K: 2012. pp. 107–124.
- 31.Cismesia MA, Yoon TP. Chem Sci. 2015;6:5426–5434. doi: 10.1039/c5sc02185e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.For reviews of photoinduced SRN1 reactions, see: Buden ME, Martin SE, Rossi RA. In: CRC Handbook of Organic Photochemistry and Photobiology. Griesbeck A, Oelgemoller M, Ghetti F, editors. CRC Press; Boca Raton, Florida: 2012. pp. 347–368.Penenory AB, Argüello JE. In: Handbook of Synthetic Photochemistry. Albini A, Fagnoni M, editors. Chapter 10 Wiley–VCH; Weinheim, Germany: 2010.
- 33.The Hatchard–Parker method was employed. See: Murov SL, Carmichael I, Hug GL. Handbook of Photochemistry. CRC Press; New York: 1993. pp. 298–313.Bolton JR, Stefan MI, Shaw PS, Lykke KR. J Photochem Photobiol A: Chem. 2011;222:166–169.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.