

Abstract
RhoPDE is a type I rhodopsin/phosphodiesterase gene fusion product from the choanoflagellate Salpingoeca rosetta. The gene was discovered around the time that a similar type I rhodopsin/guanylyl cyclase fusion protein, RhoGC, was shown to control phototaxis of an aquatic fungus through a cGMP signaling pathway. RhoPDE has potential as an optogenetic tool catalyzing the hydrolysis of cyclic nucleotides. Here we provide an expression and purification system for RhoPDE, as well as a crystal structure of the C-terminal phosphodiesterase catalytic domain. We show that RhoPDE contains an even number of transmembrane segments, with N- and C-termini both located on the cytoplasmic surface of the cell membrane. The purified protein exhibits an absorption maximum at 490 nm in the dark state, which shifts to 380 nm upon exposure to light. The protein acts as a cGMP-selective phosphodiesterase. However, the activity does not appear to be modulated by light. The protein is also active with cAMP as a substrate, but with a roughly 5–7-fold lower kcat. A truncation consisting solely of the phosphodiesterase domain is also active with a kcat for cGMP roughly 6–9-fold lower than that of the full-length protein. The isolated PDE domain was crystallized, and the X-ray structure showed the protein to be a dimer similar to human PDE9. We anticipate that the purification system introduced here will enable further structural and biochemical experiments to improve our understanding of the function and mechanism of this unique fusion protein.
Graphical abstract
Retinylidene proteins make up a family of light-sensitive integral membrane proteins consisting of an opsin domain, characterized by a seven-transmembrane α-helical fold, and a retinal chromophore covalently bound to a conserved lysine residue in the seventh helix.1,2 The family is divided into two groups based on amino acid sequence similarity: type I rhodopsins are microbial in origin and usually function as light-activated ion pumps or channels, while type II rhodopsins are G protein-coupled receptors (GPCRs) that function as photoreceptors in metazoan visual systems.1,2
Rhodopsin, the photoreceptor protein in vertebrate rod cells, is the prototypical member of the type II group.1,2 Photoexcitation of the 11-cis-retinal chromophore causes isomerization to the all-trans-form to generate the active form of rhodopsin, metarhodospin II.3 Metarhodopsin II activates the G protein transducin, which activates a cGMP phosphodiesterase. Following a drop in cellular cGMP concentration, cyclic nucleotide-gated (CNG) cation channels close and cause hyperpolarization of the rod cell.4 Rhodopsin then releases the all-trans-retinal (ATR) and must bind a new molecule of 11-cis-retinal to begin the cycle again.
In contrast to the multicomponent GPCR signaling cascade, type I rhodopsins, such as bacteriorhodopsin, function as single-component proteins that move ions (protons, in the case of bacteriorhodopsin) directly across the membrane upon light activation.1,2 Bacteriorhodopsin binds ATR in the dark state, which isomerizes to 13-cis-retinal after exposure to light. Unlike rhodopsin, bacteriorhodopsin can thermally convert 13-cis-retinal back into the all-trans form to regenerate the dark state.
A new type I rhodopsin was recently discovered in the aquatic fungus Blastocladiella emersonii. This newly discovered protein is an unusual fusion between a type I rhodopsin domain and a guanylyl cyclase catalytic domain that is homologous to the guanylyl cyclase RetGC found associated with type II rhodopsins of photoreceptor cell outer segments.5 This single-component, light-activated enzyme, RhoGC, controls a cGMP signaling system responsible for phototaxis in zoospores of the fungus5 and has been shown to be an optogenetic tool for the control of cyclic nucleotide signaling systems.6,7 A gene for a similar protein (RhoPDE) was recently identified in the genome of the choanoflagellate Salpingoeca rosetta where a type I rhodopsin domain was found to be fused to a phosphodiesterase catalytic domain (NCBI Gene ID 16078606).
In this paper, we present an expression and purification system for full-length RhoPDE in a detergent solution. The procedure makes use of two epitope tags, one at the N-terminus and the other at the C-terminus of the protein (Figure 1), that we exploit with an immunoaffinity purification procedure. We use the two epitopes to show that RhoPDE contains an even number of transmembrane segments, with N- and C-termini both located on the cytoplasmic surface of the cell membrane. The protein forms a pigment with ATR characterized by an absorbance maximum at 490 nm and a prominent shoulder at 460 nm. Exposure to light shifts the maximum to 380 nm, consistent with deprotonation of the Schiff base. The protein displays robust cGMP-specific phosphodiesterase activity that is, surprisingly, independent of light; the enzyme is active in the dark with no detectable change in activity after exposure to light. We also express, purify, and present the X-ray crystal structure of the isolated PDE domain of the protein and show that it is constitutively active, with a kcat roughly 6–9-fold lower than that of the full-length protein.
Figure 1.
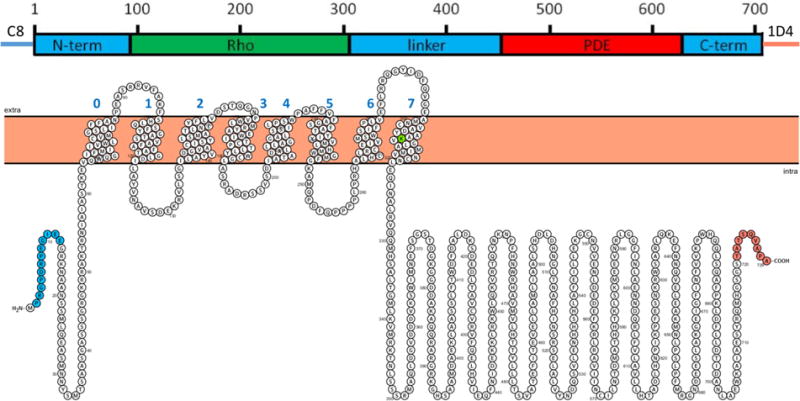
Predicted domain structure and transmembrane topology for RhoPDE. Domain nomenclature: N-term, N-terminal domain; Rho, microbial type I rhodopsin domain; linker, sequence connecting the Rho and PDE domains; PDE, phosphodiesterase domain; and C-term, short C-terminal domain. Transmembrane helices were predicted and drawn with Protter (wlab.ethz.ch/protter/start).8 Amino acids are colored: green, Lys296 (numbering of the native protein), the presumed covalent attachment site for the retinal chromophore; blue, the C8 epitope tag; and orange, the 1D4 epitope tag.
EXPERIMENTAL PROCEDURES
Materials
All-trans-retinal (ATR), cGMP, and cAMP were purchased from Sigma-Aldrich. n-Decyl β-D-maltopyranoside (DM) was from Anatrace. Bovine growth serum and Dulbecco’s phosphate-buffered saline (PBS) were purchased from HyClone Laboratories. Lipofectamine was from Invitrogen. Oligonucleotides were purchased from Integrated DNA Technologies. The C8 and 1D4 peptides were purchased from GenScript. The C89 and 1D410,11 antibodies were from the National Cell Culture Center, and the C8- and 1D4-Sepharose 4B immunoaffinity supports used for purification of RhoPDE were prepared as previously described.12,13
Expression and Purification of Full-Length RhoPDE
The gene sequence for RhoPDE was taken from S. rosetta (NCBI Gene ID 16078606) and codon optimized for expression in Escherichia coli (Genewiz Inc., South Plainfield, NJ). The gene was cloned into mammalian expression vector pMT314 between restriction sites EcoRI and NotI. An N-terminal C8 epitope tag was added between the first two amino acids of the protein sequence, and a C-terminal E8′A mutant of the 1D4 epitope tag, following a GSGS linker, was added after the last amino acid of the protein sequence.
HEK293-GnT1− cells were transfected using the calcium phosphate precipitation method for transient expression of RhoPDE and harvested 72 h after transfection, as previously described.14,15 The protein was reconstituted with the retinal chromophore by incubating the cell suspension with 10 μM ATR for 30 min in the dark at 4 °C. All procedures following the addition of retinal were performed in the dark under dim red lights. Cells were solubilized in 1% (w/v) DM in PBS for 1 h at 4 °C. The cell lysate was centrifuged at 3500 rpm and 4 °C for 10 min, and the postnuclear supernatant fraction was applied to a C8 or 1D4 immunoaffinity matrix. Batch binding of the protein to the matrix took place for 2 h at 4 °C before the resin was poured into a spin column for removal of the unbound fraction. The column was washed at 4 °C with 1% (w/v) DM in PBS. Protein was eluted at room temperature with 80 μM corresponding C8 or 1D4 peptide.
Immunofluorescence Microscopy
Immunofluorescence microscopy was performed as previously described.16 COS-7 cells (5 × 104 cells/cm2) were plated on 15 mm glass coverslips (Deckgläser) coated with 0.01% poly-L-lysine (Sigma) and grown in 1 mL of fortified DMEM in a 12-well polystyrene plate for 24 h at 37 °C. After cells had grown for 24 h, the DMEM was aspirated and replaced with 1 mL of Opti-MEM for transfection. RhoPDE with an N-terminal C8 epitope and C-terminal 1D4 epitope was used for transfections with Lipofectamine 2000. After 5 h at 37 °C, the Opti-MEM was aspirated and replaced with 1 mL of fortified DMEM for overnight expression at 37 °C. To fix the cells to the coverslips, the cells were washed three times with cold PBS and treated with 3% paraformaldehyde (EMS) for 15 min at room temperature. After being washed with PBS, two cell samples were left intact, and two samples were permeabilized with 0.25% Triton X-100 (Sigma) for 10 min at room temperature and washed again with cold PBS. All samples were treated with blocking buffer consisting of 10% BSA for 30 min at room temperature. One sample each of intact and permeabilized cells was incubated with 2 μg/mL 1D4 primary antibody, and the other pair with C8 antibody, for 1 h at room temperature. After being washed with cold PBS, each sample was incubated with 2 μg/mL fluorescent secondary antibody, Alexa-fluor 488 goat anti-mouse IgG (Life Technologies), for 1 h at room temperature in the dark. After a final wash with cold PBS, the coverslips were mounted on 3 in. × 1 in. × 1 mm glass slides (Fisher) with ProLong Diamond Antifade mountant (Life Technologies).
The cells were imaged using an automated Olympus IX81 epi-fluorescence microscope (Olympus, Waltham, MA). Alexa-fluor 488 was excited using a mercury vapor short-arc lamp (Excelitas Tech) via a 473/31 nm bandpass filter (Semrock, Rochester, NY) and the emission collected through a 520/25 nm bandpass filter (Semrock). Images were captured using an EM-CCD camera (Hamamatsu) controlled by Metamorph (Molecular Devices, Sunnyvale, CA) and analyzed with ImageJ software (National Institutes of Health, Bethesda, MD).
Absorption Spectroscopy
Ultraviolet–visible (UV–vis) absorption spectra were recorded with a Cary 50 UV–vis spectrometer at room temperature with a path length of 1.0 cm. The molar extinction coefficient for RhoPDE (ε490 = 45500 M−1 cm−1) was determined by acid denaturation of the pigment in the dark with concentrated HCl (ε440 = 31000 M−1 cm−1), as described previously.17
Preparation of HEK293 Cell Membranes
HEK293-GnT1− cell membranes containing RhoPDE were isolated by sucrose flotation.18 Cells expressing RhoPDE were harvested 72 h after transfection, lysed in a hypotonic solution of 10 mM Tris buffer (pH 7.4) and 0.1 mg/mL phenylmethanesulfonyl fluoride (PMSF), and extruded four times through a 25 gauge needle. The homogenate was layered onto a 37% (w/v) sucrose solution in a buffer containing 10 mM Tris (pH 7.4), 150 mM NaCl, 1 mM MgCl2, 1 mM CaCl2, and 0.1 mM EDTA and centrifuged at 15000 rpm for 20 min in a Beckman SW 28.1 rotor. Membranes were extracted from the resulting interface, diluted 10-fold in 10 mM Tris (pH 7.4), and pelleted by centrifugation at 33000 rpm for 45 min in a Beckman 50.2 Ti rotor. The membranes were resuspended in 1 mL of 10 mM Tris (pH 7.4), 150 mM NaCl, 1 mM MgCl2, 1 mM CaCl2, and 0.1 mM EDTA and aliquoted (25 μL) before being frozen on dry ice and stored at −80 °C. The concentration of RhoPDE was estimated from the yield of the protein purified from equivalent numbers of transfected HEK293 cells, corrected for losses during the purification.
In one case (Figure 6), membranes were prepared from transfected HEK293-GnT1− cells grown in the presence of ATR. The cells were kept in the dark and supplemented with fresh DMEM containing 10 μM ATR every 24 h after transfection. A small aliquot of membranes was used for activity assays, with the remainder used for purification of RhoPDE. Control membranes from cells grown in the absence of ATR were prepared alongside and treated with ATR before use.
Figure 6.
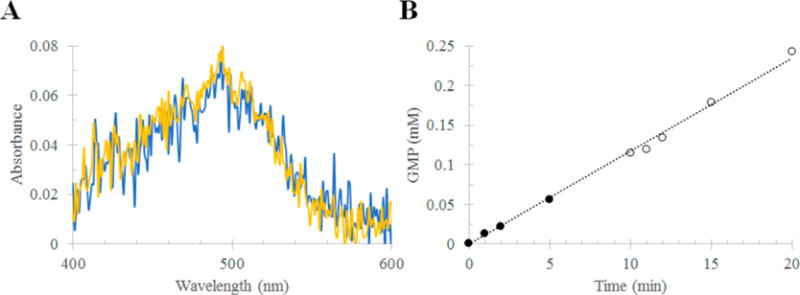
Purification and assay of RhoPDE from HEK293 cells grown in the presence of ATR. (A) Absorption spectra of RhoPDE after purification (C8 antibody column) from HEK293 membranes. The figure compares the yield of RhoPDE from cells grown in the presence and absence of ATR: yellow, cells grown in the presence of ATR; and blue, cells grown in the absence of ATR. Membranes from cells grown in the absence of ATR were treated with retinal before detergent solubilization. No ATR was added to membranes from cells grown in the presence of ATR. (B) cGMP phosphodiesterase activity of RhoPDE in membranes isolated from cells grown in the presence of ATR. No additional ATR was added. The assay buffer for this reaction consisted of 50 mM HEPES (pH 6.5), 50 mM NaCl, 10 mM MgCl2, and 0.5 mM EDTA.
Preparation of Reconstituted Liposomes
Liposomes containing RhoPDE were prepared as follows. Detergent-solubilized postnuclear extracts of transfected HEK293 cells were applied to a 1D4 matrix. The resin was washed eight times with 1% DM and twice with a solution of 10 mg/mL soybean asolectin (Sigma) in 16 mM CHAPS. RhoPDE was eluted at room temperature with 80 μM 1D4 peptide in the CHAPS/asolectin buffer. The eluate was applied to a Sephadex G-50 gel filtration resin (Sigma) to remove detergent and form liposomes. RhoPDE-containing liposomes were monitored by absorbance at 490 nm and sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE).
Expression and Purification of the Isolated Phospho-diesterase Domain
The portion of the RhoPDE gene corresponding to the phosphodiesterase domain alone (amino acids 386–702), with an added C-terminal six-His tag, was amplified from the original vector using flanking BamHI and NcoI restriction sites and cloned into a pET15b vector for bacterial expression. The plasmid was transformed into T7 Express Competent cells (New England BioLabs). Cells with ampicillin resistance were selected for and grown in 1 L cultures at 37 °C with orbital shaking until an OD of 0.6–0.8 was reached, at which point expression was induced with 1 mM isopropyl β-D-1-thiogalactopyranoside. After being expressed for 3 h at 37 °C, cells were pelleted by centrifugation and resuspended in 50 mL of lysis buffer [50 mM Tris (pH 7.5), 100 mM NaCl, and 20 mM imidazole] containing 15 mg of PMSF and 0.5 μL of DNaseI (250 units/μL; Pierce Universal Nuclease). The cells were lysed by sonication on ice using a protocol with 20 s on and 20 s off for a total of 4 min with a Misonix Sonicator 3000 instrument at a power setting of 7, and the soluble fraction was isolated by centrifugation and filtered through a 0.22 μm syringe filter before being loaded onto a prepacked 5 mL HiTrapFF Ni-Sepharose column for purification. Protein was monitored by absorbance at 280 nm, and purity was assessed by SDS–PAGE. The protein was concentrated on a 10 kDa Amicon centrifugal filter and further purified for crystallography by size exclusion chromatography using an AKTA FPLC system equipped with a Superdex-200 10/300 GL column.
Phosphodiesterase Activity Assay: Nucleotide Detection by High-Performance Liquid Chromatography (HPLC)
A discontinuous HPLC-based assay was used to measure the enzymatic activity of HEK293 cell membranes and liposomes containing RhoPDE as well as the soluble PDE domain purified from E. coli. Membranes were incubated with 10 μM ATR on ice for 30 min in the dark before being diluted to a final concentration of 50 nM in assay buffer [50 mM Tris (pH 7.5), 50 mM NaCl, 10 mM MgCl2, and 0.5 mM EDTA]. The reactions took place at room temperature, and were initiated in the dark with the addition of 5 mM cGMP or cAMP. Aliquots (20 μL) were withdrawn at the indicated time points and reactions quenched with 20 μL of 1 N HCl. The remaining reaction mixture was then exposed to light from a 300 W tungsten bulb with a 435 nm cut-on filter [475 nm cut-on filter in the case of liposomes (Figure 5D,E)], and 20 μL aliquots were again withdrawn and reactions quenched with 20 μL of 1 N HCl. In some cases, the mixture was returned to the dark, and additional time points were taken for analysis. Precipitated protein was removed from the samples by filtration through 0.22 μm Corning Costar Spin-X centrifugal filters (Fisher Scientific), and the remaining mixture was neutralized with 20 μL of 1 M potassium phosphate buffer (pH 8.0). Separation and detection of nucleotides were performed on an Agilent 1260 Infinity HPLC system. Samples (5 μL each) were injected onto a 250 mm × 2.1 mm ACE 5 C18-AR reverse-phase column and separated using a 100 mM potassium phosphate (pH 6.2) mobile phase at a flow rate of 0.4 mL/min. Nucleotides were detected by absorbance at 254 nm and quantified by peak integration using OpenLab CDS Chem-Station software. The activity assays depicted in Figure 5 were performed with RhoPDE containing only the C-terminal 1D4 tag (i.e., no C8 tag). The membrane assay in Figure 6B was performed with the C8/1D4 doubly tagged protein from cells grown in the presence of ATR and did not require additional ATR. The soluble PDE domain was assayed in the same manner but also without the need for addition of a chromophore or time points taken in the dark.
Figure 5.
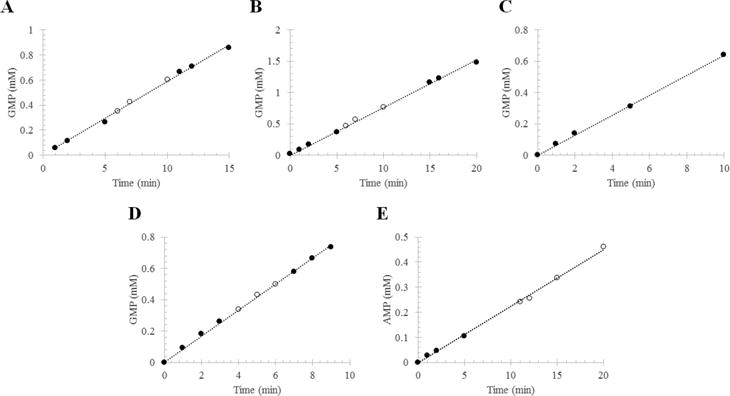
Phosphodiesterase activity of RhoPDE from transiently transfected HEK293-GnT1− cells. The reaction mixture contained RhoPDE in membranes isolated from transfected cells or in liposomes reconstituted with protein that had been purified from transfected cells. The RhoPDE used in this figure contained only the C-terminal 1D4 tag (i.e., no C8 tag). The formation of 5′-GMP from cGMP (or 5′-AMP from cAMP) was followed by HPLC: (●) reaction in the dark and (○) reaction in the light. Integrated peaks corresponding to GMP (or AMP) at each time point were converted into concentrations with a standard curve and plotted vs time. Panels A and B show two representative reactions with RhoPDE (50 nM) in HEK293 membranes reconstituted with ATR. (A) kcat = 19.6 ± 0.2 s−1; (B) kcat = 25.4 ± 0.2 s−1. (C) Reaction with RhoPDE (50 nM) in HEK293 membranes not reconstituted with ATR. This reaction was performed in the absence of light from the 300 W tungsten source. kcat = 21.3 ± 0.2 s−1. (D) cGMP phosphodiesterase actvity of RhoPDE (50 nM) in reconstituted liposomes. kcat = 27.7 ± 0.2 s−1. (E) cAMP phosphodiesterase actvity of RhoPDE (100 nM) in reconstituted liposomes. kcat = 3.7 ± 0.1 s−1.
Crystallization of the Phosphodiesterase Domain
Protein in elution buffer [50 mM Tris (pH 7.5), 100 mM NaCl, and 2 mM sodium azide] was concentrated to 10 mg/mL and mixed with Hampton sparse matrix screens (Hampton Research) in a 1:1 ratio. Crystallization trials were performed with a Phenix robot (Art Robbins Instruments) using the sitting drop vapor diffusion method in a 96-well INTELLI-PLATE. After two months, a needle-shaped crystal appeared under 100 mM succinic acid and 15% (w/v) polyethylene glycol 3350 at 4 °C and grew to maximum dimensions of 0.315 mm × 0.105 mm × 0.075 mm.
Data Collection, Processing, and Refinement
The crystal was transferred briefly to fresh crystallization solution containing 15% glycerol before being frozen under liquid nitrogen. Data were collected at beamline 8.2.1 at the Advanced Light Source (Lawrence Berkeley National Laboratory, Berkeley, CA) using ADSC Q315R CCD detectors (Area Detector Systems Corp.) at 100 K. The crystal diffracted to 2.3 Å resolution. The data were integrated using iMosflm version 7.219 and scaled using SCALA version 3.320 from the CCP4 software suite version 7.0.21,22 The crystal diffraction data were processed in the P212121 space group with the following unit cell dimensions: a = 74.7 Å, b = 96.4 Å, c = 108.6 Å, and α = β = γ = 90°. Complete data collection statistics are listed in Table 1.
Table 1.
Crystallographic Data Collection and Refinement Statistics (PDB entry 5VYD)
| Data Collectiona | |
| space group | P212121 |
| resolution range (Å) | 20–2.3 |
| highest-resolution shell (Å) | 2.42–2.30 |
| unit cell parameters (Å) | a = 74.7 |
| b = 96.4 | |
| c = 108.6 | |
| total no. of reflections | 425946 |
| no. of unique reflections | 34788 |
| completeness (%) | 98.0 (94.8) |
| CC1/2 | 0.99 (0.93) |
| Rmerge (%) | 12.9 (79.4) |
| I/σ(I) | 12.7 (2.8) |
| redundancy | 12.2 (11.6) |
| Refinement | |
| resolution range (Å) | 20–2.3 |
| no. of reflections used | 34395 |
| Rcryst (%) | 21.6 |
| Rfree (%) | 24.6 |
| no. of protein atoms | 5148 |
| no. of metal atoms | 4 |
| no. of water molecules | 288 |
| root-mean-square deviation for bond lengths (Å) | 0.002 |
| root-mean-square deviation for bond angles (deg) | 0.5 |
| Molprobity score | 1.92 |
Values for the highest-resolution shell are given in parentheses.
The structure of the PDE domain was determined by molecular replacement using BALBES23 from the CCP4 software suite version 7.0.21,22 The best solution was based on PDE 1B from Homo sapiens [Protein Data Bank (PDB) entry 1TAZ] and the PDE amino acid sequence (NCBI Gene ID 16078606). The molecular replacement solution found two protein monomers in the asymmetric unit. The structure was initially refined to starting R and Rfree values of 0.311 and 0.362, respectively. Refinements were performed using the function phenix.refine24 in the PHENIX software suite version 1.1125 with the initial two positional refinements preceded by rigid body refinement. Simulated annealing was also included in earlier refinements to minimize the initial model bias. All model rebuilding was performed using COOT version 0.8.26 Spherical solvent peaks greater than 3σ Fo − Fc and 1σ 2Fo − Fc were identified, and water molecules were modeled and included in the final rounds of refinement. The final structure was refined to R and Rfree values of 0.216 and 0.246, respectively. The refinement statistics are listed in Table 1. Coordinates and structure factors have been deposited as PDB entry 5VYD. All crystal structure figures were prepared using PyMol version 1.8 (Schrödinger LLC, Portland, OR).
RESULTS
Expression and Purification of RhoPDE
RhoPDE was expressed in transiently transfected HEK293 GnT1−1 cells and purified from detergent-solubilized cell extracts by immunoaffinity chromatography. The RhoPDE gene construct employed two epitope tags, an N-terminal C8 tag (PRGPDRPEGIEE)9 and a mutant C-terminal 1D4 tag (TATSQVAPA),10,11 which could be exploited for purification using immunoaffinity chromatography with either antibody alone or in tandem to isolate the full-length protein free of proteolysis products. Purification of the protein was followed by SDS–PAGE and Western blot analysis using the C8 antibody (Figure 2).
Figure 2.
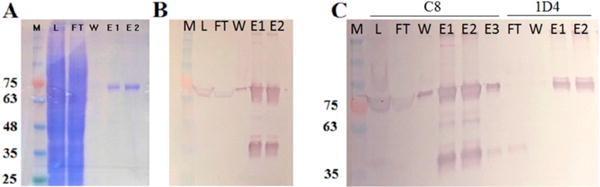
SDS–PAGE and Western blot analysis of fractions from immunoaffinity purification of RhoPDE from HEK293 cells. (A) Coomassie-stained gel and (B) C8-probed Western blot of C8-purified RhoPDE: lane M, molecular mass markers in kilodaltons; lane L, load, postnuclear supernatant fraction; lane FT, flow-through, not bound to the C8 column; lane W, final wash before the first elution with the C8 peptide; lane E1, first elution with the C8 peptide; lane E2, second elution with the C8 peptide. (C) C8-probed Western blot of tandem C8- and 1D4-purified RhoPDE. Lanes from left to right: lane M, molecular mass markers; lane L, load, postnuclear supernatant fraction; lane FT, flow-through, not bound to the C8 column; lane W, final wash before the first elution with the C8 peptide; lane E1, first elution with the C8 peptide; lane E2, second elution with the C8 peptide; lane E3, third elution with the C8 peptide; lane FT, flow-through, nonbound fraction from E1–E3 from the C8 column applied to the 1D4 column; lane W, final wash before the first elution with the 1D4 peptide; lane E1, first elution with the 1D4 peptide; lane E2, second elution with the 1D4 peptide. Note that these experiments were optimized to yield a high concentration of RhoPDE in the final eluate and not for a high yield of protein from the HEK293 cells. Thus, the C8 column was overloaded in these examples where excess RhoPDE can be observed in Western blots of the FT lanes. In addition, it must be kept in mind that the Western blots in this figure are significantly overloaded with protein levels well outside of the linear detection range (the same amount of material was loaded on the Western blots as was loaded on the Coomassie-stained gel). This can be seen most readily from the truncated Rho domain (migrating at approximately 35–40 kDa) observed in both Western blots of panels B and C, but not visible in the Coomassie-stained gel in panel A.
The purified RhoPDE protein contains a major component that migrates on gels with an electrophoretic mobility corresponding to a molecular mass of ~75 kDa, consistent with that expected for the FL protein (Figure 2A, lanes E1 and E2). This band also reacts with the C8 antibody on Western blots (Figure 2B, lanes E1 and E2), confirming that it is RhoPDE. The C8-purified protein contains small amounts of a contaminant with an apparent molecular weight of 35–40 kDa observed in C8 Western blots (Figure 2B, lanes E1 and E2) that is likely a proteolytic fragment corresponding to the rhodopsin domain truncated along its C-terminus such that it is missing the PDE domain.
While the truncated rhodopsin domain is a minor impurity (it is not observed in the Coomassie-stained gel of Figure 2A, lanes E1 and E2), we can remove it and ensure purification of the full-length RhoPDE by running sequentially the two immunoaffinity columns (Figure 2C). When purification on the C8 column is followed by purification on the 1D4 column, the sequential protocol results in highly purified full-length protein (Figure 2C, 1D4 lanes E1 and E2). The fact that the purified protein reacts with both the C8 and 1D4 antibodies ensures that the purified product is full-length RhoPDE.
In a typical protein preparation, we begin with ten 15 cm plates of transfected HEK293 cells containing an estimated 40 μg of RhoPDE per plate (this is roughly the same molar expression level that we see for bovine rhodopsin14 and half the level we observe for RhoGC15) for a total of 0.4 mg of RhoPDE in the postnuclear supernatant fraction from the detergent-solubilized cells. The yield of protein from the first C8 column is ~200 μg of RhoPDE (based upon absorbance at 490 nm). This is fairly typical in that the yield from either the 1D4 or C8 column for either RhoGC or RhoPDE is generally ~50%, with most of the lost material remaining bound to the column in a state recalcitrant to elution with the synthetic peptides.15 Application of the C8-purified sample to a 1D4 column provides roughly 100 μg of RhoPDE, again with a yield from the antibody column of ~50% of the material applied.
Absorption Spectroscopy
The UV–vis absorption spectrum of purified RhoPDE reconstituted with ATR exhibits a visible absorption maximum at 490 nm (extinction coefficient of 45500 M−1 cm−1) with shoulder at 460 nm (Figure 3A–D). The ratio of absorbance at 280 nm to that at 490 nm is 4. After the sample had been exposed to room lights for 30 s, the intensity of the peak at 490 nm decreases and a new peak appears at 380 nm (Figure 3E), consistent with deprotonation of the Schiff base. When the protein is denatured with concentrated HCl, both peaks shift to 440 nm, characteristic of a protonated Schiff base, indicating that each peak represents a form of RhoPDE with a covalently bound chromophore.17
Figure 3.
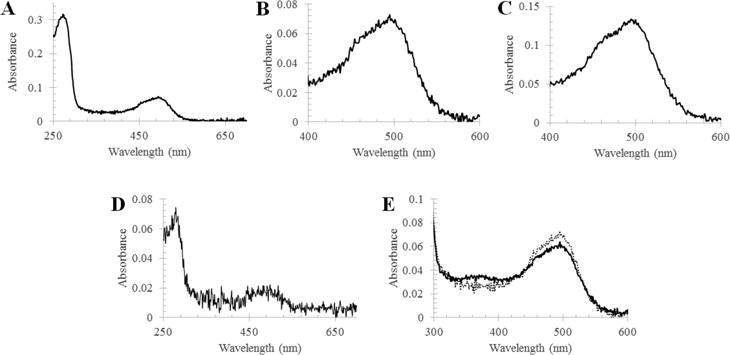
UV–vis absorption spectra of purified RhoPDE from HEK293 cells. (A) Spectrum of the C8-purified pigment with ATR in the dark state. (B) Expanded visible region of the dark state spectrum in panel A. (C) Visible region spectrum of the 1D4-purified pigment with ATR in the dark state. (D) Spectrum of the tandem C8- and 1D4-purified pigment with ATR in the dark state. (E) Visible region of the spectrum in panel A before (dotted line) and after (solid line) a 30 s exposure to room lights.
Membrane Orientation of RhoPDE
Though opsins typically have seven transmembrane α-helices,1,2 with the N-terminal domain on the extracellular face, hydropathy analysis of RhoPDE predicted an eighth helix with both N- and C-termini being intracellular (Figure 1). To differentiate between these two topologies, we expressed the C8- and 1D4-tagged RhoPDE construct in COS-7 cells immobilized on glass slides for immunofluorescence microscopy experiments. As shown in Figure 4, probing intact cells with the C8 or 1D4 primary antibody and a fluorescent secondary antibody gives no signal, indicating that neither epitope is accessible. Conversely, when the cells were first permeabilized with Triton X-100 and treated with the same antibodies, strong fluorescence is observed in the cell membranes. This result is consistent with an even number of transmembrane segments with N- and C-termini of the protein both located on the cytoplasmic surface of the plasma membrane.
Figure 4.
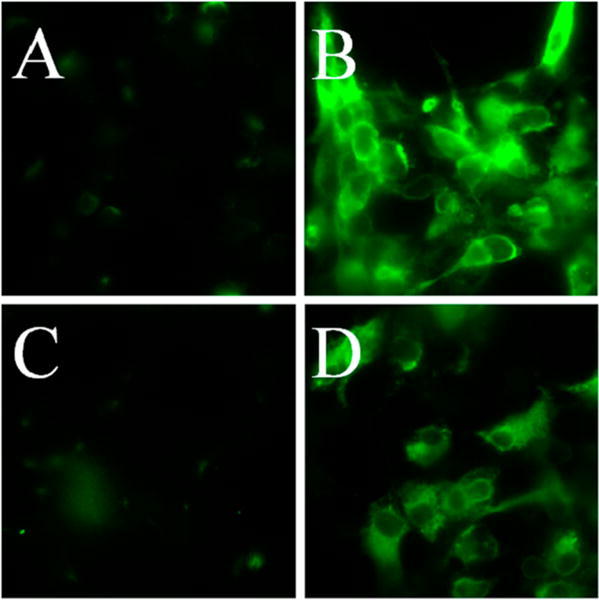
Determination of the RhoPDE transmembrane topology and orientation. Immobilized COS-7 cells were transfected with the dual (C8 and 1D4) epitope-tagged RhoPDE on glass coverslips and incubated with the antibodies under permeabilized or unpermeabilized conditions for immunofluorescent staining, as indicated in the figure and described in Experimental Procedures. Panels A and B show intact and detergent-permeabilized cells, respectively, probed with the 1D4 antibody. Panels C and D show intact and detergent-permeabilized cells, respectively, probed with the C8 antibody. Fluorescence from either probe is observed only in permeabilized cells, indicating both epitopes are intracellular. The COS-7 cells in panels B and D show a diffuse staining pattern typical of proteins localized to the plasma membrane.
Phosphodiesterase Activity of RhoPDE in Membranes
Membrane extracts of HEK293 cells expressing RhoPDE were assayed for cGMP phosphodiesterase activity. As shown in panels A and B of Figure 5, the enzyme is active in the dark, and no change in rate was observed after exposure to light. The fact that the enzyme displays robust constitutive phosphodiesterase activity independent of light is reinforced by the results depicted in Figure 5C, where the same membranes are assayed for cGMP phosphodiesterase activity but in the absence of added ATR. The membranes display activity similar to the activity of those in the presence of ATR, indicating clearly that the retinal chromophore in the type I rhodopsin domain is not regulating the activity of the phosphodiesterase domain. Similar results were obtained with liposomes reconstituted with purified RhoPDE and assayed for both cGMP (Figure 5D) and cAMP (Figure 5E) phosphodiesterase activity. RhoPDE is roughly 5–7 times less active with cAMP as a substrate than with cGMP.
Membranes from Cells Grown in the Presence of ATR
Membranes were also isolated from cells grown in the presence of ATR and compared to those grown in the absence of the chromophore. As shown in Figure 6A, the yield of RhoPDE purified from the two sets of membranes was the same, indicating that there is no need to express the protein in the presence of ATR. In addition, the cGMP phosphodiesterase with membranes from cells grown in the presence of ATR displayed the same robust constitutive activity with no change upon exposure to light (Figure 6B) as observed for membranes from cells grown in the absence of ATR (Figure 5).
Purification and Activity of the Isolated Phosphodiesterase Domain
Initial efforts to understand the interaction of domains within RhoPDE focused on expression and purification of the isolated PDE domain of the protein. The PDE domain was strongly overexpressed in E. coli and purified by Ni column affinity chromatography. As shown in the Coomassie-stained SDS–PAGE gel of Figure 7A, the protein was purified to near homogeneity in high yield, migrating as a protein slightly larger than 35 kDa, consistent with the expected molecular mass of 37808 Da. The protein in solution is a dimer, as shown in the FPLC profile of Figure 7B, as is typical of cyclic nucleotide phosphodiesterases.27 In a representative purification, 15 mg of the purified PDE domain is obtained from 1 L of cell culture. The isolated domain was then assayed for constitutive cGMP phosphodiesterase activity using the HPLC protocol described above for HEK293 cell membrane preparations (Figure 7C). While the isolated domain displayed significant activity, the kcat calculated from this experiment (3.13 ± 0.12 s−1) is roughly 6–9-fold lower than that recorded for full-length RhoPDE (Figure 5).
Figure 7.
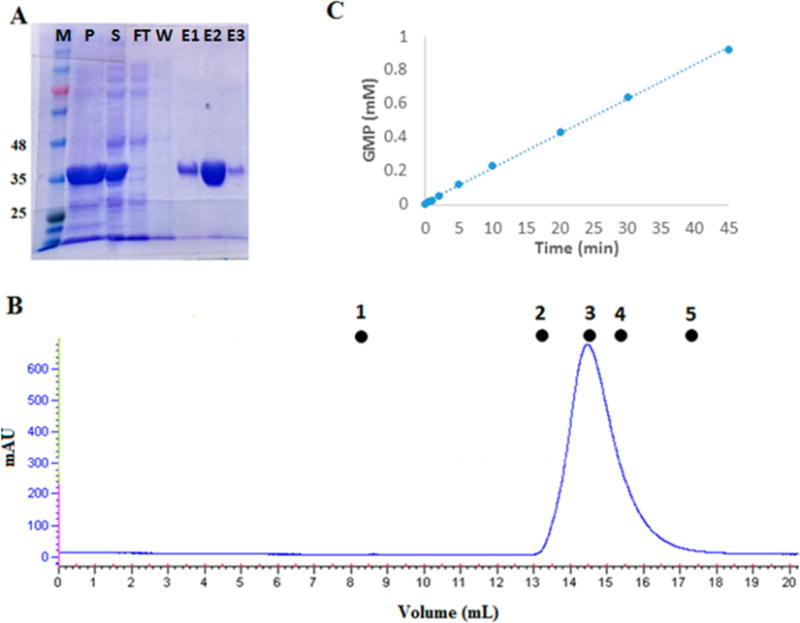
Phosphodiesterase activity of the isolated PDE domain purified from transformed E. coli T7 Express cells. (A) Coomassie-stained SDS–PAGE gel showing fractions from purification of the isolated PDE domain from transformed E. coli T7 Express cells using a six-His tag with a Ni affinity column: lane M, molecular mass markers in kilodaltons; lane P, pellet from the cell extract; lane S, soluble fraction from the cell extract; lane FT, flow-through, nonbound fraction from the Ni affinity column; lane W, last wash before elution with an imidazole gradient; and lanes E1–E3, three representative fractions from the imidazole gradient eluate. (B) AKTA FPLC profile for size exclusion chromatography of the isolated PDE domain on a Superdex-200 10/300 GL column. Molecular mass standards: (1) blue dextran, 2000 kDa, 8.5 mL (void volume); (2) aldolase, 158 kDa, 13.15 mL; (3) albumin, 67 kDa, 14.57 mL; (4) ovalbumin, 43 kDa, 15.41 mL; and (5) chymotrypsinogen, 25 kDa, 17.35 mL. The isolated PDE domain elutes at 14.5 mL, giving a mass of 74 kDa, consistent with a homodimer. (C) HPLC assay of the cGMP phosphodiesterase activity of the isolated PDE domain. The concentration of PDE in this assay was 100 nM, which gives an apparent kcat of 3.13 ± 0.12 s−1.
Crystal Structure of the Phosphodiesterase Domain Dimer
The isolated PDE domain was crystallized in space group P212121 from a PEG 3350 solution. The structure of the enzyme was determined to 2.3 Å resolution by molecular replacement using PDE 1B (PDB entry 1TAZ) as a search model. Two molecules were found in the asymmetric unit (Figure 8), with clear electron density observed for 314 residues (L22–H335) in monomer A and 317 residues (D18–M334) in monomer B. Both monomers are composed of 16 α-helices with highly similar overall structure [root-mean-square deviation (rmsd) of Cα atoms of 0.2 Å].
Figure 8.
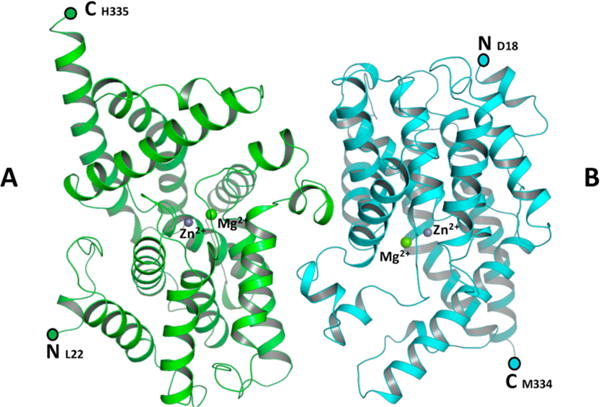
Structure of the PDE domain homodimer. The figure shows a ribbon diagram of molecules A and B of the dimer, colored green and cyan, respectively. Zn2+ and Mg2+ ions are modeled as purple and green spheres, respectively. N- and C-termini are marked.
Although no metal ion was included in any of the purification buffers, two difference Fourier peaks (18σ and 8σ in the Fo − Fc map) were observed in the active site of each monomer after the initial refinement. The highest peak was modeled as a zinc ion, while the second peak was modeled as a magnesium ion (Figure 9). The B factors of the metal ions were comparable to those of the coordinating ligands in the active site. Water molecules coordinating with metal ions were modeled before the final refinement. Both Zn2+ and Mg2+ ions display octahedral coordination geometry, similar to other previously reported PDE structures.28–30
Figure 9.
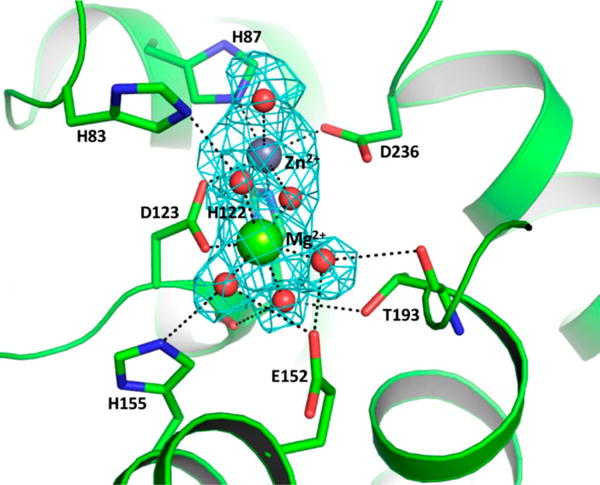
Active site of PDE monomer A, showing metal ion coordination, water molecules, and hydrogen bonds to active site residues. The figure shows an omit map (Fo − Fc = 3σ) with electron density for the Zn2+ and Mg2+ ions in the active site of monomer A. The water molecules and Zn2+ and Mg2+ ions are modeled as red, purple, and green spheres, respectively.
Comparison with the Human PDE9 Structure
The sequence and overall structure of the isolated PDE domain closely resemble those of human cGMP-specific PDE9 (PDB entry 2HD1) in complex with the nonselective inhibitor 3-isobutyl-1-methylxanthine (IBMX).29 While superposition of the two structures (Figure 10) reveals that the rmsd of Cα atoms between respective monomers is 0.9 Å, a few differences are observed; in particular, helices H5, H7, and H12–H14 are tilted outward slightly in the isolated PDE domain, compared to PDE9, as they extend from the active site to the outermost surface of the protein.
Figure 10.
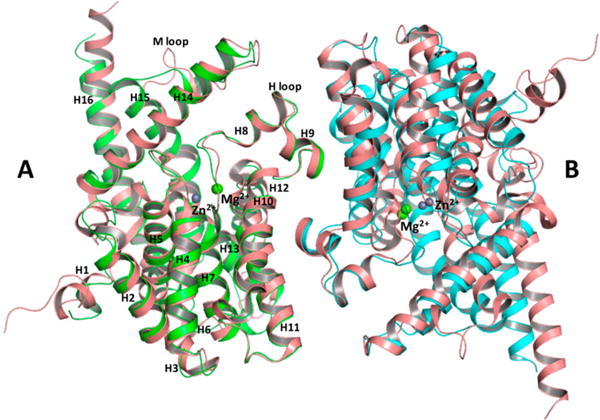
Superposition of PDE and PDE9 (PDB entry 2HD1) dimers using monomer A as a reference molecule. Monomers A and B of PDE are colored green and cyan, respectively. Both monomers of PDE9 are colored salmon. Zn2+ (purple) and Mg2+ (green) ions are modeled as spheres. Helices of monomer A are labeled according to the PDE9 model.
Dimer Interface
Residues of the dimer interface are highly conserved between PDE and PDE9. The isolated PDE domain forms a homodimer with a total contact area of 4018 Å2 for monomer A and 4109 Å2 for monomer B. PDE9 forms a similar interface with a total contact area of 4397 Å2 for monomer A and 4410 Å2 for monomer B. Superimposition of the two monomers reveals a 15.5° rotation and a 6.2 Å displacement of monomer B of PDE with respect to PDE9, causing slight differences in interfacial interactions between the two subunits in the dimer interface. The isolated PDE domain dimer interface has many of the same interactions that are found in PDE9 but has six additional hydrogen bonds (Table S1) and six additional van der Waals interactions. At this point, we do not know if the rotation of subunits seen in a comparison of the dimer interface between the two proteins is a result of inherent differences in the two proteins, a result of differences in crystal packing, or a result of binding of IBMX to PDE9.
DISCUSSION
We have developed an expression and purification system for RhoPDE to facilitate further structural and biochemical studies of this unique fusion protein in the hope that such studies will help realize the full potential of RhoPDE as an optogenetic tool for the control of cyclic nucleotide levels in cell signaling systems. RhoPDE was expressed in HEK293-GnT1− cells and purified in a detergent solution using two different epitope tags: an N-terminal C8 tag and a C-terminal 1D4 tag. The protein was purified in high yield from the C8 tag alone with trace amounts of proteolytic contaminants. These were then removed with subsequent purification on a 1D4 column. The yield of protein from the 1D4 column was increased significantly by utilizing a E8′A mutant 1D4 epitope tag, which lowers the protein’s affinity for the antibody enough that it can be eluted efficiently from the resin with the wild-type peptide. Tandem purification with both antibodies, which we have used previously15 to purify a rhodopsin/guanylyl cyclase fusion protein (RhoGC) found in the aquatic fungus B. emersonii, ensures that the full-length protein is purified.
The doubly tagged construct was also used to determine the transmembrane topology of RhoPDE expressed in COS-7 cells. While most type I and type II rhodopsins are composed of seven transmembrane α-helical segments, hydropathy analysis in Protter8 suggested that RhoPDE has eight transmembrane helices, with N- and C-termini both on the cytoplasmic side of the membrane (Figure 1). Immunofluorescence staining of the COS-7 cells provided strong evidence in support of this model. No immunofluorescence was detected with either the 1D4 or C8 antibodies until cells expressing RhoPDE were first permeabilized with Triton X-100. The antibodies then reacted with the protein, displaying a pattern typical of cell surface expression in the plasma membrane. These data are similar to those obtained for the related fusion protein RhoGC from B. emersonii, which is also predicted to have eight transmembrane helices by hydropahy analysis and has been shown experimentally to have an even number of transmembrane segments with N- and C-termini on the cytoplasmic surface of the plasma membrane.6,15 Thus, both fusion proteins share this structural feature.
The purified protein reconstituted with ATR has a light orange color with a dark state absorption maximum at 490 nm and a shoulder at 460 nm. The absorption spectrum is reminiscent of those for channelrhodopsin31 and sensory rhodopsin II32,33 and interestingly is highly similar to the spectrum of the RhoGC E254D spectral tuning mutant.15 Upon being exposed to light, RhoPDE forms a species with a maximum at 380 nm, but it is unclear at this point how exactly this species relates to the activity of the protein (vide infra).
While the spectrum of RhoPDE undergoes a shift with light, surprisingly the phosphodiesterase activity of the protein appears not to be modulated by illumination. The protein displays robust cGMP phosphodiesterase activity in the light, in the dark, and in fact even without addition of the retinal chromophore. The isolated PDE domain also displays constitutive cGMP phosphodiesterase activity, although in this case the activity is roughly 6–9-fold lower than that observed with the full-length protein. The lack of dependence on light is certainly unexpected for this unusual fusion protein and raises a number of questions about what the underlying explanation could be. It seems unlikely that nature would produce a rhodopsin/phosphodiesterase fusion protein in which the phosphodiesterase domain was not in some way modulated by light. Two possible explanations immediately suggest themselves. (1) The enzyme lacks a post-translational modification that functionally couples the rhodopsin and phosphodiesterase domains. (2) The heterologously expressed and purified RhoPDE is missing an interaction partner from S. rosetta that confers light dependence on the PDE domain. Future efforts will be directed at resolving this issue and developing RhoPDE to its full potential as an optogenetic tool.
Very recently, Yoshida et al.34 published a study of this same protein from S. rosetta, the only other paper published on this protein. There are several similarities between their study and ours. Yoshida et al.34 report heterologous expression of the protein in HEK293 cells and purification by immunoaffinity chromatography using an epitope tag and the 1D4 antibody; the absorption spectrum after addition of ATR, with a maximum at 490 nm and a shoulder at 460 nm; the phosphodiesterase activity that is selective for cGMP over cAMP; and, finally, the phosphodiesterase activity, for both cGMP and cAMP, that is constitutive and present without exposure to light. These results are also reported here. Yoshida et al.34 also report a small amount of light-dependent phosphodiesterase activity under in vitro conditions with membranes isolated from transfected HEK293 cells. While we were unable to detect light dependence, even under conditions identical to those of Yoshida et al. (Figure 6), we do not view this as a significant difference between the two studies. We emphasize that their reported amount of light activation is very small (1.4-fold), and the dominant phenotype in both studies is of a protein that displays high constitutive cGMP phosphodiesterase activity.
In addition, we show that the protein is composed of an even number (likely eight) of transmembrane segments resulting in the N- and C-termini both being located on the cytoplasmic surface of the plasma membrane, and we determined the X-ray crystal structure of the isolated PDE domain, opening an avenue for a structure–function exploration of the mechanism for regulation of phosphodiesterase activity in this domain of the protein.
In conclusion, we successfully expressed and purified a type I rhodopsin/cGMP-selective phosphodiesterase fusion protein, RhoPDE, described originally in the choanoflagellate S. rosetta. We show that the protein has an even number of transmembrane segments, placing the N- and C-termini on the cytoplasmic surface of the plasma membrane. When reconstituted with ATR, the protein displays a visible spectrum with a maximum at 490 nm and a shoulder at 460 nm. The protein is constitutively active, showing robust phosphodiesterase activity with undetectable regulation by light under the in vitro conditions of our experiments. Interestingly, the isolated PDE domain was also active, albeit significantly less so than the full-length protein. The X-ray crystal structure of the isolated PDE domain is a dimer that is significantly similar to that of the human phosphodiesterase PDE9. The two monomers of the isolated PDE domain are rotated slightly at the dimer interface in comparison to the PDE9 structure, but it is not known at this time if that is an inherent difference in the two proteins or whether it reflects a conformational change induced by the IBMX ligand in the PDE9 structure. Future studies will focus on regulation of the activity of the PDE domain, expanding to include domain constructs containing the linker region connecting the PDE and rhodopsin domains. We anticipate that additional work will help realize the full optogenetic potential of this unique and interesting enzyme.
Supplementary Material
Acknowledgments
We thank George Cohen for help with the experiments depicted in Figure 6. We are grateful to the staff at the Advanced Light Source-Berkeley Center for Structural Biology for their assistance during X-ray data collection. The Advanced Light Source is funded by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract DE-AC02-05CH11231. The Berkeley Center for Structural Biology is supported in part by grants from the National Institute of General Medical Sciences.
Funding
This work was supported by National Institutes of Health Grants T32GM007596 (E.L.D.), GM094468 and GM096053 (D.L.T.), and EY007965 (D.D.O.). A special thanks goes to The Adar Family and Friends for their continued support throughout this work. We also acknowledge support from The Brandeis University Provost’s Research Fund.
ABBREVIATIONS
- RhoPDE
rhodopsin/phosphodiesterase fusion protein
- PDE
phosphodiesterase domain
- ATR
all-trans-retinal
- DM
n-decyl β-D-maltopyranoside
- PBS
phosphate-buffered saline (pH 7.0)
- cGMP
3′,5′-cyclic guanosine monophosphate
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b00519.
Interfacial hydrogen bonding distances between monomer atoms (Table S1) (PDF)
Accession Codes
The atomic coordinates and structure factors have been deposited as Protein Data Bank entry 5VYD.
ORCID
Lindsey B. Lamarche: 0000-0002-7200-3150
Ramasamy P. Kumar: 0000-0002-6555-8289
Daniel D. Oprian: 0000-0002-6520-5459
Notes
The authors declare no competing financial interest.
References
- 1.Ernst OP, Lodowski DT, Elstner M, Hegemann P, Brown LS, Kandori H. Microbial and animal rhodopsins: structures, functions, and molecular mechanisms. Chem Rev. 2014;114:126–163. doi: 10.1021/cr4003769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spudich JL, Yang CS, Jung KH, Spudich EN. Retinylidene proteins: Structures and functions from archaea to humans. Annu Rev Cell Dev Biol. 2000;16:365–392. doi: 10.1146/annurev.cellbio.16.1.365. [DOI] [PubMed] [Google Scholar]
- 3.Smith SO. Structure and Activation of the Visual Pigment Rhodopsin. Annu Rev Biophys. 2010;39:309–328. doi: 10.1146/annurev-biophys-101209-104901. [DOI] [PubMed] [Google Scholar]
- 4.Palczewski K. Chemistry and Biology of Vision. J Biol Chem. 2012;287:1612–1619. doi: 10.1074/jbc.R111.301150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Avelar GM, Schumacher RI, Zaini PA, Leonard G, Richards TA, Gomes SL. A rhodopsin-guanylyl cyclase gene fusion functions in visual perception in a fungus. Curr Biol. 2014;24:1234–1240. doi: 10.1016/j.cub.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao S, Nagpal J, Schneider MW, Kozjak-Pavlovic V, Nagel G, Gottschalk A. Optogenetic manipulation of cGMP in cells and animals by the tightly light-regulated guanylyl-cyclase opsin CyclOp. Nat Commun. 2015;6:8046. doi: 10.1038/ncomms9046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scheib U, Stehfest K, Gee C, Korschen H, Fudim R, Oertner T, Hegemann P. The rhodopsin-guanylyl cyclase of the aquatic fingus Blastocladiella emersonii enables fast optical control of cGMP signaling. Sci Signaling. 2015;8:RS8. doi: 10.1126/scisignal.aab0611. [DOI] [PubMed] [Google Scholar]
- 8.Omasits U, Ahrens CH, Muller S, Wollscheid B. Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics. 2014;30:884–886. doi: 10.1093/bioinformatics/btt607. [DOI] [PubMed] [Google Scholar]
- 9.Strassmaier T. HIV Membrane Fusion: Analysis of Novel Inhibitors Ph D Thesis. Brandeis University; Waltham, MA: 2002. [Google Scholar]
- 10.Molday RS, MacKenzie D. Monoclonal antibodies to rhodopsin: characterization, cross-reactivity, and application as structural probes. Biochemistry. 1983;22:653–660. doi: 10.1021/bi00272a020. [DOI] [PubMed] [Google Scholar]
- 11.Oprian DD, Molday RS, Kaufman RJ, Khorana HG. Expression of a synthetic bovine rhodopsin gene in monkey kidney cells. Proc Natl Acad Sci U S A. 1987;84:8874–8878. doi: 10.1073/pnas.84.24.8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D’Antona AM, Xie G, Sligar SG, Oprian DD. Assembly of an activated rhodopsin-transducin complex in nanoscale lipid bilayers. Biochemistry. 2014;53:127–134. doi: 10.1021/bi4012995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie G, D’Antona AM, Edwards PC, Fransen M, Standfuss J, Schertler GF, Oprian DD. Preparation of an activated rhodopsin/transducin complex using a constitutively active mutant of rhodopsin. Biochemistry. 2011;50:10399–10407. doi: 10.1021/bi201126r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Devine EL, Oprian DD, Theobald DL. Relocating the active-site lysine in rhodopsin and implications for evolution of retinylidene proteins. Proc Natl Acad Sci U S A. 2013;110:13351–13355. doi: 10.1073/pnas.1306826110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trieu MM, Devine EL, Lamarche LB, Ammerman AE, Greco JA, Birge RR, Theobald DL, Oprian DD. Expression, Purification, and Spectral Tuning of RhoGC, a Retinylidene/Guanylyl Cyclase Fusion Protein and Optogenetics Tool from the Aquatic Fungus Blastocladiella emersonii. J Biol Chem. 2017;292:10379–10389. doi: 10.1074/jbc.M117.789636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plant LD, Dementieva IS, Kollewe A, Olikara S, Marks JD, Goldstein SA. One SUMO is sufficient to silence the dimeric potassium channel K2P1. Proc Natl Acad Sci U S A. 2010;107:10743–10748. doi: 10.1073/pnas.1004712107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fasick JI, Lee N, Oprian DD. Spectral tuning in the human blue cone pigment. Biochemistry. 1999;38:11593–11596. doi: 10.1021/bi991600h. [DOI] [PubMed] [Google Scholar]
- 18.Robinson PR, Cohen GB, Zhukovsky EA, Oprian DD. Constitutively active mutants of rhodopsin. Neuron. 1992;9:719–725. doi: 10.1016/0896-6273(92)90034-b. [DOI] [PubMed] [Google Scholar]
- 19.Battye TGG, Kontogiannis L, Johnson O, Powell HR, Leslie AGW. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr, Sect D: Biol Crystallogr. 2011;67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Evans P. Scaling and assessment of data quality. Acta Crystallogr, Sect D: Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 21.Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta Crystallogr, Sect D: Biol Crystallogr. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 22.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr, Sect D: Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Long F, Vagin AA, Young P, Murshudov GN. BALBES: a molecular-replacement pipeline. Acta Crystallogr, Sect D: Biol Crystallogr. 2008;64:125–132. doi: 10.1107/S0907444907050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr, Sect D: Biol Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, MOriarty NW, Oeffner R, Read RJ, Richardson DJ, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emsley P, Lohkamp B, Scott W, Cowtan K. Features and Development of Coot. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481–511. doi: 10.1146/annurev.biochem.76.060305.150444. [DOI] [PubMed] [Google Scholar]
- 28.Hou J, Xu J, Liu M, Zhao RZ, Luo HB, Ke HM. Structural Asymmetry of Phosphodiesterase-9, Potential Protonation of a Glutamic Acid, and Role of the Invariant Glutamine. PLoS One. 2011;6:e18092. doi: 10.1371/journal.pone.0018092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huai Q, Wang H, Zhang W, Colman RW, Robinson H, Ke H. Crystal structure of phosphodiesterase 9 shows orientation variation of inhibitor 3-isobutyl-1-methylxanthine binding. Proc Natl Acad Sci U S A. 2004;101:9624–9629. doi: 10.1073/pnas.0401120101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verhoest PR, Proulx-Lafrance C, Corman M, Chenard L, Helal CJ, Hou XJ, Kleiman R, Liu SP, Marr E, Menniti FS, Schmidt CJ, Vanase-Frawley M, Schmidt AW, Williams RD, Nelson FR, Fonseca KR, Liras S. Identification of a Brain Penetrant PDE9A Inhibitor Utilizing Prospective Design and Chemical Enablement as a Rapid Lead Optimization Strategy. J Med Chem. 2009;52:7946–7949. doi: 10.1021/jm9015334. [DOI] [PubMed] [Google Scholar]
- 31.Ritter E, Stehfest K, Berndt A, Hegemann P, Bartl FJ. Monitoring light-induced structural changes of Channelrhodopsin-2 by UV-visible and Fourier transform infrared spectroscopy. J Biol Chem. 2008;283:35033–35041. doi: 10.1074/jbc.M806353200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ren L, Martin CH, Wise KJ, Gillespie NB, Luecke H, Lanyi JK, Spudich JL, Birge RR. Molecular mechanism of spectral tuning in sensory rhodopsin II. Biochemistry. 2001;40:13906–13914. doi: 10.1021/bi0116487. [DOI] [PubMed] [Google Scholar]
- 33.Shimono K, Ikeura Y, Sudo Y, Iwamoto M, Kamo N. Environment around the chromophore in pharaonis phoborhodopsin: mutation analysis of the retinal binding site. Biochim Biophys Acta, Biomembr. 2001;1515:92–100. doi: 10.1016/s0005-2736(01)00394-7. [DOI] [PubMed] [Google Scholar]
- 34.Yoshida K, Tsunoda SP, Brown LS, Kandori H. A unique choanoflagellate enzyme rhodopsin exhibits light-dependent cyclic nucleotide phosphodiesterase activity. J Biol Chem. 2017;292:7531–7541. doi: 10.1074/jbc.M117.775569. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.