

Abstract
Bioassay-guided phytochemical investigation of a commercially available maqui berry (Aristotelia chilensis) extract used in botanical dietary supplement products led to the isolation of 16 compounds, including one phenolic molecule, 1, discovered for the first time from a natural source, along with several known compounds, 2–16, including three substances not reported previously in A. chilensis, 2, 14, and 15. Each isolate was characterized by detailed analysis of NMR spectroscopic and HRESIMS data, and tested for their in vitro hydroxyl radical scavenging and quinone-reductase inducing biological activities. A sensitive and accurate LC-DAD-MS method for the quantitative determination of the occurrence of six bioactive compounds, 6, 7, 10–12, and 14, was developed and validated using maqui berry isolates purified in the course of this study as authentic standards. The method presented can be utilized for dereplication efforts in future natural product research projects, or to evaluate chemical markers for quality assurance and batch-to-batch standardization of this botanical dietary supplement component.
Keywords: Aristotelia chilensis, maqui berry, antioxidants, hydroxyl radical-scavenging assay, quinone reductase-inducing assay, phloroglucinaldehyde 2-O-β-D-glucopyranoside, content determination, HPLC-DAD-ESI/MS
TABLE OF CONTENTS GRAPHIC
INTRODUCTION
According to the U.S. Centers for Disease Prevention, cancer is now the second leading cause of mortality in the United States, narrowly trailing the annual mortality attributed to heart disease. Cancer is already the most prevalent cause of deaths of Americans aged 45–64.1 However, many authors have expressed the potential of reducing future cancer deaths, the disease burden, and the economic impact of oncotherapy by the chemoprevention of cancer initiation or progression.2,3 In fact, several drugs have already been approved by the U.S. FDA for the purposeful prevention of some forms of cancers, although these agents have incompletely known mechanisms of action.4,5 However, the induction of quinone reductase has been shown to correlate with other cytoprotective phase II enzymes, making it a suitable in vitro bioassay or “reasonable biomarker for the potential chemoprotective effect of test agents against cancer initiation.”6
Among several health benefits correlated with the incorporation of berry fruits in the diet, positive impacts have been reported on various human health afflictions that include some forms of cancer.7 The bioactive constituents in berries further have been shown to possess several possible roles in cancer chemoprevention, including protection against oxidative DNA damage by scavenging reactive oxygen species, enhancement of carcinogen detoxification processes via induction of phase II detoxifying enzymes, and modulation of signaling pathways involved with cellular inflammation, proliferation, apoptosis, angiogenesis and cell cycle arrest.8 Additionally, the extractives from several berries have been validated in vivo as cancer chemopreventive agents against N-nitrosomethylbenzylamine-induced esophageal cancer in rats, using a standardized protocol, providing additional evidence for the potential health benefits of berry consumption by humans.9
It has been reported that approximately one in six Americans used or had used non-vitamin non-mineral dietary supplements for the general improvement of their diet and health in the year 2012.10 Indeed, the popularity of these products contributed to the observed increase in financial scope of the U.S. dietary supplements market by ten-fold from an estimated $4 billion in 1994 to about $40 billion per year in 2014, marking just two decades since the Dietary Supplements Health and Education Act was passed by the U.S. Congress.11 Included in this economic boom has been the new commercial availability of exotic and/or “super” fruit products, such as those derived from açaí, maqui berry, noni and pomegranate, to name a few. These ingredients are often perceived by consumers to have potent antioxidant capacities, and some have been studied scientifically in a quantitative and/or a qualitative fashion either as complex mixtures or through natural product isolation investigations.
One such dietary supplement ingredient is maqui berry [Aristotelia chilensis (Molina) Stuntz (Elaeocarpaceae)], the fruit of a South American evergreen shrub. The fruit itself is a small dark purple to black berry that can be eaten fresh or used in the preparation of jam, wine, and powdered extracts for consumption.12 Furthermore, A. chilensis fruits and leaves have reported uses in folk medicine, “to treat a variety of ailments, including sore throat, kidney pains, ulcers, fever, hemorrhoids, inflammation, diarrhea, lesions, migraines, and scars”.12 Accordingly, several studies have investigated the in vitro or in vivo biological activities of A. chilensis extracts. For example, a methanolic extract of these fruits was shown to have in vitro antioxidant and in vivo murine cardioprotective activities.13 Later, a polyphenolic-rich fraction of the methanolic extract of A. chilensis was shown to have in vivo antidiabetic activity in a hyperglycemic and obese mouse model.14 The range of in vitro effects observed from maqui berry extracts, as well as the known phytochemical profile of these samples has been extensively reviewed recently.15 However, it is important to recognize that these bioactivities typically have been attributed to anthocyanin and non-anthocyanin polyphenols present in the berries.15,16 Accordingly, HPLC analysis has been previously used to show the presence of bioactive phytochemicals, especially focusing on anthocyanins in maqui berry extract or crude subfractions thereof.16–22 There has only been one report of natural product isolation from A. chilensis fruits, in which the purification of unspecified “individual flavonoids, phenolic acids, anthocyanins and proanthocyanins” was performed, and the biological activity test results for these molecules were not disclosed.23
In the work presented herein, a maqui berry (Aristotelia chilensis) fruit extract used for formulation in dietary supplement products was studied by bioassay-guided fractionation to examine its potential cancer chemopreventive activity. This research yielded 16 pure compounds, including one molecule not previously isolated from any natural source, 1, along with a closely related phloroglucinol glycoside, 2, and two furanaldehydes, 14 and 15, not previously detected in or isolated from maqui berries. Six phenolic compounds, 6, 7, 10–12, and 14, were selected as marker compounds and utilized as authentic standards for the development and validation of an LC-DAD-MS method, because they were relatively abundant in the sample and active in the in vitro hydroxyl radical-scavenging or quinone reductase- inducing bioassays. This analytical method was designed to be complementary to the work of others, described above, and allows for qualitative and quantitative analysis of the selected molecules in maqui berry extracts or other phytochemical samples. These results can thus be utilized for extract standardizations, quality assurance and quality control protocols, and dereplication efforts in natural product drug discovery.
MATERIALS AND METHODS
Instrumentation
Optical rotations were measured on a model 343 polarimeter (PerkinElmer, Waltham, MA) with a path length of 10 mm. A U-2910 spectrophotometer (Hitachi, Tokyo, Japan) was used to record UV spectra, and a Nicolet 6700 FT-IR spectrometer (Thermo Scientific, Waltham, MA) was used to obtain IR spectra. All NMR spectroscopic data were recorded on Avance DRX-400 and 600 MHz spectrometers (Bruker, Billerica, MA) operating at room temperature with standard built-in experiments. A Q-TOF II (Micromass, Wythenshawe, UK) mass spectrometer was operated in the positive-ion mode to obtain high-resolution electrospray ionization mass spectra (HRESIMS), and sodium iodide was used for mass calibration. Column chromatography was performed using Sephadex LH-20 (Supelco, Bellefonte, PA), normal phase silica gel of 65 × 250 or 230 × 400 mesh size (Sorbent Technologies, Atlanta, GA), and 40–63 μm particle size C18-RP silica gel (Acros Organics, Geel, Belgium). Analytical thin-layer chromatography (TLC) was performed using pre-coated 250 μm thickness Partisil Si gel 60F254 glass plates, while preparative TLC was conducted on pre-coated 500 or 1000 μm thickness Partisil Si gel 60F254 glass plates (Whatman, Clifton, NJ). The column used for analytical HPLC was a 150 mm × 4.6 mm i.d., 5 μm, XBridge C18, with a 10 mm × 4.6 mm i.d., 5 μm, guard column of the same material (Waters), while the column used for semi-preparative HPLC was a 150 mm × 10 mm i.d., 5 μm, Sunfire PrepC18, with a 10 mm × 10 mm i.d., 5 μm, guard column of the same material (Waters). These columns were operated with a system (Hitachi, Tokyo, Japan) composed of an L-2130 prep pump, an L-2200 autosampler, and an L-2450 diode array detector.
Chemicals
All solvents used for chromatographic separations (ACS reagent, HPLC, and LC-MS grade) were purchased from Fisher Scientific (Fair Lawn, NJ). Bovine serum albumin (BSA), 2′,7′-dichlorodihydrofluorescin diacetate (H2DCF-DA), digitonin, DMSO, EDTA, esterase, FeSO4, flavin adenine dinucleotide phosphate (FAD), glucose-6-phosphate (G-6-P), glucose-6-phosphate dehydrogenase (G-6-P-D), H2O2, menadione, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), nicotinamide adenine dinucleotide phosphate (NADP), quercetin, L-sulforaphane, Trizma base, and Tween 20, were purchased from Sigma-Aldrich (St. Louis, MO). Cell culture media and supplements were obtained from Life Technologies, Inc. (Grand Island, NY). Deuterated NMR solvents were purchased from Cambridge Isotope Laboratories (Andover, MA).
Plant Material
Air-dried maqui berry (A. chilensis) fruit powder (lot # 09559; sample # 3410), originally collected in Chile, was obtained from Nature’s Sunshine Products, Inc. (Spanish Fork, UT) for this study. A representative sample (OSUADK-CCP0024) has been deposited in the Division of Medicinal Chemistry and Pharmacognosy, College of Pharmacy, The Ohio State University.
Extraction and Isolation
Milled dried fruits of A. chilensis (500 g) were repeatedly extracted by soaking with an excess of MeOH (3 × 1 L) at room temperature. The crude extract (ca. 181 g) was suspended in H2O (1 L) and sequentially partitioned with hexanes (3 × 1 L), CHCl3 (3 × 1 L), and n-BuOH (3 × 1 L) to afford hexanes (53.5 g), CHCl3 (21.9 g), EtOAc (7.0 g), n-BuOH (10.0 g), and H2O (ca. 89 g) partitions, respectively, after drying under vacuum. The EtOAc and n-BuOH partitions were both found to be active in the in vitro hydroxyl radical scavenging assay (ED50 = 0.6 and 1.0 μg/mL, respectively), and were subjected to chromatographic purification.
The EtOAc partition (D3) was first passed over a coarse silica gel open column (7.2 cm i.d.) with 280 g of coarse silica gel and eluted with a step gradient of CHCl3/MeOH mixtures (40:1, 30:1, 20:1, 15:1, 8:1, 6:1, 4:1, 2:1, 1:1, 1:2, and 0:1). The eluent was collected in 250 mL aliquots that were combined, after analysis by TLC, to afford eight fractions (D3F01–D3F08). The n-BuOH partition (D4) was initially passed over an RP C18 open column using MeOH/H2O mixtures (0:1, 1:4, 1:1, 4:1, and 1:0) as eluting solvents, to afford five fractions (D4F01–D4F05). Fractions showing activities in either or both of the hydroxyl radical-scavenging and quinone reductase-inducing assays were selected for further purification.
Fraction D3F02 (333 mg) was separated on a silica gel column with a solvent system of CHCl3/MeOH (15:1, 10:1, 7:1, 3:1, and 1:1) to produce five subfractions (D3F0201–D3F0205). The subfraction D3F0202 was further refined by preparative TLC developed with CHCl3/MeOH/AcOH (12:1:0.1), to yield compounds 14 (Rf = 0.29; 14.2 mg) and 15 (Rf = 0.71; 3.6 mg). Fraction D3F0205 was chromatographed on a Sephadex LH-20 column, with elution by MeOH/H2O (1:1), to purify compound 16 (4.4 mg).
Fraction D3F03 (588 mg) was chromatographed over a silica gel column with a CHCl3/MeOH solvent system (15:1, 10:1, 8:1, 5:1, and 2:1) to produce five subfractions (D3F03F01–D3F03F05). Subfraction D3F0303 was further purified by preparative TLC, developed using CHCl3/MeOH/AcOH (7:1:0.1), to yield compounds 12 (Rf = 0.20; 16.8 mg) and 13 (Rf = 0.66; 2.2 mg). The fourth subfraction D3F0304 was further chromatographed over a Sephadex LH-20 column, with elution by MeOH/H2O (40:60), to afford compounds 10 (19.8 mg), 11 (5.6 mg), and a further subfraction D3F030401. This subfraction D3F030401 was purified by semi-preparative RP-HPLC, using a CH3CN/H2O (35:65) isocratic elution at a flow rate of 4.0 mL/min, to yield compound 6 (tR = 20.1 min; 8.2 mg).
Fraction D3F06 (416 mg) was subjected to Sephadex LH-20 column chromatography, with elution by MeOH/H2O (1:3, 1:1, 3:1, and 1:0), to afford five subfractions (D3F0601–D3F0605). Of these subfractions, D3F0602 (eluted with MeOH/H2O, 50:50) was further purified by RP-HPLC with isocratic elution (MeOH/H2O, 3:7), at a flow rate of 4.0 mL/min, to yield compounds 1 (tR = 25.2 min; 1.6 mg) and 2 (tR = 32.7 min; 3.8 mg). In addition, D3F0603 (eluted with MeOH/H2O, 75:25) was also purified by RP-HPLC, with isocratic elution (CH3CN/H2O, 26:74) at a flow rate of 4.0 mL/min, to yield compounds 7 (tR = 22.8 min; 4.1 mg), 8 (tR = 31.3 min; 2.1 mg), and 9 (tR = 36.5 min; 3.0 mg).
Fraction D4F02 (669 mg) was purified initially over a Sephadex LH-20 column, with elution by a MeOH/H2O step-gradient (0:1, 1:3, 1:1, 3:1, and 1:0), to afford five subfractions (D4F0201–D4F0205). Subfraction D4F0202 was then separated by RP-HPLC with a MeOH/H2O gradient (flow rate 4.0 mL/min, 20–50% MeOH with 0.05% TFA from 0 to 60 min), to yield compounds 3 (tR = 42.6 min; 11.9 mg), 4 (tR = 30.3 min; 7.2 mg), and 5 (tR = 18.5 min; 5.8 mg).
2-O-β-D-Glucopyranosyl-4,6-dihydroxybenzaldehyde (1)
isolated (ca. 0.0003% of dry weight) as a colorless resin; [α]20D –78 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 219.5 (4.30), 285.0 (4.33) nm; IR (film) νmax 3333, 2919, 1642, 1608, 1524, 1466, 1446, 1379, 1315, 1267, 1222, 1166, 1080, 895 cm−1; 1H and 13C NMR data (Table 1); HRESIMS m/z 339.0679 [M+Na]+ (calcd for C13H16O9Na, 339.0687).
Table 1.
Comparison of the 1H (mult., J in Hz) and 13C NMR Spectroscopic Data of Compounds 1 and 2 in CD3OD.a,b,c
| position | 1 | 2 | ||
|---|---|---|---|---|
|
| ||||
| δH | δC | δH | δC | |
| 1 | – | 106.8 | – | 106.7 |
| 2 | – | 163.5 | – | 162.6 |
| 3 | 6.17 (d, 2.2) | 95.9 | 6.18 (d, 2.2) | 95.4 |
| 4 | – | 166.8 | – | 166.3 |
| 5 | 5.91 (d, 2.2) | 100.0 | 5.94 (d, 2.2) | 98.1 |
| 6 | – | 169.3 | – | 167.7 |
| 7 | 10.11 (s) | 193.2 | – | 204.8 |
| 1′ | 4.96 (d, 7.4) | 102.1 | 5.02 (d, 7.4) | 102.0 |
| 2′ | 3.50 (t, 7.4) | 74.7 | 3.54 (t, 7.4) | 74.7 |
| 3′ | 3.46 (m) | 78.4 | 3.46 (m) | 78.5 |
| 4′ | 3.41 (m) | 71.1 | 3.42 (m) | 71.1 |
| 5′ | 3.46 (m) | 77.9 | 3.46 (m) | 78.3 |
| 6′ | 3.91, (dd, 12.0, 1.8) | 62.3 | 3.92, (dd, 12.0, 1.8) | 62.4 |
| 3.72, (dd, 12.0, 4.9) | 3.72, (dd, 12.0, 4.9) | |||
| CH3 | – | – | 2.69 (s) | 33.5 |
Measured at 400 MHz for 1H NMR with residual signals of CD3OD at δ 3.31 ppm used as reference.
Measured at 100 MHz for 13C NMR with residual signals of CD3OD at δ 49.0 ppm used as reference.
Assignments supported by 2D NMR experiments.
Evaluation of in vitro Hydroxyl Radical-scavenging Activity
A hydroxyl radical-scavenging activity bioassay was performed according to a method described previously.24–26
Evaluation of in vitro Quinone Reductase (QR)-inducing and Cytotoxicity Activities
The potential QR-inducing activity and cytotoxicity of the extracts, fractions, and pure isolates was assayed in vitro using murine Hepa1c1c7 cells, as described previously.25,27
Optimized Extraction Procedure
To complement previous reports on the determination of anthocyanins in A. chilensis fruits,16–23 the present study focused on the determination of other major phenolic and flavonoid constituents that were shown to be active in the hydroxyl radical-scavenging and QR-inducing assays. Thus, on the basis of previously reported extraction methods for phenolic and flavonoid components,28,29 with minor modification, the extraction method was optimized and further confirmed by an orthogonal test. Briefly, 2 g of milled dry fruits of A. chilensis were accurately weighed and extracted with 10 mL of MeOH/H2O (95:5) by sonication at room temperature for 15 min. After vacuum filtration, the residue was extracted with 10 mL EtOH/H2O (80:20) by sonication at ambient temperature for 15 min, followed by filtration. Both filtrates were combined and dried under vacuum at 40 °C, then re-dissolved in 200 μL of MeOH. This MeOH extract was passed over a Waters Sep-Pak Vac 20cc C18 cartridge to remove the very polar substances present (eluted by MeOH/H2O, 5:95), followed by elution with MeOH/H2O (95:5) to afford the pre-treated extract. This pre-treated extract was dried and transferred into a 25 mL volumetric flask and brought up to the full volume in MeOH to prepare the extract solution for HPLC-DAD and LC-MS analysis.
HPLC-DAD Separation and Analysis
The HPLC-DAD analysis was performed using a 150 mm × 4.6 mm i.d., 5 μm, XBridge C18 column, with a 10 mm × 4.6 mm i.d., 5 μm, guard column of the same material (Waters) in a system composed of an L-2130 pump and an L-2450 diode array detector (Hitachi). The mobile phase consisted of 0.05% TFA in H2O (A) and CH3CN (B), using a gradient program of 5–20% B from 0 to 60 min and 20–100% B from 60 to 70 min. The mobile phase flow rate and the injection volume were 1 mL/min and 10 μL, respectively. The column temperature was set at 30 °C, and the chromatograms were recorded at wavelengths from 200–550 nm.
LC-IT-MS Analysis
The LC-IT-MS analysis procedure employed the same separation conditions as used for the HPLC-DAD analysis mentioned above, on an Alliance 2690 Separation Module (Waters) using the same XBridge C18 analytical column as above (Waters). The injection volume and mobile phase flow rate were 10 μL and 1 mL/min, respectively. Approximately 2% of the column eluent was split to the MS using a microsplitter valve 203 (Upchurch Scientific, Oak Harbor, WA). A dual funnel amaZon ETD Ion Trap mass spectrometer (Bruker, Bremen, Germany) equipped with an orthogonal electrospray source was used for electrospray ionization ion trap mass spectrometry (ESI-IT-MS), and this was operated in positive-ion mode with sodium iodide being used for mass calibration in the range of m/z 100–1000. The optimal ESI conditions used were: capillary voltage 4500 V, source temperature 250 °C, N2 was used as the ESI drying gas at 4.0 L/min and as the nebulizer gas at 10 psi. The ion trap was set to UltraScan mode with a target mass of m/z 500 pass ions from m/z 100–1000.
Quantitation of the Major Bioactive Constituents of A. chilensis Fruits
Stock solutions of the six standards (compounds 6, 7, 10–12, and 14) were prepared in MeOH, each at 2500 mg/L. Work solutions were obtained as a mixture of these stock solutions after series dilutions with methanol to achieve five concentration levels in the range of 2–250 mg/L. The work solutions were filtered through a 13 mm, 0.2 μm pore size, syringe filter (Fisher Scientific, Fair Lawn, NJ) prior to HPLC injection. The linearity was plotted using linear regression analysis by the integrated peak areas (Y) vs. concentration of each standard (X, mg/L) at five different concentrations.
Validation of the Analysis
The validation for limit of detection (LOD), limit of quantitation (LOQ), precision, accuracy, and recovery was implemented under the present analytical procedures.30 The LOD and LOQ were determined at the signal-to-noise ratio of 3:1 and 10:1, respectively. The inter-day and intra-day precisions were evaluated by relative standard deviation (RSD) under six repeated injections using work standard solution within 1 d and 3 d, respectively. The repeatability was assessed by extracting and analyzing the same batch of A. chilensis fruit powder six times, independently from each other. Recovery tests were carried by spiking an amount of standards with known content to the same batch of samples to calculate the ratio of the detected and added samples.
Statistical Analysis
All experimental data of the hydroxyl radical-scavenging and quinone reductase-inducing activities were determined as means ± standard deviations (SD).
RESULTS AND DISCUSSION
Bioactivity-guided Isolation of Chemical Constituents
Among the hexanes, CHCl3, EtOAc, n-BuOH, and H2O partitions of the A. chilensis fruit extract, the EtOAc (D3) and n-BuOH (D4) partitions were found to be most potent in the in vitro hydroxyl radical-scavenging assay (ED50, concentration scavenging hydroxyl radicals by 50% = 0.6 and 1.0 μg/mL, respectively), so therefore these two samples were selected for further fractionation. Afterward, subfractions D3F03, D3F06, and D4F02 showed hydroxyl radical-scavenging activity with ED50 = 0.5, 0.4, and 0.3 μg/mL, respectively. Additionally, fractions D3F02 and D3F03 exhibited quinone reductase (QR)-inducing activity (CD, concentration doubling QR activity = 19.5 and 9.6 μg/mL, respectively). Accordingly, these fractions were subjected to further purification steps. In this way, the bioactivity-guided fractionation of A. chilensis fruit extract led to the isolation of a new natural product, 1, together with fifteen known compounds, 2–16. As shown in Figure 1, this set of molecules comprises twelve phenolic compounds, 1–13, two furan derivatives, 14 and 15, and an organic acid, 16. Compounds 6, 7, 10–12, and 14 were found to be the major non-anthocyanin constituents. Furthermore, this is the first time that the phloroglucinol glycosides, 1 and 2, and furan compounds, 14 and 15, were reported to be present in A. chilensis fruits.
Figure 1.

Structures of compounds isolated from the fruits of maqui berry (A. chilensis).
Structure Elucidation of the Purified Natural Product 1
The new compound 1 was obtained as a colorless resin. The molecular formula of 1 was determined as C13H16O9 on the basis of the sodiated molecular ion peak in the HRESIMS. The IR spectrum exhibited absorptions of hydroxy (3333 cm−1), conjugated carbonyl (1642 cm−1), and phenyl groups (1608, 1524, and 1446 cm−1). The UV spectrum showed a peak at 285 nm typical of an aryl conjugated carbonyl, such as the chromophore of a phloroglucinol glycoside.31 The 1H NMR spectrum collected in CD3OD exhibited a downfield signal at δH 10.11 (1H, s, CH-7), which was further confirmed by the observation of a downfield carbon at δC 193.2, indicating the presence of a formyl group. Additionally, a pair of weak-coupled downfield signals in the 1H-1H COSY spectrum suggested the presence of an isolated 1,2,3,5-tetrasubstituted aromatic AX spin system at δH 6.17 (1H, d, J = 2.2 Hz; δC 95.9, CH-3) and 5.91 (1H, d, J = 2.2 Hz; δC 100.0, CH-5). A second isolated spin system, observed in the mid-field region of the 1H NMR spectrum, was especially characteristic of a glucosyl moiety on the basis of the observation of appropriately deshielded carbons in the 13C NMR spectrum {δH 4.96 (1H, d, J = 7.4 Hz; δC 102.1, CH-1′), [3.91 (1H, dd, J = 12.0, 1.8 Hz) and 3.72 (1H, dd, J = 12.0, 4.9 Hz); δC 62.3, CH2-6′], 3.50 (1H, t; δC 74.7, CH-2′), 3.46 (1H, m; δC 78.4, CH-3′), 3.46 (1H, m; δC 77.9, CH-5′), and 3.41 (1H, m; δC 71.1, CH-4′)}. The coupling constant of 7.4 Hz observed for the anomeric proton (H-1′) indicated a β-configuration of the glucose linkage. The connection of the three structural subunits was determined based on key correlations from the 1H-13C HMBC spectrum (Figure 2). For example, the oxygenated aromatic carbon at δC 163.5 (C-2) was determined to be the point of glycosylic substitution, because it showed an HMBC correlation with the anomeric (H-1′) proton. Furthermore, one aromatic proton (H-3) and the formyl (H-7) proton also both had HMBC correlations to C-2. Another oxygenated aromatic carbon, at δC 169.3 (C-6), had an HMBC correlation with the formyl proton and the other aromatic proton in the molecule (H-5). The last oxygenated aromatic carbon was observed at δC 166.8 (C-4), which had HMBC correlations only with H-3 and H-5. Finally, both of the aromatic protons (H-3 and H-5) and the formyl proton (H-7) all exhibited correlations in the HMBC spectrum to a relatively upfield non-protonated aromatic carbon at δC 106.8 (C-1), corresponding to the point of connection of the formyl group to the aromatic ring, and thus concluding the planar structure of 1. The 1H NMR spectrum for this molecule was found to be closely comparable to that of a second substance isolated in the course of this investigation, compound 2 (Table 1). The identity of compound 2 was determined as 4,6-dihydroxy-2-O-(β-D-glucopyranosyl)acetophenone by comparison of its physical data and chromatographic behavior with an authentic standard.32 Since the specific rotation values of 1 and 2 were consistent with each other {[α]20D –78 and –84 (c 0.1, MeOH), respectively}, the absolute configuration was assigned to match, which established 1 as 2-O-β-D-glucopyranosyl-4,6-dihydroxybenzaldehyde. The structure of 1 was previously reported, without characterization data for comparison, as a synthetic intermediate in the generation of anthocyanin.33
Figure 2.
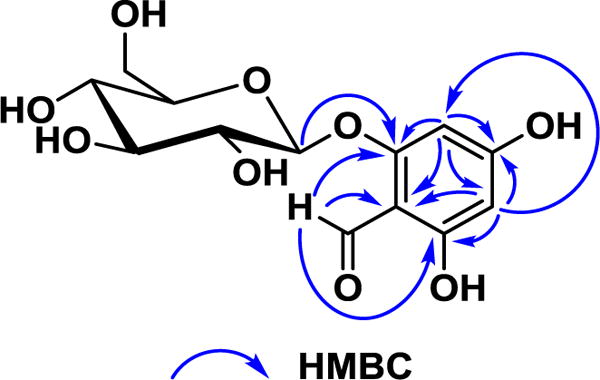
Key HMBC correlations of the new natural product 1 isolated from maqui berry.
In addition to 1 and 2, the identities of another 14 isolated compounds were determined by comparison of their spectroscopic, spectrometric, and specific rotation data with literature values. These were established as being cyanidin 3-O-β-D-glucopyranoside, 3,34 delphinidin 3-O-β-D-glucopyranoside, 4,34 cyanidin 3-O-β-D-sambubioside, 5,35 caryatin, 6,36 hyperoside, 7,37 quercetin 3-O-α-L-arabinofuranoside, 8,37 quercetin 3-O-β-D-xylopyranoside, 9,37 gallic acid, 10,38 gallic acid methyl ester, 11,39 protocatechuic acid, 12,32 protocatechuic acid methyl ester, 13,40 hydroxymethylfurfural, 14,41 acetyloxymethylfurfural, 15,42 and 1,5-dimethyl citrate, 16.43
Hydroxyl-radical Scavenging and Quinone Reductase-inducing Activities of the Pure Compounds
Although previous studies have linked the phenolic and anthocyanin fractions of A. chilensis fruits to their observed high antioxidant activity,21,22 no pure constituents of A. chilensis have been isolated for biological evaluation and reported previously. Thus, all sixteen pure compounds isolated in the present investigation were evaluated for their hydroxyl radical-scavenging and quinone reductase induction activities using standard procedures. As shown in Table 2, except for 15 and 16, which showed activity in neither of these two assays, compounds 1–14 were all found to be active in one or both of the tests. Among them, compounds 1–13 showed demonstrable hydroxyl radical-scavenging activity, with cyanidin 3-O-β-D-glucopyranoside, 3 (ED50 = 0.13 μM), caryatin, 6 (ED50 = 0.18 μM), and hyperoside, 7 (ED50 = 0.17 μM) exhibiting the greatest potencies. Remarkably, the majority of these compounds, 3–11, each showed a higher potency than the positive control, quercetin, which is known to be a quite potent hydroxyl radical scavenger. In addition, four compounds, 7, 11, 12, and 14, exhibited quinone reductase-inducing activity, with protocatechuic acid, 12 (CD = 4.1 μM) being the most potent.
Table 2.
Hydroxyl Radical-Scavenging and Quinone Reductase-Inducing Activities of the Isolated Compounds, 1 – 16, from the Fruits of Maqui Berry (A. chilensis).
| compound | hydroxyl radical scavenging ED50a (μM) |
quinone reductase (QR) induction | ||
|---|---|---|---|---|
| CDb (μM) | IC50c (μM) | CId | ||
| 1 | 1.3 ± 0.12 | >20 | >100 | N/Ae |
| 2 | 1.4 ± 0.16 | >20 | >100 | N/A |
| 3 | 0.13 ± 0.02 | >20 | >100 | N/A |
| 4 | 0.64 ± 0.06 | >20 | >100 | N/A |
| 5 | 0.69 ± 0.08 | >20 | >100 | N/A |
| 6 | 0.18 ± 0.02 | >20 | >100 | N/A |
| 7 | 0.17 ± 0.02 | 19.2 ± 2.8 | >100 | >5.2 |
| 8 | 0.46 ± 0.05 | >20 | >100 | N/A |
| 9 | 0.46 ± 0.04 | >20 | >100 | N/A |
| 10 | 0.41 ± 0.05 | >20 | 55.3 ± 7.8 | N/A |
| 11 | 0.33 ± 0.04 | 14.1 ± 2.3 | 82.4 ± 11.9 | 5.8 ± 0.82 |
| 12 | 1.9 ± 0.21 | 4.1 ± 0.78 | >100 | >24.4 |
| 13 | 1.8 ± 0.16 | >20 | >100 | N/A |
| 14 | >20 | 18.7 ± 2.9 | >100 | >5.1 |
| 15 | >20 | >20 | >100 | N/A |
| 16 | >20 | >20 | >100 | N/A |
| quercetinf | 1.2 ± 0.11 | |||
| L-sulforaphaneg | 0.77 ± 0.11 | 16.7 ± 1.9 | 21.7 ± 2.8 | |
ED50, concentration reducing scavenging hydroxyl radicals by 50%. Each value represents the mean ± SD (n = 3). Compounds with ED50 values of <20 μM are considered active.
CD, concentration required to double quinone reductase activity. Each value represents the mean ± SD (n = 3). Compounds with CD values of <20 μM are considered active.
IC50, concentration inhibiting cell growth by 50%.
CI, chemoprevention index (= IC50/CD).
NA, not applicable.
Positive control for hydroxyl radical-scavenging assay.
Positive control for quinone reductase-induction assay.
Utilization of the Major Bioactive Non-anthocyanin Phenolics as Markers for Quantitative Analysis
Since previous phytochemical analysis of A. chilensis fruits have focused on anthocyanins and have established some HPLC methods for quantitation of several major anthocyanin constituents,16–22 the present study aimed to investigate the chemical profile and biological activity of the non-anthocyanin phenolics of A. chilensis fruits. Although the total phenolic contents of A. chilensis fruits have been studied and linked to antioxidant activity,17,21,23 the composition of these components has not yet been quantitated. As mentioned above, some of these non-anthocyanin phenolics were found to be present in A. chilensis fruits in high quantities during the isolation process and showed direct hydroxyl-radical scavenging and quinone reductase-inducing activities in the assays. Thus, the major bioactive non-anthocyanin phenolics 6, 7, and 10–12 along with the major bioactive furan compound 14, which has not been previously identified from A. chilensis, were selected as markers for the development of a quantitative analysis method to reflect simultaneously the chemical profile and biological activity of A. chilensis fruits.
Quantitative Analysis of the Major Bioactive Non-anthocyanin Components
An HPLC–DAD–MS analysis method, a proven effective tool for botanical dietary supplement identification,44 was developed to achieve good separation and detection of the selected major chemical constituents of A. chilensis fruits, with a representative chromatogram shown in Figure 3. By comparing the retention times, UV profiles, and [M+H]+ and [M+Na]+ molecular ions of the peaks with those of authentic standards (Table 3), the six selected major bioactive non-anthocyanin components could be identified unambiguously as gallic acid, 10, hydroxymethylfurfural, 14, protocatechuic acid, 12, gallic acid methyl ester, 11, hyperoside, 7, and caryatin, 6, respectively. After comparing the chromatograms of the extract solution recorded at wavelengths within 200–550 nm, it was found that 300 nm could best represent the profile of the analytes. At this observation wavelength, a good linearity was achieved for each of the quantitated constituents (R2 >0.999), and the contents of these constituents were determined using the developed quantitative method (Table 4).
Figure 3.
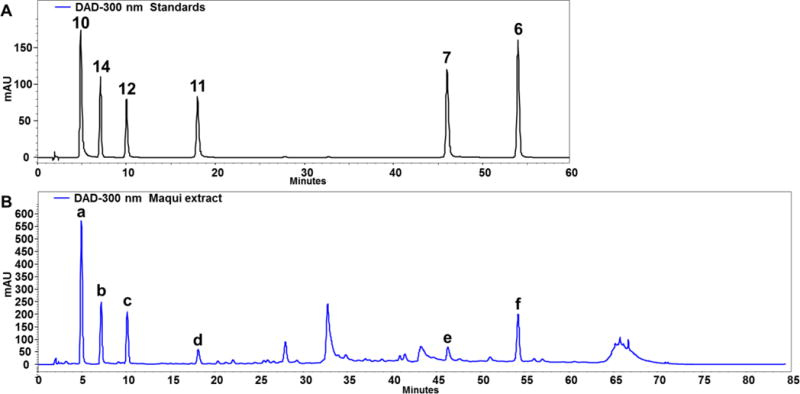
A representative HPLC chromatogram of chemical constituents of maqui berry (A. chilensis). (A). Major bioactive non-anthocyanin components isolated as standards with bold compound numbers corresponding to structures presented in Figure 1. (B). Maqui berry fruit extract with identified peaks labeled a-f described in Table 3.
Table 3.
LC-MS Data of the Major Bioactive Constituents Identified in Maqui Berry (A. chilensis) Extract.
| peak | tR (min) | UV λmax (nm) | MF | [M+H]+ | [M+Na]+ | identification |
|---|---|---|---|---|---|---|
| a | 5.0 | 216, 272 | C7H6O5 | 171.1 | 193.1 | gallic acid (10) |
| b | 7.1 | 229, 284 | C6H6O3 | 127.1 | 149.1 | hydroxymethylfurfural (14) |
| c | 10.1 | 218, 260, 294 | C7H6O4 | 155.1 | 177.1 | protocatechuic acid (12) |
| d | 18.0 | 216, 272 | C8H8O5 | 185.1 | 207.1 | gallic acid methyl ester (11) |
| e | 46.1 | 255, 355 | C21H20O12 | 465.1 | 487.1 | hyperoside (7) |
| f | 54.1 | 252, 348 | C17H14O7 | 331.1 | 353.1 | 3,5-dimethoxyquercetin (6) |
Table 4.
Quantitative Analysis of the Major Bioactive Constituents in Maqui Berry (A. chilensis) Fruits (per dry weight).
| constituent | retention time (min) |
regression equationa | linear range | contentb |
|---|---|---|---|---|
| (Y = aX + b, R2) | (mg/L) | (mg/kg) | ||
| 10 | 5.0 | Y = 33283X + 3049, 0.9996 | 2–250 | 202 ± 1.1 |
| 14 | 7.1 | Y = 66525X + 544211, 0.9993 | 2–250 | 98 ± 0.6 |
| 12 | 10.1 | Y = 23960X + 121915, 0.9995 | 2–250 | 108 ± 0.6 |
| 11 | 18.0 | Y = 22844X + 229491, 0.9993 | 2–250 | 28 ± 0.2 |
| 7 | 46.1 | Y = 35226X + 115364, 0.9996 | 2–250 | 20 ± 0.1 |
| 6 | 54.1 | Y= 58063X + 139757, 0.9998 | 2–250 | 68 ± 0.4 |
Y = peak area and X = concentration.
Mean ± SD (n = 3).
Validation
A comprehensive validation of the quantitative method described above was conducted. The LOD and LOQ were in suitable low ng ranges for the major bioactive compounds to be analyzed. The values of RSD for the precision, repeatability, and recovery tests were all <3%. The validation data for quantitation of the selected major bioactive constituents of A. chilensis are shown in Table 5. Altogether, these results indicated that the method was satisfactory for sample analysis.
Table 5.
Method Validation (Limits of Detection, Accuracy, and Precision) for Quantitation of the Major Bioactive Constituents of Maqui Berry (A. chilensis) Fruits.
| constituent | LOD | LOQ | precision (RSD, n = 6) | repeatability (n = 6) | recovery (n = 3) | ||
|---|---|---|---|---|---|---|---|
| (ng) | (ng) | intra-day | inter-day | RSD | mean | RSD | |
| 10 | 2.2 | 7.5 | 0.98% | 2.17% | 2.28% | 92.06% | 2.84% |
| 14 | 2.0 | 6.7 | 0.88% | 2.38% | 1.82% | 93.08% | 2.99% |
| 12 | 2.1 | 7.1 | 1.11% | 2.21% | 1.62% | 90.32% | 1.96% |
| 11 | 2.4 | 7.8 | 0.86% | 2.03% | 1.42% | 95.20% | 2.54% |
| 7 | 3.5 | 9.9 | 0.69% | 1.98% | 1.95% | 91.49% | 2.19% |
| 6 | 2.9 | 9.1 | 0.77% | 1.66% | 1.11% | 92.29% | 1.88% |
In the work presented herein, a maqui berry (Aristotelia chilensis) fruit extract, used as a dietary supplement ingredient, was investigated by bioassay-guided fractionation. This yielded 16 natural product isolates, including one molecule not previously disclosed from any natural source, 1, together with a closely related phloroglucinol glycoside, 2, and two furanaldehydes, 14 and 15, that were never before reported or detected in maqui berry samples. All isolates, 1–16, were evaluated for potential cancer chemopreventive activity using hydroxyl-radical scavenging and quinone reductase-induction bioassays. Six bioactive phenolic compounds, 6, 7, 10–12, and 14, were selected as marker compounds and utilized as authentic standards for the development and validation of an LC-DAD-MS method because they were present in high abundance, demonstrated in vitro bioactivities, and were not previously used in existing methods designed for analysis of maqui berry samples. Since this analytical method allows for qualitative and quantitative determination of the analytes in maqui berry extracts or other phytochemical samples, it can be utilized for extract standardizations, quality assurance and control protocols, and dereplication efforts in natural product drug discovery. This study has thus provided three of the recommended components of proposed best practices in compliance with the FDA good manufacturing practice (GMP) guidelines intended to produce consistent, effective and safe botanical dietary supplements, notably the identification of bioactive constituents of the botanical of interest, their development as chemical markers for chemical standardization, and the use of in vitro bioassays pertinent to cancer chemoprevention as preliminary biological standards.45,46 These results also represent the first direct evidence that partially correlate the reported beneficial health effects of A. chilensis fruit consumption to the identified chemical constituents, and indicate that A. chilensis may be of interest for further investigation of cancer chemopreventive agents or as a possible natural functional food ingredient.
Supplementary Material
Acknowledgments
We thank Mr. John Fowble and Dr. Craig McElroy (College of Pharmacy, The Ohio State University) for facilitating the acquisition of NMR data. Mrs. Nanette Kleinholz and Mr. Mark Apsega (Mass Spectrometry & Proteomics Facility, The Ohio State University) are acknowledged for MS training and facilitating the acquisition of LC-MS data.
Funding Sources
This study was supported in part by Nature’s Sunshine Products. C.B.N. gratefully acknowledges the financial support of pre-doctoral fellowships from the American Foundation for Pharmaceutical Education (AFPE), the Ohio State University and NIH Chemistry-Biology Interface Training Program (T32 GM008512), and the Jack L. Beal Graduate Scholarship in Medicinal Chemistry and Pharmacognosy from the College of Pharmacy, The Ohio State University. J.L. acknowledges financial support from the Jack L. Beal and Raymond W. Doskotch pre-doctoral fellowships in Medicinal Chemistry and Pharmacognosy from the College of Pharmacy, The Ohio State University.
Footnotes
Supporting Information.
1H NMR, 13C NMR, HMBC, HRESIMS, IR and UV spectra for compound 1; 1H NMR, 13C NMR, DEPT 135, HSQC and HMBC for compound 2; UV and MS data of major metabolites identified in the LC-DAD-MS evaluation of maqui berry (A. chilensis) fruit extract. This material is available free of charge via the Internet at http://pubs.acs.org.
ORCID
A. Douglas Kinghorn: 0000-0002-6647-8707
C. Benjamin Naman: 0000-0002-4361-506X
Notes
The authors declare no competing financial interest.
References
- 1.Heron M. Deaths: Leading causes for 2014. Natl Vital Stat Rep. 2016;65:1–95. [PubMed] [Google Scholar]
- 2.Steward WP, Brown K. Cancer chemoprevention: a rapidly evolving field. Br J Cancer. 2013;109:1–7. doi: 10.1038/bjc.2013.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Landis-Piwowar KR, Iyer NR. Cancer chemoprevention: current state of the art. Cancer Growth Metastasis. 2014;7:19–25. doi: 10.4137/CGM.S11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gravitz L. Chemoprevention: first line of defence. Nature. 2011;471:S5–S7. doi: 10.1038/471S5a. [DOI] [PubMed] [Google Scholar]
- 5.Meyskens FL, Jr, Curt GA, Brenner DE, Gordon G, Herberman RB, Finn O, Kelloff GJ, Khleif SN, Sigman CC, Szabo E. Regulatory approval of cancer risk-reducing (chemopreventive) drugs: moving what we have learned into the clinic. Cancer Prev Res. 2011;4:311–323. doi: 10.1158/1940-6207.CAPR-09-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cuendet M, Oteham CP, Moon RC, Pezzuto JM. Quinone reductase induction as a biomarker for cancer chemoprevention. J Nat Prod. 2006;69:460–463. doi: 10.1021/np050362q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seeram NP. Berry fruits: compositional elements, biochemical activities, and the impact of their intake on human health, performance, and disease. J Agric Food Chem. 2008;56:627–629. doi: 10.1021/jf071988k. [DOI] [PubMed] [Google Scholar]
- 8.Stoner GD, Wang L-S, Casto BC. Laboratory and clinical studies of cancer chemoprevention by antioxidants in berries. Carcinogenesis. 2008;29:1665–1674. doi: 10.1093/carcin/bgn142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stoner GD, Wang L-S, Seguin C, Rocha C, Stoner K, Chiu S, Kinghorn AD. Multiple berry types prevent N-nitrosomethylbenzylamine-induced esophageal cancer in rats. Pharm Res. 2010;27:1138–1145. doi: 10.1007/s11095-010-0102-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clarke TC, Black LI, Stussman BJ, Barnes PM, Nahin RL. Trends in the use of complementary health approaches among adults: United States, 2002–2012. Natl Health Stat Rep. 2015;79:1–16. [PMC free article] [PubMed] [Google Scholar]
- 11.Supplement Business Report. Nutrition Business Journal. 2016 [Google Scholar]
- 12.Schreckinger ME, Lotton J, Lila MA, Gonzalez de Mejia E. Berries from South America: a comprehensive review on chemistry, health potential, and commercialization. J Med Food. 2010;13:233–246. doi: 10.1089/jmf.2009.0233. [DOI] [PubMed] [Google Scholar]
- 13.Céspedes CL, El-Hafidi M, Pavon N, Alarcon J. Antioxidant and cardioprotective activities of phenolic extracts from fruits of Chilean blackberry Aristotelia chilensis (Elaeocarpaceae), maqui. Food Chem. 2008;107:820–829. [Google Scholar]
- 14.Rojo LE, Ribnicky D, Logendra S, Poulev A, Rojas-Silva P, Kuhn P, Dorn R, Grace MH, Lila MA, Raskin I. In vitro and in vivo anti-diabetic effects of anthocyanins from maqui berry (Aristotelia chilensis) Food Chem. 2012;131:387–396. doi: 10.1016/j.foodchem.2011.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Romanucci V, D’Alonzo D, Guaragna A, Di Marino C, Davinelli S, Scapagnini G, Di Fabio G, Zarrelli A. Bioactive compounds of Aristotelia chilensis Stuntz and their pharmacological effects. Curr Pharm Biotechnol. 2016;17:513–523. doi: 10.2174/1389201017666160114095246. [DOI] [PubMed] [Google Scholar]
- 16.Genskowsky E, Puente LA, Pérez-Álvarez JA, Fernández-López J, Muñoz LA, Viuda-Martos M. Determination of polyphenolic profile, antioxidant activity and antibacterial properties of maqui [Aristotelia chilensis (Molina) Stuntz] a Chilean blackberry. J Sci Food Agric. 2016;96:4235–4242. doi: 10.1002/jsfa.7628. [DOI] [PubMed] [Google Scholar]
- 17.Brauch JE, Buchweitz M, Schweiggert RM, Carle R. Detailed analyses of fresh and dried maqui (Aristotelia chilensis (Mol.) Stuntz) berries and juice. Food Chem. 2016;190:308–316. doi: 10.1016/j.foodchem.2015.05.097. [DOI] [PubMed] [Google Scholar]
- 18.Brauch JE, Reuter L, Conrad J, Vogel H, Schweiggert RM, Carle R. Characterization of anthocyanins in novel Chilean maqui berry clones by HPLC-DAD-ESI/MSn and NMR-spectroscopy. J Food Compos Anal. 2017;58:16–22. [Google Scholar]
- 19.Escribano-Bailón MT, Alcalde-Eon C, Muñoz O, Rivas-Gonzalo JC, Santos-Buelga C. Anthocyanins in berries of maqui (Aristotelia chilensis (Mol.) Stuntz) Phytochem Anal. 2006;17:8–14. doi: 10.1002/pca.872. [DOI] [PubMed] [Google Scholar]
- 20.Fredes C, Yousef GG, Robert P, Grace MH, Lila MA, Gómez M, Gebauer M, Montenegro G. Anthocyanin profiling of wild maqui berries (Aristotelia chilensis [Mol.] Stuntz) from different geographical regions in Chile. J Sci Food Agric. 2014;94:2639–2648. doi: 10.1002/jsfa.6602. [DOI] [PubMed] [Google Scholar]
- 21.Miranda-Rottmann S, Aspillaga AA, Pérez DD, Vasquez L, Martinez ALF, Leighton F. Juice and phenolic fractions of the berry Aristotelia chilensis inhibit LDL oxidation in vitro and protect human endothelial cells against oxidative stress. J Agric Food Chem. 2002;50:7542–7547. doi: 10.1021/jf025797n. [DOI] [PubMed] [Google Scholar]
- 22.Ruiz A, Hermosín-Gutiérrez I, Mardones C, Vergara C, Herlitz E, Vega M, Dorau C, Winterhalter P, von Baer D. Polyphenols and antioxidant activity of calafate (Berberis microphylla) fruits and other native berries from Southern Chile. J Agric Food Chem. 2010;58:6081–6089. doi: 10.1021/jf100173x. [DOI] [PubMed] [Google Scholar]
- 23.Céspedes CL, Valdez-Morales M, Avila JG, El-Hafidi M, Alarcón J, Paredes-López O. Phytochemical profile and the antioxidant activity of Chilean wild black-berry fruits, Aristotelia chilensis (Mol) Stuntz (Elaeocarpaceae) Food Chem. 2010;119:886–895. [Google Scholar]
- 24.Chin Y-W, Chai H-B, Keller WJ, Kinghorn AD. Lignans and other constituents of the fruits of Euterpe oleracea (açai) with antioxidant and cytoprotective activities. J Agric Food Chem. 2008;56:7759–7764. doi: 10.1021/jf801792n. [DOI] [PubMed] [Google Scholar]
- 25.Chin YW, Jung H-A, Chai H, Keller WJ, Kinghorn AD. Xanthones with quinone reductase-inducing activity from the fruits of Garcinia mangostana (mangosteen) Phytochemistry. 2008;69:754–758. doi: 10.1016/j.phytochem.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 26.LeBel CP, Ischiropoulos H, Bondy SC. Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol. 1992;5:227–231. doi: 10.1021/tx00026a012. [DOI] [PubMed] [Google Scholar]
- 27.Su BN, Parka EJ, Vigo JS, Graham JG, Cabieses F, Fong HHS, Pezzuto JM, Kinghorn AD. Activity-guided isolation of the chemical constituents of Muntingia calabura using a quinone reductase induction assay. Phytochemistry. 2003;63:335–341. doi: 10.1016/s0031-9422(03)00112-2. [DOI] [PubMed] [Google Scholar]
- 28.Do QD, Angkawijaya AE, Tran-Nguyen PL, Huynh LH, Soetaredjo FE, Ismadji S, Ju Y-H. Effect of extraction solvent on total phenol content, total flavonoid content, and antioxidant activity of Limnophila aromatica. J Food Drug Anal. 2014;22:296–302. doi: 10.1016/j.jfda.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stalikas CD. Extraction, separation, and detection methods for phenolic acids and flavonoids. J Sep Sci. 2007;30:3268–3295. doi: 10.1002/jssc.200700261. [DOI] [PubMed] [Google Scholar]
- 30.Xie J, Li J, Liang J, Luo P, Qing L Sen, Ding LS. Determination of contents of catechins in oolong teas by quantitative analysis of multi-components via a single marker (QAMS) method. Food Anal Methods. 2017;10:363–368. [Google Scholar]
- 31.Tian L-W, Zhang Y-J, Qu C, Wang Y-F, Yang C-R. Phloroglucinol glycosides from the fresh fruits of Eucalyptus maideni. J Nat Prod. 2010;73:160–163. doi: 10.1021/np900530n. [DOI] [PubMed] [Google Scholar]
- 32.Li J, Deng Y, Yuan C, Pan L, Chai H, Keller WJ, Kinghorn AD. Antioxidant and quinone reductase-inducing constituents of black chokeberry (Aronia melanocarpa) fruits. J Agric Food Chem. 2012;60:11551–11559. doi: 10.1021/jf303712e. [DOI] [PubMed] [Google Scholar]
- 33.Robinson R. Über die synthese von anthocyaninen. Ber Dtsch Chem Ges A. 1934;67:85–105. [Google Scholar]
- 34.Chorfa N, Savard S, Belkacemi K. An efficient method for high-purity anthocyanin isomers isolation from wild blueberries and their radical scavenging activity. Food Chem. 2016;197:1226–1234. doi: 10.1016/j.foodchem.2015.11.076. [DOI] [PubMed] [Google Scholar]
- 35.Du Q, Jerz G, Winterhalter P. Isolation of two anthocyanin sambubiosides from bilberry (Vaccinium myrtillus) by high-speed counter-current chromatography. J Chromatogr A. 2004;1045:59–63. doi: 10.1016/j.chroma.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 36.Wenkert E, Gottlieb HE. Carbon-13 nuclear magnetic resonance spectroscopy of flavonoid and isoflavonoid compounds. Phytochemistry. 1977;16:1811–1816. [Google Scholar]
- 37.Mabry TJ, Markham KR, Thomas MB. The Systematic Identification of Flavonoids. Springer; Berlin, Heidelberg: 1970. The NMR spectra of flavonoids; pp. 274–343. [Google Scholar]
- 38.Gottlieb HE, Kumar S, Sahai M, Ray AB. Ethyl brevifolin carboxylate from Flueggea microcarpa. Phytochemistry. 1991;30:2435–2438. [Google Scholar]
- 39.Madikizela B, Aderogba MA, Finnie JF, Van Staden J. Isolation and characterization of antimicrobial compounds from Terminalia phanerophlebia Engl. & Diels leaf extracts. J Ethnopharmacol. 2014;156:228–234. doi: 10.1016/j.jep.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 40.Iqbal P, Mayanditheuar M, Childs LJ, Hannon MJ, Spencer N, Ashton PR, Preece JA. Preparation of novel banana-shaped triple helical liquid crystals by metal coordination. Materials. 2009;2:146–168. [Google Scholar]
- 41.Li J, Pan L, Naman CB, Deng Y, Chai H, Keller WJ, Kinghorn AD. Pyrrole alkaloids with potential cancer chemopreventive activity isolated from a goji berry-contaminated commercial sample of African mango. J Agric Food Chem. 2014;62:5054–5060. doi: 10.1021/jf500802x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khalil AT, Chang F-R, Lee Y-H, Chen C-Y, Liaw C-C, Ramesh P, Yuan S-SF, Wu Y-C. Chemical constituents from the Hydrangea chinensis. Arch Pharmacal Res. 2003;26:15–20. doi: 10.1007/BF03179924. [DOI] [PubMed] [Google Scholar]
- 43.Han YN, Choo Y, Lee Y-C, Moon Y-I, Kim S-D, Choi J-W. Monoamine oxidase B inhibitors from the fruits of Opuntia ficus-indica var. saboten. Arch Pharmacal Res. 2001;24:51–54. doi: 10.1007/BF02976493. [DOI] [PubMed] [Google Scholar]
- 44.Qing L-S, Xue Y, Zhang J-G, Zhang Z-F, Liang J, Jiang Y, Liu Y-M, Liao X. Identification of flavonoid glycosides in Rosa chinensis flowers by liquid chromatography-tandem mass spectrometry in combination with 13C nuclear magnetic resonance. J Chromatogr A. 2012;1249:130–137. doi: 10.1016/j.chroma.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fu PP, Xia Q. Assessment of safety and quality assurance of herbal dietary supplements. In: Bagchi D, editor. Nutraceutical and Functional Food Regulations in the United States and Around the World. 2nd. Elsevier; Amsterdam: 2014. pp. 151–168. [Google Scholar]
- 46.van Breemen RB. Development of safe and effective botanical dietary supplements. J Med Chem. 2015;58:8360–8372. doi: 10.1021/acs.jmedchem.5b00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.