

SUMMARY
The mechanisms by which immune checkpoint blockade modulates tumor evolution during therapy are unclear. We assessed genomic changes in tumors from 68 patients with advanced melanoma, who progressed on ipilimumab or were ipilimumab-naive, before and after nivolumab initiation (CA209-038 study). Tumors were analyzed by whole-exome, transcriptome, and/or T-cell receptor (TCR) sequencing. In responding patients, mutation and neoantigen load were reduced from baseline, and analysis of intratumoral heterogeneity during therapy demonstrated differential clonal evolution within tumors and putative selection against neoantigenic mutations on-therapy. Transcriptome analyses before and during nivolumab therapy revealed increases in distinct immune cell subsets, activation of specific transcriptional networks, and upregulation of immune checkpoint genes that were more pronounced in patients with response. Temporal changes in intratumoral TCR repertoire revealed expansion of T-cell clones in the setting of neoantigen loss. Comprehensive genomic profiling data in this study provide insight into nivolumab mechanism of action.
Keywords: Melanoma, Tumor microenvironment, Tumor immune evasion, Immunotherapy, Nivolumab, Ipilimumab, Tumor mutation load/Tumor mutation burden, Neoantigen load, Clonal evolution/Clonal selection, T-cell receptor repertoire
eTOC
Mutation burden decreases with successful checkpoint blockade therapy in patients with melanoma, suggesting that selection against protective mutant neoepitopes may be a critical mechanism of action of Nivolumab
INTRODUCTION
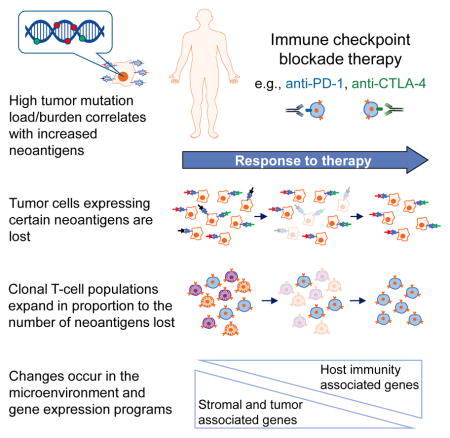
Immune checkpoint inhibitors have demonstrated improved overall survival (OS) and progression-free survival (PFS) in the treatment of many different tumor types (Brahmer et al., 2015; Ferris et al., 2016; Hodi et al., 2016; Motzer et al., 2015; Robert et al., 2015). The underlying genomic features of a tumor can contribute to its response to checkpoint blockade, and increased tumor mutation load associates with survival benefit from both anti-CTLA-4 and anti-PD-1 therapy in multiple malignancies (Hugo et al., 2016; Le et al., 2015; Rizvi et al., 2015; Rosenberg et al., 2016; Snyder et al., 2014; Van Allen et al., 2015). High tumor mutation load may increase the probability of generating immunogenic neoantigens, which facilitate recognition of a tumor as foreign (Riaz et al., 2016a; Schumacher and Schreiber, 2015). Thus, tumors with a high number of clonal neoantigens may be more likely to elicit effective immune responses (McGranahan et al., 2016).
Features of the tumor microenvironment (TME) also associate with response to checkpoint inhibitor therapy. Expression of PD-L1 in the TME associates with clinical response to anti-PD-1/PD-L1 therapies in multiple tumor types (Herbst et al., 2014; Topalian et al., 2012). Baseline levels of tumor-infiltrating CD8+ T cells correlate with the likelihood of response, and may increase during therapy in responding but not progressing tumors (Topalian et al., 2016; Tumeh et al., 2014). Further, the location of CD8+ T cells at the invasive margin of tumors may indicate an effective immune response (Chen et al., 2016; Spranger et al., 2015; Tumeh et al., 2014). The TME may limit extravasation of effector T cells into the tumor, diminish T-cell expansion, or reduce the viability of tumor-infiltrating lymphocytes (TILs) (Joyce and Fearon, 2015).
How checkpoint inhibitor-mediated immune activation modulates the mutational landscape of the tumor and the TME remains poorly understood. To characterize genomic changes, we performed comprehensive genomic analyses on melanoma samples pre- and post-nivolumab (Nivo; anti-PD-1 agent) therapy.
RESULTS
Genomic Characteristics of Tumors before Nivo Treatment
Pre-therapy biopsies from 68 patients were assessed by whole-exome sequencing (WES) at 150× (mean depth: 168; range: 121–237) (Table S1). Thirty-five patients had previously progressed on ipilimumab (Ipi) therapy (Ipi-P); 33 patients were Ipi-naive (Ipi-N) (Table S2). In the patients with WES data, rates of response (RECIST v1.1-defined complete response [CR] or partial response [PR]) to Nivo were comparable in Ipi-N (21%) and Ipi-P (22%) patients. Median mutation load in the patient cohort was 183 mutations (range: 1–7360; interquartile range: 44–433; Table S2,3) and did not differ significantly between Ipi-N and Ipi-P patients (Figure 1A). Mutational subtypes of melanoma as defined by The Cancer Genome Atlas (TCGA) (Cancer Genome Atlas Network, 2015) did not differ in response to Nivo. There were more triple wild type (WT) patients in the Ipi-P cohort than the Ipi-N cohort (57% versus 33%, p = 0.06; Fisher’s exact test; Figure 1A).
Figure 1. Genomic Features and Sculpting of the Tumor Mutational Landscape by Immunotherapy.

(A) Baseline genomic characteristics of melanoma tumors from patients treated with immune checkpoint therapy. An OncoPrint image of WES data for the cohort sorted by response group (CR/PR, SD, PD). The OncoPrint displays genes recurrently mutated in melanoma and genes that have been recently associated with response to therapy.
B) Left: Analysis of clonality in pre-therapy samples identifies a trend toward more subclonal mutations in Ipi-P patients (p = 0.08; Mann–Whitney test; see also Figure S1A). Right: OS in Ipi-N patients by mutation load (high mutation load defined as >100 mutations).
(C) Waterfall plot of change in mutation (non-synonymous) and putative neoantigen load between pre-therapy biopsy and cycle 1, day 29 on-therapy biopsy by response status.
Tumor and Clonal Mutation Load Are Associated with OS and Response in Nivo-Treated Ipi-N Patients
Tumor mutation load associated with OS in Ipi-N but not Ipi-P patients (Figures 1B and S1A), and Ipi-P patients tended to have lower numbers of clonal mutations (p = 0.08; Figure S1B). Stratification of patients by the number of clonal mutations improved the ability to predict survival and response of Ipi-N but not Ipi-P patients (Figures 1B, S1C and S1D). Mutation signatures may play a role in response to checkpoint blockade in NSCLC (Rizvi et al., 2015); however, here, no relationship between the proportion of mutations due to any of the known melanoma mutation signatures (e.g., UV or aging) (Alexandrov et al., 2013) and response to therapy was observed (Figure 1A).
No single gene mutations were significantly associated with response or resistance to therapy. SERPINB3/B4 gene mutations in melanoma samples associate with response to anti-CTLA-4 therapy (Riaz et al., 2016b), and here, five of six patients with SERPINB3/B4 mutations had disease control (CR/PR or stable disease [SD]); however, this was not statistically significant, likely due to small numbers (p = 0.21; Fisher’s exact test). One patient with PR had a frame-shift alteration in B2M with corresponding loss of heterozygosity, alterations previously associated with acquired resistance to anti-PD-1 therapy (Zaretsky et al., 2016). JAK1 and JAK2 mutations were not associated with resistance in this cohort (Figure 1A). No recurrent copy-number alterations as determined by TCGA associated with response; nor did copy-number alterations in interferon (IFN) genes on chromosome 9p (Figures S1E and S1F). However, the frequency of global genomic instability (Davoli et al., 2017) associated with OS in Ipi-P but not Ipi-N patients (Figure S1G).
Evolution of Tumors during Nivo Treatment
To determine if Nivo therapy affects tumor mutation load and intratumor heterogeneity, WES was performed on a subset (n = 41) of paired pre- and on-therapy biopsy samples (Table S4). Significant differences between pre- and on-therapy biopsies correlated with response (p = 5.87e–5; Mann–Whitney test for CR/PR versus progressive disease [PD]) (Figures 1C, S1H, and S1I). Among Ipi-N and Ipi-P responders, a reduction in mutation and neoantigen load was observed 4 weeks after initiation of Nivo, perhaps consistent with immunoediting (Tables S5A and S5B). To ensure that these observations were not solely due to changes in tumor purity after therapy, further deep sequencing (300×) was performed on responding tumors. In each case, many of the mutations seen in the pre-therapy samples were still detectable in on-therapy samples (Figures S2A–S2C). The proportion of mutations that remained detectable varied depending on response: the mean fraction of variants in on-therapy samples was 19% for CR/PR (range: 1%–99%), 82% for SD (range: 2%–140%) and 101% for PD (range: 33%–205%) (values greater than 100% indicate additional mutations beyond those in pre-therapy samples were detected). Power calculations, assuming a global five-fold decrease in the variant allele frequency on-therapy (see Methods), demonstrated that the magnitude in change of mutation load could not be explained by changes in tumor purity alone (Figures S2D–S2F). There were four cases of focal loss of CDKN2A that appeared in on-therapy samples, all in patients with PD. In three of these patients, chromosome 9p deletions also included the nearby IFN gene cluster.
Subsequently, we examined association of changes in the clonal compositions of paired pre- and on-therapy tumor samples with response. The fraction of tumor cells carrying a variant (cancer cell fraction, CCF) in both pre- and on-therapy samples was estimated (see Methods) (Roth et al., 2014). The temporally related changes that occurred in the clonal composition of tumors differed between response groups. Patients with CR/PR had markedly high frequencies of tumor clonal and subclonal variants that decreased in prevalence after Nivo therapy and, in many cases, were not detected on-therapy (mutational contraction; see Methods; Figure 2A). The relative frequencies of single nucleotide variations (SNVs) undergoing mutational contraction were significantly less in patients with PD than SD (p = 0.01). In addition, the relative frequencies of novel SNVs detectable on-therapy were significantly higher on a per-sample basis in patients with PD than SD (p = 0.02), consistent with presumed mutational expansion and/or genetic drift (Figure 2B). Net genomic changes (defined as the difference of variants representing mutational contraction and mutational persistence [see Methods]) per sample strongly associated with response and OS, and this metric was superior to the temporal change in mutation load in predicting response (Figure 2C). Tumors from patients with SD were identified as an intermediate molecular phenotype between those with CR/PR and PD: 26% (5 of 19) of PD samples had >50% of SNVs with variant gain (SD: 0 of 13), while 38% (5 of 13) of SD samples had >50% of SNVs under variant loss (PD: 0 of 19) (Figure 2A).
Figure 2. Changes in Tumor Clonal Composition after Treatment with Nivo Therapy.

(A) Changes in CCF of mutations (synonymous and non-synonymous, clonal/subclonal) from pre- to on-therapy samples. Similar CCFs in both pre- and on-therapy samples (genomic persistence) in gray; increased CCF or novel in on-therapy samples (genomic expansion) in pink; decreased CCF/lost in on-therapy samples (genomic contraction) in blue.
(B) Lost mutations indicating genomic contraction were ubiquitous in CR/PR samples, and significantly more frequent in patients with SD than PD. Persistent mutations were less common in samples without response and not significantly different between patients with SD and PD. Variant gains (genomic expansion) were significantly more frequent in patients with PD than SD. Data are presented as median and interquartile range (IQR).
(C) Left: Waterfall plot of net change between fraction of mutations representing genomic contraction and genomic persistence. Right: OS and PFS by genomic contraction and genomic persistence (p = 0.003; log-rank test and p = 3.34e–4; log-rank test, respectively).
(D) Changes of CCF in representative cases from patients with CR/PR (patient 53), SD (patient 10) and PD (patient 27). Tree diagrams illustrate the relationships between the clones. Colored lines and circles denote specific clones.
Excluding patient 3, all patients with CR/PR consistently lost one or more clones on-therapy; conversely, patients with SD and PD gained novel sets of mutations on-therapy (Figures 2A and 2D). For example, patient 27 (PD) and patient 10 (SD) both had a dominant clone, at least one smaller subclone at initiation of therapy, and the emergence of a novel subclone in the on-therapy sample. In addition, patient 10 had a subclone that was lost after treatment (Figure 2D). Losses were more common in patients with SD than PD. On aggregate, novel subclonal variants were often due to mutational signature 11, which has previously been associated with melanoma and exposure to temozolomide (Alexandrov et al., 2013), and in this setting, suggests that a specific type of repair process becomes dysfunctional due to immunotherapy-mediated stress (Figure S2G).
Transcriptome Analysis and Changes during Treatment
Pre-therapy Expression Analysis Identified Pre-existing Immune Programs in Responders and Expression Footprints of “Hot” versus “Cold” Tumors Based on Prior Immunotherapy Exposure
Baseline transcriptional programs using RNA sequencing (RNA-seq) were characterized and associations with clinical response investigated (n = 45; Table S4). Analysis of differentially expressed genes (DEGs) between patients with CR/PR and PD identified 189 DEGs (q < 0.20; Figure 3A). Highly expressed genes were immune-related, which suggests pre-existing immune recognition of the tumor; however, Gene Ontology (GO) analysis using the Reactome database only identified high-level categories such as T-cell activation and lymphocyte aggregation as enriched (q < 0.1; Figure S3A). Notably, this group included IL17RE, IL17RC and FGFR3 (Table S6A), which modulate the immune environment (Sweis et al., 2016). Next, pre-existing signatures of immune response were evaluated by immune deconvolution (see Methods). A pre-existing immunologically active or “hot tumor” environment was observed in all Ipi-P patients with CR/PR, whereas variable immunological activity was observed in Ipi-N patients with CR/PR (Figures 3B and S3B). No association between the previously reported IPRES gene signature and response was seen in this cohort or in another cohort previously described (Hugo et al., 2016) (Figure S3C).
Figure 3. Pre-therapy Tumor Gene Expression Analysis.

(A) Hierarchal clustering analysis of DEGs in tumors from pre-therapy biopsies.
(B) Heatmap associations of gene expression signatures in the Ipi-P and Ipi-N cohorts.
C) Analysis of DEGs in tumors with genomic contraction versus those with genomic persistence (n = 26).
(D) Left: Clustering of the entire cohort of patients (n = 45) by DEGs identified in (C) clusters patients into two groups in entire cohort and into four groups in combined Hugo et. al. and Van Allen et. al. cohorts. Right: Long-term OS associates with clustered groups of patients from the entire cohort and from the combined Hugo et al. and Van Allen et al. cohorts.
On-therapy Genomic Contraction Phenotype Correlated with Pre-existing Immunity in Responders
We hypothesized that a molecular phenotype of response, such as tumor genomic contraction/persistence, ascertained at an early time-point, such as 4 weeks (Figure 2C), may more strongly correspond to underlying biologic changes than would clinical assessments of response. Differential gene expression analysis between patients who had genomic contraction and genomic persistence was performed to examine pre-existing differences in immunity pre-therapy. In the cohort of 26 patients with paired WES and RNA-seq, 695 DEGs were observed (q < 0.10; Figure 3C and Table S6B), of which 565 had a fold change greater than two. Clustering analysis of all patients and other checkpoint-blockade-treated cohorts demonstrated that this set of immune-related genes stratified patient survival (Figure 3D).
Gene set enrichment analysis demonstrated enrichment of genes involved in PD-1 signaling, co-stimulation of the CD28 family, downstream T-cell receptor (TCR) signaling, IFN-γ, and IL-2 signaling (q < 0.1; Figure S3D). Although PD-1 signaling was enriched, neither PDCD1 (PD-1) nor CD274 (PD-L1) were differentially expressed. However, components of the TCR immunological synapse were enriched (e.g., CD3D/E/G, PTPN6, CD247, CD28, CD86). Notably, several HLA class II alleles were differentially regulated between the two groups, in addition to genes associated with PI3K-γ signaling by G protein–coupled receptors (Figures S3D and S3E).
On-therapy Analysis of Tumor Transcriptome
We subsequently hypothesized that anti-PD-1 therapy can induce tumor transcriptional and microenvironmental sculpting associated with response and therefore evaluated how the expression landscape of melanoma is altered during Nivo therapy. To identify expression changes indicative of a “pharmacologic response” to Nivo, expression of genes that significantly change on-therapy, regardless of response, were analyzed. 475 DEGs were identified in on-therapy samples (q < 0.20; Figure 4A and Table S6C), most of which were associated with immune regulation as determined by Ingenuity Pathway Analysis (IPA) (Figure S4A). Many immune checkpoint genes increased in expression, regardless of response to therapy, including PDCD1 (PD-1), CD274 (PD-L1), CTLA-4, CD80 (CTLA-4-L), ICOS, LAG3, and TNFRSF9 (4-1BB).
Figure 4. Changes in Gene Expression Following Nivo Therapy.

(A) Left: Analysis of ratio of DEGs and selected genes between pre- and on-therapy samples. Right: Examples of genes that change after initiation of Nivo.
(B) Analysis of changes in gene expression (on-therapy compared with pre-therapy) that are altered in tumors that respond or do not respond to Nivo.
(C) Graphical illustration of key pathways differentially expressed in (B).
(D) Immune deconvolution of RNA-seq data comparing pre- and on-therapy samples. Data are presented as median and IQR.
Matched pre- and on-therapy samples were examined to determine whether relative differences in gene expression changes on-therapy could distinguish between patients whose disease was controlled and patients with PD. 2670 DEGs were identified between pre- and on-therapy samples of responders and non-responders (q < 0.20; Figure 4B; Table S6D). Upregulated genes in responders involved a broader spectrum of immune-related genes than genes solely identified in pre-therapy samples (Figures S4B and S4C); including additional checkpoint-related genes (TNFRSF4 [OX40], TIGIT, HAVCR2 [TIM-3], and C10orf54 [VISTA]), and genes involved in lymphocyte activation, chemotaxis and cytokine signaling, and immune cytolytic activity, consistent with previous findings (Das et al., 2015). Downregulated genes in responders involved pathways related to tumor growth, including neural and melanin pathways, cell-cycle regulation, mitotic division, and translation (Figure 4C). Significantly more immune-related genes were selectively upregulated in responders than in non-responders (p = 6.17e–3 and p = 4.61e–4 for depletion in the inflammatory response and cytokine-mediated signaling pathways from GO, respectively).
Changes in immune subpopulations between pre-therapy and on-therapy samples were assessed by immune-deconvolution analysis (see Methods) and numerous changes in immune response among different tumors were observed. An increase in number of CD8+ T cells and NK cells, and a decrease in M1 macrophages, associated with response to therapy (Figure 4D).
T-Cell Repertoire Analysis and Immune Checkpoint Therapy
How Nivo therapy influences T-cell repertoires was examined by analysis of changes in the T-cell abundance, activation, and diversity. We also evaluated how these anti-PD-1-induced changes are affected by prior immunotherapy exposure. To study the dynamics of T-cell infiltration and repertoire diversity in response to Nivo, next-generation deep sequencing of TCR β-chain complementarity determining regions (CDR3s) (TCR-seq) was performed on tumor samples pre- and 4 weeks post-Nivo initiation (n = 34; Table S4). From these nucleotide sequences, the repertoire of amino acid motifs that determine the specificity of antigen-binding and their relative abundances were tabulated (see Methods). Due to the limited number of samples for which TCR-seq data was available, we grouped patients as those with benefit (CR, PR, and SD) or no benefit (PD).
Changes in T-Cell Tumor Infiltration and Activation Associated with Prior Treatment Status and Clinical Response
The fraction of TILs present within each tumor (the proportion of sample that is infiltrating lymphocytes) was assessed by both TCR-seq and immunohistochemical (IHC) staining for CD3 on mostly non-overlapping cohorts of patients (Table S4). By both approaches, an increase in the fraction of TILs upon Nivo therapy was significantly greater among benefiting than non-benefiting Ipi-N but not Ipi-P patients (TCR-seq, p = 0.040; IHC, p = 0.023). Despite the differences in T-cell infiltration, increased cytolytic pathway genes (Rooney et al., 2015) as measured by RNA-seq were associated with benefit to Nivo in both Ipi-P and Ipi-N cohorts (p = 0.043 and p = 0.005; Figure 5A). Notably, there was no significant difference in TIL abundance pre-therapy between patients with benefit and no benefit, or between Ipi-N and Ipi-P cohorts (Figure S5A).
Figure 5. T-Cell Infiltrate and Repertoire Association with Response to Nivo.

Due to the reduced number of cases with paired TCR-seq data, patients with CR/PR and SD were grouped as having “benefit”, and patients with PD were considered to have “no benefit”.
(A) Change in TIL abundance and activity as measured by multiple methods (DNA-based TCR-seq, IHC, and RNA-based cytolytic score). Data are presented as median and IQR.
(B) Change in richness and evenness of intratumoral T-cell repertoires. *Two outliers were removed per Grubbs’ test, alpha = 0.1 (see Methods). Data are presented as median and IQR.
(C) Median richness and evenness of CDR3s per VJ combinations pre-therapy and on-therapy (see also Figure S6E).
(D) Kernel density plots of CDR3 evenness versus number of unique CDR3s for every observed VJ pair in selected patients.
(E) Comparison of on-therapy TIL levels with changes in T-cell repertoire evenness (D90, defined as the minimum fraction of total unique CDR3 sequences that constitutes 90% of all sequencing reads).
(F) Fraction of on-therapy TCR repertoire utilizing V-segments associated with CD8 or CD4 T cells. Data are presented as mean ± SEM.
On-therapy Changes in Intratumoral T-Cell Repertoire Diversity Associated with Response Differentially between Ipi-P and Ipi-N Patients
The diversity of the CDR3 repertoire can be characterized by the Shannon entropy metric, which has two components: the number of unique CDR3s (richness) and their equality of distribution (evenness) (see Methods). Pre-therapy, no significant difference in either of these metrics was observed between cohorts or response status (Figures S5B and S5C). On-therapy, the median fold change in the number of unique CDR3 sequences (richness) was significantly associated with benefit in Ipi-P but not Ipi-N patients (p = 0.016 versus p = 0.489; Figure 5B). In contrast, the median change in T-cell evenness on-therapy was associated with benefit in Ipi-N but not Ipi-P patients (p = 0.036 versus p = 0.594; Figure 5B).
To refine interpretation of these findings with respect to the antigen-binding properties of the TCR repertoire, the diversity of the CDR3 amino acid sequences encoded by a single VJ cassette combination was analyzed individually for every observed VJ combination (see Methods). Notably, a significant decrease in median CDR3 evenness per VJ group on-therapy was observed in Ipi-N but not Ipi-P patients (p = 0.006 versus p = 0.600 Figures 5C and 5D) and in benefiting but not non-benefiting patients (p = 0.003 versus p = 0.636; Figures 5D and S5D). When stratified by both prior treatment status and response, the fold change in CDR3 richness per VJ combination was associated with benefit in Ipi-P but not Ipi-N patients (p = 0.014 versus p = 0.287; Figure S5E), while a significant decrease in CDR3 evenness per VJ pair was observed in Ipi-N but not Ipi-P benefiting patients (p = 0.020 versus p = 0.131; Figure S5D). These results, derived from CDR3 subsets grouped by VJ combination, are mostly consistent with the trends of bulk CDR3 populations (Figure 5B). This suggests, due to association with CDR3 amino acid sequences rather than VJ cassette identities, that T-cell population diversity dynamics are driven substantially by antigen recognition.
To visualize these changes in CDR3 diversity within each VJ combination, CDR3 evenness per VJ versus number of CDR3s per VJ were plotted for every VJ cassette group pre- and on-therapy as kernel density plots (Figure 5D). In the Ipi-N cohort, rightward shifts along the x-axis represent increases in CDR3 richness per VJ combination on-therapy and downward shifts along the y-axis represent decreases in CDR3 evenness per VJ combination on-therapy. Notably, the shifts in evenness and richness per VJ are less prominent in the Ipi-P cohort.
Integrated T-Cell Abundance and Diversity Metrics Associated with Response
Due to the diversity of the TCR repertoire in circulating blood (Zarnitsyna et al., 2013), increased T-cell infiltration is usually concomitant with an increase in the observed diversity of that TIL repertoire. To understand whether the changes observed in the TIL CDR3 repertoires during Nivo therapy were primarily a function of the degree of infiltration or were due to clonotype distribution changes, indicative of selection and expansion of T-cell clonotypes, the minimum percentage of unique CDR3 sequences accounting for 90% of the sequencing reads (D90) was calculated for each TIL sample. D90 is an indicator of evenness in which lower values indicate a more skewed distribution. While the evenness of most Ipi-P TIL repertoires did not change on-therapy, evenness varied widely among Ipi-N patients (Figure 5E). D90 value versus the level of TIL infiltration showed that in Ipi-N patients, disease control was greater among patients with lower TIL D90, including several with low TIL infiltration, such as patients 10, 89, and 94 (Figure 5E). This likely reflects the expansion of specific clonal populations in Nivo responders. This behavior contrasted with that of the Ipi-P cohort, in which high fractional TIL levels during Nivo therapy were a strong indicator of disease control, but CDR3 diversity varied relatively little (Figures 5E and S5A). Furthermore, usage of CD8-associated V-segments (Emerson et al., 2013) was significantly correlated with response (p = 0.005; ordinal regression), while CD4-associated V-segments were not (p = 0.329; ordinal regression; Figure 5F).
Integrative Analysis of Tumor and T-Cell Dynamics
Changes in T-Cell Diversity Associate with Tumor Neoantigen Landscape
We next assessed how the underlying changes in T-cell clonal dynamics related to changes in the clonal dynamics of the tumor. Changes in T-cell repertoire evenness were directly proportional to changes in the fraction of clonal mutations in patients with CR/PR and PD. Notably, there was a trend for a positive relationship in responders (p = 0.07), but a negative relationship in patients with PD (p = 0.08; Figure 6A). We therefore evaluated the relationship between the number of T-cell clones that expanded on-therapy and the number of neoantigens that became undetectable on-therapy. In patients with CR/PR there was a linear relationship between the number of expanded T-cell clones and the number of neoantigens that became undetectable on-therapy (p = 0.03) (Figure 6B; Table S5B). Notably, this did not occur in patients with SD or PD, suggesting a qualitative difference in the T-cell response in these tumors. Similar results were obtained when considering clonal mutations that became undetectable on-therapy, pre-therapy mutation load, or genomic contraction/persistence cases, supporting the view that T-cell expansion is related to the underlying genetic profile of the tumor (Figures S6A–S6C).
Figure 6. T Cells Expand in Proportion to Depletion of Neoantigens.

(A) Changes in T-cell population distribution (i.e., evenness) and changes in tumor mutation clonality by response.
(B) Relationship between the number of predicted neoantigens lost and the number of T-cell clones expanded on-therapy.
(C) Neoantigen ratios (mutations predicted to generate neoantigens per mutations not predicted to generate neoantigens) from mutations solely identified in pre-therapy samples compared with those identified solely in on-therapy samples. Data are presented as mean ± SEM.
(D) Graphical model depicting changes during anti-PD-1 therapy. (a) Changes in mutations and neoantigens during therapy. (b) Changes in TCR repertoire depend on exposure to prior immunotherapy. (c) Changes in immune landscape and checkpoints during therapy.
Selective Depletion of Antigenic Mutations On-therapy in Responding Patients
To investigate whether mutations that were undetectable on-therapy were more likely to be neoantigens or missense mutations than nonantigenic or synonymous mutations, the neoantigen ratios of mutations that produce predicted neoantigens to those that do not were compared between mutations detected solely on-therapy and those detected solely pre-therapy. We hypothesized that the ratio of neoantigen-producing mutations would be higher pre-therapy versus on-therapy in patients with an active immune response, indicating selective pressure against the generation of antigenic mutations. Patients with CR/PR had lower neoantigen ratios on-therapy than patients with PD (p = 0.03; Wilcoxon rank-sum test), and patients with SD had a borderline association (p = 0.11; Wilcoxon rank-sum test; Figures 6C and S6D).
To evaluate the possibility of selective depletion of putative neoantigens within each individual patient, the pre-therapy number of neoantigens per synonymous mutation was determined, and the expected number of neoantigens on-therapy was computed using the measured number of on-therapy synonymous mutations (see Methods). In patients with CR/PR, the observed number of neoantigens was lower than the expected value (p < 0.05; Figure S6E), suggesting that T cells were effective in eliminating tumor cells expressing immunogenic neoantigens.
DISCUSSION
Previous reports indicate that increased tumor mutation load is associated with response to immune checkpoint therapy and that this relationship improves with assessment of clonality (McGranahan et al., 2016; Rizvi et al., 2015; Snyder et al., 2014). We observed an association of pre-therapy tumor mutation load and clonal mutation load with survival and response; however, this observation was limited to the Ipi-N subset of patients, consistent with previous findings (Weber et al., 2016a). Ipi-P patients had significantly more subclonal mutations present, and neither of these genomic markers correlated with response to therapy in these patients. These findings suggest that current biomarkers used to determine which patients will respond to immunotherapy may be more useful for Ipi-N patients than Ipi-P patients, though larger studies will be necessary to confirm these findings.
After 4 weeks of Nivo therapy, we observed a marked decrease in detectable mutations among patients with CR/PR and a moderate decrease in patients with SD. Clonality analysis identified that Nivo therapy affects the evolutionary landscape of tumors in patients with CR/PR, leading to the collapse of whole clonal populations, while in patients with SD, Nivo may shift the landscape in favor of specific subclones. Notably, in several patients with SD, some subclones became undetectable while others remained. In addition, we demonstrated genomic evidence of effective immune elimination of tumor cells containing non-synonymous mutations and neoantigens on-therapy in responding patients as a group, and in a subset of responding patients individually. Moreover, T-cell clones expanded in proportion to the number of neoantigenic mutations that became undetectable on-therapy. These observations agree with evidence of immune-mediated genetic loss of patient-specific mutations, such as after adoptive T-cell therapy (Verdegaal et al., 2016). Our genetic data are consistent with immunoediting, and are derived from a larger group of patients than previously studied (Verdegaal et al., 2016). However, we cannot make definitive conclusions due to computational limitations; namely, unambiguous identification of specific neoantigens driving this effect.
Therapy-induced clonal evolution has been reported following other cancer therapies such as cytotoxic treatments in glioblastoma (GBM) and chronic lymphocytic leukemia, and our observation of CDKN2A loss in four patients with PD agrees with reported changes in GBM tumors at progression (Johnson et al., 2014; Landau et al., 2015). The appearance of new mutations on-therapy could represent genetic drift, or alternatively could be consistent with a model in which certain subclonal populations are selected under immunologic pressure. However, on-therapy expression analysis indicates that an immune ignorance or immune exclusion mechanism of resistance is likely operative rather than the evolution of an intrinsic genetic mechanism mediating resistance. The early collapse in clonal populations among responding patients is consistent with previous findings (Landau et al., 2015; Wang et al., 2016), and suggests that clonal composition undergoes significant changes after cytotoxic therapy. Our results may potentially be affected by variation in the anatomic location from which the tissue was taken and by intratumoral heterogeneity, and by decreased purity in on-therapy biopsies; however, biopsies targeted the same site and, although purity did decrease on-therapy, the magnitude of change was not large enough to explain observations herein.
Expression analysis of pre-therapy tumor samples identified a small set of upregulated immune-related genes in responders, consistent with prior reports demonstrating an IFN-γ signature associated with response (Taube et al., 2012; Taube et al., 2015). However, these expression changes were only marginally predictive of response. Notably, consistent with previous findings (Larkin et al., 2017; Weber et al., 2016b), of patients who received prior immunotherapy, only those with PD-L1-expressing or immunologically “hot tumors” appeared to respond whereas significant responses were observed among PD-L1 low to no expression subgroups among Ipi-N patients. These findings may again be limited due to the size of the cohort but, like mutation load and tumor clonality, suggest that the importance of a “hot tumor” may depend on prior therapy received.
Surprisingly, stratification based on a molecular phenotype of response (i.e., genomic contraction/persistence) demonstrated a more pronounced difference in pre-existing immunity between molecular responders and non-responders. Genes differentially expressed between groups could predict survival in other patients in this dataset and in other immunotherapy-treated cohorts, signifying their biologic relevance and suggesting that delays in clinical response may be secondary to immune infiltration (Wolchok et al., 2009). This highlights the difficulty in relying solely on clinical and radiographic response to understand the underlying biologic mechanisms of response to therapy in human tumors. Notably, this analysis identified numerous HLA class II genes, along with other genes, as differentially expressed, similar to a recently reported signature indicating differences in regulation of macrophages (Kaneda et al., 2016). Changes in macrophages on-therapy also associated with clinical response (Figure 4D), suggesting that macrophages may play an important role in response (Gordon et al., 2017).
Analysis of on-therapy expression changes revealed marked upregulation of a multitude of immune pathways that were more pronounced in responding patients, and included additional checkpoints and a broader set of DEGs than previously reported (Chen et al., 2016). Some of these newly identified gene products may be considered as candidate targets for future combination immunotherapy trials. Most patients with PD had minimal immune response on-therapy, and differential gene analysis did not identify a dominant or single method of tumor-intrinsic immune evasion, suggesting an immune ignorance or immune exclusion mechanism rather than an adaptive resistance mechanism (Salerno et al., 2016; Spranger et al., 2013) (Figure 4B). In contrast, many patients with SD appeared to have modest immune response induction, suggesting that an adaptive mechanism of resistance may have a more important role in these patients (Taube et al., 2012; Taube et al., 2015).
Increased cytolytic activity, indicative of T-cell activation, associated with response. Both cytolytic activity and response were observed at similar rates regardless of prior immune therapy exposure. However, analysis of T-cell repertoires suggested that anti-PD-1 response is associated with different patterns of T-cell diversity dynamics in Ipi-N versus Ipi-P patients. We infer that decreased evenness without a significant change in the total number of CDR3s (richness) observed in Ipi-N responders is consistent with expansion and accumulation of specific T-cell clonotypes in response to the detection of tumor antigens. We speculate that TILs of Ipi-P patients may represent a binding repertoire already differentially selected by the tumor antigenic landscape during Ipi treatment, but subsequently exhausted through PD-1/PD-L1 signaling. The predictive value of exhaustion marker expression for responsiveness in Ipi-P but not Ipi-N patients is consistent with this model (Figure 3B). Thus, we speculate that in Ipi-P patients, anti-PD-1 therapy functions mainly by alleviating exhaustion among the extant distribution of TIL clonotypes, while in Ipi-N patients, anti-PD-1 therapy facilitates selective intratumoral expansion of tumor-reactive clonotypes (Figure 6D).
We hypothesize that changes in T-cell repertoire diversity are associated with anti-PD-1-induced intratumoral clonal architecture changes, reflecting the cytolytic anti-tumor effect of expanded tumor antigen-specific T-cell clonotypes. Of note, changes in T-cell repertoire evenness were directly proportional to changes in the fraction of clonal mutations in responders and patients with PD. There was a trend for a positive and negative relationship in responders and non-responders, respectively, suggesting that in responders, as T-cell clones expand, clonal mutations are targeted and eliminated. Possible explanations for the negative relationship in non-responders include: (1) T-cell clonotypes that expand may target subclonal mutations instead of clonal mutations, resulting in a futile T-cell response (Figure S6F), or (2) decreased T-cell repertoire evenness could be due to clonal expansion of regulatory T cells, which may enhance an immunosuppressive environment.
In conclusion, we performed extensive immunogenomic analyses on melanoma samples treated with anti-PD-1 therapy and characterized how tumor genomic and microenvironmental features changed over time. Assessment of the genomic landscape on-therapy demonstrated clonal evolution consistent with therapy-dependent immunoediting. T-cell repertoire analysis identified that T-cell clonotypes expand in proportion to the number of neoantigens that become undetectable in responding patients. Gene expression analysis revealed the changing transcriptional and microenvironmental alterations induced by anti-PD-1 therapy and identified a broad spectrum of immune checkpoint-related genes that were upregulated. These data have important implications for understanding the mechanism of action of checkpoint inhibitors and for the design of future immune checkpoint blockade trials. Based on our observations, we propose a model of tumor evolution and its microenvironment in response to anti-PD-1 therapy (Figure 6D).
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Timothy Chan (chant@mskcc.org). Sequencing data may be accessed, on approval, by individual investigators upon request (https://fasttrack.bms.com and christine.horak@bms.com).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Eighty-five patients were accrued to a multi-arm, multi-institutional, institutional-review-board-approved, prospective study (CA209-038; NCT01621490) to investigate the pharmacodynamic activity of Nivo. All patients received Nivo (3 mg/kg every 2 weeks) until progression or for a maximum of 2 years. Radiographic assessment of response was performed approximately every 8 weeks until progression. Progression was confirmed with a repeat CT scan typically 4 weeks later. Tumor response for patients was defined by RECIST v1.1. Response to therapy indicates best overall response unless otherwise indicated. All patients underwent biopsy before commencing therapy (1–7 days before first dose of therapy) and underwent a repeat biopsy, collected from the same site, on cycle 1, day 29 (between days 23–29). Tumor tissue was divided for FFPE and storage with RNAlater® (Ambion) for subsequent RNA/DNA extraction. Of 85 patients, 68 had sufficient material for genomic analysis. 35 of 68 patients were Ipi-P and 33 were Ipi-N. Different subsets of patients had sufficient material for the different genomic analysis described (Tables S2 and S4). Of the 68 patients studied, the median patient age was 55 years (range 22–89) and consisted of 1 M0, 13 M1a, 10 M1b, 36 M1c and 8 patients with unknown stage at study entry.
METHOD DETAILS
Whole-Exome Sequencing
DNA was extracted from samples stored with RNAlater® and sequenced with next-generation sequencing. Sequencing libraries were generated using Illumina’s in-solution DNA probe based hybrid selection method as previously described (Fisher et al., 2011). Exonic sequences were enriched using Agilent SureSelect All Exon V2. Pooled libraries were normalized to 2 nM and denatured using 0.2 M NaOH prior to sequencing. Sequencing was performed per manufacturer’s protocols using either the HiSeq 2000 V3 or HiSeq 2500. Each run was a 76bp paired-end with a dual eight-base index barcode read. Alignment and removal of duplicates was performed as previously described (Riaz et al., 2016b). A combination of four different mutation callers (MuTect 1.1.4, SomaticSniper 1.0.4, VarScan 2.3.7, and Strelka 1.0.13) were used to identify SNVs. SNVs with an allele read count of less than 5 or with corresponding normal coverage of less than 7 reads were filtered out as previously described (Riaz et al., 2016b). Small insertions and deletions (indels) were determined using VarScan 2.3.7 and Strelka 1.0.13. Mutation load was determined as the number of remaining non-synonymous mutations. A previously published cutoff of 100 mutations was used to segregate high from low mutation-load tumors (Snyder et al., 2014). Only high- or moderate-impact indels determined by both callers were selected. Mutational subtypes of melanoma were determined using the TCGA classification system. Previously described signatures of mutational processes were determined in each sample using non-negative least squares regression as provided by the R package deconstructSigs v1.8.0 using the COMISC signatures as the mutational signature matrix.
Neoantigen Analysis
High-resolution HLA typing was performed computationally using SOAP-HLA from exome sequencing (exome-seq) data. Each non-synonymous SNV was translated into a 17-mer peptide sequence, centered on the mutated amino acid. Adjacent SNVs (such as those induced by UV irradiation) were first corrected for using MAC. Subsequently, the 17-mer was then used to create 9-mers via a sliding window approach for determination of MHC class I binding. NetMHC v4.0 was used to determine the binding strength of mutated peptides to patient-specific HLA alleles. All peptides with a rank <2% were considered for further analysis. If one mutation generated multiple 9-mer peptides that bound to patient-specific HLA alleles, it was only counted as one neoantigen (i.e., one mutation could only generate a single predicted neoantigen). We considered neopeptides as the total number of predicted 9-mers that bound to patient-specific HLA alleles (i.e., one mutation can generate multiple neopeptides).
To test whether the observed number of neoantigens was different from the expected count in each on-therapy tumor sample, we empirically calculated the following baseline rates pre-therapy:
The average number of missense mutations per silent mutation across tumors.
The number of neoantigens per missense mutation. Of note, since computational tools that predict neoantigens are dependent on patient-specific HLA class I alleles and some HLA-I alleles can bind greater numbers of peptides than others, we calculated this rate specific to the patient, rather than an average across all patients.
Using these two baseline rates, we used the number of silent mutations in each on-therapy tumor sample to calculate the expected number of neoantigens. Hence, the expected number of neoantigens in an on-therapy tumor sample is equal to the number of silent mutations in an on-therapy tumor sample multiplied by the number of missense mutations per silent mutation pre-therapy, multiplied by the number of neoantigens per missense mutation in the patient’s tumor sample pre-therapy. The chi-squared test was used to determine whether the observed number was different from the expected value. All the calculations were done using R statistical software
Copy-Number Analysis
Allele-specific copy-number analysis was performed from exome-seq data using FACETS v0.5.0 with a critical value of 300 to increase the strictness of segments. The fraction of the copy-number-altered genome was defined as the fraction of the genome with either non-diploid copy-number or evidence of loss of heterozygosity. Homozygous copy-number alterations in CDKN2A, NF1, PTEN, and B2M were manually reviewed and, when available, confirmed with corresponding expression changes. Recurrent copy-number alterations for melanoma as determined by GISTIC were downloaded from The Broad Firehose on January 28, 2016, and evaluated in each sample for presence or absence using copy-number data generated by FACETS.
We additionally assessed genomic instability as previously described (Davoli et al., 2017). Each genomic segment in the FACETS output was assigned a somatic copy-number alteration (SCNA) level on a five-tier scale based on the total copy-number (tcn) and sample-ploidy values. Specifically, a ploidy-adjusted tcn value, T, was computed by rounding the sample’s ploidy value to the nearest whole number and subtracting it from the tcn.em value (calculated by FACETS software EM algorithm). Genomic segments were classified as below:
| SCNA Level | SCNA Event | Ploidy-Adjusted tcn |
|---|---|---|
| 2 | Deep amplification | T ≥ 3 |
| 1 | Amplification | 3 > T ≥ 1 |
| 0 | Neutral | 1 > T > −1 |
| −1 | Deletion | −1 ≥ T |
| −2 | Deep deletion | Ploidy = −T |
Chromosomal, chromosome-arm and focal SCNA scores were derived for each sample. SCNA events were determined to be chromosomal if segments with the same SCNA level accounted for at least 75% of the total length of FACETS segments for a given chromosome. Segments determined not to be part of a chromosomal event were next evaluated as potentially contributing to chromosome-arm events by checking if genomic segments sharing the same SCNA level overlapped with at least 75% of the chromosome arm’s coordinates. Segments determined not to be contributing to chromosomal events or to chromosome-arm events were defined as focal SCNA events if their individual lengths were less than half the length of chromosome arms on which they were located. SCNA scores for each level were computed by individually summing the absolute values of the chromosome, arm and focal events. SCNA scores were normalized across all samples using the mean and standard deviation. An additional SCNA score was computed by adding the normalized chromosome and arm level SCNA scores.
Tumor Clonality Analysis
We determined the CCF of mutations identified in each tumor sample using PyClone v0.13.0, as previously described (Morris et al., 2016). For each SNV, CCF was estimated based on reference and variant allele read counts as well as allele-specific copy-number information determined by FACETS as described above. Tumor purity was also estimated using FACETS and provided as an input to PyClone. The infinite beta binomial mixture model with default priors was fit after 10,000 iterations. As in previous work, we considered mutations to be clonal if the 95% confidence interval (CI) of the CCF contained 95% (McGranahan et al., 2015). The CCF was calculated for every SNV in a sample and represents an estimate of the fraction of cancer cells carrying that mutation. The CCF was calculated for each SNV separately based on variant allele frequencies (VAFs), copy-number status (amplifications/deletions), ploidy of the genome, and purity of the sample, and can be interpreted as an adjusted VAF, which allows comparison of SNVs across different samples. We classified SNVs into clonal and subclonal variants depending on the CI of the CCF estimation. SNVs for which the lower bound of the CI exceeded 95% were considered clonal mutations, others subclonal. A comparison based on CCFs determined by ABSOLUTE provided similar results (data not shown) (Carter et al., 2012). High versus low clonal mutation load was split at the median of the clonal mutation load for the cohort.
SNVs were considered to indicate genomic expansion if their subclonal CCF increased by at least 10% from pre- to on-therapy sample or if they were novel to the on-therapy sample and did not occur in pre-therapy sample. SNVs indicating genomic contraction decreased by ≥10% CCF or were only identified in the pre-therapy sample but not in the on-therapy sample. SNVs that occurred at clonal or subclonal CCFs within 10% of pre- and on-therapy samples were not considered to indicate mutational changes and referred to as genomic persistence. A comparison of the selective pressure per sample, was defined as the fraction of mutations undergoing genomic contraction subtracted from the fraction of mutations undergoing genomic persistence. This metric was used for subsequent survival analysis and RNAseq differential gene expression analysis.
RNA Sequencing
RNA was extracted from tissue specimens stored using RNAlater®. Raw FASTQ files were aligned on the hg19 genome using the STAR aligner with default parameters. Aligned fragments were then counted and annotated using Rsamtools v3.2 and the TxDb.Hsapiens.UCSC.hg19.knownGene transcript database, respectively. Normalized fragments per kilobase per million mapped reads (FPKM) were obtained using the robust FPKM estimate function of DESeq2 v1.12.4. Raw reads were normalized with regularized-logarithm transformation function with robust estimation and used to construct a PCA plot, which was manually reviewed. Patient 3 was considered an outlier (data not shown) and thus removed from subsequent transcriptome analysis. All differential gene analysis was conducted using the DESeq2 package.
Differential gene expression analysis was performed using a generalized linear model with the Wald statistical test, with the assumption that underlying gene expression count data were distributed per a negative binomial distribution with DESeq2. DEGs were considered for further analysis with a q-value <0.20. Clustering of DEGs was conducted with hierarchal clustering (Euclidean distance followed by Ward agglomeration algorithm) primarily to segregate upregulated and downregulated genes.
For pre-therapy samples, we considered DEGs between responders (CR/PR) compared with non-responders (PD) or patients with SD without adjusting for any other parameters. For analysis of pharmacologic response, we compared the subset of samples with paired on- and pre-therapy RNA-seq to identify genes that had changes in expression on-therapy. We grouped patients as those who benefited (CR/PR/SD) and those who had no benefit (PD) to identify genes that changed differentially. Design matrices for each analysis are available in supplementary code available on GitHub.
GO enrichment analysis was performed with the R package clusterProfiler, with a Bonferroni correction and an adjusted p-value of 0.20 (Yu et al., 2012). Fold changes from analysis of RNA-seq data were inputted to IPA. The IPA downstream effects analysis was used to identify differentially regulated pathways.
Cytolytic activity was determined as the geometric mean of genes GZMA and PRF1 as previously described (Rooney et al., 2015). Similarly, a signature of IFN-γ was determined for each sample set using a set of genes previously described (Chiappinelli et al., 2015). To further refine subsets of immune cells that were present in each sample, CIBERSORT was used to conduct immune deconvolution on the FPKM values of each patient.
TCR β-Chain Sequencing and Analysis
Tumor biopsy samples were collected prior to and 4 weeks after initiation of Nivo therapy and stored in RNAlater®. DNA was extracted and submitted to Adaptive Biotechnologies for survey level TCR β-chain sequencing (Carlson et al., 2013; Robins et al., 2009), where targeted amplicon libraries were prepared by multiplex PCR targeting all TCR β-chain V and J gene segments, and sequenced using the Illumina HiSeq system. Data for individual TCR sequences, including V and J gene segment identification and CDR3 sequences, were obtained from Adaptive Biotechnologies for customized analysis of T-cell repertoire diversity dynamics.
Population diversity of the T-cell repertoire can be characterized and quantified by two separate factors: richness (i.e., the number of unique elements in a population), and evenness (i.e., the distribution of the frequencies of those elements). Shannon entropy (H) takes into account both richness and evenness, but gives more weight to evenness (Arnaud-Haond et al., 2007). Evenness is defined as the observed H divided by the maximum possible H, given the number of unique elements in a population. High evenness (on a scale of 0 to 1) implies that all elements are nearly equally distributed, while low evenness is indicative of population skewing, or, in the case of TCR repertoire analysis, biased expansion of individual T-cell clonotypes (Arnaud-Haond et al., 2007; Six et al., 2013). Clonality is defined as the complement of evenness (i.e., 1 – evenness). As such, these terms are used somewhat interchangeably, but with reversed scales. Note that for analysis of evenness, two outlier cases were removed as determined by Grubbs’ Test, alpha = 0.1.
The protein-binding characteristics of the CDR3 antigen-binding region of the TCR can be studied in a more targeted way by analyzing the repertoire of amino acid CDR3 sequences encoded by each combination of V and J cassettes. To explore whether the global TCR diversity dynamics described above could also be detected at the level of individual VJ cassette combinations, we determined CDR3 richness and evenness for every VJ pair, as previously described (Sims et al., 2016). This approach, although more rigorous, is an in-silico descendant of traditional spectratyping, in which specific V and J cassette primers are used to amplify all CDR3s encoded by specific VJ cassette combinations and the products are analyzed by electrophoresis (Arstila et al., 1999). The benefits of this in-silico approach are two-fold: 1) by examining diversity dynamics at the VJ cassette level, bias potentially introduced by preferential V and J cassette primer binding is diminished, and 2) a greater understanding of the extent to which antigen-binding properties (CDR3 amino acid sequences) versus VJ cassette usage biases influence global changes in TCR diversity is gained (Sims et al., 2016). Kernel density estimate plots of the number of CDR3s per VJ versus evenness of CDR3s per VJ were generated using the seaborn statistical data visualization platform for Python.
Significantly expanded T-cell clones were identified using the immunoSEQ® Analyzer from Adaptive biotechnologies®, per the method previously described (DeWitt et al., 2015); briefly, Fisher’s exact test was used to generate a p-value by for every T cell clone, and P-values were corrected using a positive false discovery rate method to identify significantly expanded T-cell clones.
Immunohistochemistry
PD-L1 staining was evaluated centrally by IHC using a rabbit monoclonal anti-human PD-L1 antibody (clone 28-8) and an automated staining procedure developed by Dako (Dako, 2016). The percentage PD-L1 expression was scored by a qualified pathologist in samples with a minimum of 100 viable tumor cells. Positive PD-L1 staining was defined as complete circumferential or partial linear plasma membrane staining of tumor cells. Expression of lymphocytic markers within tumor tissue sections was centrally assessed by IHC using antibodies against CD3, CD4, CD8 and FOXP3: clone LN10 (Leica), clone 4B12 (Dako), clone C8/144B (Dako) and clone 236A/E7 (Abcam), respectively. Duplex chromogenic IHC assays consisting of CD3 and CD8, CD3 and CD4, and CD3 and FOXP3 anti-human antibodies were established. Percentage expression of each marker was estimated using digital image analysis algorithms from multispectral images of representative 20 fields in a region of interest that included cancer cells.
Tumor Purity Assessment
Tumor purity was assessed computationally in all paired samples using estimates derived from exome-seq data and from RNA-seq data independently using OncoSNP-SEQ and ESTIMATE. There was a modest decrease in tumor purity in samples with a more significant clinical response than those with PD using any of the three methods for estimation of purity (Figure S2D).
We performed a simulation to determine our power to detect a mutation, assuming a five-fold decrease in purity. We assumed that to accurately call a mutation, five reads with the mutation needed to be present and reads were distributed according to a binomial distribution (B(n,p)), where n corresponds to the coverage of sequencing and p the true VAF of the mutation. At 150× coverage we had 95% power to detect a mutation with a VAF of 6% or greater and at 300× coverage we had 95% power to detect a mutation of 3% VAF or greater in any one sample (Figure S2E). We then computed the proportion of mutations we would anticipate to detect using 300× coverage and a fivefold decrease in purity and observed a significant depletion in the number of anticipated mutations in most cases (Figure S2F). In all cases, the expected number of mutations was larger than the observed, which is unlikely given a five-fold decrease in purity (p = 0.004; Wilcoxon rank-sum test).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics and Survival Analysis
Survival analysis was conducted with the Kaplan–Meier method, and continuous data were in general split at the median, unless prior thresholds had been established. The log-rank test was used to determine statistical significance. Comparison of values between response groups was performed with the Wilcoxon rank-sum test unless otherwise specified. Comparisons in Figures 5 (except 5F) and S6 were Mann–Whitney test by GraphPad Prism for unpaired data or Wilcoxon matched-pairs signed rank test by GraphPad Prism for paired data. All other statistical analyses were performed in the R Statistical Computing environment v3.3.1 (http://www.r-project.org).
DATA AND SOFTWARE AVAILABILITY
See key resources table. The human sequencing data (WES, RNA-seq, TCR-seq) are deposited into a controlled-access database managed by Bristol-Myers Squibb. Sequencing data may be accessed, on approval, by individual investigators upon request (https://fasttrack.bms.com and christine.horak@bms.com). The online version of the paper will be updated to include the accession numbers once they are available. Code and additional processed data to reproduce key results contained within the manuscript are available at https://github.com/riazn/bms038_analysis.
Supplementary Material
Table S1: Exome Sequencing Data (Pre-therapy Samples), Related to Figure 1
Table S2: Clinical Characteristics, Related to Figure 1
Table S3: Somatic Mutations, Related to Figure 1
(A) High mutation load (>100 mutations as previously reported) is not associated with OS in Ipi-P patients (log-rank test; p = NS).
(B) More subclonal mutations in Ipi-P patients (p = 0.08). Data are presented as median and IQR.
(C) Clonal mutation load corresponds with response in Ipi-N but not Ipi-P patients. Data are presented as median and IQR.
(D) High clonal mutation load is not associated with OS in Ipi-P patients (data split at median; log-rank test; p = NS).
(E) Association of TCGA-determined recurrent tumor copy-number alterations and response.
(F) Copy-number alterations in IFN-γ gene cluster and response.
(G) Copy-number changes split at the median and determined as previously described (Davoli et al., 2017) influence outcome in the Ipi-P cohort (p = 0.0008; log-rank test) but not in the Ipi-N cohort (p = 0.49; log-rank test).
(H) Change in mutation load is associated with response (p = 5.87e–5; Mann–Whitney test for CR/PR versus PD; red = Ipi-N; blue = Ipi-P). Data are presented as median and IQR.
(I) Change in mutation load is associated with PFS (p < 0.001; log-rank test; data split at median).
(A) Variant allele frequencies (VAFs) for all mutations detected in patients with CR/PR. In each case a moderate number of mutations remain present in on-therapy samples.
(B) VAFs for patients with SD.
(C) VAFs for patients with PD.
(D) Change in purity between on-therapy and pre-therapy samples computationally estimated from exome data and RNA-seq data (see Methods). Data are presented as median and IQR.
(E) The probability of detecting a mutation determined by sequencing coverage in a tumor.
(F) Expected number of mutations detected in CR/PR cases assuming a five-fold decrease in median VAF (results similar for SD and PD cases; data not shown).
(G) Mutational signatures (aggregated from all samples). Novel subclonal mutations feature a significant frequency of signature 11-related mutations.
(A) GO and Reactome enrichment of cluster 2 genes demonstrates that most are related to immune processes.
(B) Immune cell composition from pre-therapy samples for selected populations from RNA-seq deconvolution analysis. Top: Results for Ipi-N patients. Bottom: Results for Ipi-P patients. (Limited sample numbers for patients with CR/PR.)
(C) Clustering of samples by the recently described IPRES signature and lack of association with response.
(D) GO and Reactome enrichment of DEGs between genomic contraction and genomic persistence. Processes such as PD-1 signaling, TCR signaling, co-stimulation of CD28 family, and MHC class II antigen presentation are enriched.
(E) Previously described macrophage-related gene expression signature (Kaneda et al., 2016) weakly associates with clinical response in CA209-038 cohort, and similarly a significant number of differentially expressed HLA class II alleles are identified as previously described.
(A) Beta coefficients of genes from Figure 4A and IPA of top pathways altered during treatment.
(B) GO enrichment analysis of clusters from Figure 4B demonstrate one cluster is dominated by immune processes, whereas the other appears to involve genes related to tumor growth and progression.
(C) Venn diagram of differentially expressed immune genes from different analyses. Pre-therapy immune genes are cluster 1 from Figure 3A. Anti-PD-1 pharmacologically-related genes are from Figure 4A. Differential genes on-therapy are from Figure 4B.
(A) On-therapy, the extent of T-cell infiltration was associated with response in the Ipi-P but not Ipi-N cohort (p = 0.031 versus p = 0.231). Notably, it seems that Ipi-P non-responders showed little to no increase or even a decrease in TILs upon Nivo therapy, whereas both responders and non-responders in the Ipi-N cohort showed an increase in TILs upon Nivo therapy (see also Figure 5A).
(B) In pre-therapy samples, there was no significant difference in the number of unique intratumoral CDR3 sequences between Ipi-P and Ipi-N patients, or between responders and non-responders.
(C) In pre-therapy samples, there was no significant difference in evenness between Ipi-P and Ipi-N patients, or between responders and non-responders. Data are presented as median and IQR.
(D) Changes in median CDR3 richness and evenness per VJ pair. Decreased CDR3 richness per VJ was associated with PD in Ipi-P but not Ipi-N patients (p = 0.014 versus p = 0.287). Outliers removed, Grubbs’ alpha = 01. Data are presented as median and IQR.
(E) Paired pre- and on-therapy values of median CDR3 richness and evenness per VJ for each patient. Consistently decreased median CDR3 evenness per VJ was observed in responders overall (p = 0.003). When further segregated by prior immunotherapy status, this effect was observed only in Ipi-N responders (p = 0.020).
(A) Number of clonal mutations lost per case and number of T-cell clones expanded on-therapy. A linear relationship was observed for patients with CR/PR (r2 = 0.82; p = 0.005), whereas no relationship was observed for patients with SD (r2 = 0.0003; p = 0.96) or PD (r2 = 0.07; p = 0.40).
(B) Baseline mutation load was associated with number of T-cell clones expanded on-therapy in patients with CR/PR (r2 = 0.54; p = 0.06) and SD (r2 = 0.50; p = 0.03) but not in patients with PD (r2 = 0.004; p = 0.83).
(C) T-cell clones expanded in proportion to the number of neoantigens that became undetectable on-therapy in patients whose tumors exhibited genomic contraction (r2 = 0.47; p = 0.01) but not genomic persistence (r2 = 0.01; p = 0.74).
(D) Comparison of neoantigen ratio between mutations that become undetectable on-therapy versus all mutations detected in pre-therapy samples. This alternative analysis also demonstrates depletion of neoantigens on-therapy in patients with CR/PR versus PD (p = 0.05; Mann–Whitney test).
(E) Expected versus observed number of neoantigens detected on-therapy (selected cases shown). In patients with CR/PR, the observed number of neoantigens was lower than the expected value in all cases (* p < 0.05).
(F) Patients with PD lost a higher proportion of subclonal mutations than clonal mutations on-therapy, which is different than patients with CR/PR or SD. Each response group lost approximately the same number of subclonal mutations on-therapy (data not shown). Data are presented as median and IQR.
HIGHLIGHTS.
A prospective trial reveals molecular actions of anti-PD-1 therapy
Anti-PD-1 therapy induces changes in the mutational burden of tumors
Distinct changes in gene expression programs associate with clinical response
Shifts in the TCR repertoire occur following immune checkpoint blockade
Acknowledgments
We first thank each patient and their families for participation in this study. This work was funded by Bristol-Myers Squibb, the Pershing Square Sohn Cancer Research Foundation (T.A.C.), the PaineWebber Chair (T.A.C.), Stand Up 2 Cancer (T.A.C.), the Memorial Sloan Kettering Cancer Center core grant 5P30 CA008748-50, and the STARR Cancer Consortium (T.A.C.). S.B. reports grants from Abraxis, Amgen, Bionomics, Bristol-Myers Squibb, EMD Serono, Immune Design, Immunogen, Merck, NantKwest, and OncoSec, and personal fees from Genentech. T.A.C. is a co-founder of Gritstone Oncology and reports research grants from Bristol-Myers Squibb. H.C. and C.E.H. are full-time employees and stock shareholders of Bristol-Myers Squibb. T.F.G. reports grants from Bristol-Myers Squibb, Incyte, Merck, Ono, Genentech, and Seattle Genetics, and personal fees from AbbVie, Aduro, Bayer, Jounce Therapeutics, Merck, and Genentech, and is a stock shareholder of Jounce Therapeutics. J.J.H. reports that spouse is a full-time employee of Regeneron Pharmaceuticals. F.S.H. reports grants from Bristol-Myers Squibb, and personal fees from Bristol-Myers Squibb, EMD Serono, Genentech, Merck, and Novartis. W-J.H. reports grants from Bristol-Myers Squibb, GlaxoSmithKline, MedImmune, and Merck, and personal fees from Merck. F.K. and V.M. report grants from Bristol-Myers Squibb. S.M-A. reports personal fees from Bristol-Myers Squibb and Merck. L.G.T.M. reports personal fees from Merck. N.R. reports grants from Bristol-Myers Squibb, and personal fees from MedImmune. J.P.S. reports grants and personal fees from, and is a stock shareholder of, NexImmune Inc. W.H.S. reports grants from Bristol-Myers Squibb and Merck, and personal fees from Bristol-Myers Squibb, Castle Biosciences, Merck, and Novartis. C.L.S. reports grants from Merck and Polynoma, personal fees from Immatics, and is a patent holder for the University of Virginia. W.J.U. reports grants and personal fees from Bristol-Myers Squibb and MedImmune, and personal fees from Green Peptide and eTheRNA. D.C., A.D., S.M.K., R.M., R.S., J-W.S., J.S.S., and N.W. have nothing to disclose. Medical writing and editorial assistance provided by Amrita Dervan, MSc, and Jay Rathi, MA, of Spark Medica Inc. (US), funded by Bristol-Myers Squibb.
Footnotes
AUTHOR CONTRIBUTIONS
Funding acquisition: T.A.C. Conceptualization: T.A.C., N.R., C.E.H., and W.J.U. Clinical trial implementation and accrual: W.J.U., F.S.H., S.M-A., W.H.S., S.B., W-J.H., T.F.G., C.L.S., and C.E.H. Investigation and formal analysis: N.R., V.M., and S.M.K. Analysis of pre- and on-therapy changes in mutation load and clonality: N.W., V.M., and N.R. Neoantigen analysis: D.C., V.M., N.R., and J.J.H. Analysis of RNA-seq data: A.D., F.K., H.C., R.M., and N.R. Exome-seq data from pre-therapy samples: V.M., L.G.T.M., and N.R. Analysis of TCR-seq data: J.J.H., J.S.S., J-W.S., and J.P.S. Writing (original draft): N.R., J.J.H., A.D., N.W., and T.A.C. Writing (review and editing): all authors. Supervision: N.W. and T.A.C.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud-Haond S, Duarte CM, Alberto F, Serrao EA. Standardizing methods to address clonality in population studies. Mol Ecol. 2007;16:5115–5139. doi: 10.1111/j.1365-294X.2007.03535.x. [DOI] [PubMed] [Google Scholar]
- Arstila TP, Casrouge A, Baron V, Even J, Kanellopoulos J, Kourilsky P. A direct estimate of the human alphabeta T cell receptor diversity. Science. 1999;286:958–961. doi: 10.1126/science.286.5441.958. [DOI] [PubMed] [Google Scholar]
- Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373:123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson CS, Emerson RO, Sherwood AM, Desmarais C, Chung MW, Parsons JM, Steen MS, LaMadrid-Herrmannsfeldt MA, Williamson DW, Livingston RJ, et al. Using synthetic templates to design an unbiased multiplex PCR assay. Nat Commun. 2013;4:2680. doi: 10.1038/ncomms3680. [DOI] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, Miller JP, Bassett RL, Gopalakrishnan V, Wani K, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 2016;6:827–837. doi: 10.1158/2159-8290.CD-15-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162:974–986. doi: 10.1016/j.cell.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dako. [Accessed: February 20, 2017];PD-L1 IHC 28-8 pharmDx. 2016 Available from http://www.accessdata.fda.gov/cdrh_docs/pdf15/P150027c.pdf.
- Das R, Verma R, Sznol M, Boddupalli CS, Gettinger SN, Kluger H, Callahan M, Wolchok JD, Halaban R, Dhodapkar MV, et al. Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes in vivo. J Immunol. 2015;194:950–959. doi: 10.4049/jimmunol.1401686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science. 2017:355. doi: 10.1126/science.aaf8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt WS, Emerson RO, Lindau P, Vignali M, Snyder TM, Desmarais C, Sanders C, Utsugi H, Warren EH, McElrath J, et al. Dynamics of the cytotoxic T cell response to a model of acute viral infection. J Virol. 2015;89:4517–4526. doi: 10.1128/JVI.03474-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunckley H. HLA typing by SSO and SSP methods. Methods Mol Biol. 2012;882:9–25. doi: 10.1007/978-1-61779-842-9_2. [DOI] [PubMed] [Google Scholar]
- Emerson R, Sherwood A, Desmarais C, Malhotra S, Phippard D, Robins H. Estimating the ratio of CD4+ to CD8+ T cells using high-throughput sequence data. J Immunol Methods. 2013;391:14–21. doi: 10.1016/j.jim.2013.02.002. [DOI] [PubMed] [Google Scholar]
- Ferris RL, Blumenschein G, Jr, Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375:1856–1867. doi: 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher S, Barry A, Abreu J, Minie B, Nolan J, Delorey TM, Young G, Fennell TJ, Allen A, Ambrogio L, et al. A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biol. 2011;12:R1. doi: 10.1186/gb-2011-12-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, Gupta R, Tsai JM, Sinha R, Corey D, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495–499. doi: 10.1038/nature22396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi FS, Kluger H, Sznol M, Carvajal R, Lawrence D, Atkins M, Powderly J, Sharfman W, Puzanov I, Smith D, et al. Abstract CT001: Durable, long-term survival in previously treated patients with advanced melanoma (MEL) who received nivolumab (NIVO) monotherapy in a phase I trial. Cancer Research. 2016;76:CT001–CT001. [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno B, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno K, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343:189–193. doi: 10.1126/science.1239947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348:74–80. doi: 10.1126/science.aaa6204. [DOI] [PubMed] [Google Scholar]
- Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, Woo G, Nguyen AV, Figueiredo CC, Foubert P, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature. 2016;539:437–442. doi: 10.1038/nature19834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, Kluth S, Bozic I, Lawrence M, Bottcher S, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526:525–530. doi: 10.1038/nature15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Minor D, D’Angelo S, Neyns B, Smylie M, Miller WH, Jr, Gutzmer R, Linette G, Chmielowski B, Lao CD, et al. Overall survival in patients with advanced melanoma who received nivolumab versus investigator’s choice chemotherapy in CheckMate 037: a randomized, controlled, open-label phase III trial. J Clin Oncol. 2017 doi: 10.1200/JCO.2016.71.8023. JCO2016718023 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, Ley TJ, Mardis ER, Wilson RK, Ding L. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28:311–317. doi: 10.1093/bioinformatics/btr665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N, Favero F, de Bruin EC, Birkbak NJ, Szallasi Z, Swanton C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med. 2015;7:283ra254. doi: 10.1126/scitranslmed.aaa1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris L, Riaz N, Desrichard A, Şenbabaoğlu Y, Hakimi A, Makarov V, Reis-Filho J, Chan TA. Pan-cancer analysis of intratumor heterogeneity as a prognostic determinant of survival. Oncotarget. 2016;7:10051–10063. doi: 10.18632/oncotarget.7067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373:1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Lamberth K, Buus S, Brunak S, Lund O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003;12:1007–1017. doi: 10.1110/ps.0239403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riaz N, Havel JJ, Kendall SM, Makarov V, Walsh LA, Desrichard A, Weinhold N, Chan TA. Recurrent SERPINB3 and SERPINB4 mutations in patients who respond to anti-CTLA4 immunotherapy. Nat Genet. 2016b;48:1327–1329. doi: 10.1038/ng.3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riaz N, Morris L, Havel JJ, Makarov V, Desrichard A, Chan TA. The role of neoantigens in response to immune checkpoint blockade. Int Immunol. 2016a;28:411–419. doi: 10.1093/intimm/dxw019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320–330. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, Riddell SR, Warren EH, Carlson CS. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood. 2009;114:4099–4107. doi: 10.1182/blood-2009-04-217604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, Dawson N, O’Donnell PH, Balmanoukian A, Loriot Y, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387:1909–1920. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31. doi: 10.1186/s13059-016-0893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth A, Khattra J, Yap D, Wan A, Laks E, Biele J, Ha G, Aparicio S, Bouchard-Cote A, Shah SP. PyClone: statistical inference of clonal population structure in cancer. Nat Methods. 2014;11:396–398. doi: 10.1038/nmeth.2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salerno EP, Bedognetti D, Mauldin IS, Deacon DH, Shea SM, Pinczewski J, Obeid JM, Coukos G, Wang E, Gajewski TF, et al. Human melanomas and ovarian cancers overexpressing mechanical barrier molecule genes lack immune signatures and have increased patient mortality risk. Oncoimmunology. 2016;5:e1240857. doi: 10.1080/2162402X.2016.1240857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims JS, Grinshpun B, Feng Y, Ung TH, Neira JA, Samanamud JL, Canoll P, Shen Y, Sims PA, Bruce JN. Diversity and divergence of the glioma-infiltrating T-cell receptor repertoire. Proc Natl Acad Sci U S A. 2016;113:E3529–3537. doi: 10.1073/pnas.1601012113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Six A, Mariotti-Ferrandiz ME, Chaara W, Magadan S, Pham HP, Lefranc MP, Mora T, Thomas-Vaslin V, Walczak AM, Boudinot P. The past, present, and future of immune repertoire biology - the rise of next-generation repertoire analysis. Front Immunol. 2013;4:413. doi: 10.3389/fimmu.2013.00413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic [bgr]-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–235. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. 2013;5:200ra116. doi: 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweis RF, Spranger S, Bao R, Paner GP, Stadler WM, Steinberg G, Gajewski TF. Molecular drivers of the non–T-cell-inflamed tumor microenvironment in urothelial bladder cancer. Cancer Immunol Res. 2016;4:563–568. doi: 10.1158/2326-6066.CIR-15-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4:127ra137. doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taube JM, Young GD, McMiller TL, Chen S, Salas JT, Pritchard TS, Xu H, Meeker AK, Fan J, Cheadle C, et al. Differential expression of immune-regulatory genes associated with PD-L1 display in melanoma: implications for PD-1 pathway blockade. Clin Cancer Res. 2015;21:3969–3976. doi: 10.1158/1078-0432.CCR-15-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275–287. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MH, Goldinger SM, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdegaal EM, de Miranda NF, Visser M, Harryvan T, van Buuren MM, Andersen RS, Hadrup SR, van der Minne CE, Schotte R, Spits H, et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature. 2016;536:91–95. doi: 10.1038/nature18945. [DOI] [PubMed] [Google Scholar]
- Wang J, Cazzato E, Ladewig E, Frattini V, Rosenbloom DI, Zairis S, Abate F, Liu Z, Elliott O, Shin YJ, et al. Clonal evolution of glioblastoma under therapy. Nat Genet. 2016;48:768–776. doi: 10.1038/ng.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber J, Gibney G, Kudchadkar R, Yu B, Cheng P, Martinez AJ, Kroeger J, Richards A, McCormick L, Moberg V, et al. Phase I/II study of metastatic melanoma patients treated with nivolumab who had progressed after ipilimumab. Cancer Immunol Res. 2016b;4:345–353. doi: 10.1158/2326-6066.CIR-15-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber J, Horak C, Hodi FS, Chang H, Woods D, Sanders C, Robins H, Yusko E. Baseline tumor T cell receptor (TcR) sequencing analysis and neo antigen load is associated with benefit in melanoma patients receiving sequential nivolumab and ipilimumab. Ann Oncol. 2016a;27:1047O. [Google Scholar]
- Wei L, Liu LT, Conroy JR, Hu Q, Conroy JM, Morrison CD, Johnson CS, Wang J, Liu S. MAC: identifying and correcting annotation for multi-nucleotide variations. BMC Genomics. 2015;16:569. doi: 10.1186/s12864-015-1779-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolchok JD, Hoos A, O’Day S, Weber JS, Hamid O, Lebbe C, Maio M, Binder M, Bohnsack O, Nichol G, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15:7412–7420. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- Yau C. OncoSNP-SEQ: a statistical approach for the identification of somatic copy number alterations from next-generation sequencing of cancer genomes. Bioinformatics. 2013;29:2482–2484. doi: 10.1093/bioinformatics/btt416. [DOI] [PubMed] [Google Scholar]
- Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, Trevino V, Shen H, Laird PW, Levine DA, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612. doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375:819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarnitsyna VI, Evavold BD, Schoettle LN, Blattman JN, Antia R. Estimating the diversity, completeness, and cross-reactivity of the T cell repertoire. Front Immunol. 2013;4:485. doi: 10.3389/fimmu.2013.00485. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Exome Sequencing Data (Pre-therapy Samples), Related to Figure 1
Table S2: Clinical Characteristics, Related to Figure 1
Table S3: Somatic Mutations, Related to Figure 1
(A) High mutation load (>100 mutations as previously reported) is not associated with OS in Ipi-P patients (log-rank test; p = NS).
(B) More subclonal mutations in Ipi-P patients (p = 0.08). Data are presented as median and IQR.
(C) Clonal mutation load corresponds with response in Ipi-N but not Ipi-P patients. Data are presented as median and IQR.
(D) High clonal mutation load is not associated with OS in Ipi-P patients (data split at median; log-rank test; p = NS).
(E) Association of TCGA-determined recurrent tumor copy-number alterations and response.
(F) Copy-number alterations in IFN-γ gene cluster and response.
(G) Copy-number changes split at the median and determined as previously described (Davoli et al., 2017) influence outcome in the Ipi-P cohort (p = 0.0008; log-rank test) but not in the Ipi-N cohort (p = 0.49; log-rank test).
(H) Change in mutation load is associated with response (p = 5.87e–5; Mann–Whitney test for CR/PR versus PD; red = Ipi-N; blue = Ipi-P). Data are presented as median and IQR.
(I) Change in mutation load is associated with PFS (p < 0.001; log-rank test; data split at median).
(A) Variant allele frequencies (VAFs) for all mutations detected in patients with CR/PR. In each case a moderate number of mutations remain present in on-therapy samples.
(B) VAFs for patients with SD.
(C) VAFs for patients with PD.
(D) Change in purity between on-therapy and pre-therapy samples computationally estimated from exome data and RNA-seq data (see Methods). Data are presented as median and IQR.
(E) The probability of detecting a mutation determined by sequencing coverage in a tumor.
(F) Expected number of mutations detected in CR/PR cases assuming a five-fold decrease in median VAF (results similar for SD and PD cases; data not shown).
(G) Mutational signatures (aggregated from all samples). Novel subclonal mutations feature a significant frequency of signature 11-related mutations.
(A) GO and Reactome enrichment of cluster 2 genes demonstrates that most are related to immune processes.
(B) Immune cell composition from pre-therapy samples for selected populations from RNA-seq deconvolution analysis. Top: Results for Ipi-N patients. Bottom: Results for Ipi-P patients. (Limited sample numbers for patients with CR/PR.)
(C) Clustering of samples by the recently described IPRES signature and lack of association with response.
(D) GO and Reactome enrichment of DEGs between genomic contraction and genomic persistence. Processes such as PD-1 signaling, TCR signaling, co-stimulation of CD28 family, and MHC class II antigen presentation are enriched.
(E) Previously described macrophage-related gene expression signature (Kaneda et al., 2016) weakly associates with clinical response in CA209-038 cohort, and similarly a significant number of differentially expressed HLA class II alleles are identified as previously described.
(A) Beta coefficients of genes from Figure 4A and IPA of top pathways altered during treatment.
(B) GO enrichment analysis of clusters from Figure 4B demonstrate one cluster is dominated by immune processes, whereas the other appears to involve genes related to tumor growth and progression.
(C) Venn diagram of differentially expressed immune genes from different analyses. Pre-therapy immune genes are cluster 1 from Figure 3A. Anti-PD-1 pharmacologically-related genes are from Figure 4A. Differential genes on-therapy are from Figure 4B.
(A) On-therapy, the extent of T-cell infiltration was associated with response in the Ipi-P but not Ipi-N cohort (p = 0.031 versus p = 0.231). Notably, it seems that Ipi-P non-responders showed little to no increase or even a decrease in TILs upon Nivo therapy, whereas both responders and non-responders in the Ipi-N cohort showed an increase in TILs upon Nivo therapy (see also Figure 5A).
(B) In pre-therapy samples, there was no significant difference in the number of unique intratumoral CDR3 sequences between Ipi-P and Ipi-N patients, or between responders and non-responders.
(C) In pre-therapy samples, there was no significant difference in evenness between Ipi-P and Ipi-N patients, or between responders and non-responders. Data are presented as median and IQR.
(D) Changes in median CDR3 richness and evenness per VJ pair. Decreased CDR3 richness per VJ was associated with PD in Ipi-P but not Ipi-N patients (p = 0.014 versus p = 0.287). Outliers removed, Grubbs’ alpha = 01. Data are presented as median and IQR.
(E) Paired pre- and on-therapy values of median CDR3 richness and evenness per VJ for each patient. Consistently decreased median CDR3 evenness per VJ was observed in responders overall (p = 0.003). When further segregated by prior immunotherapy status, this effect was observed only in Ipi-N responders (p = 0.020).
(A) Number of clonal mutations lost per case and number of T-cell clones expanded on-therapy. A linear relationship was observed for patients with CR/PR (r2 = 0.82; p = 0.005), whereas no relationship was observed for patients with SD (r2 = 0.0003; p = 0.96) or PD (r2 = 0.07; p = 0.40).
(B) Baseline mutation load was associated with number of T-cell clones expanded on-therapy in patients with CR/PR (r2 = 0.54; p = 0.06) and SD (r2 = 0.50; p = 0.03) but not in patients with PD (r2 = 0.004; p = 0.83).
(C) T-cell clones expanded in proportion to the number of neoantigens that became undetectable on-therapy in patients whose tumors exhibited genomic contraction (r2 = 0.47; p = 0.01) but not genomic persistence (r2 = 0.01; p = 0.74).
(D) Comparison of neoantigen ratio between mutations that become undetectable on-therapy versus all mutations detected in pre-therapy samples. This alternative analysis also demonstrates depletion of neoantigens on-therapy in patients with CR/PR versus PD (p = 0.05; Mann–Whitney test).
(E) Expected versus observed number of neoantigens detected on-therapy (selected cases shown). In patients with CR/PR, the observed number of neoantigens was lower than the expected value in all cases (* p < 0.05).
(F) Patients with PD lost a higher proportion of subclonal mutations than clonal mutations on-therapy, which is different than patients with CR/PR or SD. Each response group lost approximately the same number of subclonal mutations on-therapy (data not shown). Data are presented as median and IQR.