

Abstract
Lipopolysaccharide (LPS) from the cell envelope of Gram-negative bacteria is a principal cause of the symptoms of sepsis. LPS has been reported to modulate the function of platelets although the underlying mechanisms of LPS action in these cells remain unclear. Platelets express the Toll-like receptor 4 (TLR4) which serves as a receptor for LPS, although the potential role of TLR4 and associated cell signalling in controlling platelet responses to LPS has not been extensively explored. In this study, we therefore investigated the actions of LPS prepared from different strains of Escherichia coli on platelet function, the underlying signalling mechanisms, and the potential role of TLR4 in orchestrating these. We report that LPS increased the aggregation of washed platelets stimulated by thromboxane (U46619) or GPVI collagen receptor agonists, effects that were prevented by a TLR4 antagonist. Associated with this, LPS enhanced fibrinogen binding, P-selectin exposure and reactive oxygen species (ROS) release. Increase of ROS was found to be important for the actions of LPS on platelets, since these were inhibited in the presence of superoxide dismutase or catalase. The effects of LPS were associated with phosphorylation of Akt, ERK1/2 and PLA2 in stimulated platelets, and inhibitors of PI3-kinase, Akt and ERK1/2 reduced significantly LPS enhanced platelet function and associated ROS production. Furthermore, inhibition of platelet cyclooxygenase or the thromboxane receptor, revealed an important role for thromboxane A2. We therefore conclude that LPS increases human platelet activation through a TLR4-PI3K-Akt-ERK1/2-PLA2 -dependent pathway that is dependent on ROS and TXA2 formation.
Introduction
Platelets are required for haemostasis and respond to tissue injury to form an aggregate or platelet plug that limits the loss of blood. Inappropriate activation of platelets in diseased blood vessels is, however, a major trigger for thrombosis and platelets are increasingly implicated in inflammation and the symptoms of sepsis [1]. Lipopolysaccharide (LPS) from the cell envelope of Gram-negative bacteria is a principal cause of the symptoms of sepsis and this can result in platelet sequestration in the lungs and liver, thrombocytopenia and disseminated intravascular coagulation (DIC) [2, 3]. LPS has been reported to modulate the function of platelets [4, 5, 6], although the underlying mechanisms of LPS action remain unclear. Indeed, some studies have indicated that exposure of platelets to LPS in vitro potentiates platelet function [7, 8], while others have indicated that it supresses their activity [9]. Furthermore, LPS from different sources and at distinct concentrations are able to promote variable effects in a range of cells types [10,11].
LPS has been demonstrated to induce excessive secretion of pro-inflammatory mediators such as cytokines, tumor necrosis factor alpha (TNFα) and reactive oxygen species (ROS) in different cells [5, 12], and overproduction of ROS has been associated with damage to vascular endothelium and multiple organ dysfunction [13]. Previous studies have suggested that stimulated and unstimulated platelets are capable of releasing ROS that participate in the control of platelet function [14, 15]. The mechanisms that lead the release of ROS by platelets are, however, incompletely understood.
Platelets, express the pathogen recognition receptor, Toll-like receptor 4 (TLR4) that serves as a receptor for LPS on a range of cells types [3, 16, 17]. The role in platelets that this receptor may play in stimulating the signalling that controls platelet responses to LPS has yet to be explored. Therefore, in the present study we sought to explore the signalling pathway(s) through which LPS modulates platelets regulation.
Ligation of TLR4 induces recruitment of multiple adaptor proteins such as TRIF, MyD88, TIRAP, TRAM and SARM through interactions with Toll-interleukin-1 receptor domains [18]. Previous studies have identified a number of proteins implicated in TRIF dependent signalling (eg. TBK-1, IRAK-1, JNKs, MAPK, TRAF3, TRAF6, IRF-3, Ikk-I, IκB-α, NK-κB) that are present in platelets [7, 19, 20]. In vascular smooth muscle, LPS- mediated signalling promotes activation of the mitogen-activated kinases (MAPKs) p38, ERK1/2 and JNK1/2 and also the phosphoinositide- 3 kinase (PI3-kinase) dependent signalling [21]. Furthermore, Brown and McIntyre showed that in platelets, MyD88, TRAF6, JNK and AKT are required in IL-1β production stimulated by LPS [22].
Platelet activation by agonists such as collagen or thromboxane A2 is associated with the activation of a complex network of cell signalling [23, 24] in which several molecules implicated in TLR4 signalling are critically involved, including PI3-kinase (PI3K), Akt, and ERK1/2 [25, 26, 27]. While these signalling proteins have been shown to mediate some actions of LPS on different cells [25, 26], their potential involvement in TLR4 signalling in platelets has not been established.
The aim of this study was to elucidate whether components of the TLR4 signalling pathway are implicated in the functional responses of platelets to LPS and the involvement of ROS production in these responses. Here, we found that LPS increases human platelet activation through a TLR4-PI3K-Akt-ERK1/2-PLA2-dependent pathway that is dependent on ROS and TXA2 formation.
Materials and methods
Isolation and purification of Lipopolysaccharide from Gram-negative bacteria
LPS from E. coli K12 was isolated and purified using methodology described by Davis and Goldberg, 2012 [28]. The purity LPS samples prepared were visualized by direct staining following separation on 12% SDS-polyacrilamide gel using Pro-Q Emerald 300 Lipopolysaccharide Gel Stain Kit (Sigma Aldrich, UK) (S1 Fig). LPS purified from E. coli O111:B4 was purchased from Sigma Aldrich.
Platelet preparation
Blood was drawn from healthy donors that had given informed consent. The study and procedures for obtaining informed consent were approved by the University of Reading Research Ethics Committee. Blood was collected into 50mL syringes containing 4% (w/v) sodium citrate and acid citrate dextrose (ACD, 2.5% sodium citrate, 2% D-glucose and 1.5% citric acid). Platelet preparation was according to the methodology developed previously to limit the levels of leukocyte or erythrocyte contamination to no more than one contamination cell per 6500 platelets [29]. The blood was centrifuged for 20 min at 102× g at room temperature to obtain platelet-rich plasma (PRP). Further centrifugation of PRP [with 125 ng·mL–1 prostaglandin I2 (PGI2)] at 1413× g for 10 min resulted in a platelet pellet that was re-suspended in modified Tyrodes-HEPES buffer (134 mM NaCl, 2.9 mM KCl, 0.34 mM Na2HPO4·12H2O, 12 mM NaHCO3, 20 mM HEPES and 1 mM MgCl2, pH 7.3) and washed by centrifuging in the presence of PGI2 again at the same speed for 10 min. The resulting platelet pellet was re-suspended in modified Tyrodes-HEPES buffer to a final density of 4 × 108 cells·mL−1 for optical aggregometry, flow cytometry and ROS release assays, and 8 × 108 cells·mL−1 for immunoblot analysis.
Aggregation
Platelet aggregation assays were performed by optical aggregometry stimulated with the thromboxane A2 receptor agonist U46619 (Cayman Chemical) or the collagen receptor glycoprotein VI agonist, cross-linked collagen-related peptide (CRP-XL) (supplied by Prof R. Farndale, University of Cambridge, UK) [30]. Platelets were stimulated in this way in the presence or absence of LPS from E. coli O111:B4 (Sigma Aldrich, UK) or from E. coli K12 (purified as described above), superoxide dismutase (SOD, Sigma Aldrich, UK), catalase (Sigma Aldrich, UK), inhibitor of PI3K- LY294002 (Selleckchem, UK), inhibitor of PI3K-δ- Cal (Selleckchem, UK), Akt inhibitor IV (Calbiochem, UK), inhibitor of MEK- Cobimetinib (Selleckchem, UK), indomethacin (Sigma Aldrich, UK), the TXA2 receptor antagonist- GR32191 (Sigma Aldrich, UK), the TLR4 antagonist- LPS RS ultrapure (InvivoGen, UK) or vehicle used to dissolve each of the above (containing 0.01% (v/v) dimethyl sulphoxide, which did not affect platelet function, or distilled water). Aggregation assays were performed with 250μL of washed platelet suspension after incubation for 3 minutes with respective inhibitors followed by stimulation with agonist or agonist plus LPS from Escherichia coli- K12 or from Escherichia coli- O111:B4 for 5 minutes. Aggregation was monitored using Chrono-Log Model 700/ Optical-Lumi Aggregometer.
Immunobloting assay
SDS-PAGE and immunoblotting were performed using standard protocols as described previously [31]. Rabbit anti-human 14-3-3ζ (Santa Cruz Biotechnology, USA) was used to detect 14-3-3ζ to ensure equivalent levels of protein loading on immunoblots. The phospho-specific antibodies against various signalling proteins (Akt, ERK1/2 and PLA2) were obtained from Cell signalling technology, USA (catologue numbers: 9271, 4370 and 2831 respectively). The secondary antibodies for immunoblotting; Cy5 goat anti-rabbit IgG and Cy3 goat anti-mouse IgG antibodies were obtained from Invitrogen, UK. Blots were visualised using a Typhoon FLA 9500 fluorimager (GE healthcare, UK). Image Quant TL software (GE Healthcare) was used for the fluorescence visualisation and analysis of protein bands.
Alpha granule secretion and fibrinogen binding
Flow cytometric assays were performed in 96-well plates. U46619 or CRP-XL-stimulated fibrinogen binding and P-selectin exposure were measured in PRP using FITC labelled rabbit anti-human fibrinogen antibodies (Dako UK Ltd) and PE/Cy5 labelled mouse anti-human anti-CD62P antibody (BD Biosciences, UK) respectively in the presence or absence of different antagonists or inhibitors for 3 minutes prior to activation (e.g. superoxide dismutase- SOD, catalase, inhibitor of PI3-Kinase- LY294002, inhibitor of PI3K-δ (Cal), Akt inhibitor IV, inhibitor of MEK (cobimetinib), Indomethacin, TXA2 antagonist (GR32191) or TLR4 antagonist (LPS RS ultrapure) or vehicle (containing 0.01% dimethyl sulphoxide or distilled water)). Platelets were stimulated with U46619 or CRP in the presence or absence of LPS from Escherichia coli- O111:B4 or from Escherichia coli- K12 for 5 minutes at room temperature and then fixed in 0.2% (v/v) formyl saline prior to analysis by BD Accuri flow cytometry (BD Biosciences, Oxford, UK) equipped with a 488 nm wavelength argon laser, 510–540 nm band pass filter. Data were acquired from 10000 cells and recorded as percentage of cells positive or median fluorescence intensity (MFI).
Platelet ROS-production measured by flow cytometry
Production of ROS by platelets was measured by flow cytometry following the loading of platelets with the fluorescent dye, 2’,7’-dichlorofluorescin diacetate (DCFH-DA) (Sigma Aldrich, UK). DCFH is membrane-impermeable and rapidly oxidizes into the highly fluorescent 2´,7`-dichlorofluorescin (DCF) in the presence of intracellular hydrogen peroxide (H2O2), peroxynitrite (ONOO-), peroxides and hydroxyl radicals (·OH) [32]. Briefly, 50μL of washed platelets (4 x 108/ml) were preincubated with 10μM DCFH-DA in the presence or absence of inhibitors (e.g. superoxide dismutase, catalase, inhibitor of PI3-Kinase (LY294002), inhibitor of PI3K (Cal), Akt inhibitor IV, inhibitor of MEK- (cobimetinib), indomethacin, TXA2 receptor antagonist (GR32191) or TLR4 antagonist (LPS RS ultrapure), or vehicle (containing 0.01% (v/v) dimethyl sulphoxide) for 10 minutes at 37°C in the dark in a 24 well plate before stimulation with U46619 or CRP-XL, together with LPS from Escherichia coli- O111:B4 or LPS from Escherichia coli- K12 for 10 minutes. The samples were then fixed with 200μL of sodium azide (0.1% w/v) at room temperature for 10 minutes in the dark. Samples were analysed by BD Accuri flow cytometry (BD Biosciences, Oxford, UK) equipped with a 488 nm wavelength argon laser, 510–540 nm band pass filter. Data were acquired from 10000 cells and recorded as percentage of cells positive or median fluorescence intensity (MFI).
Statistical analysis
Data are presented as mean ± standard deviation of the mean (SEM). Statistical analyses were performed using PRISM 5 GRAPHPAD software (GraphPad Software Inc, La Jolla, CA, USA). Data were analysed to confirm normal distribution and compared using a Student’s t-Test or One-way ANOVA- and Bonferroni post-test analysis as appropriate.
Results
LPS in vitro potentiates activation of stimulated platelets
The effect of LPS on platelet function remains uncertain, although previous work has shown that different preparations of LPS in vitro induce platelet aggregation [7, 33]. To analyse the effects of LPS on platelet aggregation, washed human platelets were incubated with LPS from E. coli O111:B4 or alternatively purified from E. coli K12 (0.5, 1, 5 or 7.5µg/mL) in the presence or absence of the thromboxane receptor agonist U46619 (0.25μM) or the GPVI collagen receptor agonist CRP-XL (0.25μg/mL) and analysed for 5 min in an optical aggregometer. Both sources of LPS increased significantly the aggregation of platelets stimulated with U46619 (Fig 1A and 1B (LPS: E. coli O111:B4); S2A Fig (LPS: E. coli K12)) and CRP-XL Fig 1E and 1F (LPS: E. coli O111:B4); S2E Fig (LPS: E. coli K12)) by 42% and 21% respectively. To confirm that the effects of LPS on platelets were stimulated by binding to TLR4, LPS from Rhodobacter sphaeroides (LPS-RS) (1μg/mL), a TLR4 antagonist, was used. LPS-RS prevented the increase in aggregation stimulated by LPS from E.coli O111:B4 in platelets stimulated with U46619 (Fig 1B) or CRP-XL (Fig 1F).
Fig 1. LPS potenciates U46619-stimulated platelet activation.

Human-washed platelet aggregation was performed by optical aggregometry following stimulation with U46619 (0.25μM) or CRP-XL (0.25μg/ml) in the presence or absence of LPS from E. coli O111:B4 (0.5, 1, 5 or 7.5μg/mL) or LPS RS (1 or 7.5 μg/mL) (A, B, E, F). The effect of U46619 or CRP-XL and LPS- on fibrinogen binding and P-selectin exposure was measured in PRP by flow cytometry (C, D, G, H). Cumulative data represent mean values ± SEM (n = 4). (Anova-Bonferroni test, * P≤ 0.05; ** P≤ 0.01; *** P≤ 0.001).
Platelet inside-out signalling leads to a conformational change in integrin αIIbβ3 converting it from a low to high affinity state, thereby allowing activation dependent binding of its ligands fibrinogen and Von Willebrand Fator (VWF) [34]. This is also associated with the secretion of α-granules which contain adhesion proteins such as fibrinogen, VWF, coagulation and fibrinolytic factors [35]. To analyse the effects of LPS on the modulation of integrin αIIbβ3 affinity and α-granule secretion, fibrinogen binding and P-selectin exposure on the surface of platelets were measured by flow cytometry. Although both LPS from E. coli O111:B4 or the alternative LPS from E. coli K12 (1 and 7.5 μg/mL) caused a slight decrease in optical density of platelet suspensions (5%), LPS from E. coli O111:B4 raised approximately two-fold the level of fibrinogen binding and P-selectin exposure on unstimulated platelets. Following stimulation with U46619 (Fig 1C and 1D (LPS: E. coli O111:B4)); S2B and S2C Fig (LPS: E. coli K12)) or CRP-XL (Fig 1G and 1H (LPS: E. coli O111:B4); S2F and S2G Fig (LPS: E. coli K12)), both source of LPS were able to significantly increase fibrinogen binding (LPS from E. coli O111:B4: 70% and 200% respectively with U46619 and CRP-XL; LPS from E. coli K12: 170% and 145% respectively with U46619 and CRP-XL) and P-selectin exposure (LPS from E. coli O111:B4: 130% and 100% respectively with U46619 and CRP-XL; LPS from E. coli K12: 170% and 120% respectively with U46619 and CRP-XL).
LPS potentiates aggregation through an increase of platelet-ROS production
Platelets are widely recognised to contribute to inflammation and themselves can generate endogenous inflammatory mediators and ROS [14, 15]. To investigate the capacity of LPS to induce platelet ROS-generation by TLR4 binding and to explore the cell signalling that may control this, ROS production was measured in stimulated and unstimulated platelets by flow cytometry using the DCFH-DA fluorescent ROS probe (10μM). Stimulation of platelets with U46619 was associated with an increase in platelet- derived ROS. Both sources of LPS (1 and 7.5 μg/mL) promoted approximately three-fold increase of ROS release in platelets stimulated with U46619 (0.25μM) (Fig 2A and 2B (LPS: E. coli O111:B4); S2D Fig (LPS: E. coli K12). LPS from E. coli O111:B4 and from E. coli K12 were able also to raise by approximately 70% the ROS release of unstimulated platelets (Fig 2A (LPS: E. coli O111:B4 S2D and S2H Fig (LPS: E. coli K12)). Incubation with TLR4 antagonist- LPS RS ultrapure decreased substantially the production of ROS raised by LPS from E. coli O111:B4 in platelets stimulated with U46619 (0.25μM, reduction of 60%) (Fig 2A), suggesting that LPS potentiates ROS generation in platelets through TLR4 binding.
Fig 2. ROS release induced by LPS promotes potentiation of aggregation.
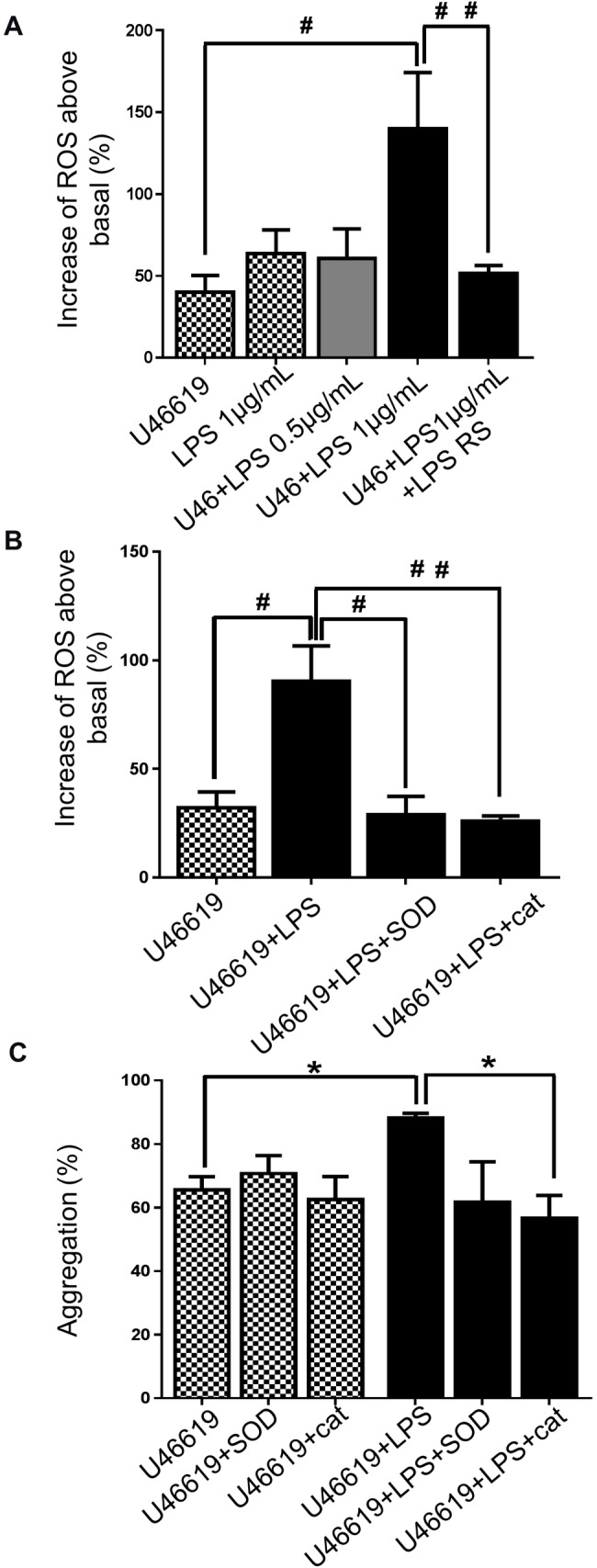
Washed platelets (4 x 108/ml) were pre-incubated with 10μM DCFH-DA in the presence or absence of vehicle, LPS from E. coli O111:B4 (0.5 or 1μg/mL) or LPS RS (1μg/mL), superoxide dismutase- SOD (30U/mL) or catalase (300U/mL) before being activated with U46619 (0.25μM). Samples were analysed by flow cytometry and the levels of ROS released were detected and expressed as % increase above levels detected in unstimulated platelets (A and B). Human-washed platelet aggregation was performed by optical aggregometry following stimulation with U46619 (0.25μM) in the presence or absence of LPS from E. coli (1μg/mL) after 3 min of incubation with superoxide dismutase- SOD (30U/mL) or catalase (300U/mL) (C). Cumulative data represent mean values ± SEM (n = 4). (Anova-Bonferroni test, * P≤ 0.05; Test t student # P≤ 0.01; # # P≤ 0.01).
Platelet activation can be modulated by ROS derived from sources including stimulated platelets themselves, such as superoxide anions (O-2) and hydrogen peroxide (H2O2) [36]. To analyse whether LPS-potentiated platelet aggregation was associated with the modulation of O-2 and H2O2 production, platelets were incubated with superoxide dismutase- SOD (30U/mL) or catalase (300U/mL) (inhibitors of O-2 and H2O2 respectively), prior to being stimulated with U46619 (0.25μM). Both SOD and catalase were able to reduce the LPS- E. coli O111:B4 -induced increases in aggregation and ROS generation to a similar level to that seen in the absence of LPS (Fig 2B and 2C). These analyses indicate that H2O2 and O-2 are likely to contribute to the potentiation of aggregation mediated by LPS in vitro.
LPS potentiates platelets responses via PI3K/Akt signalling
Phosphoinositide 3-kinase (PI3K)/Akt-dependent signalling is involved in activation of integrin αIIbβ3 and regulates platelet secretion [23]. Previous studies have shown the involvement of the PI3K-Akt pathway in LPS-mediated effects in various cells [25, 26]. To assess the effect of LPS in vitro on PI3K-Akt dependent signalling in platelets, we employed western blotting to measure Akt phosphorylation on Ser 473. LPS (1μg/mL) raised by three-fold the phosphorylation of Akt in platelets stimulated with U46619 (0.25μM) (Fig 3A). Platelets were incubated with a pan- PI3K isoform inhibitor LY294002 (20μM), an inhibitor of PI3K-δ- Cal (60μM) or the Akt inhibitor IV (5μM) and platelet function was measured. The increase of aggregation induced by LPS in platelets stimulated with U46619 (0.25μM, (Fig 3B) or CRP-XL (0.25μg/mL, (S3A Fig)) was reduced significantly by the three inhibitors (approximately 50% inhibition for LY294002 and 70% for Cal or Akt inhibitor). Consistent with their effects on aggregation, the three inhibitors decreased substantially fibrinogen binding and P-selectin exposure of platelets stimulated with U46619 (0.25μM) and LPS (1μg/mL) (Fig 3C and 3D). LY294002 and Cal reduced the increase of ROS produced by LPS treatment to the level observed in the absence of LPS in platelets stimulated with U46619 (Fig 3E), although CRP-XL-stimulated ROS production was not inhibited by these inhibitors (S3B Fig). Together these data suggest that the PI3K/Akt pathway cooperates with LPS signalling which promotes enhanced platelet activation through the release and actions of ROS.
Fig 3. Exposure of platelets to LPS potentiates PI3K/Akt signalling.

Washed human platelets with or without treatment with U46619 (0.25μM) in the presence or absence of LPS from E. coli O111:B4 (1μg/mL) were analysed by immunoblotting using antiphospho-Akt (Ser473) antibody. Total levels of 14-3-3 ζ were measured on each sample as a loading control (A). Human-washed platelet aggregation was performed by optical aggregometry activated with U46619 (0.25μM) in the presence or absence of LPS from E. coli O111:B4 (1μg/mL) after 3 min of incubation with LY294002 (20μM), Cal (60μM) or Akt inhibitor IV (5μM) (B). The effect of U46619 and LPS- induced fibrinogen binding and P-selectin exposure after incubation with LY294002 (20μM), Cal (60μM) or Akt inhibitor IV (5μM) were measure in PRP by flow cytometry (C and D). Washed platelets (4 x 108/ml) were pre-incubated with 10μM DCFH-DA in the presence or absence of LY294002 (20μM), Cal (60μM) or Akt inhibitor IV (5μM) before being activated with U46619 (0.25μM) in the presence or absence of LPS from E. coli O111:B4 (1μg/mL) and ROS levels were analysed by flow cytometry (E). Cumulative data represent mean values ± SEM (n = 4). (Anova-Bonferroni test, * P≤ 0.05; ** P≤ 0.01; *** P≤ 0.001; Test t student # # P≤ 0.01).
Akt mediates LPS pathway through ERK1/2 and PLA2- phosphorylation
ERK activation plays an essential role in mediating platelet granule release by amplifying platelet responses and promoting a second wave of platelet aggregation [24]. It has previously been suggested that cell signalling promoted through the binding of LPS with TLR4 involves ERK1/2 phosphorylation and activation [31]. The ability of LPS (1μg/mL) to modulate ERK1/2 phosphorylation was therefore tested using immunoblot analysis. In stimulated platelets (U46619- 0.25μM), ERK1/2 phosphorylation was increased by a factor of three in the presence of LPS (1μg/mL) (Fig 4A). The inhibitor of ERK1/2, Cobimetinib (100μM) reduced dramatically (approximately 95%) the aggregation of platelets induced by U46619 (Fig 4B) or CRP-XL (S4A Fig) in the presence or absence of LPS. Additionally, Cobimetinib did not significantly change fibrinogen binding and P-selectin exposure in U46619-stimulated platelets but reduced by 65% both functions in platelets stimulated in the presence of LPS (Fig 4C and 4D). The inhibitor of ERK1/2 was also able to decrease by 75% the rise of ROS production by LPS in platelets stimulated with U46619 (Fig 4E) and CRP (S4B Fig).
Fig 4. The role of ERK1/2 in LPS-mediated platelet potentiation.

Washed human platelets stimulated or not with U46619 (0.25μM) in the presence or absence of LPS from E. coli O111:B4 (1μg/mL) were analysed by immunoblotting using antiphospho- ERK1/2 antibody. Total levels of 14-3-3 ζ were measured on each sample as a loading control (A). Human-washed platelet aggregation was performed by optical aggregometry following stimulation with U46619 (0.25μM) in the presence or absence of LPS from E. coli O111:B4 (1μg/mL) after 3 min of incubation with Cobimetinib (100μM) before activation with U46619 (0.25μM) (B). The effect of U46619 (0.25μM) and LPS- induced fibrinogen binding and P-selectin exposure after incubation with Cobimetinib (100μM) were measured in PRP by flow cytometry (C and D). Washed platelets (4 x 108/mL) were pre-incubated with 10μM DCFH-DA in the presence or absence of Cobimetinib (100μM) before being activated with U46619 (0.25μM) in the presence or absence of LPS from E. coli O111:B4 (1μg/mL) and ROS levels were analysed by flow cytometry (E). Cumulative data represent mean values ± SEM (n = 4). (Anova-Bonferroni test, * P≤ 0.05; ** P≤ 0.01; *** P≤ 0.001; Test t student # P≤ 0.05; # #P≤ 0.01).
Previous research has shown that phosphorylation of ERK1/2 is associated with PLA2 activity [37] and in platelets this link is poorly understood. Given the significant effect of ERK1/2 on LPS signalling in platelet function, PLA2 activation was explored. Through immunoblotting we verified that platelets stimulated with U46619 (0.25μM) and LPS (1μg/mL) exhibited an increase of PLA2 phosphorylation of approximately 2.3-fold compared with platelets in the absence of LPS (Fig 5A and 5C). The TLR4 antagonist did not have any effect on PLA2 activation in platelets stimulated with U46619 but this antagonist was able to decrease PLA2 phosphorylation of platelets stimulated with U46619 and LPS to levels observed in the absence of LPS (Fig 5A and 5C). Indeed, in platelets stimulated with U46619 plus LPS, TLR4 antagonist reduced ERK1/2 phosphorylation to the levels observed in the absence of LPS (Fig 5A and 5B). These data suggest that ERK1/2 and PLA2 contribute to platelet signalling stimulated by LPS.
Fig 5. LPS stimulates ERK1/2 and PLA2- phosphorylation via TLR4 and Akt.

Washed human platelets were incubated for 3 minutes with LPS RS (1μg/mL) or Akt inhibitor IV (5μM) before activation with U46619 (0.25μM) in the presence or absence of vehicle or LPS from E. coli (1μg/mL) and were analysed by immunoblotting using antiphospho- ERK1/2 (A and B) and antiphospho-PLA2 (A and C) antibodies. Total levels of 14-3-3ζ were measured on each sample as a loading control. Cumulative data represent mean values ± SEM (n = 4). (Anova-Bonferroni test, * P≤ 0.05; ** P≤ 0.01).
In order to analyse the involvement of Akt on ERK1/2 and PLA2 in LPS treated platelets, immunoblotting was performed with platelets stimulated by U46619 (0.25μM) in the presence or absence of Akt inhibitor IV (5μM). ERK1/2 and PLA2 phosphorylation induced by LPS were decreased to levels observed in absence of LPS by Akt inhibitor IV (Fig 5A–5C). These data indicate the potential effect of Akt in mediating platelet signalling through ERK1/2 and PLA2 activation.
LPS signalling is associated with TXA2 production
Thromboxane A2 (TXA2) has been suggested to contribute in the development of sepsis [38, 39] and PLA2 activation may be expected to result in TXA2 production [40]. To determine whether TXA2 is a downstream molecule involved in effects of TLR4 signalling in platelets, the cyclooxygenase inhibitor indomethacin (10μM) was incubated with washed platelets before activation with U46619 (0.25μM) or CRP-XL (0.25μg/mL). Indomethacin decreased significantly the LPS induced increase in platelet function: aggregation was reduced by 42% and 73% (in platelets stimulated with U46619 and CRP-XL respectively (Fig 6A; S5A Fig); fibrinogen binding and P-selectin exposure were reduced to levels seen in the absence of LPS (Fig 6B and 6C; S5B and S5C Fig); ROS production was decreased by 55% (with both antagonists plus LPS) (Fig 6D; S5D Fig).
Fig 6. LPS- mediated platelet activation is associated with TXA2 production.

Human-washed platelet aggregation was performed by optical aggregometry following stimulation with U46619 (0.25μM) in the presence or absence of LPS from E. coli O111:B4 (1g/mL) after 3 min of incubation with indomethacin (10μM) or GR32191 (100ng) (A and E). The effect of U46619 (0.25μM) and LPS- induced fibrinogen binding and P-selectin exposure after incubation with indomethacin (10μM) or GR32191 (100ng) were measure in PRP by flow cytometry (B, C, F and G). Washed platelets (4 x 108/mL) were pre-incubated with 10μM DCFHDA in the presence or absence of Indomethacin (10μM) or GR32191 (100ng) before being activated with U46619 (0.25μM) in the presence or absence of LPS from E. coli O111:B4 (1μg/mL) and ROS levels were analysed by flow cytometry (D and H). Cumulative data represent mean values ± SEM (n = 4). (Anova-Bonferroni test, * P≤ 0.05; ** P≤ 0.01; *** P≤ 0.001; Test t student # P≤ 0.05; # # P≤ 0.01; # # # P≤ 0.001).
Consistent with this, the TXA2 receptor antagonist- GR32191 (100ng) prevented the potentiation of aggregation induced by LPS in platelets stimulated with both U46619 (0.25μM, (Fig 6E)) or CRP-XL (0.25μg/ml, (S5E Fig)). The pre-incubation of platelets with GR32191 reduced fibrinogen binding and P-selectin exposure to the levels observed in the absence of LPS (in platelets stimulated with both agonists) (Fig 6F and 6G, S5F and S5G Fig). GR32191 also decreased by approximately 70% ROS production in platelets stimulated by U46619 and CRP-XL in the presence of LPS (Fig 6H; S5H Fig). Thus, these data indicate that the effects of LPS are mediated via TXA2.
ROS released by LPS modulates ERK1/2 and PLA2- phosphorylation
We have demonstrated that LPS activates ERK1/2 and PLA2 in platelets. To examine the role of ROS in modulating the activation of these enzymes, the levels of ERK1/2 and PLA2 phosphorylation in LPS-stimulated platelets were measured in the presence or absence of SOD or catalase. SOD treatment prevented LPS-induced phosphorylation of both ERK1/2 and PLA2 in platelets stimulated with U46619 (0.25μM), and catalase strongly inhibited ERK1/2 phosphorylation (Fig 7A–7C).
Fig 7. LPS induced ROS modulates ERK1/2 and PLA2 phosphorylation.

Washed human platelets were incubated for 3 minutes with SOD (30U/mL) or catalase (300U/mL) before activation with U46619 (0.25μM) in the presence or absence of vehicle or LPS from E. coli O111:B4 (1μg/mL) and were analysed by immunoblotting using antiphospho- ERK1/2 (A and B) or antiphospho-PLA2 (A and C) antibodies. Total levels of 14-3-3ζ were measured on each sample as a loading control. Cumulative data represent mean values ± SEM (n = 4). (Anova-Bonferroni test, * P≤ 0.05; *** P≤ 0.001).
Discussion
In the present work we have demonstrated that PI3K and Akt mediate an increase of platelet activation by LPS in vitro via ERK1/2/PLA2 pathway and our data also suggest this mechanism to involve TXA2 production and ROS release.
The treatment of platelets with LPS in vitro promoted platelet stimulation by inducing fibrinogen binding and the secretion of α-granule contents, both important markers of platelet activation. The action of LPS in platelets is a matter of some debate in the literature. The results of our study are consistent with previous studies in which LPS-induced enhancement of platelet function was observed in vitro [7, 8]. We found LPS to induce P-selectin secretion and activation of fibrinogen in both stimulated and unstimulated platelets, as seen in previous studies [7, 31]. We also confirmed that LPS in vitro enhanced aggregation of stimulated platelets, but not in resting platelets, in agreement with the work of Zhang et al (2009) and Rivadeneyra et al (2014) [7,8]. It is interesting that LPS alone did not induce substantial platelet activation, but it had an additive effect with established platelets agonists. An important limitation of existing literature in which the potential roles of LPS on platelets has been considered is that LPS derived from different bacteria are used, and the purity and quality are rarely presented. This is likely to explain some divergent results, including studies that suggest that LPS causes inhibition of platelet function following acute exposure in vitro. To overcome this action, in this study we employed LPS from two different strains of E.coli and TLR4-antogonising LPS. Notably, LPS purified from the two E. coli strains exerted similar effects. In addition to releasing classic inflammatory mediators, LPS promotes the generation of reactive oxygen and nitrogen species (ROS and RNS) by a range of cell types [5, 12]. Reports indicate that ROS can modulate the pathogenesis of sepsis through modulation of innate immune signalling and causing pathologic damage to cells and organs [13].
In this study we have shown that LPS promotes an increase in ROS release both in resting and in stimulated platelets. Studies have shown that O2- reduces the threshold for platelet activation to collagen [14]. In addition, Dong et al showed that catalase in vitro reduces the likelihood of platelet dysfunction during LPS-induced sepsis [41]. In the present study, using SOD and catalase, we demonstrated that reactive oxygen species, such as O2- and H2O2 do indeed contribute to enhancement of platelet function followed acute exposure to LPS.
The involvement of platelets in innate immune responses is partly due to the expression of Toll-like receptors (TLRs). Each TLR responds to a different set of ligands or PAMPs (pathogen-associated molecular patterns), lipids, lipoproteins, proteins or nucleic acids derived from bacteria, viruses and fungi [42]. Although platelets express several TLR family members [6], TLR2 and 4 have been most extensively studied because they recognise ligands in Gram-positive and negative bacteria, respectively [19] and because platelets contribute significantly to the pathophysiology of sepsis and are also involved in the high mortality levels associated with this disease [1]. After engagement, each TLR triggers its own distinctive biological response, which is specific for the PAMP recognized [6, 43]. Our study focused on the mechanisms triggered by LPS from E. coli.
Although PI3-kinase/Akt-dependent signalling has been shown to mediate some effects derived from LPS action in different cells [28, 29], it has not been established whether TLR4 signalling involves PI3-kinase/Akt activation. Here, we demonstrate that LPS in vitro increased Akt phosphorylation and the inhibitors of PI3K and Akt prevented the increase of platelet function and ROS production induced by LPS. Beyond the PI3K/Akt pathway, LPS stimulation also triggers the activation of small GTPases and the ERK, JNK and p38 MAPK pathways [44]. Here we found that LPS promoted ERK1/2 phosphorylation. Additionally, our findings demonstrated that TLR4 signals promote platelet stimulation and ROS release via MAPK- ERK1/2, and that is regulated by PI3K/Akt. These data are consistent with previous work in other cell types which showed PI3K/Akt and ERK1/2 to mediate a range of biological effects [21, 45, 46].
The binding of platelet integrin αIIbβ3 to ligands such as fibrinogen and VWF during aggregation/adhesion contributes to platelet thrombus formation by promoting the secretion of internal granules and the stimulation of a secondary wave of aggregation, together with the formation of membrane vesicles with procoagulant activities [47]. These result in the phosphorylation of tyrosine and serine/threonine residues of several proteins including ERK, myosin light chain (MLC) and cytoplasmic phospholipase A2 (cPLA2) [48]. In this context, our data show that in stimulated platelets, LPS increases PLA2 activation via the actions of Akt.
During activation by various agonists, platelets undergo a cascade of events that result in the enzymatic metabolism of arachidonic acid (AA) [48]. AA is derived from the plasma membrane by the action of PLA2, and undergoes further metabolism by COX and TXA2 synthase to form eicosanoid products such as prostaglandins (PGs), thromboxane (TX), and other oxygenated derivatives [49]. One of the downstream consequences of TLR signalling is cyclooxygenase activation and the formation of pro-inflammatory cytokines. In the context of platelets a notable prostaglandin produced by COX-1 is TXA2 [50]. Various studies have reported the increased release of TXA2 in the liver [37] and kidneys [27] in endotoxemia. Here, we sought to verify if TXA2 is associated with the in vitro effects of LPS in platelets. Indeed, our results demonstrated that indomethacin, an inhibitor of COX-1 and GR32191, an antagonist of the TXA2 receptor decreased the effect of LPS on platelet function. Our results suggest that TXA2 produced by platelets is necessary for effective LPS actions. Consistent with this, Nocella et al shows that the effects of LPS are inhibited by a TXA2 antagonist [46]. Kassouf et al report that thrombin-induced TXB2 formation (a stable TXA2 metabolite) in platelets is suppressed by Akt inhibitor IV [51]. Here we propose that the pathway PI3K/Akt/ERK1/2/PLA2 leads to TXA2 formation following the exposure of platelets to LPS.
Gram-negative pathogens such as E.coli induce platelet hyperactivity and ROS release, which may add to the potentiation of the risk of vascular pathology. ROS can modulate LPS-TLR4 signalling at different levels and promote an increase of consecutive stimuli in immune cell [52]. Indeed, our results demonstrate that SOD was able to reduce ERK1/2 and PLA2 phosphorylation and catalase reduced ERK1/2 activation, showing that ROS release acts to modulate the signalling pathway following the binding of LPS to TLR4.
The involvement of PI3K, Akt and MAPK in the stimulation of platelets by agonists is established. In this work we showed that LPS in vitro promotes an increase of platelet function via proteins associated with the activation of platelets including Akt, ERK and PLA2. The effects of LPS in vivo are likely to be more complex, since the range of cytokines released by platelets and other cells, such as TNFα, interleukins and TGFβ may result in a systemic inflammatory state [53, 54, 55].
Disseminated intravascular coagulation associated with sepsis is present in approximately 35% of severe cases, developing to microvascular dysfunction and death [56]. Platelet activation is an essential event involved in the pathophysiological processes of the coagulation system during sepsis [57]. PLA2 inhibitors have gained particular attention since this enzyme is activated during inflammatory events and its expression is increased in several diseases [58] including septic shock [59]. In equine platelets and leukocytes, LPS stimulation in vivo increases TXA2 production and p38 MAPK phosphorylation to levels similar that reported in clinical endotoxaemia [60]. Additionally, a study performed ex vivo has reported that LPS stimulation of TLR4 potentiates platelet activation and TXA2 production in patients with community-acquired pneumonia [46]. The impact of increase secretion of factors from platelets and endothelial cells in response to LPS is also likely to have complex effects on homeostasis and potentially thrombosis, with some factors enhancing this, while others such as thrombomodulim, serving to inhibit coagulation [61].
In summary, our findings demonstrate that TLR4 triggered by acute exposure of platelets to LPS promotes platelet stimulation via ERK1/2 and PLA2 activation and that is co-ordinately regulated by PI3K/Akt, and provide evidence that this signalling pathway involves TXA2 production and ROS generation.
Supporting information
LPS standard from E. coli serotype 055:B5 and LPS from E. coli K12 were separated by 12% acrylamide gel electrophoresis and stained using Pro-Q Emerald 300 Lipopolysaccharide Gel Stain Kit.
(TIF)
Human-washed platelet aggregation was performed by optical aggregometry following stimulation with U46619 (0.25μM) or CRP-XL (0.25μg/mL) in the presence or absence of LPS from E.coli K12 (1 or 7.5μg/mL) (A and E). The effects of U46619 (0.25μM) or CRP (0.25μg/mL) and LPS on fibrinogen binding and P-selectin exposure were measured in PRP by flow cytometry (B, C, F and G). Washed platelets (4 x 108/mL) were pre-incubated with 10μM DCFHDA before being activated with U46619 (0.25μM) or CRP-XL (0.25μg/mL) in the presence or absence of LPS from E.coli K12 (1 or 7.5μg/mL) and ROS levels were analysed by flow cytometry (D and H). Cumulative data represent mean values ± SEM (n = 4). (Anova-Bonferroni test, * P≤ 0.05; ** P≤ 0.01; *** P≤ 0.001; Test t student # P≤ 0.05).
(TIF)
Human-washed platelet aggregation was performed by optical aggregometry activated with CRP-XL (0.25μg/mL) in the presence or absence of LPS (7.5μg/mL) after 3 min of incubation with LY294002 (20μM), Cal (60μM) or Akt inhibitor IV (5μM) (A). Washed platelets (4 x 108/mL) were pre-incubated with 10μM DCFH-DA in the presence or absence of LY294002 (20μM), Cal (60μM) or Akt inhibitor IV (5μM) before being activated with CRP-XL (0.25μg/mL) in the presence or absence of LPS from E. coli O111:B4 (7.5μg/mL) and ROS levels were analysed by flow cytometry. Cumulative data represent mean values ± SEM (n = 4). (Anova-Bonferroni test, * P≤ 0.05; ** P≤ 0.01; *** P≤ 0.001; Test t student # P≤ 0.05).
(TIF)
Aggregation of human washed platelet was measured by optical aggregometry following stimulation with CRP-XL (0.25μg/mL) in the presence or absence of LPS from E. coli O111:B4 (7.5μg/mL) after 3 min of incubation with Cobimetinib (100μM) (A). Washed platelets (4 x 108/mL) were pre-incubated with DCFH-DA (10μM) in the presence or absence of Cobimetinib (100μM) before being activated with CRP-XL (0.25μg/mL) in the presence or absence of LPS from E. coli O111:B4 (7.5μg/mL) and ROS levels were analysed by flow cytometry. Cumulative data represent mean values ± SEM (n = 4). (Anova-Bonferroni test, ** P≤ 0.01; *** P≤ 0.001; Test t student # # P≤ 0.01).
(TIF)
Aggregation of human washed platelet was measured by optical aggregometry following stimulation with CRP-XL (0.25μg/mL) in the presence or absence of LPS from E. coli O111:B4 (7.5g/mL) after 3 min of incubation with Indomethacin (10μM) or GR32191 (100ng) (A and E). The effect of CRP-XL (0.25μg/ml) and LPS- on fibrinogen binding and P-selectin exposure after incubation with indomethacin (10μM) or GR32191 (100ng) were measure in PRP by flow cytometry (B, C, F and G). Washed platelets (4 x 108/mL) were pre-incubated with DCFHDA (10μM) in the presence or absence of Indomethacin (10μM) or GR32191 (100ng) before being stimulated with CRP-XL (0.25μg/ml) in the presence or absence of LPS from E. coli O111:B4 (7.5μg/mL) and ROS levels were analysed by flow cytometry (D and H). Cumulative data represent mean values ± SEM (n = 4). (Anova-Bonferroni test, * P≤ 0.05; ** P≤ 0.01; *** P≤ 0.001; Test t student # P≤ 0.05; # # # P≤ 0.001).
(TIF)
Acknowledgments
This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnologico (CNPQ) Brazil and by British Heart Foundation (RG/15/2/31224).
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
MELP received grants from Conselho Nacional de Desenvolvimento Científico e Tecnologico (CNPQ), Brazil. Jonathan Gibbins received grants from British Heart Foundation: Programme Grant RG/15/2/31224. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Li Z, Yang F, Dunn S, Gross AK, Smyth SS. Platelets as immune mediators: their role in host defense responses and sepsis. Thromb Res. 2011; 127:184–188. doi: 10.1016/j.thromres.2010.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science. 1990; 249: 1431–1433. [DOI] [PubMed] [Google Scholar]
- 3.Andonegui G, Kerfoot SM, McNagny K, Ebbert KV, Patel KD, Kubes P. Platelets express functional Toll-like receptor-4. Blood. 2005; 106: 2417–2423. doi: 10.1182/blood-2005-03-0916 [DOI] [PubMed] [Google Scholar]
- 4.Casarin AL, Lopes-Pires ME, Morganti RP, Antunes E, Marcondes S. Reactive oxygen and nitrogen species modulate the ex-vivo effects of LPS on platelet adhesion to fibrinogen. Life Sci. 2011; 89: 773–778. doi: 10.1016/j.lfs.2011.09.004 [DOI] [PubMed] [Google Scholar]
- 5.Lopes-Pires ME, Casarin AL, Pereira-Cunha FG, Lorand-Metze I, Antunes E, Marcondes S. Lipopolysaccharide treatment reduces rat platelet aggregation independent of intracellular reactive-oxygen species generation. Platelets. 2012; 23: 195–201. doi: 10.3109/09537104.2011.603065 [DOI] [PubMed] [Google Scholar]
- 6.Cognasse F, Hamzeh H, Chavarin P, Acquart S, Genin C, Garraud O. Evidence of Toll like receptor molecules on human platelets. Immunol Cell Biol. 2005; 83:196–198. doi: 10.1111/j.1440-1711.2005.01314.x [DOI] [PubMed] [Google Scholar]
- 7.Zhang G, Han J, Welch EJ, Ye RD, Voyno-Yasenetskaya TA, Malik AB, et al. Lipopolysaccharide stimulates platelet secretion and potentiates platelet aggregation via TLR4/MyD88 and the cGMP-dependent protein kinase pathway. J Immunol. 2009; 182: 7997–8004. doi: 10.4049/jimmunol.0802884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rivadeneyra L, Carestia A, Etulain J, Pozner RG, Fondevila C, Negrotto S, et al. Regulation of platelet responses triggered by Toll-like receptor 2 and 4 ligands is another non-genomic role of nuclear factor-kappa B. Thromb res. 2014; 133: 235–243. doi: 10.1016/j.thromres.2013.11.028 [DOI] [PubMed] [Google Scholar]
- 9.Morganti RP, Cardoso MH, Pereira FG, Lorand-Metze I, De Nucci G, Marcondes S, et al. Mechanisms underlying the inhibitory effects of lipopolysaccharide on human platelet adhesion. Platelets. 2010; 21: 260–269. doi: 10.3109/09537101003637240 [DOI] [PubMed] [Google Scholar]
- 10.Gangloff SC, Hijiya N, Haziot A, Goyert SM. Lipopolysaccharide structure influences the macrophage response via CD14-independent and CD14-dependent pathways. Clin Infect Dis. 1999; 28: 491–496. doi: 10.1086/515176 [DOI] [PubMed] [Google Scholar]
- 11.Mathiak G, Kabir K, Grass G, Keller H, Steinringer E, Minor T, et al. Lipopolysaccharides from different bacterial sources elicit disparate cytokine responses in whole blood assays. Int J Mol Med. 2003; 11: 41–44. [PubMed] [Google Scholar]
- 12.Soromou LW, Zhang Z, Li R, Chen N, Guo W, Huo M, et al. Regulation of inflammatory cytokines in lipopolysaccharide RAW 264.7 murine macrophage by 7-O-methyl-naringenin. Molecules. 2012; 17: 3574–3585. doi: 10.3390/molecules17033574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keen RR, Stella L, Flanigan DP, Lands WE. Differential detection of plasma hydroperoxides in sepsis. Crit Care Med. 1991; 19: 1114–1119. [DOI] [PubMed] [Google Scholar]
- 14.Krötz F, Sohn HY, Gloe T, Zahler S, Riexinger T, Schiele TM, et al. NAD(P)H-oxidase-dependent platelet superoxide anion release increases platelet recruitment. Blood. 2002; 100: 917–924. [DOI] [PubMed] [Google Scholar]
- 15.Bakdash N, Willians MS. Spatially disticty production of reactive oxygen species regulates platelet activation. Free Radic Biol Med. 2008; 45: 158–166. doi: 10.1016/j.freeradbiomed.2008.03.021 [DOI] [PubMed] [Google Scholar]
- 16.Hornef MW, Normark BH, Vandewalle A, Normark S. Intracellular recognition of lipopolysaccharide by toll-like receptor 4 in intestinal epithelial cells. J Exp Med. 2003; 198: 1225–1235. doi: 10.1084/jem.20022194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tavener SA, Long EM, Robbins SM, McRae KM, Van Remmen H, Kubes P. Immune cell Toll-like receptor 4 is required for cardiac myocyte impairment during endotoxemia. Circ Res. 2004; 95:700–707. doi: 10.1161/01.RES.0000144175.70140.8c [DOI] [PubMed] [Google Scholar]
- 18.O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007; 7: 353–364. doi: 10.1038/nri2079 [DOI] [PubMed] [Google Scholar]
- 19.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010; 11: 373–384. doi: 10.1038/ni.1863 [DOI] [PubMed] [Google Scholar]
- 20.Kawasaki T and Kawai T. Toll-Like Receptor Signaling Pathways. Front Immunol. 2014; 5: 1–8. doi: 10.3389/fimmu.2014.00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee GL, Wu JY, Tsai CS, Lin CY, Tsai YT, Lin CS, et al. TLR4-Activated MAPK-IL-6 Axis Regulates Vascular Smooth Muscle Cell Function. Int J Mol Sci. 2016; 17: 1394 doi: 10.3390/ijms17091394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown GT, McIntyre TM. Lipopolysaccharide signaling without a nucleous: kinase cascades stimulate plateles hedding of proinflammatory IL-1β-rich microparticles. J Immunol. 2011; 186: 5489–5496. doi: 10.4049/jimmunol.1001623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woulfe DS. Akt signaling in platelets and thrombosis. Expert Rev Hematol. 2010; 3: 81–91. doi: 10.1586/ehm.09.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flevaris P, Li Z, Zhang G, Zheng Y, Liu J, Du X. Two distinct roles of mitogen-activated protein kinases in platelet and a novel Rac1-MAPK-dependent integrin outside-in retractile signaling pathway. Blood. 2009; 113: 893–901. doi: 10.1182/blood-2008-05-155978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou PH, Hu W, Zhang XB, Wang W, Zhang LJ. Protective Effect of Adrenomedullin on Rat Leydig Cells from Lipopolysaccharide-Induced Inflammation and Apoptosis via the PI3K/Akt Signaling Pathway ADM on Rat Leydig Cells from Inflammation and Apoptosis. Mediators Inflamm. 2016; 2016: 7201549 doi: 10.1155/2016/7201549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saponaro C, Cianciulli A, Calvello R, Dragone T, Iacobazzi F, Panaro MA. The PI3K/Akt pathway is required for LPS activation of microglial cells. Immunopharmacol Immunotoxicol. 2012; 34: 858–865. doi: 10.3109/08923973.2012.665461 [DOI] [PubMed] [Google Scholar]
- 27.Li X, Jiang S, Tapping RI. Toll-like receptor signaling in cell proliferation and survival. Cytokine. 2010; 49:1–9. doi: 10.1016/j.cyto.2009.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis MR Jr, Goldberg JB. Purification and visualization of lipopolyssacharide from Gram-negative bacteria by hot aqueous-phenol extraction. J Vis Exp. 2012; 63:3916 doi: 10.3791/3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaiser WJ, Holbrook LM, Tucker KL, Stanley RG, Gibbins JM. A functional proteomic method for the enrichment of peripheral membrane proteins reveals the collagen binding protein Hsp47 is exposed on the surface of activated human platelets. J Proteome Res. 2009. 8(6):2903–14. doi: 10.1021/pr900027j [DOI] [PubMed] [Google Scholar]
- 30.Morton LF, Hargreaves PG, Farndale RW, Young RD, Barnes MJ. Integrin alpha 2 beta 1-independent activation of platelets by simple collagen-like peptides: collagen tertiary (triple-helical) and quaternary (polymeric) structures are sufficient alone for alpha 2 beta 1-independent platelet reactivity. Biochem J. 1995;306 (Pt 2):337–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gibbins JM, Briddon S, Shutes A, van Vugt MJ, van de Winkel JG, Saito T, et al. The p85 subunit of phosphatidylinositol 3-kinase associates with the Fc receptor gamma-chain and linker for activitor of T cells (LAT) in platelets stimulated by collagen and convulxin. J Biol Chem. 1998; 273: 34437–34443. [DOI] [PubMed] [Google Scholar]
- 32.Myhre O, Andersen JM, Aarnes H, Fonnum F. Evaluation of the probes 2',7'-dichlorofluorescin diacetate, luminol, and lucigenin as indicators of reactive species formation. Biochem Pharmacol. 2003; 65: 1575–1582. [DOI] [PubMed] [Google Scholar]
- 33.Kälvegren H, Majeed M, Bengtsson T. Chlamydia pneumoniae binds to platelets and triggers P-selectin expression and aggregation: a causal role in cardiovascular disease? Arterioscler Thromb Vasc Biol. 2003; 23: 1677–1683. doi: 10.1161/01.ATV.0000084810.52464.D5 [DOI] [PubMed] [Google Scholar]
- 34.Li Z, Denaley MK, O’Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler thromb Vasc Biol. 2010; 30: 2341–2349. doi: 10.1161/ATVBAHA.110.207522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ren Q, Ye S, Whiteheart SW. The platelet release reaction: just when you thought platelet secretion was simple. Curr Opin Hematol. 2008; 15: 537–541. doi: 10.1097/MOH.0b013e328309ec74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krötz F, Sohn HY, Pohl U. Reactive oxygen species: players in the platelet game. Arterioscler Thromb Vasc Biol. 2004; 24: 1988–1996. doi: 10.1161/01.ATV.0000145574.90840.7d [DOI] [PubMed] [Google Scholar]
- 37.Burkholder TJ. Stretch-induced ERK2 phosphorylation requires PLA2 activity in skeletal myotubes. Biochem Biophys Res Commun. 2009; 386: 60–64. doi: 10.1016/j.bbrc.2009.05.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boffa JJ, Just A, Coffman TM, Arendshorst WJ. Thromboxane receptor mediates vasoconstriction and contributes to acute renal failure in endotoxemic mice. J Am Soc Nephrol. 2004; 15: 2358–2365. doi: 10.1097/01.ASN.0000136300.72480.86 [DOI] [PubMed] [Google Scholar]
- 39.Katagiri H, Ito Y, Ishii K, Hayashi I, Suematsu M, Yamashina S, et al. Role of thrombox thromboxane derived from COX-1 and -2 in hepatic microcirculatory dysfunction during endotoxemia in mice. Hepatology. 2004; 39: 139–150. doi: 10.1002/hep.20000 [DOI] [PubMed] [Google Scholar]
- 40.Moncada S, Gryglewski R, Bunting S, Vane JR. An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature. 1976; 263: 663–665. [DOI] [PubMed] [Google Scholar]
- 41.Dong HP, Chunag IC, Wang DC, Huang LJ, Lee CI, Tsai JH, et al. Lipopolysaccharide-stimulated leukocytes contribute to platelet aggregative dysfunction, which is attenuated by catalase in rats. Kaohsiung J Med Sci. 2010; 26: 5845–92. doi: 10.1016/S1607-551X(10)70090-5 [DOI] [PubMed] [Google Scholar]
- 42.Mogensen TH. Pathogen recognition and inflammatory signaling in innate immunedefenses. Clin Microbiol Rev. 2009; 22: 240–273. doi: 10.1128/CMR.00046-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cognasse F, Nguyen KA, Damien P, McNicol A, Pozzetto B, Hamzeh-Cognasse H, et al. The inflammatory Role of Platelets via their TLRs and Siglec Receptors. Front Immunol. 2015; 6:83 doi: 10.3389/fimmu.2015.00083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006; 124: 783–801. doi: 10.1016/j.cell.2006.02.015 [DOI] [PubMed] [Google Scholar]
- 45.Li X, Liu Y, Wang L, Li Z, Ma X. Unfractionated heparin attenuates LPS-induced IL-8 secretion via PI3K/Akt/NF-κB signaling pathway in human endothelial cells. Immunobiology. 2015; 220: 399–405. doi: 10.1016/j.imbio.2014.10.008 [DOI] [PubMed] [Google Scholar]
- 46.Nocella C, Carnevale R, Bartimoccia S, Novo M, Cangemi R, Pastori D, et al. Lipopolysaccharide as trigger of platelet aggregation via eicosanoid over-production. Thromb Haemost. 2017, 117(8): 1558–1570. doi: 10.1160/TH16-11-0857 [DOI] [PubMed] [Google Scholar]
- 47.Phillips DR, Nannizzi-Alaimo L, Prasad KS. Beta3 tyrosine phosphorylation in alphaIIbbeta3 (platelet membrane GP IIb-IIIa) outside-in integrin signaling. Thromb Haemost. 2001; 86: 246–258. [PubMed] [Google Scholar]
- 48.Lewis TS, Shapiro PS, Ahn NG. Signal transduction through MAP kinase cascades. Adv Cancer Res. 1998; 74: 49–139. [DOI] [PubMed] [Google Scholar]
- 49.Siess W. Molecular mechanisms of platelet activation. Physiol Rev. 1989; 69: 58–178. [DOI] [PubMed] [Google Scholar]
- 50.El-Achkar TM, Plotkin Z, Marcic B, Dagher PC. Sepsis induces an increase in thick ascending limb Cox-2 that is TLR4 dependent. Am J Physiol Renal Physiol. 2007; 293: 1187–1196. doi: 10.1152/ajprenal.00217.2007 [DOI] [PubMed] [Google Scholar]
- 51.Kassouf N, Ambily A, Watson S, Hassock S, Authi HS, Srivastava S, et al. Phosphatidylinositol-3,4,5-trisphosphate stimulates Ca(2+) elevation and Akt phosphorylation to constitute a major mechanism of thromboxane A2 formation in human platelets. Cell Signal. 2015; 27: 1488–1498. doi: 10.1016/j.cellsig.2015.03.008 [DOI] [PubMed] [Google Scholar]
- 52.Powers KA, Szaszi K, Khadaroo RG, Tawadros PS, Marshall JC, Kapus A, et al. Oxidative stress generated by hemorrhagic shock recruits Toll-like receptor 4 to the plasma membrane in macrophages. J Exp Med. 2006; 203: 1951–1961. doi: 10.1084/jem.20060943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frost RA, Nystrom GJ, Lang CH. Lipopolysaccharide regulates proinflammatory cytokine expression in mousemyoblasts and skeletal muscle. Am J Physiol Regul Integr Comp Physiol. 2002 Sep;283(3):R698-709. Am J Physiol Regul Integr Comp Physiol. 2002;283(3):R698–709. doi: 10.1152/ajpregu.00039.2002 [DOI] [PubMed] [Google Scholar]
- 54.Cognasse F, Hamzeh-Cognasse H, Lafarge S, Delezay O, Pozzetto B, McNicol A, et al. Toll-like receptor 4 ligand can differentially modulate the release of cytokines by human platelets. Br J Haematol. 2008;141(1):84–91. doi: 10.1111/j.1365-2141.2008.06999.x [DOI] [PubMed] [Google Scholar]
- 55.Rossol M, Heine H, Meusch U, Quandt D, Klein C, Sweet MJ, et al. LPS-induced cytokine production in human monocytes and macrophages. Crit Rev Immunol. 2011;31(5):379–446. [DOI] [PubMed] [Google Scholar]
- 56.Iba T, Ito T, Maruyama I, Jilma B, Brenner T, Muller MC, et al. Potential diagnostic markers for disseminated intravascular coagulation of sepsis. Blood Rev. 2016;30:149–155. doi: 10.1016/j.blre.2015.10.002 [DOI] [PubMed] [Google Scholar]
- 57.Yin H, Stojanovic-Terpo A, Xu W, Corken A, Zakharov A, Qian F, et al. Role for platelet glycoprotein Ib-IX and effects of its inhibition in endotoxemia-induced thrombosis, thrombocytopenia, and mortality. Arterioscler. Thromb. Vasc. Biol. 2013;33:2529–2537. doi: 10.1161/ATVBAHA.113.302339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Quach ND, Arnold RD2, Cummings BS. Secretory phospholipase A2 enzymes as pharmacological targets for treatment of disease. Biochem Pharmacol. 2014;90(4):338–48. doi: 10.1016/j.bcp.2014.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakae H1, Endo S, Inada K, Yaegashi Y, Takakuwa T, Yamada Y, et al. Nitrite/nitrate (NOX) and type II phospholipase A2, leukotriene B4, and platelet-activating factor levels in patients with septic shock. Res Commun Mol Pathol Pharmacol. 1996; 92(2):131–9. [PubMed] [Google Scholar]
- 60.Brooks AC1, Menzies-Gow NJ, Wheeler-Jones C, Bailey SR, Cunningham FM, Elliott J. Endotoxin-induced activation of equine platelets: evidence for direct activation of p38 MAPK pathways and vasoactive mediator production. Inflamm Res. 2007; 56(4):154–61. doi: 10.1007/s00011-006-6151-6 [DOI] [PubMed] [Google Scholar]
- 61.Ikezou T. Thrombomodulin/activated protein C system in septic disseminatedintravascular coagulation. J Intensive Care. 2015; 3(1):1 doi: 10.1186/s40560-014-0050-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
LPS standard from E. coli serotype 055:B5 and LPS from E. coli K12 were separated by 12% acrylamide gel electrophoresis and stained using Pro-Q Emerald 300 Lipopolysaccharide Gel Stain Kit.
(TIF)
Human-washed platelet aggregation was performed by optical aggregometry following stimulation with U46619 (0.25μM) or CRP-XL (0.25μg/mL) in the presence or absence of LPS from E.coli K12 (1 or 7.5μg/mL) (A and E). The effects of U46619 (0.25μM) or CRP (0.25μg/mL) and LPS on fibrinogen binding and P-selectin exposure were measured in PRP by flow cytometry (B, C, F and G). Washed platelets (4 x 108/mL) were pre-incubated with 10μM DCFHDA before being activated with U46619 (0.25μM) or CRP-XL (0.25μg/mL) in the presence or absence of LPS from E.coli K12 (1 or 7.5μg/mL) and ROS levels were analysed by flow cytometry (D and H). Cumulative data represent mean values ± SEM (n = 4). (Anova-Bonferroni test, * P≤ 0.05; ** P≤ 0.01; *** P≤ 0.001; Test t student # P≤ 0.05).
(TIF)
Human-washed platelet aggregation was performed by optical aggregometry activated with CRP-XL (0.25μg/mL) in the presence or absence of LPS (7.5μg/mL) after 3 min of incubation with LY294002 (20μM), Cal (60μM) or Akt inhibitor IV (5μM) (A). Washed platelets (4 x 108/mL) were pre-incubated with 10μM DCFH-DA in the presence or absence of LY294002 (20μM), Cal (60μM) or Akt inhibitor IV (5μM) before being activated with CRP-XL (0.25μg/mL) in the presence or absence of LPS from E. coli O111:B4 (7.5μg/mL) and ROS levels were analysed by flow cytometry. Cumulative data represent mean values ± SEM (n = 4). (Anova-Bonferroni test, * P≤ 0.05; ** P≤ 0.01; *** P≤ 0.001; Test t student # P≤ 0.05).
(TIF)
Aggregation of human washed platelet was measured by optical aggregometry following stimulation with CRP-XL (0.25μg/mL) in the presence or absence of LPS from E. coli O111:B4 (7.5μg/mL) after 3 min of incubation with Cobimetinib (100μM) (A). Washed platelets (4 x 108/mL) were pre-incubated with DCFH-DA (10μM) in the presence or absence of Cobimetinib (100μM) before being activated with CRP-XL (0.25μg/mL) in the presence or absence of LPS from E. coli O111:B4 (7.5μg/mL) and ROS levels were analysed by flow cytometry. Cumulative data represent mean values ± SEM (n = 4). (Anova-Bonferroni test, ** P≤ 0.01; *** P≤ 0.001; Test t student # # P≤ 0.01).
(TIF)
Aggregation of human washed platelet was measured by optical aggregometry following stimulation with CRP-XL (0.25μg/mL) in the presence or absence of LPS from E. coli O111:B4 (7.5g/mL) after 3 min of incubation with Indomethacin (10μM) or GR32191 (100ng) (A and E). The effect of CRP-XL (0.25μg/ml) and LPS- on fibrinogen binding and P-selectin exposure after incubation with indomethacin (10μM) or GR32191 (100ng) were measure in PRP by flow cytometry (B, C, F and G). Washed platelets (4 x 108/mL) were pre-incubated with DCFHDA (10μM) in the presence or absence of Indomethacin (10μM) or GR32191 (100ng) before being stimulated with CRP-XL (0.25μg/ml) in the presence or absence of LPS from E. coli O111:B4 (7.5μg/mL) and ROS levels were analysed by flow cytometry (D and H). Cumulative data represent mean values ± SEM (n = 4). (Anova-Bonferroni test, * P≤ 0.05; ** P≤ 0.01; *** P≤ 0.001; Test t student # P≤ 0.05; # # # P≤ 0.001).
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.