

Abstract
Sporulation of budding yeast is a developmental process in which cells undergo meiosis to generate stress-resistant progeny. The dynamic nature of the budding yeast meiotic transcriptome has been well established by a number of genome-wide studies. Here we develop an analysis pipeline to systematically identify novel transcription start sites that reside internal to a gene. Application of this pipeline to data from a synchronized meiotic time course reveals over 40 genes that display specific internal initiations in mid-sporulation. Consistent with the time of induction, motif analysis on upstream sequences of these internal transcription start sites reveals a significant enrichment for the binding site of Ndt80, the transcriptional activator of middle sporulation genes. Further examination of one gene, MRK1, demonstrates the Ndt80 binding site is necessary for internal initiation and results in the expression of an N-terminally truncated protein isoform. When the MRK1 paralog RIM11 is downregulated, the MRK1 internal transcript promotes efficient sporulation, indicating functional significance of the internal initiation. Our findings suggest internal transcriptional initiation to be a dynamic, regulated process with potential functional impacts on development.
Introduction
The generation of gametes through meiosis is the foundation for sexual reproduction in eukaryotes. During this process, diploid cells generate haploid gametes by undergoing one round of DNA replication followed by two rounds of chromosome segregation, namely meiosis I and II. These germ cells later undergo fertilization to restore diploidy, completing the cycle of sexual reproduction. In budding yeast Saccharomyces cerevisiae, gametogenesis (also called sporulation) involves the exit of cells from mitotic growth, followed by meiosis and spore morphogenesis, producing four haploid spores from one diploid mother. The coordination between these events during sporulation requires precise transcriptional and translational control of hundreds of genes [1–3].
Initiation of sporulation requires convergence of nutritional and mating type signals at the promoter of the master inducer, IME1 [4]. Ime1 complexes with the DNA-binding protein Ume6 to activate the expression of early genes through a conserved site named Upstream Regulatory Sequence 1 (URS1) in the upstream regions of these genes [5]. The expression of early genes is essential for pre-meiotic DNA replication, chromosome remodeling and homologous recombination [4, 6]. One of the Ime1/Ume6 complex targets, NDT80, encodes a transcription factor which plays a central role in activating genes required for the meiotic divisions during middle sporulation [7, 8]. Ndt80 binds specifically to a site called the middle sporulation element (MSE), which is found in the upstream region of most middle genes as well its own promoter [9]. The induction of NDT80 and upregulation of middle genes then drives cells to exit the pachytene stage of meiotic prophase and enter the meiotic divisions [7].
Genome-wide approaches like high-resolution tiling arrays and RNA sequencing have revealed unexpected complexity of the transcriptional landscape in eukaryotic cells [10–14]. Global expression profiling found not only splice variants, intergenic and antisense transcripts, but a large number of variants of transcripts with diversified 5’- and 3’- UTRs [15–17]. One common mechanism underlying UTR heterogeneity is alternative transcription start site (TSS) selection, which is prevalent in eukaryotes [10, 15]. Changes in TSS can alter mRNA stability, splicing, and in some cases localization through a variety of mechanisms [18]. At the translational level, variation in mRNA isoforms often leads to changes in translation efficiency [19, 20]. Additionally, although most TSS changes do not influence the coding capacity of the transcript, mRNA isoforms with extended 5'-UTRs often include upstream open reading frames (uORFs) which can function as translational regulators [21]. Alternative TSSs may also occur within the coding region, where the internally initiated transcript can produce a truncated protein isoform with potential implications for the function of the encoded protein. In yeast, for example, alternative TSSs in the SUC2 gene lead to protein products that do or do not include an ER targeting signal. Thus, one TSS leads to a secreted form of the protein and the other a cytoplasmic form [22, 23]. Alternative initiation of SUC2 is a normal process but misregulation of initiation can have pathological consequences. In humans, an alternative TSS in an intron of ALK creates a truncated protein expressing only the protein kinase domain of the enzyme which promotes tumorigenesis [24]. Thus, misregulated internal initiations can have dramatic functional outcomes like alterations of cell fate.
Sporulation in budding yeast provides a useful system to study alternative TSS selection in a developmental context. Multiple studies of the budding yeast transcriptome during sporulation have documented dynamic changes in transcript architecture including extended 5'-UTRs and intronic TSSs [2, 10, 25]. Additionally, ribosome profiling revealed that many meiosis-specific extended 5’-UTRs include uORFs that may regulate translation of the major reading frame [3]. The 5’-UTR extension of one such gene SPO24, which is required for efficient sporulation, is regulated by Ndt80 and enhances the translation efficiency of the main ORF [26]. While hundreds of meiotic genes with extended meiotic 5’UTRs have been described, internal TSSs that might occur during sporulation are less well documented [3, 10].
One documented internal initiation occurs in the MRK1 gene [2]. After NDT80 expression, the MRK1 locus expresses not only a properly spliced full-length transcript, but also an alternative isoform that initiates within the intron and spans the rest of the gene [2]. MRK1 encodes one of four GSK-3 family serine/threonine protein kinase homologs in budding yeast. The four GSK-3 homologs: MCK1, MRK1, RIM11 and YGK3 share around 70% similarity with human GSK-3β [27]. MCK1 functions in sporulation to enhance transcription of IME1, phosphorylate Ume6, and promote spore maturation as well having important roles in mitotic cells including regulation of cell cycle progression, chromosomal stability and stress signaling [28–34]. RIM11 is essential for meiotic entry as its product phosphorylates both Ime1 and Ume6 to promote complex formation between the two factors [35–38]. MRK1 and YGK3 have been shown to have redundant functions with MCK1 and/or RIM11 in various stress responses [34, 39, 40], but except for some redundancy with Mck1 and Rim11 in phosphorylation of Ume6, little is known about the role and regulation of MRK1 in sporulation [28].
In order to detect sporulation-specific internal initiation sites, we developed an analytic pipeline that enables sensitive, unbiased detection for novel internal TSSs, and applied it to an RNA-Seq dataset from a highly synchronized meiotic time-course. Different genes with internal TSSs are seen in early and mid-sporulation, suggesting that internal initiation is highly dynamic. MSE sites are highly enriched in the upstream regions of mid-sporulation specific internal TSSs, and internal initiation for several of these genes, including MRK1, is NDT80-dependent. Furthermore, an MSE site within the annotated intron directly regulates MRK1 internal initiation, which results in the expression of an N-terminally truncated isoform that contributes to sporulation efficiency. These findings suggest that internal initiations are highly regulated and likely represent a common mechanism for gene regulation during development.
Materials and methods
Yeast media and strains
Strains used are listed in Table 1. Unless otherwise noted, standard yeast genetic techniques, media and growth conditions were used [41]. MRK1 was C-terminally tagged with three copies of the HA epitope by PCR-based integration into strain A14201 using the oligos tg-MRK1-F and -R and the plasmid pFA6a-3HA-HisMX6 [42] as template to generate ySZ68. To construct strains ySZ23 and ySZ102, the open reading frame of MCK1 or MRK1, respectively, were deleted by PCR-based gene deletion using the oligos ko-MCK1-F and -R or ko-MRK1-F and -R and the plasmid pFA6a-kanMX6 [42] as template in the haploids AN117-16D and AN117-4B and subsequent mating of the transformants. Two of these transformants, ySZ159 (mrk1Δ) and ySZ21 (mck1Δ), were crossed and segregants from non-parental ditype tetrads were mated to generate strain ySZ46. Similarly, RIM11 was deleted from AN117-16D and AN117-4B using oligos ko-RIM11-F and -R and the plasmid pFA6a-kanMX6 to create ySZ18 and ySZ19. A meiotic depletion allele of RIM11 (rim11-md) consisting of RIM11 under control of the CLB2 promoter, was constructed by PCR-based gene replacement of 50bp directly upstream of the RIM11 ATG codon with a PCLB2-3HA cassette in strain AN117-4B using oligos prms-RIM11-F and -R and the plasmid pFA6a-PCLB2-3HA-kanMX6 as template [43] to create strain ySZ34. ySZ34 was then mated with ySZ18 to obtain ySZ39. ySZ18 and ySZ34 were each mated with ySZ159 and mrk1Δ rim11Δ or mrk1Δ rim11-md double mutants, respectively, were obtained by dissection of the diploids. These haploids were crossed to generate ySZ135.
Table 1. Yeast strains.
| Strain name | Genotype | Reference |
|---|---|---|
| A14154 | MATa his3::hisG leu2::hisG trp1::hisG lys2 ho::LYS2 GAL-NDT80::TRP1 ura3::PGPD1-GAL4(848).ER::URA3 | Berchowitz, Gajadhar et al., 2013 |
| A14155 | MATα his3::hisG leu2::hisG trp1::hisG lys2 ho::LYS2 GAL-NDT80::TRP1 ura3:: PGPD1-GAL4(848).ER::URA3 | Berchowitz, Gajadhar et al., 2013 |
| A14201 | MATa/MATα his3::hisG/his3::hisG leu2::hisG/leu2::hisG trp1::hisG/trp1::hisG lys2/lys2 ho::LYS2/ho::LYS2 GAL-NDT80::TRP1/GAL-NDT80::TRP1 ura3:: PGPD1-GAL4(848).ER::URA3/ura3:: PGPD1-GAL4(848).ER::URA3 | Berchowitz, Gajadhar et al., 2013 |
| AN120 | MATa/MATα ura3/ura3 his3ΔSK/his3ΔSK leu2/leu2 trp1::hisG/trp1::hisG lys2/lys2 arg4-NSP1/ARG4 hoΔ::LYS2/hoΔ::LYS2 rme1Δ::leu2/RME1 | Neiman, Katz et al., 2000 |
| AN117-16D | MATa ura3 his3ΔSK leu2 trp1::hisG lys2 ho::LYS2 | Neiman, Katz et al., 2000 |
| AN117-4B | MATα ura3 his3ΔSK leu2 trp1::hisG lys2 ho::LYS2 arg4-NSP1 rme1::LEU2 | Neiman, Katz et al., 2000 |
| AN262 | MATa/MATα ura3/ura3 his3ΔSK/his3ΔSK leu2/leu2 trp1Δ::hisG/trp1Δ::hisG lys2/lys2 ARG4/arg4-NspI RME1/rme1Δ::LEU2 hoΔ::LYS2/hoΔ::LYS2 chs3Δ::His3MX6/chs3Δ::His3MX6 | Coluccio et al., 2004 |
| ySZ18 | As AN117-16D, plus rim11Δ::kanMX6 | this study |
| ySZ19 | As AN117-4B, plus rim11Δ::kanMX6 | this study |
| ySZ21 | As AN117-4B, plus mck1Δ::kanMX6 | this study |
| ySZ23 | As AN120, plus mck1Δ::kanMX6/mck1Δ::kanMX6 | this study |
| ySZ34 | As AN117-4B, plus PCLB2-RIM11::kanMX6 | this study |
| ySZ39 | As AN120, plus rim11Δ::kanMX6/ PCLB2-RIM11::kanMX6 | this study |
| ySZ44 | As A14154, plus mrk1-mMSE | this study |
| ySZ46 | As AN120, plus mrk1Δ::kanMX6/mrk1Δ::kanMX6 mck1Δ::kanMX6/mck1Δ::kanMX6 | this study |
| ySZ48 | As AN117-16D, plus mrk1-mMSE | this study |
| ySZ68 | As A14201, plus MRK1-3HA::His3MX6/MRK1 | this study |
| ySZ69 | As A14201, plus mrk1-mMSE/mrk1-mMSE | this study |
| ySZ76 | As A14155, plus mrk1-mMSE | this study |
| ySZ98 | As AN120, plus mrk1-mMSE/mrk1-mMSE rim11Δ::kanMX6/ PCLB2-RIM11::kanMX6 | this study |
| ySZ102 | As AN120, plus mrk1Δ::kanMX6/mrk1Δ::kanMX6 | this study |
| ySZ135 | As AN120, plus mrk1Δ::kanMX6/mrk1Δ::kanMX6 rim11Δ::kanMX6/ PCLB2-RIM11::kanMX6 | this study |
| ySZ159 | As AN117-16D, plus mrk1Δ::kanMX6 | this study |
| ySZ311 | As A14155, plus mrk1-mMSE-3HA::His3MX6 | this study |
| ySZ313 | As A14201, plus mrk1-mMSE-3HA::His3MX6/mrk1-mMSE | this study |
| ySZ315 | As ySZ135, plus ura3::sMRK1::URA3/ura3 | this study |
| ySZ317 | As ySZ39, plus ura3::URA3/ura3 | this study |
| ySZ318 | As ySZ135, plus ura3::URA3/ura3 | this study |
To construct the mrk1-mMSE strains ySZ48, ySZ44 and ySZ76, 9 nucleotides containing an MSE consensus within the MRK1 intron (CGACACAAA) were replaced with the kanMX6 cassette by PCR mediated gene replacement in AN117-16D, A14154 and A14155 using oligos ko-MRK1_MSE-F and -R and the plasmid pFA6a-kanMX6. CRISPR/Cas9 was then used as described earlier [44] to replace the kanMX6 cassette with the sequence CGGAATTCA, restoring the original length of the intron but mutating the MSE and creating an EcoRI restriction site. Briefly, the strain was co-transformed with the plasmid pRS425-Cas9-kan280 (gift of G. Zhao) and a double stranded rescue oligonucleotide containing the mutated MSE sequence flanked by 35 nucleotides identical to the chromosomal sequences on each side of the wild-type MSE (oligos rp-MRK1_mMSE-F and -R). Correct introduction of the mutated MSE (mMSE) was confirmed by DNA sequencing. ySZ44 and ySZ76 were crossed to generate ySZ69. To create mrk1-mMSE tagged with 3xHA on the C-terminus, PCR-based integration was performed using the oligos tg-MRK1-F and -R and the plasmid pFA6a-3HA-HisMX6 [42] as template and transformed into ySZ76 to generate ySZ311. ySZ311 was then crossed with ySZ44 to obtain the diploid ySZ313.
To obtain an mrk1-mMSE homozygous diploid in the rim11-md background, ySZ19 and ySZ34 were each crossed with ySZ48 and dissected. The double mutants were then screened from the G418 resistant segregants by PCR amplification of the MSE region followed by EcoRI digestion, and the appropriate haploids were mated to generate ySZ98. To construct the integration plasmid carrying only the internally initiated, short version of MRK1 (sMRK1), the second exon of MRK1 with 534 nucleotides of upstream sequence and 299 nucleotides of downstream sequence was PCR amplified using oligos cl-sMRK1-306-F and -R with genomic SK1 DNA as template, and cloned into the BamHI/XhoI digested integration plasmid pRS306 (ref) via Gibson assembly to generate pKZ116. This plasmid was linearized by digestion with StuI and transformed into ySZ39 to generate ySZ314. pRS306 was linearized the same way and transformed into ySZ39 and ySZ135, to generate strains ySZ317 and ySZ318, respectively. To construct the integration plasmid carrying only the internally initiated, short version of MRK1 (sMRK1), the second exon of MRK1 with 534 nucleotides of upstream sequence containing the intron and 3’ end of the first exon and 299 nucleotides of downstream sequence was PCR amplified using oligos cl-sMRK1-306-F and -R with genomic SK1 DNA as template, and cloned into the BamHI/XhoI digested integration plasmid pRS306 via Gibson assembly to generate pKZ116. This plasmid was linearized by digestion with StuI and transformed into ySZ135 to generate ySZ315. pRS306 was linearized the same way and transformed into ySZ39 and ySZ135, to generate strains ySZ317 and ySZ318, respectively.
All deletion and tagged alleles were confirmed by PCR, and all point mutation alleles were confirmed by sequencing. For oligo sequences, see Table 2.
Table 2. Oligos.
| tg-MRK1-F | CCATTGAAGAACGGTTGAAACATTTTGTTTCTGCACCTTCATCGTCTTTGCGGATCCCCGGGTTAATTAA |
| tg-MRK1-R | ATGTAATAGTAATGATACAATAGACTAAGAAATTTGAAGGATGAGATATAgaattcgagctcgtttaaac |
| ko-MCK1-F | AATTTTCTTTTTATTTTCCGAAACCCCCACTCCATCACATTCTAGTACATcggatccccgggttaattaa |
| ko-MCK1-R | TTGTTCATTAAATTTTCCGAGGGGAAAGAGAACAAATTAATAGAAAATTAgaattcgagctcgtttaaac |
| ko-MRK1-F | TTGAAAATAATCTCAAGATTGAAGAGGTGAAAATTGTAAAATAGTCTACTcggatccccgggttaattaa |
| ko-MRK1-R | TGTAATAGTAATGATACAATAGACTAAGAAATTTGAAGGATGAGATATATgaattcgagctcgtttaaac |
| ko-RIM11-F | TACATTACAACACGGCAACACTAATCACGCAACGCTAGAACTCGCGCAGGcggatccccgggttaattaa |
| ko-RIM11-R | GTTCCTTCCTTCTCCCATTATTCTTGCCTGGGCTCCCTCCGGTGCTATCAgaattcgagctcgtttaaac |
| prms-RIM11-F | ACCACCGACGTCTGATTCACACATTACTGGGCAAGATCTTGACATAGCATGAATTCGAGCTCGTTTAAAC |
| prms-RIM11-R | TTTGACACTATGTTATTACTGAGATTCGGAGAATTATTGCTTTGAATATTCATCTATAAGATCAATGAAGAGAGAGAGGG |
| ko-MRK1_MSE-F | TTCGTCACAACCGGAGTGAATAGTGGAACTACTGTCAGTTCATAGTATCGcggatccccgggttaattaa |
| ko-MRK1_MSE-R | TCATAAATCTTTATTTTACACTCTCTTTTTTTTTCTTTTAATTATTTTTTgaattcgagctcgtttaaac |
| rp-MRK1_mMSE-F | GTGAATAGTGGAACTACTGTCAGTTCATAGTATCGCGGAATTCAAAAAAATAATTAAAAGAAAAAAAAAGAGAGTGTAA |
| rp-MRK1_mMSE-R |
TTACACTCTCTTTTTTTTTCTTTTAATTATTTTTTTGAATTCCGCGATACTATGAACTGACAGTAGTTCCACTATTCAC |
| cl-sMRK1-306-F | GCGGCCGCTCTAGAACTAGTGGATCCGTACCTAATTTCAATAACAGTTCA |
| cl-sMRK1-306-R | AGCTGGGTACCGGGCCCCCCCTCGAGATCGCAAGTTTCATCAATTTTTCT |
Sporulation conditions
To sporulate strains containing the estradiol-inducible version of NDT80 [45, 46], cells were grown in yeast extract-peptone-dextrose (YPD) medium overnight at 30°C until saturation, then diluted in yeast extract-peptone-potassium acetate (YPA) medium to an optical density at 660nm (OD660) of 0.25. After incubation at 30°C until the culture reached an OD660 of ~1.2, cells were centrifuged at 3,000 x g for 2 min, washed with dH2O, and resuspended in sporulation medium (SPO, 2% potassium acetate) at a final OD660 of 1.6. The cells were incubated in SPO for 6 h to allow for entry into meiosis and arrest in meiotic prophase and then β-estradiol was added to a final concentration of 1 μM to induce NDT80 expression. For assays of sporulation efficiency, ascal type distribution and ether sensitivity in strains carrying native NDT80, sporulation conditions were similar except there was no addition of β-estradiol, and for sporulation at 16°C, cells were cultured in a refrigerated shaker after shift to SPO. After incubation in SPO medium (> 48 h for cells at 30°C; > 96 h for cells at 16°C), cells were fixed in 3.7% formaldehyde and analyzed under a Nikon Eclipse E200 microscope.
Computational analysis of RNA-seq data for internal TSS identification
RNA-Seq data obtained from a highly synchronized yeast meiotic culture (GEO database: GSE90008) was used for the computational analyses. In this culture, cells carrying copper-inducible forms of IME1 and IME4 were incubated in SPO medium for two hours before copper was added to induce sporulation (GEO database: GSE90008; [47]). Data of various time-points from this time-course dataset was analyzed in this study: the early-meiotic samples refer to samples taken 0.25 h, 0.5 h and 0.75 h after addition of copper (when early meiotic genes are highly expressed); the mid-meiotic samples refer to samples taken 3.75 h, 4h and 4.25 h after induction (when meiotic divisions start); the sample taken at 2 h in SPO medium (when copper was added) was used as the control. The sequencing reads were aligned to yeast reference genome Saccharomyces cerevisiae S288C R64.1.1 using TopHat v 2.1.0 [48]. Reads that mapped to the mitochondrial DNA, 2-micron plasmid, rRNA or tRNA genes were removed with BEDTools [49] for accurate quantification, and coverage per nucleotide position was obtained with BEDTools genomecov function.
For identification of novel meiotic TSSs (TSSs specific to the meiotic sample as compared to the control), a Python script was written for the following analysis (S3 File). First, the coverage data was smoothed and converted to Gaussian kernel density estimates (standard deviation of 50 nt, 99.5% confidence), and normalized to the sum coverage of each sample (referred to as "smoothed coverage" below). The shape of the smoothed coverage curves in each sample were then compared to the control sample in sliding windows of 150 nt with 5 nt steps along the whole genome. The root mean squared deviation of smoothed coverage values between meiotic and control sample in every window was computed after normalization of area under either curve to 1. Novel TSSs and TESs (transcription end sites) are both identified in this analysis as shape difference peaks (Fig 1). Potential TES peaks from the meiotic sample were defined as peaks with a greater average RNA-Seq coverage in the window immediately 5’ of the peak location (with respect to the direction of transcription) than the one immediately to the 3’ side. These peaks were then removed from the analysis. Possible TSS signals were further filtered by the following criteria: (1) the shape difference value at the TSS was required to be larger than median of all shape difference values; (2) the RNA-seq signal was judged to be significant in that the sum of smoothed coverage of meiotic and control samples at the TSS was between the median and 95th percentile; and (3) the difference in signal between the meiotic and control sample at the TSS was required to be larger than 90th percentile. Each TSS was then placed into a contig to define the boundaries of the transcript. Positions both 5’ and 3’ of the TSS were scanned until the difference in expression between the meiotic and control sample was lower than the 90th percentile, defining 5’ and 3’ boundaries of that contig. Contigs of close proximity (those with less than 1 kb between 5’ boundaries) and similar expression levels (less than 3-fold variation of averaged difference of smoothed coverage) were merged. TSS locations were adjusted to be the 5’ end of each contig. Contigs were ranked by a confidence score calculated by multiplying the averaged difference of smoothed coverage and the size of the contig. After contig identification in all three meiotic samples was done, those that were only identified in one sample and those longer than 4 kb were discarded (as really long contigs are likely fusions instead of independent transcripts). The remaining contigs were assigned a new rank by averaging the rankings in all samples in which it was identified, a TSS position based on the average TSS locations and a TSS range based on the variation in TSS locations. Among all of the predicted meiosis specific TSSs, internal TSSs were defined as those with the 5’ boundary of the contig within an ORF and at least 75 nt from the annotated start codon or stop codon, and a contig ranking below 800. Finally, each candidate was visually examined in the genome browser with the raw data, and those likely caused by transcription of an adjacent gene, as well as those with noisy transcription that came through the previous filtering steps were removed (Table 3). Each gene in the refined list was also examined in the tiling array dataset of Lardenois et al., 2010 for indications of internal initiations. Aligned reads and all feature tracks generated from the above analyses were visualized using the Integrative Genomics Viewer v2.3.90 (IGV, https://www.broadinstitute.org/igv/) from the Broad Institute (Cambridge, MA).
Fig 1. Analytical flowchart for calling mid-sporulation specific internal TSSs from RNA-Seq data of control and mid-sporulation samples.
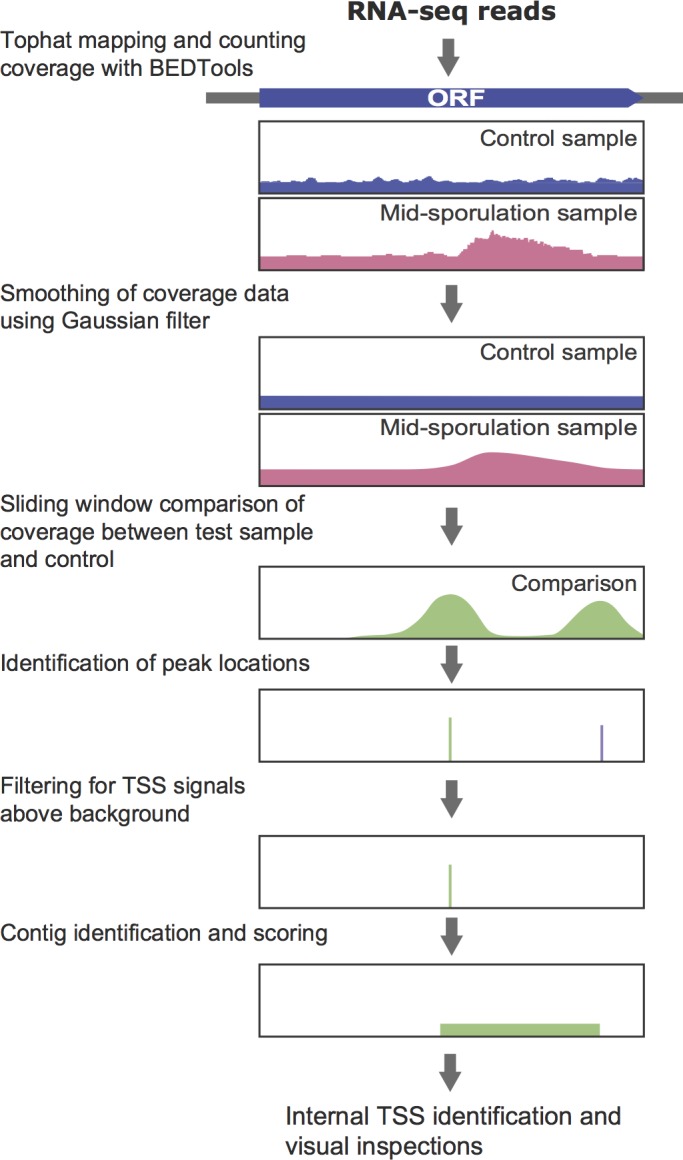
RNA-seq reads of synchronized yeast meiotic culture with PCUP1-IME1 PCUP1-IME4 strain at IME1 and IME4 induction (2 h in SPO) and mid-meiosis (3.75 h, 4 h and 4.25 h after addition of copper) (GSE90008) was used. Analysis flow for a pair of samples is shown as an illustrative example. Reads were aligned to reference assembly (Saccharomyces cerevisiae S288C R64.1.1) using TopHat, then quantified as absolute read counts with BEDTools. Coverage data smoothing was performed by the application of a Gaussian kernel on genome-wide read counts. Using sliding windows along the whole genome, the root mean squared deviation between normalized coverage curves of control and test samples reports the shape difference between the curves in each window. Shape difference peaks were filtered to remove potential TES signals (purple line) and signals from noise. For each TSS signal, transcript boundaries were defined into a contig from the difference in smoothed coverage between control and test samples. Close contigs were merged, assigned a score (product of the average of shape difference values and contig size) and ranked. Contigs identified more than once in the analysis of 3 mid-meiotic samples were further evaluated for whether they were internal to known ORFs. Finally, genes with mid-sporulation specific internal TSSs were filtered at an average contig ranking cutoff of 800 and visually examined. Detailed analysis procedures are described in Material and Methods.
Table 3. Genes identified to contain mid-sporulation specific internal TSSs.
| ORF Name | Gene Name | Start Locationa | MSEb | Lardenois et al.c | Gene Descriptiond |
|---|---|---|---|---|---|
| YAL043c | PTA1 | +1116±145 | Yes | Yes | Essential subunit of holo-CPF |
| YAR050w | FLO1 | +789±20 | No | Yes | Lectin-like cell wall protein (flocculin) |
| YBL063we | KIP1 | +497±212 | No | No | Kinesin-related motor protein |
| YBR069c | TAT1 | +585±4 | Yes | Ambiguous | Amino acid transporter |
| YBR169c | SSE2 | +1037±47 | No | Ambiguous | Nucleotide exchange factor |
| YBR245c | ISW1 | +1797±115 | No | No | ATPase subunit of chromatin remodelers |
| YBR260c | RGD1 | +481±152 | No | Yes | GTPase-activating protein for Rho3p and Rho4p |
| YDL079c | MRK1 | +535±11 | Yes | Yes | Glycogen synthase kinase 3 homolog; deletion reduces sporulation |
| YDL226c | GCS1 | +380±34 | Yes | Yes | ADP-ribosylation factor GTPase activating protein; deletion abolishes sporulation |
| YDR089w | VTC5 | +1249±36 | No | Yes | Vacuolar transmembrane protein |
| YDR143c | SAN1 | +479±232 | No | No | Ubiquitin-protein ligase |
| YDR173c | ARG82 | +218±103 | No | Ambiguous | Inositol polyphosphate multikinase (IPMK); deletion reduces sporulation |
| YDR406w | PDR15 | +884±38 | Yes | No | Plasma membrane ATP binding cassette transporter |
| YDR490c | PKH1 | +1256±112 | No | Yes | Serine/threonine protein kinase |
| YER076c | +526±92 | Yes | No | Protein of unknown function | |
| YFR005c | SAD1 | +671±45 | Yes | Yes | Conserved zinc-finger domain protein involved in pre-mRNA splicing; essential |
| YGL075c | MPS2 | +286±26 | Yes | Ambiguous | Essential membrane protein |
| YHR082c | KSP1 | +584±115 | Yes | Yes | Essential serine/threonine protein kinase |
| YHR176w | FMO1 | +703±139 | Yes | Yes | Flavin-containing monooxygenase |
| YHR182w | +1429±62 | No | Yes | Protein of unknown function | |
| YHR211w | FLO5 | +2429±7 | Yes | Yes | Lectin-like cell wall protein (flocculin) |
| YIL078w | THS1 | +1647±29 | No | Ambiguous | Essential threonyl-tRNA synthetase |
| YIL154c | IMP2' | +586±71 | Yes | Ambiguous | Transcriptional activator involved in maintenance of ion homeostasis |
| YJL092w | SRS2 | +915±72 | Yes | Yes | DNA helicase and DNA-dependent ATPase; deletion reduces spore viability |
| YLL008w | DRS1 | +1598±118 | No | Yes | Essential nucleolar DEAD-box protein |
| YLR106c | MDN1 | +11975±97 | No | Yes | Huge dynein-related AAA-type ATPase; essential |
| YLR273c | PIG1 | +706±120 | No | Yes | Putative targeting subunit for type-1 protein phosphatase Glc7p |
| YLR430w | SEN1 | +3696±90 | No | Yes | Essential splicing endonuclease |
| YML010w | SPT5 | +2633±22 | No | No | Spt4p/5p transcription elongation factor complex subunit; essential |
| YML047c | PRM6 | +271±190 | No | Yes | Pheromone-regulated membrane protein that transports potassium |
| YML072c | TCB3 | +1848±8 | Yes | Yes | Cortical ER protein involved in ER-plasma membrane tethering |
| YNL077w | APJ1 | +940±149 | No | Ambiguous | Chaperone involved in SUMO-mediated protein degradation |
| YNL083w | SAL1 | +345±82 | Yes | Yes | ADP/ATP transporter |
| YNL227c | JJJ1 | +1357±54 | No | Yes | Co-chaperone that associates with the cytosolic large ribosomal subunit |
| YOL057w | +1283±30 | No | Ambiguous | Dipeptidyl-peptidase III | |
| YOR140w | SFL1 | +454±102 | Yes | No | Transcriptional repressor and activator |
| YOR188w | MSB1 | +1443±50 | No | Yes | Protein of unknown function |
| YOR237w | HES1 | +336±132 | No | No | Protein implicated in the regulation of ergosterol biosynthesis |
| YOR330c | MIP1 | +2025±172 | No | Ambiguous | Mitochondrial DNA polymerase gamma |
| YOR355w | GDS1 | +374±85 | No | Ambiguous | Protein of unknown function |
| YPL174c | NIP100 | +1803±13 | Yes | Yes | Large subunit of the dynactin complex |
| YPL242c | IQG1 | +3452±138 | No | Yes | Essential IQGAP-related protein |
| YPR029c | APL4 | +1119±6 | No | Ambiguous | Gamma-adaptin; large subunit of the clathrin-associated protein complex |
aNucleotide position of the internal TSS relative to the first nucleotide of the annotated ORF, based on the average of TSS locations from at least two mid-meiotic samples. Estimation of initiation range is calculated from the variations in TSS locations.
bDenotes whether sequence of 300 nt upstream to the most 3' possible internal TSS position of each gene contains any MSE sites.
cDenotes whether similar pattern of transcription is observed in the tiling array dataset from another study [25] (http://sgv.genouest.org/).
dTaken from SGD (http://www.yeastgenome.org/).
eShown in bold: ORFs that display internal TSS (localized within the range of start location estimated in the current study) during sporulation in a 5'-end sequencing dataset [10].
To identify potential MSE and URS1 sites upstream of each defined internal TSS, DNA sequences 300 nt upstream to the 3' end of the possible TSS range were examined. Motif analysis was then performed on these sequences using the Motif module from the Biopython libraries (http://biopython.org) with the Position Weight Matrix (PWM) of the Ndt80 or Ime1 binding profile retrieved from ScerTF [50] (http://stormo.wustl.edu/ScerTF/). Sequences 300 nt upstream to each ORF served as negative control. Sites with a PWM score larger than 85% of the possible score range were classified as potential MSE/URS1 sites.
Analysis for MRK1 sequence homology
Multiple sequence alignment was performed using the nucleotide sequences of S. cerevisiae MRK1 and homologous regions from S. paradoxus, S. mikatae, S. bayanus and S. kuzriazevii (GenBank acessions: AABY01000135.1, AABZ01000319.1, AACG02000051.1, JH797022.1) by MAFFT version 7 (http://mafft.cbrc.jp/alignment/server/) with default settings.
Western blot assays
For western blots, strains A14201 (MRK1), ySZ68 (MRK1::3xHA) and ySZ313 (mrk1-mMSE::3xHA) were sporulated as described above. 5 ml of cell culture for protein extraction was collected before the switch to SPO medium, at the time of addition of β-estradiol, and two hours after addition of β-estradiol. At these time points, aliquots of cells were fixed in 3.7% formaldehyde, washed, and stained with 4', 6-diamidino-2-phenylindole (DAPI). These samples were examined for progression into the meiotic divisions by epifluorescence with a Zeiss Axioplan 2 microscope. Samples were only processed for western blot experiments if more than 50% of cells had entered the meiotic divisions by two hours after β-estradiol treatment. Cell samples were pelleted, washed, transferred to ice and lysed by alkaline treatment [51]. Pelleted samples were resuspended in SDS sample buffer (10% glycerol, 60 mM Tris-HCl pH 6.8, 0.005% bromophenol blue, 2% β-mercaptoethanol) followed by incubation at 100°C for 5 minutes. Cell debris was removed by centrifugation at 16,000 x g for 3 minutes, and 2 μl of each sample was loaded onto a 10% SDS polyacrylamide gel. Mrk1-3HA was detected using monoclonal anti-HA antibody 12CA5 (BAbCo) at 1:5000 dilution. Porin was detected using monoclonal anti-Por1 antibodies (Molecular Probes) at 1:4000 dilution. HRP-conjugated anti-mouse antibodies (Amersham Biosciences) were used at 1:5000 dilution for detection of Mrk1-3HA and 1:30000 for detection of porin, and proteins were visualized using Clarity-Western ECL substrate (Bio-Rad).
qPCR
For qPCR experiments, strains A14201 (wild type) and ySZ69 (mrk1-mMSE) were sporulated as described above, and 5 ml of culture was collected at the time of addition of β-estradiol and two hours after β-estradiol treatment. Total mRNA was extracted and purified with a RiboPure Yeast Kit (Ambion). cDNA was synthesized from the mRNA using the qScipt cDNA SuperMix (Quanta Bioscience) at a final RNA concentration of 50 ng/μl, and then treated with RNase A (0.5 μg/μl) at 37°C for 30 minutes. The cDNA reaction mix was diluted 10-fold, and 3 μl was used in each 10 μl reaction with LightCycler 480 DNA Sybr Green I PCR master mix (Roche) to determine the mRNA level of each gene using a Mastercycler EP Realplex (Eppendorf). Each sample was assayed in triplicate in each experiment, and all experiments were performed at least twice.
Ether sensitivity assay
To test for ether resistance, cells were sporulated as described above, and sonicated briefly to break the clumps into separate cells. For each sample, 100 cells were counted for sporulation efficiency. Cell concentration was measured with a Z1 Beckman-Coulter Counter, and with this information, each culture was diluted so that the final concentration of asci was 5x106 asci/ml, and 5-fold serial dilutions were spotted identically on two YPD plates. One plate served as a no-ether control, while the other was inverted over ether soaked filter paper (Whatman #3, 1003–090) for 1 hour. Plates were incubated at 30°C for 2–3 days and scanned.
Results
A bioinformatic pipeline for detection of meiosis-induced internal transcription start sites
Transcriptomic datasets, e.g. microarray and RNA-Seq data, can enable observations of changes in TSSs over a meiotic time-course [2, 10, 25]. We analyzed an RNA-Seq dataset of a highly synchronized meiotic time-course (GSE90008). The culture was synchronized by first arresting the cells in sporulation media and then inducing IME1 and IME4, two important regulators of meiotic entry (Yifeng Xu, personal communication). This system produces highly synchronized entry into meiosis enabling efficient sporulation at fine time-resolution [47, 52]. The cDNA library in the study was prepared with random hexamer primers, which is preferential to oligo(dT) primed libraries that generate bias towards 3' ends [53]. In order to capture novel TSSs regardless of their positions relative to genomic features, we developed a customized bioinformatic pipeline. The work flow of analysis is shown in Fig 1.
After mapping and quantifying the RNA-Seq reads from each sample to the genome, each meiotic sample was compared to the control to look for TSSs specific to the meiotic sample. To reduce fluctuations of raw read pileups between adjacent genomic positions, the histogram of sequence reads was first smoothed across the genome. Then, these smoothed coverage profiles were compared between the control and each meiotic sample. Differences in the shape of the transcription profiles were identified computationally as Root Mean Squared Deviation (RMSD) of normalized coverage values between the samples in sliding windows. Locations of shape difference peaks were then filtered to remove potential TES signals and weak TSS signals.
Because of the high sensitivity in RMSD-based detection for shape differences, TSS signals were called not only in positions representing true TSSs, but also from locations where the expression profile appeared wavy due to noise in the data. To distinguish between the two situations, for each TSS peak, a transcript contig was constructed, the boundaries of which were defined from the difference in smoothed coverage between the meiotic sample and control. Nearby contigs of similar expression levels were merged, and the final TSS predictions were defined to be the 5'-end of the contig containing that TSS. To rank the contigs by confidence level, each contig was assigned a confidence score based on two parameters: (1) expression level of the contig; (2) size of the contig, as longer contigs are more likely to represent true novel transcripts than shorter ones, which tend to result from noise. Three time-points taken from cells after the induction of NDT80 (3.75 h, 4 h, 4.25 h after addition of copper) were processed to identify contigs, and only contigs identified in at least two samples were kept. A final ranking was assigned to each contig by averaging all rankings of that contig in different samples. Similarly, the final TSS location was assigned by averaging all TSS locations in different samples, with a range estimate that reflects variation in TSS locations.
To identify internal initiation events, each transcript contig was evaluated for whether its TSS lies internal to an annotated ORF. In the top 800 ranked contigs, 100 were identified to have TSSs internal to the coding region. The corresponding ORFs were then visually examined in the original mapping profile to exclude cases where the internal initiation likely represents transcription from an adjacent gene or noisy data that came through the informatics filters. Further details, including parameters used for the analysis, are described in Material and Methods. A description of all sporulation-specific contigs is available in S1 File.
In sum, this approach identified 43 genes that display potential internal TSSs after NDT80 induction (Table 3). Visual examination of transcription data for these genes in a previously published tiling array dataset (http://sgv.genouest.org, [25]]) confirmed internal initiations of 24 of the 43, suggesting the robustness of this pipeline. From a 5'-end sequencing dataset which attempted to capture budding yeast TSSs at a genome-wide scale [10], direct evidence was found for 11 genes on our list to have meiosis specific TSS at similar positions, supporting the idea that the 5'-boundary of these transcript contigs corresponds to true initiation sites.
Mid-sporulation specific internal initiations are NDT80 dependent
This analysis identified many potential TSSs in genes transcribed after NDT80 induction. However, whether these alternative initiations are coupled to NDT80 induction or arise earlier in meiosis and persist after NDT80 induction is not clear. To address this question, we applied the same pipeline to early meiotic samples from the same time-course. The three early meiotic samples we chose to analyze (0.25 h, 0.5 h and 0.75 h after addition of copper) represent the time immediately after IME1 and IME4 are induced, when well-studied early meiotic genes like IME1, IME2 and RIM4 are highly expressed. This analysis revealed a similar number of genes displaying potential internal TSSs (S2 File). However, only two genes (MPS2, YOL057w) were identified in both the early and mid-meiotic samples. This suggests that while internal initiations are not unique to a particusslar time in meiosis the specific internal TSSs listed in Table 3 may largely be induced by Ndt80 activity.
To examine the likelihood that these internal initiations are driven by Ndt80, sequences upstream of the predicted start sites were examined. Most regulatory motifs lie within 300 bp upstream of the TSS and this is also true for MSE sites in NDT80-induced genes [54, 55]. Therefore, 300 nt of sequence upstream to the 3'-end of the predicted range of TSS position for each internal initiation was examined for MSE sites using a Position Weight Matrix (PWM) for the Ndt80 binding site (see Materials and Methods). The upstream regions of 17 mid-meiotic internal TSS were found to contain MSE sites (Table 3), an extremely significant enrichment (p<1.5x10-22, Fisher's exact test). When the same analysis was used to look for binding sites for the early meiotic transcription factor Ime1, no enrichment for URS1 sites was found in these sequences. Also, no enrichment was found for either MSE or URS1 sites in the upstream regions of early-meiotic internal TSSs. These results are consistent with the idea that induction of NDT80 results in internal transcription initiation in several of its target genes.
Internal initiation and NDT80-dependence was tested by quantitative PCR (qPCR). Synchronous progression into the meiotic divisions can be achieved by placing the NDT80 gene under the control of an estradiol-inducible promoter (NDT80-IN) [45, 46]. Cells homozygous for this allele arrest in meiotic prophase due to the absence of NDT80 expression and then proceed synchronously into the meiotic divisions upon addition of estradiol to the culture. A strain homozygous for NDT80-IN was cultured in sporulation medium for 6 hours, allowing the cells to progress to the ndt80Δ arrest point and then estradiol was added to induce NDT80 transcription. Four candidate genes; MRK1, IQG1, DRS1 and SAD1 were selected for analysis because all appear to have internally initiated transcripts after NDT80 expression in our analysis and in multiple previous datasets [2, 10, 25]. Moreover, in a ribosome profiling dataset, all four display increased ribosome binding near the predicted internal TSS after NDT80 expression [3]. CDC3 and CDC10 were included as controls since both genes are known to be up-regulated following NDT80 expression, but do not display internal start sites in any dataset examined [1, 2, 10, 25, 56].
For each gene, two pairs of primers were used, one pair in the portion of the gene 5’ of the internal start site and one pair that amplified from the 3’ side, within the internally initiated transcript. The presence or absence of transcriptional induction internal to the ORF (i.e. whether more expression is detected from the 3’-end than the 5’-end of the ORF) was determined by taking the log ratio of the level of expression in the 3’ primer pair relative to the 5' pair (log2[(3' expression)/(5' expression)]) so positive values indicate increased expression of the 3' end of the ORF. Unlike CDC3 and CDC10 where both genes display more transcription in the 5' portion of the gene, all 4 genes tested show 3' transcriptional induction specifically after NDT80 induction (Fig 2). These inductions are significant (Student's t-test; p<5.54×10−7 for MRK1, 0.03 for IQG1, 3.02×10−4 for DRS1) for all except SAD1 (p<0.32). These qPCR results confirm our bioinformatics analysis for three out of four genes and suggest that Ndt80 is directly responsible for the internally initiated transcripts.
Fig 2. qPCR examination of NDT80 dependent 3' transcriptional induction for representative genes with mid-sporulation specific internal TSS.
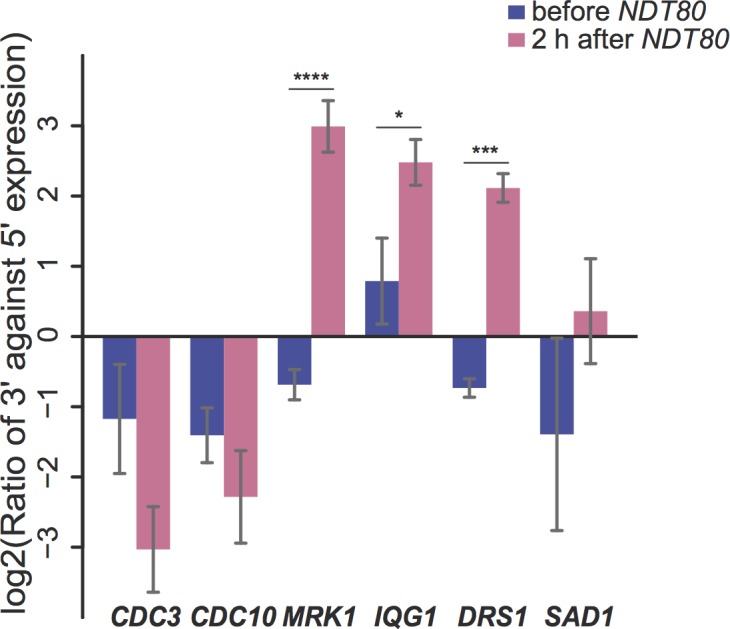
Wild-type (A14201) strain was synchronized in sporulation using β-estradiol inducible NDT80. The levels of 3' and 5' expression of control transcripts (CDC3 and CDC10) and transcripts of a subset of genes identified with mid-sporulation specific internal TSSs (MRK1, IQG1, DRS1 and SAD1) were measured by qPCR of samples harvested when β-estradiol was added to the media and 2 hours after NDT80 induction. The ratios of 3' expression to 5' expression are shown on a log2 scale. Values shown are the averages from two or more independent experiments. Error bars represent standard error of the mean. Statistical differences were examined by Student's t-test. Symbols denote statistical significance (* = p<0.05; *** = p<0.001; **** = p<0.0001).
Internal initiation expands potential proteome diversity during sporulation
The internally initiated transcripts potentially encode N-terminally truncated proteins. These shorter isoforms of the proteins could have distinct function or regulation from the full-length protein. The predicted domain structure of each protein was examined with respect to the position of the internal TSS and the first in-frame ATG codon. Several examples are shown in Fig 3. In some cases, the shorter transcript encodes a predicted catalytic domain separate from regulatory sequences, for example the GAP domains of Rgd1 and Iqg1 or the kinase domain of Mrk1. Conversely, other transcripts would express regulatory regions without catalytic activity, such as the SANT domains of Isw1 or the C-terminal region of Pkh1. These examples show the potential of internal initiations to expand the diversity of the meiotic proteome.
Fig 3. Protein domain structure of example targets from the analysis.
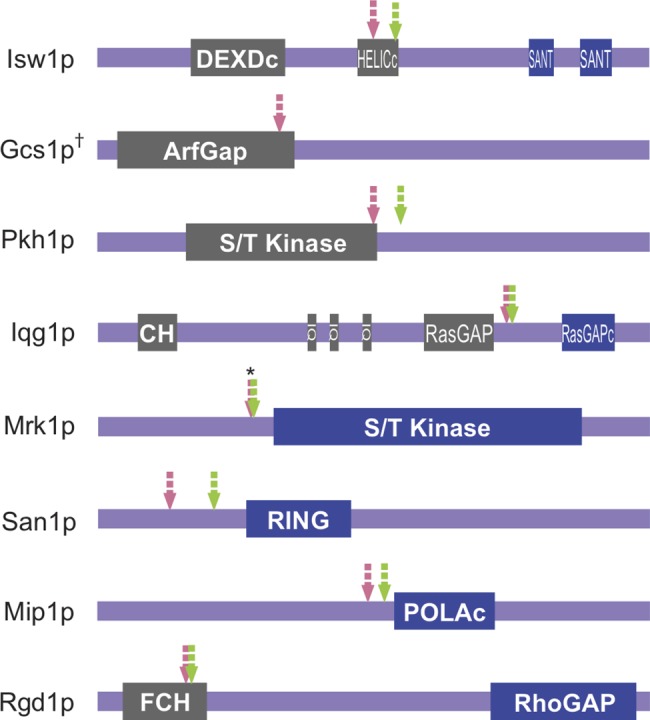
Schematic diagram showing domain organizations of gene products of 8 candidates from Table 3. Predicted motifs from Pfam (http://pfam.xfam.org/) and SMART (http://smart.embl-heidelberg.de/) are illustrated on each protein. Pink arrows denote the amino-acid position corresponding to the 5’ most position of the range for each internal TSS. Green arrows refer to the first in-frame methionine encoded by each internally initiated transcript. Motifs C-terminus to each arrow are shown in blue, while those to the N-terminus are shown in grey. Asterisk (*): for Mrk1p, since both the predicted TSS location and the first methionine encoded by its internal TSS are located inside the intron (Fig 4A), both features are marked at the exon 1 and 2 junction. †: No in-frame start codon can be found 3’ to the predicted internal TSS of GCS1.
An MSE site within the intron is required for the expression of the internally initiated MRK1
Among the genes we examined with qPCR, MRK1 has the strongest induction of the internally initiated transcript, with as high as 8-fold higher expression of the 3' end relative to the to 5' region (Fig 2). MRK1 is the only one of the four GSK-3β homologs in yeast that contain an intron ([57]], Fig 4A). The first exon encodes an extra N-terminal extension unique to Mrk1p compared with the other members of the GSK-3 family in budding yeast, while the kinase domain is contained entirely within the second exon. A consensus MSE site is found within the intron 5’ to the internal TSS. Also within the intron, 3’ of the TSS lies an ATG codon that is in frame with the second exon of MRK1. Translation beginning at this ATG would produce a protein isoform consisting of 11 amino acids encoded within intron sequences plus the entire protein kinase domain encoded by the second exon (Fig 4A). The positioning of these sequence elements strongly suggests that the internally initiated transcript of MRK1 is directly regulated by Ndt80 at the MSE site, and translated into a shorter protein.
Fig 4. Induction of internally initiated MRK1 is regulated by Ndt80.

(A) MRK1 gene structure. Exons are indicated in blue boxes. Solid arrow: TSS upstream of the ORF defined by 5'-end sequencing [10]. Dashed arrow: internal TSS (TSS-in) defined in this study. Purple dot: location of MSE motif in the intron of MRK1. Green dot: location of the first in-frame start codon of the internally initiated transcript. (B) Multiple sequence alignment of MRK1 intronic region from S. cerevisiae and the homologous regions of closely related species. Conservation of residues is indicated by shading: black- identical; grey- >60% similarity. Red boxes indicate exon sequences. Green boxes indicate the 5’ splice site and branchpoint consensus sequences. Cyan box indicates core MSE consensus and yellow box denotes the predicted start codon of the internally initiated MRK1. (C) qPCR examination of NDT80 dependent 3' transcriptional induction of MRK1 in wild-type and MSE mutant. Wild-type (A14201) strain and mrk1-mMSE strain (ySZ69) were synchronized in sporulation using β-estradiol inducible NDT80. The levels of 3' and 5' expression of MRK1 were measured by qPCR of samples harvested when β-estradiol was added to the media and 2 hours after NDT80 induction. The ratios of 3' expression to 5' expression are shown on a log2 scale. Values shown are the averages from 7 or more independent experiments. Error bars represent standard error of the mean. Statistical differences were examined by Student's t-test. Symbols denote statistical significance (**** = p<0.0001). (D) Detection of Mrk1 isoforms by western blot analysis. Wild-type (A14201), MRK1-3HA (ySZ68), and mrk1-mMSE-3HA (ySZ313) strains were synchronized in sporulation using β-estradiol inducible NDT80. For each strain, cells grown in YPA, when β-estradiol was added to sporulation media and 2 hours after NDT80 induction were analyzed.
The intron in MRK1 is only found in closely related Saccharomyces species. To investigate the conservation the MSE of we aligned the nucleotide sequences of the MRK1 intron in five Saccharomyces species (Fig 4B). Sequences within the intron are divergent in the different species, but both the MSE site and the predicted ATG codon of the internally initiated MRK1 are perfectly conserved. Taken together, the sequence analysis suggests that in the Saccharomyces cluster, the MSE site in the MRK1 intron serves as a functional element preserved during evolution.
To test the idea of direct NDT80 regulation, CRISPR/CAS9 was used to mutate the MSE site in the intron in the native chromosomal context. A diploid homozygous for this mutation (mrk1-mMSE) in the NDT80-IN background was generated and internal initiation was examined by qPCR before and after NDT80 induction. Contrary to wild-type, the mrk1-mMSE allele did not display increased 3’ transcription after NDT80 was induced (Fig 4C). To examine whether these changes in transcription ultimately affect protein expression, three tandem copies of the hemagglutinin (HA) epitope were inserted at the 3’ end of the coding sequence of both MRK1 and mrk1-mMSE and protein levels were monitored by Western blot with anti-HA antibodies. Whole cell lysates were made from cells prior to transfer to SPO medium, at the time of estradiol addition, and 2 h after induction of NDT80 by addition of estradiol. In the MRK1-HA strain, full-length Mrk1 was observed in all samples, with a lower amount in pre-NDT80 meiotic sample and somewhat elevated expression in post-NDT80 meiotic sample (Fig 4D). A faster migrating band, corresponding to the predicted size of the N-terminally truncated Mrk1 expressed from the internally initiated transcript was seen only in the post-NDT80 meiotic sample. In contrast, the mrk1-mMSE strain exhibited similar levels to wild-type of the full-length protein across growth conditions, but no expression of the short protein was observed after NDT80 induction. In sum, Ndt80 likely directly regulates internal initiation of MRK1 by binding to the MSE site present in the intron and this results in the translation of an N-terminally truncated form of Mrk1 in meiotic cells.
Internally initiated MRK1 promotes sporulation redundantly with RIM11
In a systematic screen of the S. cerevisiae knockout collection, deletion of MRK1 was found to moderately reduce sporulation efficiency [58]. However, no effect on sporulation was reported in two other genome-wide screens [59, 60]. To re-examine the effect of MRK1 knock-out on sporulation efficiency, we created a homozygous deletion of MRK1 in the SK1 strain background. Mean sporulation efficiency of mrk1Δ appeared 13% lower than that of wild-type, although the difference is not statistically significant (Fig 5A, left panel). Thus, MRK1 is not important for sporulation under these conditions.
Fig 5. The effect of MRK1 deletion or depletion of internally initiated MRK1 on sporulation and spore wall integrity.

Strains of indicated genotypes were sporulated in liquid (WT-AN120; chs3Δ-AN262; mrk1Δ-ySZ102; mck1Δ-ySZ23; mrk1Δ mck1Δ-ySZ46; rim11-md-ySZ39; mrk1Δ rim11-md-ySZ135; mrk1-mMSE rim11-md-ySZ98; rim11-md+vector-ySZ317; mrk1Δ rim11-md+vector-ySZ318; mrk1Δ rim11-md+sMRK1-ySZ315). Cells were allowed to sporulate either at 30°C for >48 h, or at 16°C for >96 h. (A) Sporulation efficiency. At least 300 cells of each strain were counted in each experiment. Bar plots represent the mean sporulation efficiency of at least 3 independent experiments. Error bars represent standard error of the mean. Statistical differences of strains in rim11-md background were examined by Student's t-test. Symbols denote statistical significance (* = p<0.05). (B) Ascal type distribution. At least 125 ascus of each strain were counted in each experiment. Bar plots represent the mean ascal type distribution of at least 3 independent experiments. (C) Ether resistance of spores. Each sporulation culture was diluted to a concentration of 106 asci/ml, and 5-fold serial dilutions were spotted onto YPD plates. Left panels, plates unexposed to ether. Right panels, plates exposed to ether vapor for 30 minutes.
The S. cerevisiae GSK-3 homologs have been shown to have overlapping functions [28, 33], and both RIM11 and MCK1 have been shown to have roles in meiosis [28, 35]. The lack of sporulation phenotype of mrk1Δ could be related to redundancy with RIM11 or MCK1. Deletion of MCK1 reduced sporulation efficiency relative to wild-type, consistent with earlier reports [29]; however, deletion of MRK1 did not exacerbate the mck1Δ phenotype (Fig 5A, left panel). This result suggests that MRK1 does not act redundantly with MCK1 in sporulation.
Deletion of RIM11 blocks entry into meiosis [35]. This defect occurs long before the induction of NDT80 and could, therefore, mask functions of RIM11 later in the meiotic program. To bypass this early role of RIM11 a meiotic depletion allele, rim11-md, was constructed, in which RIM11 expression is under the control of the meiotically inactive CLB2 promoter [43]. A diploid hemizygous for this allele (rim11-md/rim11Δ) sporulated well, indicating that one rim11-md allele provides sufficient protein to fulfill Rim11 function early in sporulation. Deletion of MRK1 in the rim11-md/rim11Δ background caused a reproducible, but not statistically significant (p<0.16, Student’s t-test) reduction in sporulation efficiency (Fig 5A, left panel). In vegetative cells, mck1Δ phenotypes are exacerbated by cold and high-temperature [61]. Sporulation was therefore examined in this same set of strains with incubation at lower (16°C) and higher (37°C) temperatures (Fig 5A, right panel). While no differences were seen at higher temperature (unpublished observations), at lower temperature a modest but significant reduction in sporulation was seen when mrk1Δ was combined with rim11-md (p<0.036 for mrk1Δ in rim11-md; p<0.040 for mrk1Δ in rim11-md carrying the vector plasmid). This same effect was seen when the mrk1-mMSE allele was present rather than mrk1Δ (p<0.021). The reduction could be rescued by ectopic expression of the internally initiated form of MRK1 alone (mrk1Δ rim11-md+sMRK). This suggests that MRK1 acts in concert with RIM11 to promote spore formation and that it is specifically the truncated form of MRK1 induced by NDT80 that is responsible.
Mutants that only modestly reduce sporulation efficiency, defined as the fraction of cells that produce asci, can have stronger effects when the number of spores present in each ascus is considered. For example, modest defects in meiotic chromosome segregation or meiotic spindle pole body function do not inhibit sporulation but shift the major ascal type from tetrads to dyads [62, 63]. For each of the strains bearing mutations in different GSK-3 family genes, the number of spores contained in the asci was assessed. No strong effect on the distribution of ascal types was seen for any of the mutants, regardless of the temperature at which sporulation occurred (Fig 5B).
NDT80 induces the expression of many genes important for spore wall assembly and mutation of genes in this class can result in defects in spore wall function without changes in the ability of the cell to produce visible spores [6]. To evaluate spore wall functionality, the viability of the spores after exposure to ether vapor, a common assay for spore wall integrity [64], was examined (see Materials and Methods). Serial dilutions of sporulated cultures were spotted on to rich media and exposed to ether vapor before incubation to allow germination and growth. A chs3Δ strain was used as a control for ether sensitivity ([64, 65], Fig 5C). We did not observe notable defects in ether resistance in either mrk1Δ or mrk1-mMSE (Fig 5C). Therefore, after examining all these aspects of sporulation, we conclude that the internally initiated MRK1 has redundant functions to RIM11 in regulating the efficiency of sporulation.
Discussion
Earlier studies have reported widespread changes in transcript initiation sites during sporulation [2, 10, 25]. Here we report an informatics analysis of RNA-Seq data to identify TSSs that occur within annotated coding regions during budding yeast meiosis. Dozens of genes are identified to have internal TSSs specific to sporulation, suggesting that internal transcription initiation is not a rare phenomenon. A previous RNA tiling array study also suggests sporulation-specific internal initiation for about half the genes identified and the presence of these shortened transcripts was confirmed by qPCR for several of the genes. Moreover, at least 11 of these genes have been found to display 5'-cap structures proximal to the predicted start locations in sporulating cells [10]. Thus, our analysis is able to efficiently and correctly identify bona fide TSSs.
Early and mid-sporulation transcriptomes reveal distinct sets of genes with internal TSSs. The dynamic switch of internal TSSs along this developmental pathway suggests a tightly regulated process. For those internal TSSs appearing in mid-sporulation, MSE sites are significantly enriched in the sequences upstream to these internal TSSs. Further, qPCR analysis of four genes that have MSE sites 5' to the internal TSSs demonstrates that Ndt80 leads to internal transcription of MRK1, IQG1 and DRS1, and mutation of an MSE site within the MRK1 intron abolished the expression of the internal transcript. Thus, the appearance of these internal TSSs is likely driven directly by Ndt80 as part of the general induction of mid-meiotic genes. NDT80 is expressed only during sporulation, so these shorter transcripts and the truncated proteins they encode are sporulation specific. Placing enhancer binding sites within the coding region may be a common mechanism to increase protein diversity during developmental processes.
How does internal initiation affect which protein variant is expressed, and how does this impact sporulation? Of 43 genes with mid-sporulation specific internal initiations, only three genes—MRK1, GCS1 and SRS2, have been previously associated with roles in sporulation or germination [58, 66, 67]. There are a number of reasons earlier studies may not have revealed sporulation phenotypes for mutations in these genes. First, 10 genes on the list out of the 43 are essential and so their possible role in sporulation cannot be assessed by deletion alleles. A second possibility is that the shorter proteins expressed from the internal transcripts may act to inhibit the function of the full-length protein. From the analysis of protein domain structures, the genes fall into two general categories. In the first instance, the predicted truncated proteins contain only a catalytic domain, such as Rgd1, Mrk1, San1 and Mip1. In the second instance, the shorter transcripts encode potential regulatory domains lacking a catalytic domain, such as Isw1, Gcs1, Pkh1 and Iqg1 (Fig 3). For the latter class, if the full-length protein itself impairs sporulation, the shorter proteins produced by internal initiations could counteract that effect. In this circumstance, deletion studies would eliminate both forms of the protein and have no effect on sporulation efficiency. As with the internal initiations that occur in essential genes, it will be necessary to identify mutations that eliminate the internal TSS without affecting expression of the intact gene in order to test the significance of each transcript for sporulation efficiency.
Redundant gene functions might also account for why no sporulation phenotypes have been reported for deletions in some of the genes with internal TSSs. Redundant functions are common amongst genes in the NDT80-regulon [68], and overlap between MRK1 and RIM11 appears to account for the lack of phenotype of the loss of MRK1. The sporulation defect seen in the mrk1Δ rim11-md diploids is quite mild; however, this might reflect insufficient depletion of the Rim11 kinase rather than a minor role for these kinases in sporulation. While the CLB2 promoter is an effective means to shut off expression, whether complete depletion is achieved depends on the stability of the mitotically expressed protein and how much protein is required for activity. An alternative strategy to inactivate the kinase is to engineer mutations into active site that render it sensitive to specific ATP analog inhibitors [69]. This has been used to acutely inhibit kinases during sporulation and can be used to separate functions of a kinase that occur early and late in the process [45, 70, 71]. Such “analog-sensitive” alleles of RIM11 were constructed, but were not effectively inhibited by the available analogs (S.Z., unpublished observations). Nonetheless, future development of an effective Rim11 shutoff system could aid in defining the functions of MRK1 and RIM11 late in sporulation.
It is also possible that some of the genes with internal initiations have no sporulation phenotype when deleted because the internal initiations represent transcriptional ‘noise’. That is, transcription triggered by random Ndt80 binding sites that are simply neutral and do not have an evolutionarily selected function. The internal initiation of MRK1, however, appears to be positively selected in a cluster of Saccharomyces species, as the intronic MSE site is much more conserved in a highly variable genomic context. Nonetheless, the proportion of functionally important internal initiations remains unclear.
The prevalence and dynamic properties of internal transcriptional initiation observed are unlikely to be specific to budding yeast sporulation. Alteration in TSSs involving internal initiations have also been observed in budding yeast under different growth conditions, when fission yeast undergo meiosis, during Drosophila embryonic development and in human cells of different tissue types and of disease states [17, 72–74]. The analysis pipeline described performs a comparison of transcript architecture on RNA-Seq data independent of genome annotations, and thus can be adapted to other RNA-Seq datasets in other systems for novel TSS detection. Such investigations could unveil the prevalence, regulation and functional importance of internal transcriptional initiation in other processes.
Supporting information
(XLSX)
(XLSX)
(TXT)
Acknowledgments
The authors wish to thank Bruce Futcher, Yuping Chen and Justin Gardin for extensive suggestions on the development of the algorithm, and Alisa Yurovski for general guidance on RNA-Seq data processing. We thank Nancy Hollingsworth for the 12CA5 anti-HA antibody, and Bruce Futcher and Gang Zhao for the CRISPR/Cas9 plasmid. We are greatly indebted to Bruce Futcher, Yifeng Xu and Folkert van Werven for the RNA-Seq data of the PCUP1-IME1 PCUP1-IME4 meiotic time-course. We also thank Janet Leatherwood, members of the Neiman and the Futcher labs for helpful discussions. This work was supported by NIH grant R01 GM072540 to A.M.N.
Data Availability
Our bioinformatic analysis was based on a publicly available RNAseq dataset (GSE90008). All additional relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by National Institutes of Health grants R01 GM072540 to A.M.N. and P01 GM088297 to R.S. and A.M.N. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown PO, et al. The Transcriptional Program of Sporulation in Budding Yeast. Science. 1998;282(5389):699–705. doi: 10.1126/science.282.5389.699 [DOI] [PubMed] [Google Scholar]
- 2.Kim Guisbert KS, Zhang Y, Flatow J, Hurtado S, Staley JP, Lin S, et al. Meiosis-induced alterations in transcript architecture and noncoding RNA expression in S. cerevisiae. RNA. 2012;18(6):1142–53. doi: 10.1261/rna.030510.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, Weissman JS. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science. 2012;335(6068):552–7. doi: 10.1126/science.1215110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Werven FJ, Amon A. Regulation of entry into gametogenesis. Philos Trans R Soc Lond B Biol Sci. 2011;366(1584):3521–31. doi: 10.1098/rstb.2011.0081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bowdish KS, Yuan HE, Mitchell AP. Positive control of yeast meiotic genes by the negative regulator UME6. Mol Cell Biol. 1995;15(6):2955–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neiman AM. Sporulation in the budding yeast Saccharomyces cerevisiae. Genetics. 2011;189(3):737–65. doi: 10.1534/genetics.111.127126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu L, Ajimura M, Padmore R, Klein C, Kleckner N. NDT80, a meiosis-specific gene required for exit from pachytene in Saccharomyces cerevisiae. Mol Cell Biol. 1995;15(12):6572–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pak J, Segall J. Role of Ndt80, Sum1, and Swe1 as targets of the meiotic recombination checkpoint that control exit from pachytene and spore formation in Saccharomyces cerevisiae. Mol Cell Biol. 2002;22(18):6430–40. doi: 10.1128/MCB.22.18.6430-6440.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chu S, Herskowitz I. Gametogenesis in yeast is regulated by a transcriptional cascade dependent on Ndt80. Mol Cell. 1998;1(5):685–96. [DOI] [PubMed] [Google Scholar]
- 10.Miura F, Kawaguchi N, Sese J, Toyoda A, Hattori M, Morishita S, et al. A large-scale full-length cDNA analysis to explore the budding yeast transcriptome. Proc Natl Acad Sci U S A. 2006;103(47):17846–51. doi: 10.1073/pnas.0605645103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Munster S, Camblong J, et al. Bidirectional promoters generate pervasive transcription in yeast. Nature. 2009;457(7232):1033–7. doi: 10.1038/nature07728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kimura K, Wakamatsu A, Suzuki Y, Ota T, Nishikawa T, Yamashita R, et al. Diversification of transcriptional modulation: large-scale identification and characterization of putative alternative promoters of human genes. Genome Res. 2006;16(1):55–65. doi: 10.1101/gr.4039406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40(12):1413–5. doi: 10.1038/ng.259 [DOI] [PubMed] [Google Scholar]
- 14.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–5. doi: 10.1038/nbt.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309(5740):1559–63. doi: 10.1126/science.1112014 [DOI] [PubMed] [Google Scholar]
- 16.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470–6. doi: 10.1038/nature07509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waern K, Snyder M. Extensive transcript diversity and novel upstream open reading frame regulation in yeast. G3 (Bethesda). 2013;3(2):343–52. doi: 10.1534/g3.112.003640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hughes TA. Regulation of gene expression by alternative untranslated regions. Trends Genet. 2006;22(3):119–22. doi: 10.1016/j.tig.2006.01.001 [DOI] [PubMed] [Google Scholar]
- 19.Rojas-Duran MF, Gilbert WV. Alternative transcription start site selection leads to large differences in translation activity in yeast. RNA. 2012;18(12):2299–305. doi: 10.1261/rna.035865.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Floor SN, Doudna JA. Tunable protein synthesis by transcript isoforms in human cells. Elife. 2016;5:e10921 doi: 10.7554/eLife.10921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris DR, Geballe AP. Upstream Open Reading Frames as Regulators of mRNA Translation. Mol Cell Biol. 2000;20(23):8635–42. doi: 10.1128/mcb.20.23.8635–8642.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carlson M, Botstein D. Two differentially regulated mRNAs with different 5′ ends encode secreted and intracellular forms of yeast invertase. Cell. 1982;28(1):145–54. doi: 10.1016/0092-8674(82)90384-1 [DOI] [PubMed] [Google Scholar]
- 23.Pelechano V, Wei W, Steinmetz LM. Extensive transcriptional heterogeneity revealed by isoform profiling. Nature. 2013;497(7447):127–31. doi: 10.1038/nature12121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wiesner T, Lee W, Obenauf AC, Ran L, Murali R, Zhang QF, et al. Alternative transcription initiation leads to expression of a novel ALK isoform in cancer. Nature. 2015;526(7573):453–7. doi: 10.1038/nature15258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lardenois A, Liu Y, Walther T, Chalmel F, Evrard B, Granovskaia M, et al. Execution of the meiotic noncoding RNA expression program and the onset of gametogenesis in yeast require the conserved exosome subunit Rrp6. Proc Natl Acad Sci U S A. 2011;108(3):1058–63. doi: 10.1073/pnas.1016459108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hurtado S, Kim Guisbert KS, Sontheimer EJ. SPO24 is a transcriptionally dynamic, small ORF-encoding locus required for efficient sporulation in Saccharomyces cerevisiae. PLoS One. 2014;9(8):e105058 doi: 10.1371/journal.pone.0105058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ali A, Hoeflich KP, Woodgett JR. Glycogen synthase kinase-3: properties, functions, and regulation. Chem Rev. 2001;101(8):2527–40. doi: 10.1021/cr000110o [DOI] [PubMed] [Google Scholar]
- 28.Xiao Y, Mitchell AP. Shared roles of yeast glycogen synthase kinase 3 family members in nitrogen-responsive phosphorylation of meiotic regulator Ume6p. Mol Cell Biol. 2000;20(15):5447–53. doi: 10.1128/MCB.20.15.5447–5453.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neigeborn L, Mitchell AP. The yeast MCK1 gene encodes a protein kinase homolog that activates early meiotic gene expression. Genes Dev. 1991;5(4):533–48. doi: 10.1101/gad.5.4.533 [DOI] [PubMed] [Google Scholar]
- 30.Ikui AE, Rossio V, Schroeder L, Yoshida S. A yeast GSK-3 kinase Mck1 promotes Cdc6 degradation to inhibit DNA re-replication. PLoS Genet. 2012;8(12):e1003099 doi: 10.1371/journal.pgen.1003099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quan Z, Cao L, Tang Y, Yan Y, Oliver SG, Zhang N. The Yeast GSK-3 Homologue Mck1 Is a Key Controller of Quiescence Entry and Chronological Lifespan. PLoS Genet. 2015;11(6):e1005282 doi: 10.1371/journal.pgen.1005282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shero JH, Hieter P. A suppressor of a centromere DNA mutation encodes a putative protein kinase (MCK1). Genes Dev. 1991;5(4):549–60. doi: 10.1101/gad.5.4.549 [DOI] [PubMed] [Google Scholar]
- 33.Hirata Y, Andoh T, Asahara T, Kikuchi A. Yeast glycogen synthase kinase-3 activates Msn2p-dependent transcription of stress responsive genes. Mol Biol Cell. 2003;14(1):302–12. doi: 10.1091/mbc.E02-05-0247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Griffioen G, Swinnen S, Thevelein JM. Feedback inhibition on cell wall integrity signaling by Zds1 involves Gsk3 phosphorylation of a cAMP-dependent protein kinase regulatory subunit. J Biol Chem. 2003;278(26):23460–71. doi: 10.1074/jbc.M210691200 [DOI] [PubMed] [Google Scholar]
- 35.Bowdish KS, Yuan HE, Mitchell AP. Analysis of RIM11, a yeast protein kinase that phosphorylates the meiotic activator IME1. Mol Cell Biol. 1994;14(12):7909–19. doi: 10.1128/mcb.14.12.7909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malathi K, Xiao Y, Mitchell AP. Interaction of yeast repressor-activator protein Ume6p with glycogen synthase kinase 3 homolog Rim11p. Molecular and cellular biology. 1997. doi: 10.1128/MCB.17.12.7230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malathi K, Xiao Y, Mitchell AP. Catalytic roles of yeast GSK3beta/shaggy homolog Rim11p in meiotic activation. Genetics. 1999;153(3):1145–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rubin-Bejerano I, Sagee S, Friedman O, Pnueli L, Kassir Y. The in vivo activity of Ime1, the key transcriptional activator of meiosis-specific genes in Saccharomyces cerevisiae, is inhibited by the cyclic AMP/protein kinase A signal pathway through the glycogen synthase kinase 3-beta homolog Rim11. Mol Cell Biol. 2004;24(16):6967–79. doi: 10.1128/MCB.24.16.6967-6979.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andoh T, Hirata Y, Kikuchi A. Yeast Glycogen Synthase Kinase 3 Is Involved in Protein Degradation in Cooperation with Bul1, Bul2, and Rsp5. Mol Cell Biol. 2000;20(18):6712–20. doi: 10.1128/mcb.20.18.6712–6720.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hilioti Z, Gallagher DA, Low-Nam ST, Ramaswamy P, Gajer P, Kingsbury TJ, et al. GSK-3 kinases enhance calcineurin signaling by phosphorylation of RCNs. Genes Dev. 2004;18(1):35–47. doi: 10.1101/gad.1159204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rose MD, Winston F, Hieter P. Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. NY: Cold Spring Harbor Press; 1990. [Google Scholar]
- 42.Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14(10):953–61. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U [DOI] [PubMed] [Google Scholar]
- 43.Lee BH, Amon A. Role of Polo-like kinase CDC5 in programming meiosis I chromosome segregation. Science. 2003;300(5618):482–6. doi: 10.1126/science.1081846 [DOI] [PubMed] [Google Scholar]
- 44.Jin L, Zhang K, Xu Y, Sternglanz R, Neiman AM. Sequestration of mRNAs Modulates the Timing of Translation during Meiosis in Budding Yeast. Mol Cell Biol. 2015;35(20):3448–58. doi: 10.1128/MCB.00189-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benjamin KR, Zhang C, Shokat KM, Herskowitz I. Control of landmark events in meiosis by the CDK Cdc28 and the meiosis-specific kinase Ime2. Genes Dev. 2003;17(12):1524–39. doi: 10.1101/gad.1101503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carlile TM, Amon A. Meiosis I is established through division-specific translational control of a cyclin. Cell. 2008;133(2):280–91. doi: 10.1016/j.cell.2008.02.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Berchowitz LE, Gajadhar AS, van Werven FJ, De Rosa AA, Samoylova ML, Brar GA, et al. A developmentally regulated translational control pathway establishes the meiotic chromosome segregation pattern. Genes Dev. 2013;27(19):2147–63. doi: 10.1101/gad.224253.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36 doi: 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–2. doi: 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spivak AT, Stormo GD. ScerTF: a comprehensive database of benchmarked position weight matrices for Saccharomyces species. Nucleic Acids Res. 2012;40(Database issue):D162–8. doi: 10.1093/nar/gkr1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kushnirov VV. Rapid and reliable protein extraction from yeast. Yeast. 2000;16(9):857–60. doi: 10.1002/1097-0061(20000630)16:9<857::AID-YEA561>3.0.CO;2-B [DOI] [PubMed] [Google Scholar]
- 52.Chia M, van Werven FJ. Temporal Expression of a Master Regulator Drives Synchronous Sporulation in Budding Yeast. G3 (Bethesda). 2016;6(11):3553–60. doi: 10.1534/g3.116.034983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hansen KD, Brenner SE, Dudoit S. Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 2010;38(12):e131 doi: 10.1093/nar/gkq224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Erb I, van Nimwegen E. Transcription factor binding site positioning in yeast: proximal promoter motifs characterize TATA-less promoters. PLoS One. 2011;6(9):e24279 doi: 10.1371/journal.pone.0024279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jolly ER, Chin CS, Herskowitz I, Li H. Genome-wide identification of the regulatory targets of a transcription factor using biochemical characterization and computational genomic analysis. BMC Bioinformatics. 2005;6(1):275 doi: 10.1186/1471-2105-6-275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kaback DB, Feldberg LR. Saccharomyces cerevisiae exhibits a sporulation-specific temporal pattern of transcript accumulation. Mol Cell Biol. 1985;5(4):751–61. doi: 10.1128/MCB.5.4.751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davis CA, Grate L, Spingola M, Ares M Jr. Test of intron predictions reveals novel splice sites, alternatively spliced mRNAs and new introns in meiotically regulated genes of yeast. Nucleic Acids Res. 2000;28(8):1700–6. doi: 10.1093/nar/28.8.1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Enyenihi AH, Saunders WS. Large-scale functional genomic analysis of sporulation and meiosis in Saccharomyces cerevisiae. Genetics. 2003;163(1):47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Deutschbauer AM, Williams RM, Chu AM, Davis RW. Parallel phenotypic analysis of sporulation and postgermination growth in Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences. 2002;99(24):15530–5. doi: 10.1073/pnas.202604399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marston AL, Tham WH, Shah H, Amon A. A genome-wide screen identifies genes required for centromeric cohesion. Science. 2004;303(5662):1367–70. doi: 10.1126/science.1094220 [DOI] [PubMed] [Google Scholar]
- 61.Puziss JW, Hardy TA, Johnson RB, Roach PJ, Hieter P. MDS1, a dosage suppressor of an mck1 mutant, encodes a putative yeast homolog of glycogen synthase kinase 3. Mol Cell Biol. 1994;14(1):831–9. doi: 10.1128/MCB.14.1.831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lo HC, Wan L, Rosebrock A, Futcher B, Hollingsworth NM. Cdc7-Dbf4 regulates NDT80 transcription as well as reductional segregation during budding yeast meiosis. Mol Biol Cell. 2008;19(11):4956–67. doi: 10.1091/mbc.E08-07-0755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bajgier BK, Malzone M, Nickas M, Neiman AM. SPO21 Is Required for Meiosis-specific Modification of the Spindle Pole Body in Yeast. Mol Biol Cell. 2001;12(6):1611–21. doi: 10.1091/mbc.12.6.1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dawes IW, Hardie ID. Selective killing of vegetative cells in sporulated yeast cultures by exposure to diethyl ether. Mol Gen Genet. 1974;131(4):281–9. doi: 10.1007/BF00264859 [DOI] [PubMed] [Google Scholar]
- 65.Pammer M, Briza P, Ellinger A, Schuster T, Stucka R, Feldmann H, et al. DIT101 (CSD2, CAL1), a cell cycle-regulated yeast gene required for synthesis of chitin in cell walls and chitosan in spore walls. Yeast. 1992;8(12):1089–99. doi: 10.1002/yea.320081211 [DOI] [PubMed] [Google Scholar]
- 66.Connolly JE, Engebrecht J. The Arf-GTPase-activating protein Gcs1p is essential for sporulation and regulates the phospholipase D Spo14p. Eukaryot Cell. 2006;5(1):112–24. doi: 10.1128/EC.5.1.112-124.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sasanuma H, Furihata Y, Shinohara M, Shinohara A. Remodeling of the Rad51 DNA strand-exchange protein by the Srs2 helicase. Genetics. 2013;194(4):859–72. doi: 10.1534/genetics.113.150615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lin CP, Kim C, Smith SO, Neiman AM. A highly redundant gene network controls assembly of the outer spore wall in S. cerevisiae. PLoS Genet. 2013;9(8):e1003700 doi: 10.1371/journal.pgen.1003700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407(6802):395–401. doi: 10.1038/35030148 [DOI] [PubMed] [Google Scholar]
- 70.Wan L, Zhang C, Shokat KM, Hollingsworth NM. Chemical inactivation of cdc7 kinase in budding yeast results in a reversible arrest that allows efficient cell synchronization prior to meiotic recombination. Genetics. 2006;174(4):1767–74. doi: 10.1534/genetics.106.064303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arguello-Miranda O, Zagoriy I, Mengoli V, Rojas J, Jonak K, Oz T, et al. Casein Kinase 1 Coordinates Cohesin Cleavage, Gametogenesis, and Exit from M Phase in Meiosis II. Dev Cell. 2017;40(1):37–52. doi: 10.1016/j.devcel.2016.11.021 [DOI] [PubMed] [Google Scholar]
- 72.Chen HM, Rosebrock AP, Khan SR, Futcher B, Leatherwood JK. Repression of meiotic genes by antisense transcription and by Fkh2 transcription factor in Schizosaccharomyces pombe. PLoS One. 2012;7(1):e29917 doi: 10.1371/journal.pone.0029917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ni T, Corcoran DL, Rach EA, Song S, Spana EP, Gao Y, et al. A paired-end sequencing strategy to map the complex landscape of transcription initiation. Nat Methods. 2010;7(7):521–7. doi: 10.1038/nmeth.1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li TW, Ting JH, Yokoyama NN, Bernstein A, van de Wetering M, Waterman ML. Wnt activation and alternative promoter repression of LEF1 in colon cancer. Mol Cell Biol. 2006;26(14):5284–99. doi: 10.1128/MCB.00105-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX)
(XLSX)
(TXT)
Data Availability Statement
Our bioinformatic analysis was based on a publicly available RNAseq dataset (GSE90008). All additional relevant data are within the paper and its Supporting Information files.