

Abstract
Background
Omecamtiv mecarbil (OM) enhances systolic function in vivo by directly binding the myosin cross-bridges (XBs) in the sarcomere. However, the mechanistic details governing OM-induced modulation of XB behavior in failing human myocardium are unclear.
Methods and results
The effects of OM on steady-state and dynamic XB behavior were measured in chemically-skinned myocardial preparations isolated from human donor and heart failure (HF) left ventricle. HF myocardium exhibited impaired contractile function as evidenced by reduced maximal force, magnitude of XB recruitment (Pdf), and a slowed rate of XB detachment (krel) at submaximal Ca2+-activations. Ca2+-sensitivity of force generation (pCa50) was higher in HF myocardium when compared to donor myocardium, both prior to and following OM incubations. OM incubation (0.5μM and 1.0μM) enhanced force generation at sub-maximal Ca2+-activations in a dose-dependent manner. Notably, OM-induced a slowing in krel with 1.0μM OM but not with 0.5μM OM in HF myocardium. Additionally, OM exerted other differential effects on XB behavior in HF myocardium as evidenced by a greater enhancement in Pdf and slowing in the time-course of cooperative XB recruitment (Trec), which collectively prolonged achievement of peak force development (Tpk), compared to donor myocardium.
Conclusions
Our findings demonstrate that OM augments force generation but also prolongs the time course of XB transitions to force-bearing states in remodeled HF myocardium which may extend the systolic ejection time (SET) in vivo. Optimal OM dosing is critical for eliciting enhanced systolic function without excessive prolongation of SET which may compromise diastolic filling.
Keywords: Omecamtiv mecarbil, contractile function, human heart failure
Progression of heart disease to end-stage heart failure (HF) is often the result of long-term maladaptive remodeling coupled with deteriorating cardiomyocyte function. Although, the fundamental pathophysiological mechanisms that lead to HF are not completely understood1, a common feature of many forms of HF is decreased contractility leading to diminished force generation at the cardiomyocyte level and diminished systolic performance at the whole-heart level. Importantly, despite significant therapeutic advances, the clinical course of HF patients remains unacceptably poor and continues to pose major clinical challenges.2, 3 Current therapies aimed at increasing cardiac contractility have not improved long-term outcomes, and carry the potential for undesirable adverse side effects related to activation of signaling pathways that enhance cardiomyocyte intracellular Ca2+ concentrations.2, 4, 5 Thus, recent efforts6, 7 have focused on designing HF therapies that circumvent complex signaling pathways, and instead directly target the force-generating contractile machinery of the myofilaments.
In this regard, omecamtiv mecarbil (OM), a molecule that selectively binds the catalytic domain of myosin, has gained significant attention due to its potentially beneficial effect on systolic function in HF patients.5, 7, 8 Early clinical trials have demonstrated that OM augments systolic indices such as systolic ejection time, stroke volume, and ejection fraction5 without altering cardiomyocyte Ca2+ transients.9 The mechanisms underlying OM-induced force enhancements have been linked to an enhanced stability of the actin-bound myosin cross-bridges (XBs)10, slowed strain-induced XB detachment from actin11, 12, and slowed shortening velocity13, which act to prolong the XB duty cycle14. Additionally, OM has been shown to accelerate phosphate release10, 15 but concomitantly delays the XB powerstroke and actin-activated ATP turnover15, thereby, maintaining XBs in a strongly-bound state. Thus, in theory, a prolonged XB duty cycle should reduce ATP utilization and improve contractile efficiency11, thereby reducing workload in failing hearts.
Initial clinical trials have demonstrated that OM induces dose-dependent increases in stroke volume and fractional shortening in healthy volunteers5 and HF subjects without adverse side-effects.2, 7 Despite these early positive results, the functional impact of OM on dynamic XB behavior at the cellular level has not been established, and there are no studies that have systematically examined the impact of OM on myofilament contractile function in human failing myocardium. This information would be valuable because it has been shown that failing human myocardium displays pathological remodeling, altered contractile protein phosphorylation, and diminished force generating capacity16, 17 –factors that could potentially modify the impact of OM on XB behavior. Thus, in this study we tested whether the dose-dependent impact of OM on XB behavior differs in ventricular myocardium isolated from donor and failing hearts.
Methods
An expanded Methods section is available in the Supplemental material.
Ethical approval and procurement of human myocardial tissue samples
Normal, non-failing (donor) and failing left ventricular (LV) human cardiac tissue samples were collected and stored until used for experiments as per the approved guidelines of the University of Sydney.
Determination of sarcomeric protein expression and phosphorylation levels
Coomassie and Pro-Q Diamond phosphoprotein staining was performed to quantify myofilament protein expression and phosphorylation in the cardiac samples as described previously18, 19 (Figures 1A and B). Quantification of β-myosin heavy chain (β-MHC) expression (n=6 per group) (Figure 1C) was performed as described previously.20 Gels were scanned using Image J software.18
Figure 1. Phosphorylation of sarcomeric proteins and MHC expression in donor and HF myocardium.

(A) Representative Coomassie stained (left) SDS gel and Pro-Q Diamond-stained (right) showing the expression and phosphorylation status of myofilament proteins in donor and HF samples. Cardiac samples isolated from 4 donor hearts and 4 failing hearts were used to analyze contractile protein expression and phosphorylation levels. (B) Quantification of phosphorylation of MyBP-C, TnT, and TnI in donor and HF samples. (C) Representative 5% Tris-HCl gel showing MHC isoform expression in donor and HF myocardium. Values are expressed as mean ± S.E.M. * P < 0.05.
Preparation of chemically-skinned multicellular myocardial preparations, Ca2+ solutions, and OM solutions
Detergent-skinned multicellular ventricular preparations (∼100μm in width and ∼400μm in length) were prepared as described earlier.18 The composition of various Ca2+ solutions21 used for experiments was calculated using a computer program22 and using established stability constants23. OM (Selleckchem, Houston, TX, USA) and was initially dissolved in DMSO11 and diluted in a relaxing solution to achieve final concentrations of 0.5μM or 1μM OM. Functional measurements (Figures 2, 3, 4, and 5) were performed at ∼24°C following a 2-minute incubation with 0.5μM or 1μM OM.
Figure 2. Effect of OM on force enhancements at variable [Ca2+] activations in donor and HF myocardium.

Baseline forces produced by the skinned myocardial preparations were first measured in Ca2+ solutions producing a range of submaximal forces. Forces were measured at baseline or following a 2-minute incubation with 0.5μM or 1.0μM OM. The net increase in force generation from the untreated baseline (pre-OM) following OM incubation was calculated and is expressed as % increase in force from baseline. Force enhancements were pronounced at submaximal Ca2+ activation but declined as the level of Ca2+ was increased. 12 skinned myocardial preparations (3 fibers each from 4 hearts) were used for both groups. *, significant force enhancement from baseline level in donor and HF myocardium following 1.0μM OM incubation; †, significant force enhancement from baseline level in donor and HF myocardium following 0.5μM OM incubation. Asterisks indicate P < 0.05.
Figure 3. Effect of OM on pCa50 in donor and HF myocardium.

Skinned myocardial preparations were exposed to a 2-minute incubation with 0.5 μM or 1.0μM OM. (A) Effect of OM on the force-pCa relationships in donor and HF myocardium. (B) Effect of OM on the force-pCa relationships in donor myocardium. (C) Effect of OM on the force-pCa relationships in HF myocardium. 12 skinned myocardial preparations (3 fibers each from 4 hearts) were used for both groups. *, pCa50 is significantly higher in HF when compared to the donor myocardium. Asterisks indicate P < 0.05.
Figure 4. Representative stretch activation responses in donor and HF myocardium prior to and following OM incubations.
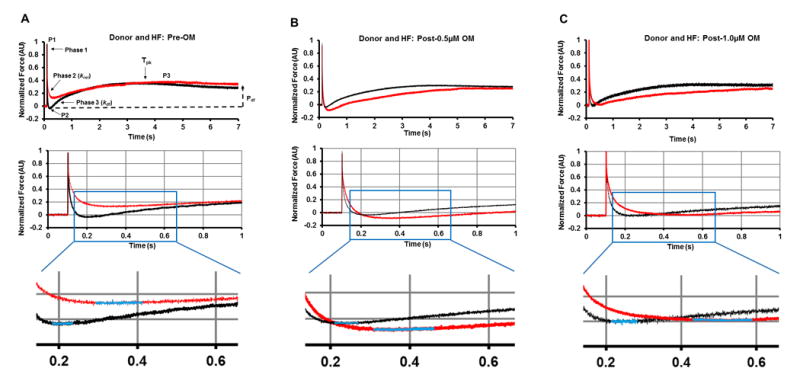
Representative force responses are shown following a sudden 2% stretch in muscle length (ML) in isometrically-contracting donor (black traces), and HF (red traces) myocardial preparations prior to (A) and following incubation with 0.5μM OM (B) or 1.0μM OM (C). Expanded views are shown below panels A to C to demonstrate that krel is slower in HF myocardium compared to donor myocardium both prior to and following OM incubation. Insets below the expanded views depict the Trec region of the trace. The highlighted parts of the stretch activation traces (blue colored regions) in the insets show that the time required for XB recruitment, Trec, is prolonged post-OM incubations in the HF myocardium. Panel A highlights the important phases of the force transients and various stretch activation parameters that are measured from force responses to 2% stretch in ML (explained in methods section).
Figure 5. Effect of OM on the time required for XB recruitment (Trec) and time required to achieve the peak of force development (Tpk) in donor and HF myocardium.

The effect of OM was measured by incubating myocardial preparations in 0.5μM or 1.0μM OM. (A) Trec prior to and following OM incubation in donor and HF myocardium. (B) Tpk prior to and following OM incubation in donor and HF myocardium. The supplement contains a description of how these parameters are measured. Values are expressed as mean ± S.E.M. 12 skinned myocardial preparations (3 fibers each from 4 hearts) were used for both groups. *, different vs. the corresponding pre-OM group; †, different vs. the corresponding donor group. Asterisks indicate P < 0.05.
Stretch activation experiments to measure dynamic XB contractile parameters
The stretch activation protocol used here was described in detail previously.11, 18 Skinned myocardial preparations were exposed to pCa solutions that generated ∼25% of maximal force prior to OM incubations (i.e., pCa 6.6 and pCa 6.3 in HF and donor myocardium, respectively). As done earlier24, a lower pCa (i.e., pCa 6.6) was chosen for HF group to account for an increased myofilament Ca2+ sensitivity when compared to the donor group. The key features of cardiac muscle stretch activation responses are depicted in Figure 4A. A mechanical slack-restretch protocol was used to measure the rate of tension redevelopment, ktr, using linear transformation of the half-time of force redevelopment, i.e., ktr = 0.693/t1/2, where t1/2 is the time (in milliseconds) required to reach half maximal force of the ktr trace.24
Data Analysis
Data were analyzed with a linear mixed (LM) effect model25 with an unstructured covariance matrix across the conditions and across the groups. The R package lme4 was used to implement the LM analysis.26, 27 Asterisks in figures and tables denote statistical significance using a Tukey post hoc multiple comparisons test, and type-I error was controlled by adjusting P-values for multiple comparisons using a false discovery rate (FDR) method. Values are reported as mean±SEM and the criterion for statistical significance was set at P < 0.05.
Results
Sarcomeric protein expression and phosphorylation levels
The basal phosphorylation levels of cardiac myosin binding protein-C (MyBP-C), troponin T (TnT) and troponin I (TnI) were reduced in HF samples compared to donor samples. Phosphorylation levels of MyBP-C, TnT, and TnI in HF were ∼65±11% (P=0.001), ∼68±10% (P=0.04), and ∼33±8% (P<0.005), respectively compared to donor myocardium (n=4; Figures 1A and 1B). Phosphorylation levels of other sarcomeric proteins such as regulatory light chain were not significantly different between the groups. Donor and HF ventricular samples expressed 100% β-MHC isoform without evidence of α-MHC expression (Figure 1C). We have previously shown that OM incubation does not affect the phosphorylation status of sarcomeric proteins.11
Effects of OM on Ca2+-activated force generation
Ca2+-activated force generation was measured in skinned ventricular myocardial preparations in a range of pCa that yielded forces below half-maximal force (i.e., in pCa's 6.6, 6.5, 6.4, and 6.3). Force measurements were collected at baseline or following a 2-minute incubation in 0.5μM or 1.0μM OM. The increase in OM-induced force enhancement gradually decreased with increasing levels of Ca2+ activations (Figure 2), as reported previously.11 Force enhancements following 0.5μM OM incubation were: ∼28±5%, 25±3%, 22±3%, and 19±4% in the donor group; and ∼24±7%, 19±3%, 13±2%, and 8±2% in the HF group at pCa's 6.6, 6.5, 6.4, and 6.3, respectively. Force enhancements following 1.0μM OM incubation were more pronounced: ∼85±11%, 63±9%, 54±7%, and 28±5% in the donor group; and ∼78±13%, 67±17%, 36±10%, and 27±6% in the HF group at pCa's 6.6, 6.5, 6.4, and 6.3, respectively. Importantly, OM-induced force enhancement was more pronounced in HF myocardium than in donor myocardium when comparisons were made at equivalent baseline levels of force production (i.e., at pCa 6.6 in HF vs. pCa 6.3 in donor). Furthermore, OM incubation did not alter either the Ca2+-independent force (i.e., Fmin measured at pCa 9.0) and maximal Ca2+-activated force (i.e., Fmax measured at pCa 4.5) in both donor11 and HF groups (Table 1).
Table 1. Steady-state parameters measured prior to and following OM incubation in donor and HF myocardium.
| Group | pCa50 | nH | Fmax (mN/mm2) | Fmin (mN/mm2) |
|---|---|---|---|---|
| Donor | ||||
| Pre-OM | 5.89±0.04 | 2.23±0.11 | 16.86±2.20 | 1.65±0.24 |
| 0.5μM OM | 5.93±0.03 | 1.52±0.10* | 16.32±1.58 | 1.62±0.14 |
| 1.0μM OM | 5.90±0.03 | 1.58±0.14* | 19.46±2.11 | 1.98±0.29 |
| HF | ||||
| Pre-OM | 6.09±0.04† | 1.95±0.14 | 8.96±0.99† | 0.87±0.07† |
| 0.5μM OM | 6.11±0.02† | 1.39±0.11* | 10.11±0.92† | 1.08±0.14 |
| 1.0μM OM | 6.12±0.04† | 1.43±0.10* | 9.37±0.90† | 1.06±0.10† |
pCa50: myofilament Ca2+ sensitivity; nH: cooperativity of force production; Fmax: Ca2+-activated maximal force measured at pCa 4.5; Fmin: Ca2+-independent force measured at pCa 9.0. Values are expressed as mean ± S.E.M. 12 skinned myocardial preparations (3 fibers each from 4 hearts) were used for both groups.
different vs. the corresponding Pre-OM group;
different vs. the corresponding donor group. Asterisks indicate P < 0.05.
Effects of OM on myofilament Ca2+ sensitivity (pCa50) and cooperativity of force development (nH)
Under basal conditions HF myocardium displayed increased pCa50 i.e., an increased responsiveness of cardiac myofilaments to Ca2+ when compared to donor myocardium, as indicated by a significant left-ward shift in the force-pCa relationships (Figure 3A), a trend that was maintained even after OM incubation (Table 1). The increase in Ca2+-sensitivity post-OM incubations was mainly confined to forces < 40% of maximal force, and hence pCa50 was unaltered in both groups (Table 1; Figures 3B and 3C). nH values decreased post-OM incubations in both groups, indicating that OM decreased the overall cooperativity of force production (Table 1).
Effects of OM on ktr
When studied at equivalent levels of steady-state force generation (i.e., at pCa 6.3 and 6.6, in donor and HF myocardium, respectively), no differences in ktr were observed between donor and HF prior to OM incubation (Table 2). OM differentially affected ktr in HF and the donor group: following 0.5μM and 1.0μM OM incubations: ktr was slowed by ∼33% and ∼32%, respectively in the HF group but not in donor group, suggesting that OM slows XB transitions from weakly-bound to strongly-bound states, or from strongly-bound to weakly-bound states, or both, resulting in an overall slowing of XB cycling in HF myocardium.
Table 2. Dynamic stretch-activation parameters measured prior to and following OM incubation in donor and HF myocardium.
| Group | ktr (s-1) | krel (s-1) | kdf (s-1) | P1 | P2 | P3 | |
|---|---|---|---|---|---|---|---|
| Donor | |||||||
| Pre-OM | 1.23±0.08 | 88.49±5.70 | 1.44±0.11 | 0.569±0.017 | -0.019±0.012 | 0.209±0.008 | 0.228±0.013 |
| 0.5μM OM | 1.03±0.05 | 84.60±8.13 | 1.04±0.06* | 0.725±0.048* | 0.019±0.027 | 0.242±0.023 | 0.223±0.026 |
| 1.0μM OM | 1.20±0.10 | 70.51±5.54 | 0.81±0.06* | 0.633±0.020 | -0.043±0.007 | 0.205±0.010 | 0.248±0.011 |
| HF | |||||||
| Pre-OM | 1.18±0.11 | 65.59±6.75† | 1.25±0.12 | 0.648±0.051 | 0.125±0.022† | 0.258±0.020 | 0.133±0.024† |
| 0.5μM OM | 0.79±0.07* | 64.55±3.98† | 0.73±0.05*† | 0.705±0.027 | 0.009±0.019* | 0.239±0.014 | 0.230±0.022* |
| 1.0μM OM | 0.80±0.06*† | 45.96±4.62*† | 0.54±0.05* | 0.693±0.051 | -0.014±0.019* | 0.205±0.018 | 0.219±0.024* |
ktr: rate of tension redevelopment; krel: rate of XB detachment; kdf: rate of XB recruitment; P1: XB stiffness; P2: magnitude of XB detachment; P3: the new steady-state force attained following a 2% stretch in muscle length; Pdf: magnitude of XB recruitment. As done in all our previous studies11, 21, 24, 30, stretch activation amplitudes were normalized to pre-stretch steady-state Ca2+-activated force, P0, which refers to the activation level (∼25% of maximal Ca2+-activated force), and is equivalent in the donor and HF groups (see methods for details). Values are expressed as mean ± S.E.M. 12 skinned myocardial preparations (3 fibers each from 4 hearts) were used for both groups.
different vs. the corresponding Pre-OM group;
different vs. the corresponding donor group. Asterisks indicate P < 0.05.
Effects of OM on dynamic stretch activation parameters
Stretch activation parameters (Figure 4) were assessed following OM incubations (0.5μM or 1.0μM) at equivalent levels of force generation (i.e., at pCa 6.3 and 6.6, respectively in donor and HF myocardium). The impact of OM on the rate of XB detachment was assessed by measuring krel. Our findings show that donor myocardium displayed accelerated krel (∼35%; p=0.03), and a greater magnitude of XB detachment (P2) as indicated by more negative P2 values compared to HF myocardium (Table 2). Interestingly, incubation with OM did not alter krel in donor myocardium, but krel was slowed following 1.0μM OM by ∼30% in HF myocardium, indicating a differential effect of OM on krel in HF myocardium (Table 2).
The impact of OM on the rate and magnitude of XB recruitment was assessed by measuring kdf and Pdf, respectively. kdf was slowed by ∼28% and ∼44%, respectively, in donor myocardium following 0.5μM and 1.0μM OM incubations. Whereas kdf was slowed by ∼42% and ∼57%, respectively, in HF myocardium following 0.5μM and 1.0μM OM incubations (Table 2). Prior to OM incubation, donor myocardium exhibited greater Pdf (∼73% higher vs. HF myocardium; p=0.02, Table 2), suggesting impairments in XB recruitment in HF myocardium. OM incubations did not affect the new steady-state force attained following the imposed stretch (i.e., P3) in either group (Table 2). Pdf was enhanced by ∼74% and ∼65% (p=0.02) following 0.5μM and 1.0μM OM incubations in HF myocardium, respectively, such that the overall number of XBs recruited into the force-bearing state was substantially enhanced, whereas no OM-induced enhancement in Pdf was observed in donor myocardium (Table 2) –indicating a differential effect of OM on Pdf in HF myocardium.
To gain further insights into the mechanisms by which OM differentially affects XB behavior in donor and HF myocardium, we measured Trec, the time lag between krel and kdf (please see supplement for a detailed explanation). Trec can be interpreted as the time required for the transition between XB detachment and the commencement of XB attachment to actin due to the recruitment of XBs into the force-bearing state. Trec was not statistically different between HF and donor myocardium (Table 2; Figure 5A) under basal conditions. Importantly, following OM incubation HF myocardium displayed ∼47% and ∼62% increases in Trec (Table 3; Figure 5A) at 0.5μM and 1.0μM OM, respectively, but no effects of OM on Trec were observed in donor myocardium. Thus, Trec in HF myocardium was ∼109% and ∼149% longer than in donor myocardium following 0.5μM and 1.0μM OM incubations (p<0.005; Table 3, Figure 5A), suggesting a differential effect of OM on Trec in HF myocardium.
Table 3. Time required for XB recruitment (Trec) and time required to reach the peak of force development (Tpk) prior to and following OM incubation in donor and HF skinned myocardium.
| Group | Trec (msec) | Tpk (sec) |
|---|---|---|
| Donor | ||
| Pre-OM | 66.25±4.03 | 2.79±0.08 |
| 0.5μM OM | 73.00±5.41 | 3.44±0.14* |
| 1.0μM OM | 67.33±6.09 | 4.84±0.24* |
| HF | ||
| Pre-OM | 103.96±13.98 | 2.75±0.23 |
| 0.5μM OM | 152.63±12.60*† | 4.55±0.27*† |
| 1.0μM OM | 167.92±17.93*† | 6.66±0.29*† |
Values are expressed as mean ± S.E.M. 12 skinned myocardial preparations (3 fibers each from 4 hearts) were used for both the groups.
different vs. the corresponding Pre-OM group;
different vs. the corresponding donor group. Asterisks indicate P < 0.05.
No differences in Tpk (i.e., time to achieve peak force development) were observed between donor and HF myocardium under basal conditions, but Tpk was ∼32% and ∼38% longer (p<0.005) (Figures 4A and 5B) in HF myocardium vs. donor myocardium following 0.5μM and 1.0μM OM incubations, respectively (Table 3). This indicates a greater delay in achievement of peak force generation in HF myocardium and thereby a differential effect of OM on Tpk in HF myocardium.
Discussion
Omecamtiv mecarbil (OM) is currently in clinical trials as a potential therapy for systolic heart failure (HF)6, 7 due to its ability to augment acto-myosin XB interactions and force generation without the undesirable secondary effects of other inotropic agents. However, despite early evidence of the utility of OM in HF patients, the detailed mechanisms underlying OM-induced effects on XB behavior at the cellular level in HF myocardium have not been established. Here we show that OM enhances force generation in HF myocardium by shifting the XB equilibrium towards a force-bearing state, however, the time course of XB recruitment is prolonged such that achievement of peak force generation is delayed when compared to donor myocardium.
HF myocardium exhibits reduced magnitude of XB recruitment
Poor systolic function is a common feature of HF patients, and the contractile deficits can often be traced to the contractile machinery's inability to elicit sufficient force-generating interactions between myosin and actin. Indeed, previous studies have reported contractile abnormalities in HF myocardium17, as evidenced by reduced maximal force generation and enhanced myofilament Ca2+ sensitivity (i.e., pCa50). However, the mechanisms underlying the changes in dynamic regulation of XB behavior in HF myocardium are unknown. Consistent with previous reports17, we observed reduced steady-state maximal force generation and increased pCa50 in HF myocardium when compared to donor myocardium (Table 1; Figure 3A). However, unlike previous studies11, 25, OM incubation did not enhance the pCa50 (Table 1). The cause of this discrepancy is unclear, but may be related to a basal temperature-dependent sensitization of thin filaments which acts to decrease the impact of OM on pCa50 (15°C25 and 22°C11 in earlier studies vs. 24°C in the present study). Results from stretch activation experiments revealed a slowed rate of XB detachment (i.e., krel, Table 2) in HF myocardium, which acts to prolong XB duty ratio and increase force generation at low Ca2+-activations. Furthermore, despite a lack of difference in basal rates of XB recruitment between HF and donor myocardium (i.e., ktr and kdf, Table 2), HF myocardium displayed impairments in the magnitude of XB recruitment, (i.e., Pdf, Table 2), thereby contributing to a reduced force generating capacity. These findings suggest that factors inherent to HF myocardium, such as contractile protein dephosphorylation, contribute to impaired force generation. Collectively, prolonged XB on time due to slowed XB detachment and a reduced magnitude of XB recruitment are likely contributors to slowed rates of diastolic relaxation and impaired systolic function displayed by HF patients in vivo.
It is unclear why HF myocardial samples generate lower maximal forces, display slower rates of XB detachment, and recruit less XBs into force-bearing states, but these effects could be related to diminished phosphorylation of key contractile proteins such as MyBP-C, TnI, and TnT (Figure 1), which have been shown to be reduced in HF17. In particular, we have previously shown that increased MyBP-C phosphorylation by protein kinase A (PKA) accelerates the rates and enhances the magnitudes of XB detachment and recruitment24, 28, whereas MyBP-C dephosphorylation slows the rates and reduces the magnitudes of XB detachment and recruitment18, 24. In addition, PKA-mediated TnI phosphorylation has been shown to accelerate XB detachment and decrease myofilament Ca2+ sensitivity29. Thus, reduced MyBP-C and TnI phosphorylation in HF myocardium would be expected to slow the rate of XB detachment (Table 2; Figure 5), and decrease the number of XBs entering the cycling pool during myofilament activation. Furthermore, our finding that TnT phosphorylation is reduced in HF myocardium (Figure 1) suggests that TnT may also be a component of β-adrenergic signaling21, 30, a pathway known to be downregulated during HF31, although the specific impact of PKA-mediated TnT phosphorylation is yet to be elucidated. Moreover, it is likely that although total TnT phosphorylation is reduced, PKC-mediated TnT phosphorylation may be upregulated in HF32, contributing to slowed XB detachment in HF myocardium. In contrast to reduced MyBP-C, TnI, and TnT phosphorylation, we did not observe changes in RLC phosphorylation which is consistent with our data demonstrating that baseline kdf is similar in HF and donor groups (Table 2), because increased RLC phosphorylation would be expected to accelerate kdf33.
Additionally, it is also possible that HF myocardium harbors an increased number of super-relaxed (SRX) myosin heads, characterized by substantially slower ATP turnover rates, which have been linked to reduced MyBP-C phosphorylation34. These SRX heads are proposed to be folded over and interacting with the thick filament backbone rather than interacting with myosin binding sites on actin, thus potentially decreasing thin filament activation and force generation in HF myocardium, although this possibility requires further investigation.
OM enhances the magnitude of XB recruitment in HF myocardium
Earlier studies have shown that OM enhances myofilament force generation by altering the kinetic properties of the β-MHC motor.11, 13, 35 As shown earlier11, we observed that OM-induced force enhancements were primarily confined to sub-maximal Ca2+-activations and that force enhancements gradually decline with increasing Ca2+ levels (Figure 2). As reported earlier11, OM treatment also reduced the cooperativity of force generation, nH (Table 1) which may be related to the fact that at low [Ca2+], thin filament activation relies more on XB-mediated cooperative XB recruitment as most of the regulatory units (RU) are in the off state.36 However, as intracellular [Ca2+] rises most of the RUs switch to the on state, effectively reducing the dependence on XB-mediated cooperative XB recruitment, which offsets the impact of OM and decreases the overall steepness of nH37.
In addition to enhanced force generation, OM slowed the rate of XB recruitment (kdf) (Table 2) which can be linked to OM's ability to shift XB equilibrium toward a force-bearing state through enhanced XB-mediated cooperative XB-recruitment to neighboring RUs, a slow and time consuming process. At higher doses (1.0μM), OM also slowed the rate of XB detachment (krel) in HF myocardium (Table 2). Thus, at higher concentrations, OM-induced force increases maybe, in part, attributed to a slowing in krel which concomitantly increases the time that myosin XBs remain bound to actin (increased XB duty ratio)11, 25, an effect which has been proposed to be due to allosteric changes in myosin heads38 and delayed actin-induced myosin light chain domain rotation.15 Furthermore, OM also traps a population of XBs in a weak actin affinity state25, and those myosin heads may not be readily available for recruitment, or they may transition towards the force-bearing state very slowly –a mechanism that contributes to a prolonged time course of cooperative XB recruitment and slowing of force generation.
Importantly, when tested at equivalent submaximal activation levels, OM elicited differential effects on XB behavior in HF myocardium. Notably, the basal time course of XB recruitment to a force-generating state, Trec, was not different between donor and HF myocardium, however, following OM incubation Trec was significantly slower in HF myocardium (Table 3). A greater OM-induced slowing of Trec in HF myocardium would be predicted to slow the overall rate of XB cycling from a weakly-bound to a strongly-bound state and vice versa (ktr).39 Indeed, OM-induced slowing in ktr is observed in HF myocardium but not in donor myocardium (Table 2). An indirect effect of slowed XB cycling in HF myocardium is a further prolongation of the XB duty ratio, which acts to prolong the open state of tropomyosin, thus permitting more XB recruitment and binding to additional open actin sites. In support of this notion, we observed that the magnitude of XB recruitment (Pdf) is enhanced to a greater extent in HF myocardium compared to donor myocardium following OM incubation (Table 2).
Our observation that OM slows XB kinetics may seem to differ from results of enzymology studies which showed that OM accelerated the rate of phosphate release10, 15, which would be expected to accelerate the rate of force generation. Interestingly, although not tested at multiple doses of OM, Rohde et al15 also showed that OM slows the force-generating myosin powerstroke step by delaying the rotation of myosin light chain domain and effectively uncouples the powerstroke from actin-activated phosphate release.15 This mechanism may resolve the seemingly contradictory findings that OM accelerates phosphate release yet slows XB kinetics, as we show here. An additional consideration in analyzing results of OM experiments, is that some studies have employed systems that lack an intact myofilament lattice10, 13, 15, in contrast to skinned myocardium utilized here. Notably, ADP release is a strain-sensitive mechanism40, a property that would be difficult to capture using unregulated systems. Nevertheless, studies using regulated systems (i.e., in vitro motility assays or skinned myocardium) reported slowed rates of shortening velocity and XB detachment11, 13, 14, suggestive of a slowed ADP release (although this was not directly measured in these studies). Collectively, these findings suggest that OM acts to slow the rate of XB transitions to force-bearing states and prolongs XB duty cycle, which together act to promote XB-mediated cooperative XB recruitment and slow the rate of force generation, as observed in our studies.
Another key finding is that despite a significant OM-induced enhancement in the magnitude of XB recruitment, HF myocardium displayed a delay in achievement of peak force development, Tpk, when compared to donor myocardium, in part, due to a prolongation of Trec in HF myocardium (Table 3; Figure 5). Collectively, these findings suggest that the beneficial effects of OM manifest more slowly in the HF myocardium. This differential response to OM may be due to inherent deficits in force generating capacity in HF myocardium such as reduced numbers of readily available XBs for recruitment, impaired XB cycling due to contractile protein dephosphorylation, or both.
Potential in vivo consequences of OM-induced effects on XB behavior
OM differentially impacts HF myocardium, in that there is a greater slowing of cooperative XB recruitment, Trec, and a greater prolongation of the time to reach peak force development, Tpk, –a cumulative impact is a substantially enhanced XB recruitment, Pdf in HF myocardium. The differential dose-response to OM may be related to inherent differences in basal contractile function in donor and HF myocardium and has significant implications for in vivo contractile function in HF patients. Specifically, during the initial phase of the isovolumic contraction, the Ca2+ levels surrounding the myofilaments is very low.41 Because the effects of OM are more prominent at low Ca2+ levels, OM treatment enhances XB recruitment and amplifies the number of XBs ultimately participating in force generation during this initial phase of isovolumic contraction thereby augmenting ventricular pressure development later in systole.10
Specifically, our data show that a higher OM dose (1.0μM), which may be greater than the effective doses administered to HF patients in clinical trials to date6, 7, results in robust enhancements in force generation, but also significantly slows XB detachment (krel) in HF myocardium. Slowed krel likely plays a major role in determining the total systolic ejection time (SET) and the commencement of ventricular relaxation and diastolic filling.42 In particular, in the late systolic ejection phase, during which a substantial amount of blood is pumped into the circulation, the Ca2+ transient wanes off43 and XBs begin to rapidly detach due to shortening-induced cooperative deactivation of the thin filaments.41 Thus, slowed krel at higher OM doses may act to prolong XB-mediated thin filament activation which is a net effect of all bound XBs, thereby prolonging the SET –potentially delaying ventricular pressure decay and reducing diastolic filling.42 In contrast, incubations of skinned myocardium with 0.5μM OM produced relatively smaller enhancements in force generation, but did not slow krel, suggesting that it is possible to optimize OM dose to elicit beneficial effects on systolic force generation without the potential adverse impact of excessive prolongation of SET that can infringe on the diastolic filling phase.
Study limitations
It is important to note that our in vitro experiments cannot fully account for a slew of complexities that are inherent to clinical HF studies such as alterations in neuro-hormonal signaling, Ca2+-cycling, and post-translational modifications in non-contractile proteins that typically occur in a remodeled HF myocardium. Furthermore, our experiments cannot specifically account for various comorbidities such as diabetes, hypertension, and peripheral arterial disease, or supplementary pharmacological interventions such as diuretics, ACE inhibitors, β-blockers, etc., that may be administered to HF patients in addition to OM.6 All the above factors may influence the in vivo impact of OM to some degree, and cannot be fully recapitulated in vitro. Thus, caution should be exercised when extrapolating our data to clinical outcomes.
Conclusions
Our novel findings demonstrate that OM promotes XB formation but prolongs the time course of cooperative XB recruitment leading to delayed peak force development in HF myocardium when compared to donor myocardium. At higher doses of OM, slower rates of force development are also accompanied by slower rates of XB cycling, which may underlie the prolongation of systolic ejection time in vivo. Of clinical significance, our results demonstrate that correct OM dosing may be critical to achieve a balance of increased systolic performance without diminished diastolic filling in vivo.
Supplementary Material
Clinical Perspective.
What is New
-
-
We investigated if omecamtiv mecarbil (OM) affects actomyosin cross-bridge (XB) behavior similarly in healthy and pathologically remodeled heart failure (HF) myocardium.
-
-
Dose-dependent impact of OM on XB function was examined in detergent-skinned myocardial preparations made from donor and HF heart samples.
-
-
OM significantly enhanced the magnitude of XB recruitment to a force-bearing state in HF myocardium, effectively reducing the basal gap in force generation compared to donor myocardium.
-
-
However, dose-dependent OM-induced force enhancement in HF myocardium was also accompanied by slowed peak force development and XB cycling compared to donor myocardium, particularly at higher OM doses.
What are the Clinical Implications
-
-
Slowed XB kinetics at the cellular level may underlie the observed prolongation of systolic ejection time (SET) at the whole-heart level following OM treatment in healthy volunteers and HF patients.
-
-
Our results highlight the importance of administering optimal OM doses to HF patients to balance increases in force generation and stroke volume while avoiding excessive prolongation of SET which can delay diastolic filling.
Acknowledgments
R.M., J.L., and J.E.S contributed to the conception and design of the experiments. R.M., J.L K.S.G., C.Y.D., C.G.D.R., A.L., S.L., and J.E.S participated in performing the experiments, data acquisition, data analysis, data interpretation, drafting, and revising the manuscript. S.V. participated in data analysis, data interpretation, drafting, and revising the manuscript. All authors approved the final version of the manuscript.
Sources of Funding: This work was supported by the National Institutes of Health (HL-114770 to J.E.S), American Heart Association (16POST30730000 to R.M), and P30 EY011373 grants.
Footnotes
Disclosures: None.
References
- 1.Triposkiadis F, Giamouzis G, Parissis J, Starling RC, Boudoulas H, Skoularigis J, Butler J, Filippatos G. Reframing the association and significance of co-morbidities in heart failure. European journal of heart failure. 2016;18:744–758. doi: 10.1002/ejhf.600. [DOI] [PubMed] [Google Scholar]
- 2.Greenberg B. Novel therapies for heart failure- where do they stand? Circulation journal. 2016;80:1882–1891. doi: 10.1253/circj.CJ-16-0742. [DOI] [PubMed] [Google Scholar]
- 3.Xie M, Burchfield JS, Hill JA. Pathological ventricular remodeling: Therapies: Part 2 of 2. Circulation. 2013;128:1021–1030. doi: 10.1161/CIRCULATIONAHA.113.001879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tardiff JC, Carrier L, Bers DM, Poggesi C, Ferrantini C, Coppini R, Maier LS, Ashrafian H, Huke S, van der Velden J. Targets for therapy in sarcomeric cardiomyopathies. Cardiovascular research. 2015;105:457–470. doi: 10.1093/cvr/cvv023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Teerlink JR, Clarke CP, Saikali KG, Lee JH, Chen MM, Escandon RD, Elliott L, Bee R, Habibzadeh MR, Goldman JH, Schiller NB, Malik FI, Wolff AA. Dose-dependent augmentation of cardiac systolic function with the selective cardiac myosin activator, omecamtiv mecarbil: A first-in-man study. Lancet. 2011;378:667–675. doi: 10.1016/S0140-6736(11)61219-1. [DOI] [PubMed] [Google Scholar]
- 6.Teerlink JR, Felker GM, McMurray JJ, Ponikowski P, Metra M, Filippatos GS, Ezekowitz JA, Dickstein K, Cleland JG, Kim JB, Lei L, Knusel B, Wolff AA, Malik FI, Wasserman SM, Investigators AA. Acute treatment with omecamtiv mecarbil to increase contractility in acute heart failure: The atomic-ahf study. Journal of the American College of Cardiology. 2016;67:1444–1455. doi: 10.1016/j.jacc.2016.01.031. [DOI] [PubMed] [Google Scholar]
- 7.Teerlink JR, Felker GM, McMurray JJ, Solomon SD, Adams KF, Jr, Cleland JG, Ezekowitz JA, Goudev A, Macdonald P, Metra M, Mitrovic V, Ponikowski P, Serpytis P, Spinar J, Tomcsanyi J, Vandekerckhove HJ, Voors AA, Monsalvo ML, Johnston J, Malik FI, Honarpour N, Investigators CH. Chronic oral study of myosin activation to increase contractility in heart failure (cosmic-hf): A phase 2, pharmacokinetic, randomised, placebo-controlled trial. Lancet. 2016;388:2895–2903. doi: 10.1016/S0140-6736(16)32049-9. [DOI] [PubMed] [Google Scholar]
- 8.Cleland JG, Teerlink JR, Senior R, Nifontov EM, Mc Murray JJ, Lang CC, Tsyrlin VA, Greenberg BH, Mayet J, Francis DP, Shaburishvili T, Monaghan M, Saltzberg M, Neyses L, Wasserman SM, Lee JH, Saikali KG, Clarke CP, Goldman JH, Wolff AA, Malik FI. The effects of the cardiac myosin activator, omecamtiv mecarbil, on cardiac function in systolic heart failure: A double-blind, placebo-controlled, crossover, dose-ranging phase 2 trial. Lancet. 2011;378:676–683. doi: 10.1016/S0140-6736(11)61126-4. [DOI] [PubMed] [Google Scholar]
- 9.Malik FI, Morgan BP. Cardiac myosin activation part 1: From concept to clinic. Journal of molecular and cellular cardiology. 2011;51:454–461. doi: 10.1016/j.yjmcc.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 10.Malik FI, Hartman JJ, Elias KA, Morgan BP, Rodriguez H, Brejc K, Anderson RL, Sueoka SH, Lee KH, Finer JT, Sakowicz R, Baliga R, Cox DR, Garard M, Godinez G, Kawas R, Kraynack E, Lenzi D, Lu PP, Muci A, Niu C, Qian X, Pierce DW, Pokrovskii M, Suehiro I, Sylvester S, Tochimoto T, Valdez C, Wang W, Katori T, Kass DA, Shen YT, Vatner SF, Morgans DJ. Cardiac myosin activation: A potential therapeutic approach for systolic heart failure. Science. 2011;331:1439–1443. doi: 10.1126/science.1200113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mamidi R, Gresham KS, Li A, Dos Remedios CG, Stelzer JE. Molecular effects of the myosin activator omecamtiv mecarbil on contractile properties of skinned myocardium lacking cardiac myosin binding protein-c. Journal of molecular and cellular cardiology. 2015;85:262–272. doi: 10.1016/j.yjmcc.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stelzer JE, Larsson L, Fitzsimons DP, Moss RL. Activation dependence of stretch activation in mouse skinned myocardium: Implications for ventricular function. The Journal of general physiology. 2006;127:95–107. doi: 10.1085/jgp.200509432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y, White HD, Belknap B, Winkelmann DA, Forgacs E. Omecamtiv mecarbil modulates the kinetic and motile properties of porcine beta-cardiac myosin. Biochemistry. 2015;54:1963–1975. doi: 10.1021/bi5015166. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Ajtai K, Burghardt TP. Analytical comparison of natural and pharmaceutical ventricular myosin activators. Biochemistry. 2014;53:5298–5306. doi: 10.1021/bi500730t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rohde JA, Thomas DD, Muretta JM. Heart failure drug changes the mechanoenzymology of the cardiac myosin powerstroke. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:E1796–E1804. doi: 10.1073/pnas.1611698114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burchfield JS, Xie M, Hill JA. Pathological ventricular remodeling: Mechanisms: Part 1 of 2. Circulation. 2013;128:388–400. doi: 10.1161/CIRCULATIONAHA.113.001878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sequeira V, Wijnker PJ, Nijenkamp LL, Kuster DW, Najafi A, Witjas-Paalberends ER, Regan JA, Boontje N, Ten Cate FJ, Germans T, Carrier L, Sadayappan S, van Slegtenhorst MA, Zaremba R, Foster DB, Murphy AM, Poggesi C, Dos Remedios C, Stienen GJ, Ho CY, Michels M, van der Velden J. Perturbed length-dependent activation in human hypertrophic cardiomyopathy with missense sarcomeric gene mutations. Circulation research. 2013;112:1491–1505. doi: 10.1161/CIRCRESAHA.111.300436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gresham KS, Mamidi R, Stelzer JE. The contribution of cardiac myosin binding protein-c ser282 phosphorylation to the rate of force generation and in vivo cardiac contractility. The Journal of physiology. 2014;592:3747–3765. doi: 10.1113/jphysiol.2014.276022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mamidi R, Gresham KS, Stelzer JE. Length-dependent changes in contractile dynamics are blunted due to cardiac myosin binding protein-c ablation. Front Physiol. 2014;5:461. doi: 10.3389/fphys.2014.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tong CW, Stelzer JE, Greaser ML, Powers PA, Moss RL. Acceleration of crossbridge kinetics by protein kinase a phosphorylation of cardiac myosin binding protein c modulates cardiac function. Circulation research. 2008;103:974–982. doi: 10.1161/CIRCRESAHA.108.177683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mamidi R, Gresham KS, Li J, Stelzer JE. Cardiac myosin binding protein-c ser302 phosphorylation regulates cardiac beta-adrenergic reserve. Science advances. 2017;3:e1602445. doi: 10.1126/sciadv.1602445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fabiato A. Computer programs for calculating total from specified free or free from specified total ionic concentrations in aqueous solutions containing multiple metals and ligands. Methods Enzymol. 1988;157:378–417. doi: 10.1016/0076-6879(88)57093-3. [DOI] [PubMed] [Google Scholar]
- 23.Godt RE, Lindley BD. Influence of temperature upon contractile activation and isometric force production in mechanically skinned muscle fibers of the frog. The Journal of general physiology. 1982;80:279–297. doi: 10.1085/jgp.80.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mamidi R, Gresham KS, Verma S, Stelzer JE. Cardiac myosin binding protein-c phosphorylation modulates myofilament length-dependent activation. Front Physiol. 2016;7:38. doi: 10.3389/fphys.2016.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swenson AM, Tang W, Blair CA, Fetrow CM, Unrath WC, Previs MJ, Campbell KS, Yengo CM. Omecamtiv mecarbil enhances the duty ratio of human beta-cardiac myosin resulting in increased calcium sensitivity and slowed force development in cardiac muscle. The Journal of biological chemistry. 2017;292:3768–3778. doi: 10.1074/jbc.M116.748780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bates D, Martin M, Ben B, Steve W. Fitting linear-mixed effects model using ime4. Journal of Statistical Software. 2015;67(1):1–48. [Google Scholar]
- 27.Core TR. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2017. [Google Scholar]
- 28.Stelzer JE, Patel JR, Moss RL. Protein kinase a-mediated acceleration of the stretch activation response in murine skinned myocardium is eliminated by ablation of cmybp-c. Circulation research. 2006;99:884–890. doi: 10.1161/01.RES.0000245191.34690.66. [DOI] [PubMed] [Google Scholar]
- 29.Solaro RJ, Moir AJ, Perry SV. Phosphorylation of troponin i and the inotropic effect of adrenaline in the perfused rabbit heart. Nature. 1976;262:615–617. doi: 10.1038/262615a0. [DOI] [PubMed] [Google Scholar]
- 30.Gresham KS, Mamidi R, Li J, Kwak H, Stelzer JE. Sarcomeric protein modification during adrenergic stress enhances cross-bridge kinetics and cardiac output. Journal of applied physiology. 2017;122:520–530. doi: 10.1152/japplphysiol.00306.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, Billingham ME, Harrison DC, Stinson EB. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982;307:205–211. doi: 10.1056/NEJM198207223070401. [DOI] [PubMed] [Google Scholar]
- 32.Bowling N, Walsh RA, Song G, Estridge T, Sandusky GE, Fouts RL, Mintze K, Pickard T, Roden R, Bristow MR, Sabbah HN, Mizrahi JL, Gromo G, King GL, Vlahos CJ. Increased protein kinase c activity and expression of ca2+-sensitive isoforms in the failing human heart. Circulation. 1999;99:384–391. doi: 10.1161/01.cir.99.3.384. [DOI] [PubMed] [Google Scholar]
- 33.Stelzer JE, Patel JR, Moss RL. Acceleration of stretch activation in murine myocardium due to phosphorylation of myosin regulatory light chain. The Journal of general physiology. 2006;128:261–272. doi: 10.1085/jgp.200609547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McNamara JW, Li A, dos Remedios GC, Cooke R. The role of super-relaxed myosin in skeletal and cardiac muscle. Biophysical Reviews. 2015;7:5–14. doi: 10.1007/s12551-014-0151-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Swenson AM, Tang W, Blair CA, Fetrow CM, Unrath WC, Previs MJ, Campbell KS, Yengo CM. Omecamtiv mecarbil enhances the duty ratio of human beta cardiac myosin resulting in increased calcium sensitivity and slowed force development in cardiac muscle. J Biol Chem. 2017;292:3768–3778. doi: 10.1074/jbc.M116.748780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiological reviews. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 37.Razumova MV, Bukatina AE, Campbell KB. Different myofilament nearest-neighbor interactions have distinctive effects on contractile behavior. Biophysical journal. 2000;78:3120–3137. doi: 10.1016/S0006-3495(00)76849-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winkelmann DA, Forgacs E, Miller MT, Stock AM. Structural basis for drug-induced allosteric changes to human beta-cardiac myosin motor activity. Nature communications. 2015;6:7974. doi: 10.1038/ncomms8974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brenner B. Effect of ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: Implications for regulation of muscle contraction. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:3265–3269. doi: 10.1073/pnas.85.9.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanner BC, Breithaupt JJ, Awinda PO. Myosin mgadp release rate decreases at longer sarcomere length to prolong myosin attachment time in skinned rat myocardium. American journal of physiology Heart and circulatory physiology. 2015;309:H2087–2097. doi: 10.1152/ajpheart.00555.2015. [DOI] [PubMed] [Google Scholar]
- 41.Hanft LM, Korte FS, McDonald KS. Cardiac function and modulation of sarcomeric function by length. Cardiovascular research. 2008;77:627–636. doi: 10.1093/cvr/cvm099. [DOI] [PubMed] [Google Scholar]
- 42.Gresham KS, Stelzer JE. The contributions of cardiac myosin binding protein c and troponin i phosphorylation to beta-adrenergic enhancement of in vivo cardiac function. The Journal of physiology. 2016;594:669–686. doi: 10.1113/JP270959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stehle R, Iorga B. Kinetics of cardiac sarcomeric processes and rate-limiting steps in contraction and relaxation. Journal of molecular and cellular cardiology. 2010;48:843–850. doi: 10.1016/j.yjmcc.2009.12.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.