

Abstract
Key points
Endothelial cells employ transient receptor potential isoform 4 (TRPV4) channels to sense ambient mechanical and chemical stimuli.
In retinal microvascular endothelial cells, TRPV4 channels regulate calcium homeostasis, cytoskeletal signalling and the organization of adherens junctional contacts.
Intracellular calcium increases induced by TRPV4 agonists include a significant contribution from calcium release from internal stores.
Activation of TRPV4 channels regulates retinal endothelial barriers in vitro and in vivo.
TRPV4 sensing may provide a feedback mechanism between sensing shear flow and eicosanoid modulators, vascular permeability and contractility at the inner retinal endothelial barrier.
Abstract
The identity of microvascular endothelial (MVE) mechanosensors that sense blood flow in response to mechanical and chemical stimuli and regulate vascular permeability in the retina is unknown. Using immunohistochemistry, calcium imaging, electrophysiology, impedance measurements and vascular permeability assays, we show that the transient receptor potential isoform 4 (TRPV4) plays a major role in Ca2+/cation signalling, cytoskeletal remodelling and barrier function in retinal microvasculature in vitro and in vivo. Human retinal MVE cells (HrMVECs) predominantly expressed Trpv1 and Trpv4 transcripts, and TRPV4 was broadly localized to the plasma membrane of cultured cells and intact blood vessels in the inner retina. Treatment with the selective TRPV4 agonist GSK1016790A (GSK101) activated a nonselective cation current, robustly elevated [Ca2+]i and reversibly increased the permeability of MVEC monolayers. This was associated with disrupted organization of endothelial F‐actin, downregulated expression of occludin and remodelling of adherens contacts consisting of vascular endothelial cadherin (VE‐cadherin) and β‐catenin. In vivo, GSK101 increased the permeability of retinal blood vessels in wild type but not in TRPV4 knockout mice. Agonist‐evoked effects on barrier permeability and cytoskeletal reorganization were antagonized by the selective TRPV4 blocker HC 067047. Human choroidal endothelial cells expressed lower TRPV4 mRNA/protein levels and showed less pronounced agonist‐evoked calcium signals compared to MVECs. These findings indicate a major role for TRPV4 in Ca2+ homeostasis and barrier function in human retinal capillaries and suggest that TRPV4 may differentially contribute to the inner vs. outer blood–retinal barrier function.
Keywords: retinal microvascular endothelial cells, adherens junctions, TRPV4, vascular permeability, calcium
Key points
Endothelial cells employ transient receptor potential isoform 4 (TRPV4) channels to sense ambient mechanical and chemical stimuli.
In retinal microvascular endothelial cells, TRPV4 channels regulate calcium homeostasis, cytoskeletal signalling and the organization of adherens junctional contacts.
Intracellular calcium increases induced by TRPV4 agonists include a significant contribution from calcium release from internal stores.
Activation of TRPV4 channels regulates retinal endothelial barriers in vitro and in vivo.
TRPV4 sensing may provide a feedback mechanism between sensing shear flow and eicosanoid modulators, vascular permeability and contractility at the inner retinal endothelial barrier.
Introduction
Healthy visual function requires tight control of the milieu within which the transduction and transmission of light stimuli take place. The blood–retinal barrier (BRB) consists of two morphologically and functionally distinct microvascular beds that regulate the exchange of water, electrolytes, oxygen and glucose, mediate the drainage of metabolic products and control entry of neurotoxic plasma‐derived proteins, pathogens and immune cells. Choriocapillaries that form the outer barrier are highly fenestrated and permeable, in contrast to the high transendothelial resistance maintained by adherens (AJ) and tight junctions (TJ) between adjacent microvascular endothelial cells (MVECs) that form the inner retinal BRB (Raviola, 1977; Bharadwaj et al. 2013). Permeability of the inner BRB is dynamically regulated through exchange of local signals [such as nitric oxide (NO), epoxyeicosatrienoic acids (EETs), vascular endothelial growth factor (VEGF)] between ECs, pericytes, astrocytes and Müller glial end‐feet. When compromised in chronic retinal diseases such as diabetic retinopathy, glaucoma, uveitis and ischaemia, the decrease in barrier function can significantly augment the pathology (Klaassen et al. 2013). Vascular permeability is believed to be regulated by calcium influx and arachidonic acid metabolites (i.e., EETs, thromboxanes and prostacyclins) (Moore et al. 1998; Brown and Davis, 2002; Komarova and Malik, 2010; Attwell et al. 2010) but neither the sources nor the sinks that control Ca2+ homeostasis in retinal microvasculature have been studied in detail.
Here, we investigate the TRPV4 channel as a source of Ca2+ and regulator of junctional remodelling in human retinal MVECs (HrMVECs). The channel is activated by a remarkably diverse set of physical–chemical stimuli that include cell swelling (Strotmann et al. 2000; Jo et al. 2015; Toft‐Bertelsen et al. 2017), moderate temperature (Güler et al. 2002), shear stress (Köhler et al. 2006) and EETs (Watanabe et al. 2003; Dunn et al. 2013; Ryskamp et al. 2014). TRPV4 has been implicated in the regulation of vascular tone (Saliez et al. 2008; Mendoza et al. 2010; Bagher et al. 2012), endothelial cell orientation, mechanosensing and contractility (Thodeti et al. 2009; Hill‐Eubanks et al. 2014; Suresh et al. 2015) but its role in barrier function is under debate (Darby et al. 2016). The channel was suggested to increase pulmonary vascular resistance (Ke et al. 2015) and strengthen cell–cell junctions (Akazawa et al. 2013) yet excessive TRPV4 activation triggers a circulatory collapse due to the breakdown of microvascular permeability barriers (Willette et al. 2008; Villalta et al. 2014). The inconsistency has been reinforced by studies in Trpv4 knockout mice, which may display protective and pathological phenotypes across different endothelia. An additional confounding aspect is that functional TRPV4 channels can be expressed across multiple cell types that constitute the neurogliovascular unit (Earley et al. 2009; Kim et al. 2016; Redmon et al. 2017) and play different functions across different vascular beds (Maishan et al. 2017). As the first step towards defining the properties of TRPV4 signalling in retinal vascular function, we applied molecular, optical imaging, electrophysiological and vascular permeability assays to study calcium regulation, cytoskeletal organization and barrier permeability in cultured retinal MVECs. We found that TRPV4 activation induces massive influx of Ca2+ that underlies cytoskeletal remodelling, reorganization of AJs together with reversible changes in barrier permeability in vitro, and loss of vascular barrier function in vivo. These findings pinpoint TRPV4 as a potential modulator of activity‐dependent modulation of vascular permeability and blood flow in healthy retinas and a target for pathological remodelling associated with vascular leakage in retinal disease.
Methods
Ethical approval
We acknowledge the ethical principles of The Journal of Physiology and confirm that the protocols were performed within these principles and in accordance with the NIH Guide for the Care and Use of Laboratory Animals, the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and the Institutional Animal Care and Use Committee at the University of Utah. The in vivo experiments were conducted with wild type C57BL/6J (Jackson Laboratory: 000664; Bar Harbor, ME, USA) and Trpv4−/− mice on C57BL/6J background obtained from Dr Wolfgang Liedtke (Duke University, Durham, NC, USA). The mice were reared in a pathogen‐free facility with a 12 h light/dark cycle and ad libitum access to food and water. An abstract containing a portion of this work was published as Ts'o et al. (2017).
Cell culture
HrMVECs were purchased from Cell Systems (Kirkland, WA, USA; ACBRI 181) and grown in human endothelial growth medium (EBM‐2, Lonza, Basel, Switzerland; CC‐3156 & CC4176) at 37°C and 5% CO2. The cells were used between the fourth and seventh passages.
Human retinas
Retinas from cadavers were obtained through the Lions Eye Bank at the Moran Eye Center at the University of Utah, approved to function as a research tissue bank accredited by the Eye Bank Association of America. The experimental procedures associated with procuring human retinas followed the tenets of the Declaration of Helsinki. The eyecups were fixed in 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS), pH 7.4, for 2 h and dehydrated in 30% sucrose in PBS overnight (Ryskamp et al. 2011). The tissue was embedded in OCT (Sakura Finetek, Leiden, The Netherlands) and stored at −80°C. Retinas were vertically sectioned at 12 μm, collected onto Superfrost slides and kept at −20°C until use.
Chemicals
The following drugs were used: GSK1016790A (hereafter GSK101, Millipore/Sigma, Billerica, MA, USA/St Louis, MO, USA) and HC067047 (hereafter HC‐06, Sigma). Typically, GSK101 and HC‐06 were aliquoted to 1 and 5 mm stocks in dimethyl sulfoxide (DMSO). Other chemicals used in this study were purchased from Sigma.
Immunoblotting
As previously described (Phuong et al. 2013; Ryskamp et al. 2016), total protein was extracted from primary cultured HrMVECs in RIPA buffer supplemented with the inhibition cocktail (Biotechnology, Inc., Santa Cruz, CA, USA). Protein concentration was estimated with the Bradford assay. Total protein was heat‐inactivated for 3 min at 95°C in Laemmli buffer (Bio‐Rad Laboratories, Hercules, CA, USA). Thirty micrograms of total protein per lane was loaded in 8% mini polyacrylamide gels. Electrophoresis was performed at 90 V for 1 h in the Mini‐PROTEAN Tetra apparatus (Bio‐Rad; running buffer 25 mm Tris, 195 mm glycine, 0.1% SDS). Proteins were transferred to a polyvinylidene fluoride membrane (PVDF, 0.2 μm, Bio‐Rad Laboratories) in transfer buffer (25 mm Tris, 195 mm glycine, 20% methanol) overnight at 4°C at 80 mV. The membrane was blocked with 5% skimmed milk for 30 min and incubated overnight with an anti‐TRPV4 antibody (1:2000, Lifespan Biosciences, Providence, RI, USA), anti‐occludin antibody (1:1000), anti‐VE‐cadherin (1:2000) (gifts from Dr E. M. Hartnett, University of Utah) or anti‐β‐catenin antibody (1:1000, Santa Cruz Biotechnology, Dallas, TX, USA). PVDF membranes were washed with TBS‐T solution for 3 × 5 min. Control membranes were incubated with anti‐rabbit‐HRP (1:2000, Santa Cruz Biotechnology) or GAPDH (1:5000, Abcam, Cambridge, MA, USA) antibodies for 2 h. Protein bands were detected on X‐ray film by using enhanced chemiluminescence (ECL; Pierce Biotechnology, Inc., Rockford, IL, USA) and developer (AFP Imaging Corp.).
Reverse transcription‐PCR amplification
RT‐PCR in HrMVECs was performed as described (Jo et al. 2015; Ryskamp et al. 2016). mRNA was isolated with TRI Reagent (Sigma) and total RNA concentrations (2 μg) were converted to cDNA using the qScript XLT cDNA Supermix (Quanta). Primers for human Trpv1‐4, Pecam‐1 were designed by using Primer‐BLAST program (NCBI) (Table 1). Semi‐quantitative PCR was performed with Hottaq polymerase (Thermo Scientific, Waltham, MA, USA) using the following protocol: 95°C for 2 min; 35 cycles of 95°C for 15 s, 60°C for 30 s and 72°C for 20 s; 72°C for 5 min. After amplification, the PCR product was loaded into a 2% agarose gel and run at 80 V for 30 min. The DNA band(s) were visualized under UV light using a ChemiDoc apparatus (Bio‐Rad).
Table 1.
List of primers
| Primer | Sequence (5′ to 3′) | PCR product (bp) | NCBI reference sequence |
|---|---|---|---|
| Trpv1 sense | GCCCAGCATGTTCCCAAATC | 169 | NM_080704.3 |
| Trpv1 antisense | TGTCCCAGTAGAGACTGACCA | ||
| Trpv2 sense | GAGGAGGTGAACTGGGCTTC | 87 | XM_005256676.1 |
| Trpv2 antisense | CTCGAGAGTTCGAGGGACAC | ||
| Trpv3 sense | CCTTTTCTCCGGTGGGGATG | 294 | NM_145068.3 |
| Trpv3 antisense | GCTTTCATGGCTGGTGAGGT | ||
| Trpv4 sense | TCCCATTCTTGCTGACCCAC | 217 | XM_011538636.2 |
| Trpv4 antisense | AGGGCTGTCTGACCTCGATA | ||
| Vegf‐a sense | CTCATGGACGGGTGAGGC | 265 | NM_001025368.2 |
| Vegf‐a antisense | CTGGGACCACTTGGCATGG | ||
| Pecam‐1 sense | GAAATGTCCAGGCCAGCAGT | 277 | XM_011524890.1 |
| Pecam‐1 antisense | ATCTGCTTTCCACGGCATCA | ||
| RhoA sense | GCGCTTTTGGGTACATGGAG | 100 | NM_001664.3 |
| RhoA antisense | TTCCCACGTCTAGCTTGCAG | ||
| Rock1 sense | CTTGAACTTCAGTGCCCTCAC | 139 | NM_005406.2 |
| Rock1 antisense | TCCCATTCCCCAAACTTGCT |
Immunofluorescence staining and quantification of junctional overlap
Cultured cells were labelled as described (Ryskamp et al. 2016); protocols for staining human retinal sections were adapted from Rentería et al. (2005). HrMVECs were plated on glass coverslips in 24 well plates and grown to 100% confluent monolayers (i.e. 5–7 days). The growth medium was changed every 2 days. AJ/TJ proteins were evaluated following treatment with GSK101 (5 nm), HC‐06 (5 μm) or GSK101 + HC‐06 for 30 min at 37°C. Samples were rinsed with three times in PBS and fixed with 4% paraformaldehyde for 10 min at room temperature. Samples were blocked for 1 h at room temperature with 5% fetal bovine serum and 0.3% Triton‐X. Primary antibodies in the dilution buffer (2% bovine serum albumin, 0.3 % Triton‐X, PBS) were added to plate wells overnight at 4°C. The primary antibodies as given in Table 2 were used.
Table 2.
List of antibodies
| Host species, primary antibodt | Concentration | Company |
|---|---|---|
| Rabbit, TRPV4 | 1:2000 | LS‐A8583‐50; Lifespan Biosciences, Seattle, WA, USA |
| Rabbit, VE‐cadherin | 1:500 | Sc‐28644; Santa Cruz Biotechnology; Santa Cruz, CA, USA; gift from M. E. Hartnett, University of Utah |
| Rabbit, Occludin | 1:1000 | Sc‐5562; Santa Cruz Biotechnology; gift from M. E. Hartnett, University of Utah |
| Mouse, β‐catenin | 1:1000 | Sc‐7199; Santa Cruz Biotechnology |
| Rabbit, horseradish peroxidase | 1:2000 | Sc‐2004; Santa Cruz Biotechnology |
| Mouse, GAPDH | 1:5000 | ab185059; Abcam, Cambridge, MA, USA |
The samples were washed three times for 5 min each with PBS and incubated with secondary antibodies: anti‐rabbit Alexa Fluor‐488, anti‐rabbit Alexa Fluor‐594, anti‐mouse Alexa Fluor‐488, anti‐mouse Alexa Fluor‐594 (all 1:1000, Life Technologies Inc., Carlsbad, CA, USA) or phalloidin‐actin conjugated to Alexa Fluor‐488 (1:1000). Vasculature in sections from human retinas was labelled with Dylight 594‐conjugated tomato (Lycopersicum esculentum) lectin (Vector, DL‐1177). Images were taken using an Olympus confocal microscope (C1100) and collected through a 40× lens (0.8 NA). Cell–cell junctional area overlaps were calculated as described (Seebach et al. 2007).
Calcium imaging
HrMVECs were seeded on glass coverslips for 48 h, loaded with Fura‐2AM (Invitrogen, Carlsbad, CA, USA) for 40–60 min, and washed with the bath solution containing (in mm): 140 NaCl, 4.7 KCl, 1.2 MgCl2, 5.6 glucose, 10 HEPES, 1.8 CaCl2 (pH 7.4, osmolarity 295–300 mOsm/L) for 5–30 min. The imaging followed recently published protocols (Ryskamp et al. 2014; Molnar et al. 2016). Excitation for 340 and 380 nm filters (Semrock, Rochester, NY, USA) was delivered by a liquid light guide from a 150 W Xenon arc lamp (DG4, Sutter Instruments, Novato, CA, USA). Fluorescence emission was high pass‐filtered at 510 nm and captured with a cooled digital CCD camera binned at 3 × 3 or 4 × 4 (Photometrics, Novato, CA, USA). Data acquisition and F 340/F 380 ratio calculations were performed by NIS Elements 3.22 (Nikon), typically on circular regions of interest encompassing the endothelial cell nucleus. Background fluorescence was measured in similarly sized regions of interest in neighbouring areas devoid of cells, and subtracted. Data in a subset of experiments are presented as background‐subtracted 340/380 nm ratios.
Electrophysiology
Membrane properties of HrMVECs were investigated as described (Ryskamp et al. 2016). Cells were plated on glass coverslips 48 h prior to the experiments. Patch pipettes were pulled from borosilicate glass on a P‐2000 micropipette puller (Sutter Instruments, Novato, CA, USA). Membrane properties were assessed using whole‐cell patch clamp by applying ramp voltage commands (−100 to +100 mV relative to the holding potential for 1 s), at a frequency of 0.2 Hz. Resting potentials were recorded in the current‐clamp mode and at zero holding current in voltage‐clamp mode. Whole‐cell currents were monitored using an Axon Multiclamp 700B and the Digidata 1550 (Molecular Devices, Sunnyvale, CA, USA) AD converter, and digitized at 10 kHz with a low pass 5 kHz Bessel filter. If not otherwise stated, the standard bath recording solution contained (in mm): 150 NaCl, 3 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES and 5.5 d‐glucose (pH was adjusted with NaOH to 7.4). The internal solution contained (in mm): 125 potassium gluconate, 10 KCl, 1 MgCl2, 4 Mg‐ATP, 0.6 Na‐GTP, 5 BAPTA (caesium salt), 10 HEPES (pH 7.3 was adjusted with KOH). The filled‐tip resistance was between 7 and 10 MΩ. All experiments were performed at room temperature (22–24°C).
Electric cell substrate impedance sensing
The permeability of HrMVECs was measured as described (Gibson et al. 2015). In brief, HrMVECs were seeded in complete medium on fibronectin‐coated 96 well electric cell substrate impedance sensing culture plate (96W10E+, Applied Biophysics, Troy, NY, USA) at a density of 4 × 104 cells per well. Cells were monitored until a stable monolayer formed with 100% confluence. For permeability measurements, cells were treated with 0.5% DMSO (positive control), 2.5, 5.0 or 10.0 nm GSK101 in the presence/absence of the selective antagonist HC‐06 (5 μm). Impedance was normalized for each well before treatment, and plotted in real time across 24 h as a ‘Normalized Cell Index’ (Sun et al. 2012; Ziegler et al. 2016)
ELISA
Cells (104 per well, six‐well plates) were cultured for 5 days to form monolayers. After 3–4 washes with growth medium EBM‐2 lacking culture supplement, 300 μl of GSK101 (final concentration range 1–5 nm) or HC‐06 (5 μm) was added for 30 min; the vehicle DMSO (0.1% in medium without supplement) was added to control wells. The supernatant was harvested into 1.5 ml tubes, centrifuged at 12,000 r.p.m. for 3 min and transferred to tubes containing protease inhibitors. VEGF was measured with a commercial ELISA kit from R & D Systems (Minneapolis, MN, USA).
In vivo vascular leakage assay
The in vivo dye leakage assay was performed using Evans Blue, a tetrasodium diazo dye that binds plasma albumin in a 10:1 ratio. The method takes advantage of extravasation of plasma from leaking blood vessels into the surrounding tissue, where it is quantified. Its sensitivity in studies of BRB breakdown is comparable to radioisotope measurements (Xu et al. 2001; Jones et al. 2008). C57BL/6J WT and Trpv4−/− mice on the C57BL/6J background were anaesthetized by intraperitoneal injection of ketamine/xylazine, followed by intravitreal injection of 1.5 μl of 5 μm GSK101 into the ipsilateral eye; the contralateral eye received 1.5 μl of the vehicle (PBS). After 5–10 min, the anaesthetized animals were injected with 50–100 μl of 60 mg ml−1 Evans Blue into the femoral vein. Two hours after the dye injection, the animals were killed via isoflurane overexposure (AB‐1 box, Braintreee Scientific, Inc., Braintree, MA, USA). Another cohort of WT/KO animals was anaesthetized with ketamine/xylazine and injected with GSK101/PBS. After 24 h, these mice were anaesthetized with ketamine/xylazine for Evans Blue injection, and then killed by isoflurane overexposure. The dye accumulated in the retina was eluted overnight in 200 μl of formamide solution in 1.5 ml tubes at 60°C. Samples were centrifuged at 14,000 r.p.m. for 5 min. One hundred microlitre samples of the eluted Evans Blue in formamide solution was measured using the Modulus II Microplate Multimode Reader (Turner BioSystems, Sunnyvale, CA, USA) at 620 nm excitation and 680 nm emission; formamide was used as the blank control. The absorbance signal from GSK101‐injected eyes was normalized to that of the contralateral PBS‐injected eye.
Analysis
Data expressed as means ± SEM were analysed by Student's t test. Values of P < 0.05 were considered to be significant.
Results
TRPV4 is expressed in HrMVECs
To gain insight into the role of TRPV4 channels in HrMVECs we analysed the expression of cognate vanilloid Trp mRNAs together with the endothelial‐specific transcript marker Pecam‐1 (CD31). The predominant transcripts were encoded by Trpv1 and Trpv4 genes whereas the levels of Trpv2 and Trpv3 were below the resolution of the RT‐PCR assay (Fig. 1 A, number of independent experiments (N) = 3). An anti‐TRPV4 antibody, validated in kidney and ocular Trpv4−/− preparations (Ryskamp et al. 2011; Jo et al. 2016), yielded the expected protein bands at ∼90 and ∼98 kDa, with human choroidal endothelial cells (HCECs) showing lower protein levels compared to HrMVECs (Fig. 1 B, N = 3). The TRPV4 antibody (Fig. 1 Ca) and Trpv4:eGFP constructs (Fig. 1 Cb) labelled the HrMVEC plasma membrane in subconfluent MVECs as indicated by colocalization with wheat germ agglutinin (WGA), a marker of glycosylated membrane proteins. The localization of Trpv4:eGFP signals in the cytosol matched the staining with the TRPV4 antibody; cytosolic expression presumably reflects low PACSIN3 (protein kinase C and casein kinase substrate in neurons 3) levels that are typical of cultured cells (Cuajungco et al. 2006).
Figure 1. TRPV4 is expressed in retinal microvascular cells.
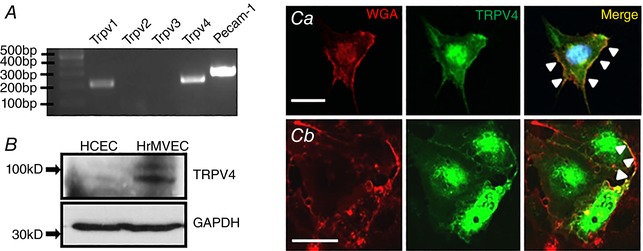
A, RT‐PCR. Trpv1, Trpv4 and Pecam‐1 but not Trpv2 & 3 transcripts are expressed in HrMVECs. B, Western blot. TRPV4 protein expression is prominent in HrMVECs but weak in HCECs. C, HrMVEC labelled by anti‐TRPV4 antibody and a plasma membrane marker (WGA; wheat germ agglutinin) (Ca). The TRPV4 signal within the plasmalemma is marked by arrowheads. Transfection with the Trpv4:eGfp construct (Cb). While TRPV4 expression is predominantly cytosolic, the TRPV4‐ir signal colocalizes with WGA at the plasma membrane (arrowheads). Scale bar = 50 μm. [Color figure can be viewed at wileyonlinelibrary.com]
We labelled human retinal sections for TRPV4, the blood vessel marker DyLight 594‐Tomato Lectin and the Müller cell marker glutamine synthetase (GS). Similar to TRPV4 immunolocalization in the mouse (Ryskamp et al. 2011, 2014; Jo et al. 2015), human retinas showed TRPV4 immunoreactivity (ir) in the retinal ganglion cell layer and presumed retinal ganglion cells (RGCs; arrowheads in Fig. 2 A), and in GS‐labelled Müller glial processes and endfeet (arrowheads in Fig. 2 B). Inner retinal microvasculature marked by the lectin was double‐labelled by the TRPV4 antibody (arrows in Fig 2), with TRPV4‐ir abolished by preadsorption with the immunizing peptide (Fig. 2 C). These data show that (i) TRPV4 localization in the human retina mirrors its expression in rodent retinas and (ii) both cultured MVECs and intact blood vessels express TRPV4.
Figure 2. Vertical section from the human retina.
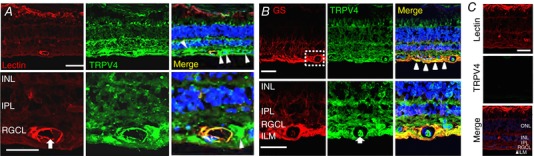
A, TRPV4 colocalizes with blood vessel marker tomato lectin (TL). The lumen of the blood vessel is marked by the arrow. Putative RGCs are labelled by arrowheads. Scale bar = 20 μm. B, double labelling for TRPV4 and glutamine synthetase (GS); the two proteins colocalize in Müller end‐feet (arrowheads) and MVECs (arrow). Scale bar = 50 μm. C, TRPV4‐ir is reduced in retinas pre‐incubated with a blocking antibody. Scale bar = 20 μm. ILM, inner limiting membrane; RGCL, retinal ganglion cell layer; IPL, inner plexiform layer; INK, inner nuclear layer. [Color figure can be viewed at wileyonlinelibrary.com]
TRPV4 is functionally expressed in human MVECs
The functional expression of TRPV4 channels was assessed using electrophysiology and optical imaging. HrMVECs were voltage‐clamped at ±100 mV and stimulated with the selective TRPV4 agonist GSK101 (3 nm). GSK101‐evoked transmembrane currents were characterized by slow onset and peak average amplitudes of −327 ± 109 pA (holding potential V h = −100 mV) and 685 ± 230 pA (V h = +100 mV) that relaxed towards a sustained plateau (circles in Fig. 3 A). Currents induced at positive and negative holding potentials were abolished by the co‐application of HC‐06 (triangles in Fig. 3 A). When the membrane potential was ramped between ±100 mV, the GSK101‐dependent conductance exhibited modest outward rectification, reversal potential of 9.9 ± 7.1 mV (number of the animals used (n) = 10) and inhibition by HC‐06 (Fig. 3 B–D), an I–V signature that is typical of ‘canonical’ TRPV4 currents (Redmon et al. 2017). The average resting membrane potential measured under current clamp conditions was −15.80 ± 0.97 mV whereas GSK101 depolarized the cells to 5.0 ± 1.3 mV (Fig. 3 E and F).
Figure 3. TRPV4 is functionally expressed in HrMVECs, and associated with non‐selective cation conductance.
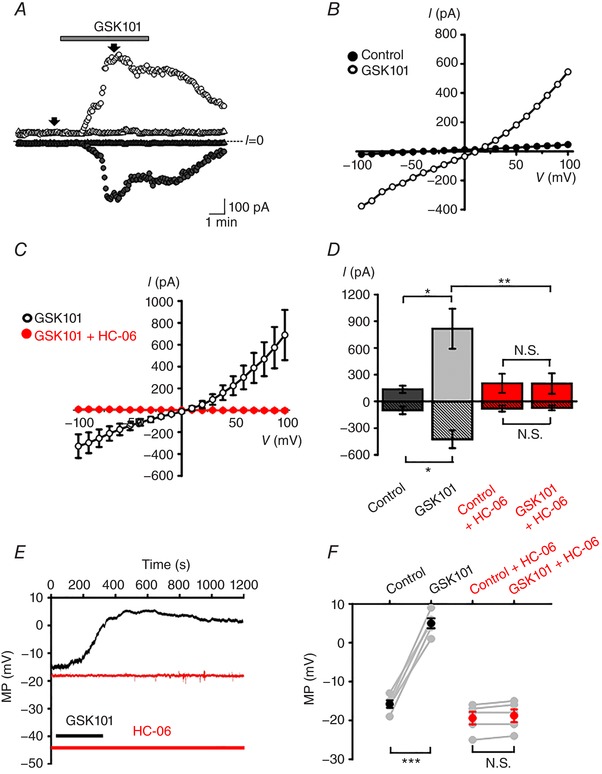
A, voltage clamp. Representative time courses of whole cell currents in control (circles) and HC‐06 (5 μm)‐treated (triangles) cells held at −100 mV (filled symbols) and +100 mV (open symbols). HC‐06 was applied 3 min prior to the stimulation with GSK101 (3 nm). Arrows indicate time points where the current amplitude values were taken for analysis. B, conductances evoked by ramping the holding potential from −100 to +100 mV. The I–V relationship for the cell shown in A was generated by subtracting controls from GSK101‐evoked responses. C, averaged I–V curves from control (black trace, n = 10 cells) and HC‐06‐treated (red trace, n = 10) cells. D, summary of amplitude values derived from I–V curves at −100 mV (stippled bars) and 100 mV (open bars). Shown are the mean ± SEM values. * P < 0.05; ** P < 0.01; N.S. P > 0.05; paired t test. E, current clamp. Representative membrane potential (MP) responses to GSK101 in control untreated (black) and HC‐06 treated (red) cells. F, summary for the depolarizing effect of GSK101 on the membrane potential (MP) in control (*** P < 0.001, paired t test, n = 5 cells) and HC‐06‐treated (N.S., P > 0.05, paired t test, n = 5) cells. [Color figure can be viewed at wileyonlinelibrary.com]
Given that TRPV4 is a nonselective cation channel with a slight preference for calcium (P Ca/P Na = 6–10; Redmon et al. 2017), we evaluated the changes in calcium homeostasis associated with its activation in MVECs. Cells were loaded with the Ca2+ indicator Fura‐2AM and stimulated with GSK101 (1–10 nm). The agonist elevated the 340/380 nm ratio with an EC50 of 1.58 ± 0.72 nm; at 1 nm, it elevated the 340/380 nm ratio from the baseline of 1.10 ± 0.02 to 2.31 ± 0.04 (n = 152, N = 3; P < 0.0001) (Fig. 4 A and B). Similar [Ca2+]i increases were observed in response to 4ɑ‐PDD, a chemically distinct TRPV4 agonist (Fig. 4 C). Confirming selectivity, GSK101‐ and 4ɑ‐PDD‐evoked signals were abolished by the selective antagonist HC‐06 (5 μm) and by the nonselective TRP antagonist Ruthenium Red (RR) (Fig. 4 C and D). Both the number of GSK101‐responding cells and the peak amplitudes of [Ca2+]i signals were significantly lower in HCECs compared to MVECs (Fig. 4 E and F).
Figure 4. TRPV4 activation elevates [Ca2+]i in HrMVECs.
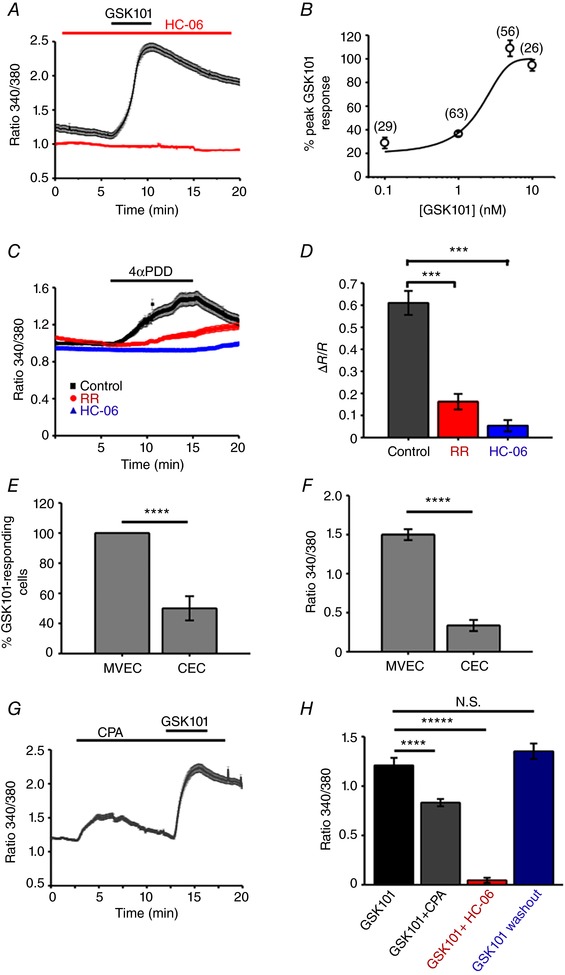
A, 340/380 Fura‐2 ratio signals. GSK101 (1 nm) elevates [Ca2+]i in a representative experiment (n = 12 MVECs), an effect that is inhibited by HC‐06 (5 μm). B, the dose–response curve for the GSC101‐induced [Ca2+]i increases. Shown in parentheses are the numbers of studied cells. C, Ruthenium Red (RR) and HC‐06 inhibit 4α‐PDD‐mediated rise in [Ca2+]i. ±SEM show the inter‐cell variability at each time point in this representative experiment. D, summary of RR and HC‐06 effects on 4α‐PDD‐induced [Ca2+]i elevations. Mean ± SEM. *** P < 0.001, n = 102, n = 61 and n = 53 cells for control, RR and HC‐06, respectively; paired t test. E, a subset of HCECs responded to GSK101 (25 nm); the number of CEC responders (37.5 ± 10%) is markedly lower compared to HrMVECs responders (∼100%) to a much lower (1 nm) GSK101 concentration (P < 0.001). F, peak amplitude GSK101‐evoked ΔR/R [Ca2+]i signals in HrMVECs are significantly larger than in HCECs (P < 0.001). G, 340/380 ratio. CPA (10 μm) elevates [Ca2+]i and reduces the absolute amplitude of GSK101‐evoked [Ca2+]i elevations. H, ΔR/R responses. Summary of the fluorescence measurements in cells stimulated with GSK101, GSK101 + CPA and GSK101 + HC‐06 (N ≥ 3 independent experiments per condition). [Color figure can be viewed at wileyonlinelibrary.com]
The peak amplitude of TRPV4‐mediated signals was attenuated by cyclopiazonic acid (CPA; 10 μm). Applied alone, CPA transiently increased [Ca2+]i due to the blockade of sarcoplasmic‐endoplasmic ATPases (SERCAs) (Križaj et al. 2003). When cells were stimulated with GSK101 in the presence of CPA, the average ΔR/R responses were reduced from 1.21 ± 0.077 in control cells to 0.83 ± 0.036 (n = 95, N = 3; paired t test, P < 0.00001) (∼31% decrease; Fig. 4 G and H). These results suggest that TRPV4 activation in HrMVECs is positively coupled to calcium‐induced calcium release and that its role in Ca2+ homeostasis may differ in inner and outer retinal microvascular beds.
TRPV4 regulates the permeability of HrMVEC monolayers
To test whether [Ca2+]i increases evoked by TRPV4 activation modulate the retinal microvascular endothelial barrier we used the ACEA xCELLigence system to measure the paracellular current flow across HrMVEC monolayers (Sun et al. 2012; Gibson et al. 2015). GSK101 triggered time‐ and concentration‐dependent reductions in monolayer impedance (Fig. 5). The ‘normalized monolayer impedance’ in the presence of 2.5, 5 and 10 nm GSK101 showed 8.0 ± 1.27, 80.4 ± 1.6 and 93.8 ± 0.45% decreases, respectively (n = 3 per concentration; n ≥ 3 wells per concentration per experiment). The effects of 2.5–5 nm GSK101 were fully reversible, returning to control levels within 1–3 h of exposure whereas monolayers stimulated with 10 nm GSK101 did not show complete recovery for up to 15 h after exposure. Applied alone, HC‐06 (5 μm) did not affect the monolayer permeability, although it inhibited the GSK101‐induced effects (P < 0.00001) (Fig. 5 D). These results demonstrate that TRPV4 activation dramatically increases the permeability of retinal microvascular endothelial monolayers.
Figure 5. TRPV4 stimulation rapidly and dramatically decreases the impedance of HrMVEC monolayers and increases the in vivo permeability of retinal blood vessels.
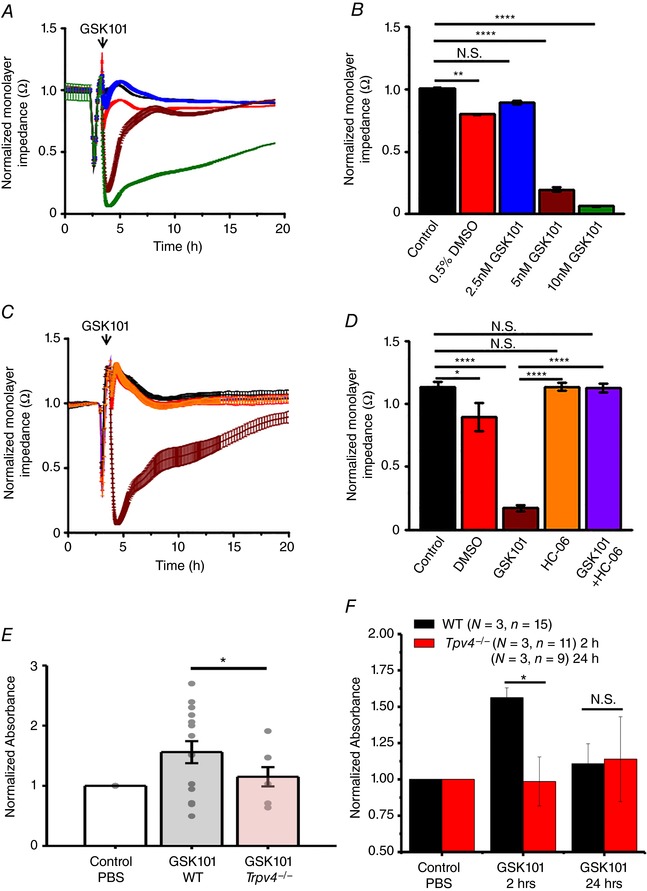
The ‘normalized monolayer impedance’ is derived by dividing the impedance value by the value at a reference time point. A, 2.5 nm (red), 5 nm (brown) and 10 nm (green trace) GSK101 dose‐dependently decrease the resistance of HrMVEC monolayers. B, cumulative data for the experiments shown in A (N = 3). C, HC‐06 (orange) inhibits 10 nm GSK101‐induced decreases in monolayer resistance (brown trace) (N = 3). D, averaged data from C. * P < 0.05, ** P < 0.001, **** P < 0.00001, paired t test. E, systemic injection of GSK101 increases the retinal extravasation of Evans Blue 2 h after dye injection, indicated by increased absorbance signal in WT (grey bar) compared to Trpv4−/− retinas (red bar). The dots represent individual retinas; values are normalized to control values of dye‐exposed retinas in the absence of the TRPV4 agonist. Retinal extravasation was measured 2 and 24 h after GSK101 treatment. F, cumulative data for three independent experiments from WT (black bars) and Trpv4−/− retinas measured 2 and 24 h after GSK101 treatment. * P = 0.033; N.S., P > 0.05; one‐way ANOVA. Mean ± SEM. [Color figure can be viewed at wileyonlinelibrary.com]
VEGF is a potent regulator of retinal barrier integrity and breakdown (Kevil et al. 1998; Miyamoto et al. 2000). We tested whether the TRPV4‐dependent increase in MVEC monolayer barrier permeability is associated with changes in VEGF release. Cells were cultured for 5 days, exposed to GSK101, HC‐06 and GSK101+HC‐06 for 30 min and the concentration of VEGF was determined using a commercial ELISA kit. Neither exposure to GSK101 (1, 2.5 and 5 nm) nor exposure to HC‐06 (5 μm) changed the amount of VEGF released into the supernatant when compared to the vehicle‐exposed control (P > 0.05; N = 3 independent experiments) (data not shown).
TRPV4 agonist induced retinal blood vessel permeability in vivo
Disruption of the endothelial barrier may result in increased permeability and vascular leakage. To determine if endothelial junctional remodelling caused by TRPV4 overactivation is associated with in vivo permeability changes, we conducted the standard vascular leakage assay based on Evans blue, an albumin‐binding azo dye (Jones et al. 2008). The lateral tail vein of WT and Trpv4−/− mice was injected with the dye, 5 min after intravitreal injection of GSK101 and spectrometric analysis of the retinal dye content. In 3/4 experiments, the TRPV4 agonist increased the vascular leakage in WT (N = 15 retinas) compared to Trpv4−/− retinas (N = 11), with an average increase of ∼30% (1.56 ± 0.068 vs. 1.10 ± 0.14; P = 0.033). The effect was detectable 2 h after injection but was not observed at 24 h (N = 9) (Fig. 5 E and F). Thus, TRPV4 activation may be sufficient to increase retinal vascular permeability.
TRPV4 activation induces remodelling of AJ and TJ proteins in HrMVECs
We next tested the hypothesis that TRPV4‐induced modulation of endothelial permeability involves the readjustment of intercellular contacts maintained by AJs (zonulae adherentes) and TJs (zonulae occludentes). The former are composed of vascular endothelial cadherin (VE‐cadherin), which binds to actin, extracellular matrix and accessory intracellular catenin proteins in a Ca2+‐dependent manner (Giampietro et al. 2012). VE‐cadherin and β‐catenin form zigzag‐like structures within overlapping cell–cell domains in confluent and non‐confluent monolayers (Fig. 6 A and B). Exposure to GSK101 ‘smoothened’ the tortuous distribution of both AJ proteins and decreased the area overlap (Fig. 6 A and B) without affecting the total expression of TRPV4 (Fig. 6 E). Quantification of GSK101‐induced changes in the junctional contact area showed ∼32.1% reduction in β‐catenin and ∼25.6% reduction in VE‐cadherin colocalization (P < 0.001) (Fig. 6 Ci, ii) whereas HC‐06‐treated cohorts and cells treated with HC‐06 + GSK101 showed little junctional remodelling (Fig. 6). TJs are another important regulator of endothelial barrier permeability (Kevil et al. 1998; Brown and Davis, 2002). GSK101 treatment triggered an ∼40% decrease in protein levels of occludin (P < 0.05), an integral component of TJs, and this effect that was rescued by the TRPV4 antagonist (Fig. 6 D and E). These data suggest that TRPV4 activation drives junctional remodelling through redistribution of VE‐cadherin and β‐catenin and downregulation of occludin.
Figure 6. TRPV4 agonist triggers reorganization of cell‐cell junctions.
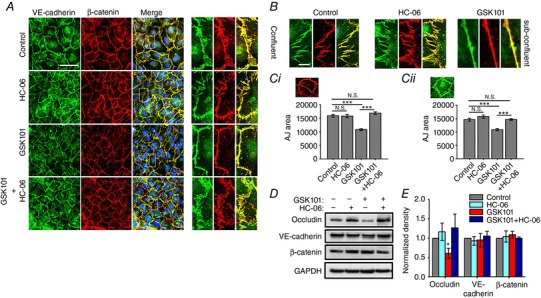
A and B, VE‐cadherin‐ir (green) and β‐catenin‐ir (red) in confluent (A) and sub‐confluent cells (B) in the presence of GSK101, HC‐06 and GSK101/HC‐06. Merged images in A include DAPI staining (blue). Scale bar = 50 μm. The panels on the right show high‐resolution images. Scale bar = 10 μm. C, summary of the effects of TRPV4 agonist/antagonist on AJ overlap. Overlap was quantified by tracing the β‐catenin‐ir boundary (Ca) in adjacent cells. Quantification of VE‐cadherin‐ir areas (Cb). Paired t test, N = 3 independent experiments. D, western blots. Protein levels of occludin, but not VE‐cadherin and β‐catenin are lowered following exposure to GSK101 (5 nm). E, relative protein density from experiments shown in D, normalized to control, *** P < 0.0001 (N = 3 independent experiments). [Color figure can be viewed at wileyonlinelibrary.com]
TRPV4 modulates F‐actin filament distribution in HrMVECs
VE‐cadherin, β‐catenin and occludin localization are regulated by the actin cytoskeleton (Wheelock & Johnson, 2003; Ratheesh & Yap, 2012), which also binds the Ser824 residue of TRPV4 (Fan et al. 2009; Shin et al. 2012). To determine whether the TRPV4‐dependent reorganization of AJ, downregulation of TJ protein and increased permeability of HrMVEC monolayers correlate with actin remodelling, we labelled the cells with phalloidin:actin in the presence of GSK101, HC‐06, GSK101+HC‐06 or the vehicle (DMSO). The TRPV4 agonist disrupted cortical F‐actin area overlap (Fig. 7 A), an effect that was blocked by the pre‐treatment with HC‐06. We visualized the remodelling in real time in cells transfected with F‐actin:mApple constructs. As shown (Fig. 7 B and Supporting Information, Video S1) for Fura‐2‐loaded cells simultaneously imaged for actin and [Ca2+]i, GSK101‐induced actin loss from the cell cortex was followed by a gradual accumulation of the actin probe within the perinuclear region (Fig. 7 B) and was associated with a massive increase in cell [Ca2+]i (Fig. 4 A). These results demonstrate that TRPV4‐mediated Ca2+ influx modulates the organization of the MVEC cytoskeleton.
Figure 7. TRPV4‐dependent remodelling of F‐actin and AJs in confluent HrMVECs.
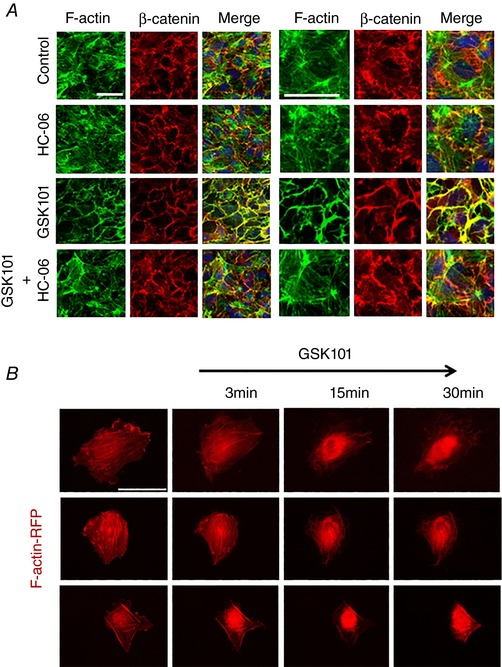
A, double labelling for β‐catenin and F‐actin. Combined images include DAPI staining (blue). GSK101‐induced disruption of central and cortical F‐actin was rescued by the TRPV4 antagonist (HC‐06). Scale bar = 50 μm. B, red fluorescent protein (RFP)‐actin‐transfected HrMVEC cells exposed to GSK101 (5 nm) show time‐dependent redistribution of F‐actin towards the cell nucleus. Scale bar = 50 μm. [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
The results of the present study show that TRPV4 regulates Ca2+ homeostasis, cytoskeletal remodelling and junctional permeability of retinal MVECs. TRPV4 agonists increased the in vitro and in vivo permeability of MVEC barriers together with increases in [Ca2+]i, disassembly of VE‐cadherin:β‐catenin‐containing junctions and reorganization of the actin cytoskeleton. Together, our data suggest that TRPV4 functions as a polymodal sensor of physical–chemical stimuli that dynamically modulate the permeability of the inner retinal endothelial barrier.
HrMVECs and blood vessels from donor retinas express TRPV4 mRNA and protein, with the two molecular weight bands in protein lysates indicating the possible presence of glycosylated and non‐glycosylated variants (Narita et al. 2015), membrane vs. intracellular pools (Vriens et al. 2005) and/or long vs. short forms of the protein (Strotmann et al. 2000). Consistent with other endothelia (Earley & Brayden, 2015), transcript analysis shows TRPV4 to be the dominant vanilloid isoform in rMVECs, which also express TRPV1 but lack TRPV2/TRPV3. Functional expression was confirmed by GSK101‐induced transmembrane currents that exhibited the ‘canonical’ signature associated with the eicosanoid signalling pathway: outward rectification, reversal at ∼0 mV and delayed onset‐to‐peak and a sustained plateau phase (Loot et al. 2008; White et al. 2016; Redmon et al. 2017). While slower from the millisecond response times reported in bovine capillary endothelial cells in which the channel may be directly sensitive to mechanical forces (Matthews et al. 2010), the rMVEC response to the TRPV4 agonist mirrors metabotropic activation of the canonical phospholipase A2/CYP450 pathway reported for the lung, kidney, cardiac and CNS endothelial barriers (Hartmannsgruber et al. 2007; Marrelli et al. 2007; Loot et al. 2008). The potency of GSK101 to induce increases in [Ca2+]i and barrier permeability in HrMVEC monolayers (EC50 ∼1.6 nm) was close to previously reported affinities of mouse and human TRPV4 channels (EC50 3–20 nm; Thorneloe et al. 2008; Willette et al. 2008), with amplitudes that exceeded the signals in other TRPV4‐expressing ocular cells by ∼10‐fold (Ryskamp et al. 2014, 2016; Jo et al. 2016). Ca2+ release from internal stores contributed ∼30% of the overall response amplitude, a relatively modest proportion compared to the ∼75% Ca2+ store component reported in bovine MVECs (Monaghan et al. 2015). HC‐06 alone had no effect on Ca2+ homeostasis, suggesting that TRPV4 and/or TRPV4–TRPC1 heteromers (Ma et al. 2011) did not contribute to baseline [Ca2+]i.
The ∼30% increase in Evans Blue extravasation in WT retinas exposed to GSK101 in vivo is congruent with the increased barrier permeability, cytoskeletal and Ca2+ signalling effects of the agonist in vitro and also with the systemic effects of GSK101 (EC retraction, vascular leakage, loss of vessel tone and fatal circulatory collapse) reported in pulmonary endothelia (Willette et al. 2008; Sokabe & Tominaga, 2010; Villalta et al. 2014). The dramatic yet reversible decreases in the microvascular transendothelial resistance induced by the selective agonist show that activation of the TRPV4 channel is sufficient to dynamically regulate the permeability of the endothelial component of the BRB. The resistance decrease was correlated with VEGF‐independent remodelling of cortical F‐actin, redistribution of VE‐cadherin from interdigitated processes between adjacent cells to a smoother membrane surface, dislocation of β‐catenin and decreased expression of occludin. We hypothesize that Ca2+ influx controls local actin remodelling (Shin et al. 2012; Ryskamp et al. 2016), organization of focal adhesions (Thodeti et al. 2009) and bonding between VE‐cadherin and β‐catenin (Brown & Davis, 2002; Giampietro et al. 2012; Giannotta et al. 2013) and that these processes feed back to dynamically adjust the strength of the local mechano‐chemical transduction and cell–cell coupling that control barrier permeability. The time course of recovery from increased transendothelial monolayer permeability was slower (∼hours) compared to the relatively rapid agonist response (∼minutes), presumably due to the compensatory activation of downstream signalling pathways (MAPKs/Src/Raf/Ras, RhoA and myosin‐light chain kinase pathways; Zeiller et al. 2009; Eyckmans et al. 2011). The focus on single cells allowed us to confine the effects of TRPV4 activation to MVECs, and evade concomitant activation of other cellular components of the neurogliovascular unit. If (as suggested by a recent study in bovine MVECs; Monaghan et al. 2015) inner retinal TRPV4 is predominantly localized to endothelial cells, vascular leakage induced by GSK101 largely reflects activation of EC‐intrinsic channels. In this scenario, the sensitivity to intraluminal pressure and shear flow, myogenic contraction and responsiveness to Ca2+‐dependent vasoactive modulators released from adjacent glial end‐feet are principally mediated through the endothelium with minor contributions from neurons, pericytes and glia. In the more likely case that the channel is functionally expressed by other components of neurogliovascular units which match the supply of metabolites to local energy needs of neuronal circuits (Attwell et al. 2010), TRPV4 activation would simultaneously take place in pericytes, glia astrocytes, Müller cells and/or RGCs (Sonkusare et al. 2012; Iuso & Križaj, 2016; Kim et al. 2016), as a part of a dynamic sensory framework that senses ambient mechanical (local osmotic gradients, shear stress and strain), chemical and thermal stimuli. We hypothesize that TRPV4 channels, expressed in MVECs, smooth muscle cells (pericytes), astrocytes and/or Müller glial end‐endfeet, modulate the vascular tone, permeability and mechano‐induced hyperaemia. Activation of this sensory circuit could additionally modulate the RGC responsiveness to mechanical stress impelled by intraocular pressure or strain. TRPV4 and Ca2+‐dependent release of EETs could induce vasorelaxation by stimulating pericytes from luminal (endothelial) and abluminal (glial) sides as eicosanoids bind the conserved K535A recognition site in the S2‐S3 linker of TRPV4 (e.g. Kim et al. 2016; Berna‐Erro et al. 2017). The differences in TRPV4 expression in cells that form the inner and outer BRB (MVECs and HCECs) are consistent with the heterogeneity of TRPV4 signalling across dissimilar vascular beds (Maishan et al. 2017), and reinforce the view that CNS endothelia and epithelia employ diverse molecular mechanisms to regulate barrier permeability. Another illustrative example of molecular heterogeneity of TRPV4‐dependent barrier regulation is the choroid epithelial barrier, in which permeability increase is subserved by the reorganization of TJs (redistribution of claudin 1 and ZO‐1; Narita et al. 2015) rather than the remodelling of AJs.
Endothelial TRPV4 transduces diverse physical stimuli that include tensile stress, shear flow, swelling, intraluminal pressure and vascular repair (Koehler et al. 2006; Troidl et al. 2009; Schierling et al. 2011; Bagher et al. 2012) and chemical stimuli that include endocannabinoids, EETs and acetylcholine (Watanabe et al. 2003; Zhang et al. 2009). Our results show that TRPV4 activation provides a net depolarizing drive to rMVECs, and link TRPV4 to modulation of actomyosin contractility and long‐term upregulation of actin stress fibres in response to mechanical stress (Ryskamp et al. 2016). Future studies will determine whether TRPV4‐associated microdomains in MVECs contribute to Ca2+‐dependent activation of nitric oxide synthase, KCa channels and/or EET release that have been reported in other endothelia (e.g. Attwell et al. 2010; Bagher et al. 2012; Sonkusare et al. 2012). Another possible modulator of these signaling mechanisms could be TRPV1 channels which, similar to the isoform 4, are expressed in MVECs, activated by derivatives of arachidonic acid (EETs, lipoxygenase products and endocannabinoids) and may contribute to endothelium‐dependent relaxation (Yang et al. 2010). Unfortunately, currently available TRPV1 antibodies are not sufficiently selective to label mammalian retinal tissue (Jo et al. 2017).
Overall, this report provides evidence that TRPV4 channels regulate retinal endothelial Ca2+ homeostasis, cytoskeletal and VE‐cadherin remodeling, and suggests a potential mechanism that modulates the permeability of MVEC barriers in response to a diverse array of mechanical and chemical stimuli in the inner retina. TRPV4 activation could contribute to use‐dependent redistribution of blood flow from superficial to deep vascular plexi by stimulating Ca2+‐dependent production of nitric oxide in response to local elevations of eicosanoids (EETs and 20‐HETE). Interactions with caveolin‐1 (Saliez et al. 2008) could modulate BRB permeability in the presence of dyslipidaemic stress (Lakk et al. 2017), with TRPV4‐dependent junctional remodelling playing a role in the loss of vascular autoregulation associated with focal fluid leakage, angiogenesis, oedema formation and transendothelial invasion of monocytes in retinal pathologies such as glaucoma, diabetic retinopathy, ischaemia and blast trauma (Navaratna et al. 2007; Klaassen et al. 2013; Narita et al. 2015). While TRPV4 activation can be modulated by mechanical trauma, hypercholesterolaemia and hyperglycaemia (Loot et al. 2008; Ma et al. 2011; Monaghan et al. 2015; Lakk et al. 2017), its effects on vascular mechanosensing are likely to be influenced by changes in the expression of CYP450 (CYP2C; CYP2J) enzymes and translocation of TRPV4 from submembrane reserve pools (White et al. 2016; Redmon et al. 2017). Conditional elimination of TRPV4 from RGCs, Müller cells, astrocytes and pericytes will be required to clarify the respective functions of TRPV4‐dependent sensory transduction to neurogliovascular function in the inner retina.
Additional information
Author contributions
T.T.T.P. and D.K. conceived the project; T.T.T.P. and D.K. designed the experiments; T.T.P., S.N.R., O.Y. and J.M.W. carried out the experiments; T.T.T.P., S.N.R., O.Y., J.M.W., D.L. and D.K. analysed the data; D.K. wrote the paper.
Supporting information
Disclaimer: Supporting information has been peer‐reviewed but not copyedited.
Video S1. MVEC transfected with the actin:mApple construct. Exposure to GSK101 reduces membrane ruffling, reduces the transcellular expression of actin stress fibres and induces redistribution of actin from cell cortex towards the nucleus.
Acknowledgements
This work was supported by the National Institutes of Health (EY022076, EY027920, P30EY014800), the University of Utah Neuroscience Initiative, the Diabetes and Metabolism Center at the University of Utah, the Willard L. Eccles Foundation and unrestricted support from Research to Prevent Blindness to the Department of Ophthalmology and the Moran Eye Institute at the University of Utah. We thank Dr Mary Beckerle (University of Utah) for the actin:mApple constructs, Dr Wolfgang Liedtke (Duke University) for Trpv4 −/− mice, Drs M. E. Hartnett and Haibo Wang (University of Utah) for the gift of antibodies, Dr Lara Carroll (University of Utah) for advice on vascular permeability assays and Drs Daniel Ts'o and Momotaz Begum (SUNY Upstate Medical College) for helpful comments on the manuscript and experimental design.
This is an Editor's Choice article from the 15 November 2017 issue.
Contributor Information
Tam T. T. Phuong, Email: tamphuong303@gmail.com
David Križaj, Email: david.krizaj@hsc.utah.edu.
References
- Akazawa Y, Yuki T, Yoshida H, Sugiyama Y & Inoue S (2013). Activation of TRPV4 strengthens the tight‐junction barrier in human epidermal keratinocytes. Skin Pharmacol Physiol 26, 15–21. [DOI] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA & Newman EA (2010). Glial and neuronal control of brain blood flow. Nature 468, 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagher P, Beleznai T, Kansui Y, Mitchell R, Garland CJ & Dora KA (2012). Low intravascular pressure activates endothelial cell TRPV4 channels, local Ca2+ events, and I KCa channels, reducing arteriolar tone. Proc Natl Acad Sci U S A 109, 18174–18179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berna‐Erro A, Izquierdo‐Serra M, Sepúlveda RV, Rubio‐Moscardo F, Doñate‐Macián P, Serra SA, Carrillo‐Garcia J, Perálvarez‐Marín A, González‐Nilo F, Fernández‐Fernández JM & Valverde MA (2017). Structural determinants of 5′,6′‐epoxyeicosatrienoic acid binding to and activation of TRPV4 channel. Sci Rep 7, 10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj AS, Appukuttan B, Wilmarth PA, Pan Y, Stempel AJ, Chipps TJ, Benedetti EE, Zamora DO, Choi D, David LL & Smith JR (2013). Role of the retinal vascular endothelial cell in ocular disease. Prog Retin Eye Res 32, 102–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RC & Davis TP (2002). Calcium modulation of adherens and tight junction function: a potential mechanism for blood–brain barrier disruption after stroke. Stroke 33, 1706–1711. [DOI] [PubMed] [Google Scholar]
- Cuajungco MP, Grimm C, Oshima K, D'Hoedt D, Nilius B, Mensenkamp AR, Bindels RJ, Plomann M & Heller S (2006). PACSINs bind to the TRPV4 cation channel. PACSIN 3 modulates the subcellular localization of TRPV4. J Biol Chem 281, 18753–18762. [DOI] [PubMed] [Google Scholar]
- Darby WG, Grace MS, Baratchi S & Mclntyre P (2016). Modulation of TRPV4 by diverse mechanisms. Int J Biochem Cell Biol 78, 217–228. [DOI] [PubMed] [Google Scholar]
- Dunn KM, Hill‐Eubanksa DC, Liedtkeb WB & Nelsona MT (2013). TRPV4 channels stimulate Ca2+‐induced Ca2+ release in astrocytic endfeet and amplify neurovascular coupling responses. Proc Natl Acad Sci U S A 110, 6157–6162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley S, Pauyo T, Drapp R, Tavares MJ, Liedtke W & Brayden JE (2009). TRPV4‐dependent dilation of peripheral resistance arteries influences arterial pressure. Am J Physiol Heart Circ Physiol 297, H1096–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley S & Brayden JE (2015). Transient receptor potential channels in the vasculature. Physiol Rev 95, 645–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyckmans J, Boudou T, Yu X & Chen CS (2011). A hitchhiker's guide to mechanobiology. Dev Cell 21, 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan HC, Zhang X & McNaughton PA (2009). Activation of the TRPV4 ion channel is enhanced by phosphorylation. J Biol Chem 284, 27884–27891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giampietro C, Taddei A, Corada M, Sarra‐Ferraris GM, Alcalay M, Cavallaro U, Orsenigo F, Lampugnani MG & Dejana E (2012). Overlapping and divergent signaling pathways of N‐cadherin and VE‐cadherin in endothelial cells. Blood 119, 2159–2170. [DOI] [PubMed] [Google Scholar]
- Giannotta M, Trani M & Dejana E (2013). VE‐cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell 26, 441–454. [DOI] [PubMed] [Google Scholar]
- Gibson CC, Zhu W, Davis CT, Bowman‐Kirigin JA, Chan AC, Ling J, Walker AE, Goitre L, Delle Monache S, Retta SF, Shiu YT, Grossmann AH, Thomas KR, Donato AJ, Lesniewski LA, Whitehead KJ & Li DY (2015). Strategy for identifying repurposed drugs for the treatment of cerebral cavernous malformation. Circulation 131, 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güler AD, Lee H, Iida T, Shimizu I, Tominaga M & Caterina M (2002). Heat‐evoked activation of the ion channel, TRPV4. J Neurosci 22, 6408–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmannsgruber V, Heyken WT, Kacik M, Kaistha A, Grgic I, Harteneck C, Liedtke W, Hoyer J & Köhler R (2007). Arterial response to shear stress critically depends on endothelial TRPV4 expression. PLoS One 2, e827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill‐Eubanks DC, Gonzales AL, Sonkusare SK & Nelson MT (2014). Vascular TRP channels: performing under pressure and going with the flow. Physiology (Bethesda) 29, 343–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iuso A & Križaj D (2016). TRPV4–AQP4 interactions ‘turbocharge’ astroglial sensitivity to small osmotic gradients. Channels 10, 172–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo AO, Ryskamp DA, Phuong TT, Verkman AS, Yarishkin O, MacAulay N & Križaj D (2015). TRPV4 and AQP4 channels synergistically regulate cell volume and calcium homeostasis in retinal Muller glia. J Neurosci 35, 13525–13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo AO, Lakk M, Frye AM, Phuong TT, Redmon SN, Roberts R, Berkowitz BA, Yarishkin O & Križaj D (2016). Differential volume regulation and calcium signaling in two ciliary body cell types is subserved by TRPV4 channels. Proc Natl Acad Sci U S A 113, 3885–3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo AO, Noel JM, Lakk M, Yarishkin O, Ryskamp DA, Shibasaki K, McCall MA & Križaj D (2017). Mouse retinal ganglion cell signalling is dynamically modulated through parallel anterograde activation of cannabinoid and vanilloid pathways. J Physiol 595, 6499–6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CA, London NR, Chen H, Park KW, Sauvaget D, Stockton RA, Wythe JD, Suh W, Larrieu‐Lahargue F, Mukouyama YS, Lindblom P, Seth P, Frias A, Nishiya N, Ginsberg MH, Gerhardt H, Zhang K & Li DY (2008). Robo4 stabilizes the vascular network by inhibiting pathologic angiogenesis and endothelial hyperpermeability. Nat Med 14, 448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke SK, Chen L, Duan HB & Tu YR (2015). Opposing actions of TRPV4 channel activation in the lung vasculature. Respir Physiol Neurobiol 219, 43–50. [DOI] [PubMed] [Google Scholar]
- Kevil CG, Payne DK, Mire E & Alexander JS (1998). Vascular permeability factor/vascular endothelial cell growth factor‐mediated permeability occurs through disorganization of endothelial junctional proteins. J Biol Chem 273, 15099–15103. [DOI] [PubMed] [Google Scholar]
- Kim KJ, Ramiro Diaz J, Iddings JA & Filosa JA (2016). Vasculo‐neuronal coupling: retrograde vascular communication to brain neurons. J Neurosci 36, 12624–12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassen I, Van Noorden CJ & Schlingemann RO (2013). Molecular basis of the inner blood‐retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog Retin Eye Res 34, 19–48. [DOI] [PubMed] [Google Scholar]
- Kohler R, Heyken WT, Heinau P, Schubert R, Si H, Kacik M, Busch C, Grgic I, Maier T & Hoyer J (2006). Evidence for a functional role of endothelial transient receptor potential V4 in shear stress‐induced vasodilatation. Arterioscler Thromb Vasc Biol 26, 1495–1502. [DOI] [PubMed] [Google Scholar]
- Komarova Y & Malik AB (2010). Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol 72, 463–493. [DOI] [PubMed] [Google Scholar]
- Križaj D, Lai FA & Copenhagen DR (2003). Ryanodine stores and calcium regulation in the inner segments of salamander rods and cones. J Physiol 547, 761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakk M, Yarishkin O, Baumann J, Iuso A & Križaj D (2017). Cholesterol regulates polymodal sensory transduction in retinal glia. Glia 65, 2038–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loot AE, Popp R, Fisslthaler B, Vriens J, Nilius B & Fleming I (2008). Role of cytochrome P450‐dependent transient receptor potential V4 activation in flow‐induced vasodilatation. Cardiovasc Res 80, 445–452. [DOI] [PubMed] [Google Scholar]
- Ma X, Cheng KT, Wong CO, O'Neil RG, Birnbaumer L, Ambudkar IS & Yao X (2011). Heteromeric TRPV4‐C1 channel contribute to store‐operated Ca2+ entry in vascular endothelial cells. Cell Calcium 50, 502–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maishan M, Lamb D, Lee WL & Kuebler WM (2017). Heterogeneity of TRPV4 expression and function in endothelial cells of different vascular beds. FASEB J 31 (Suppl. 1), 837.14. [Google Scholar]
- Marrelli SP, O'Neil R G, Brown RC & Bryan RM Jr (2007). PLA2 and TRPV4 channels regulate endothelial calcium in cerebral arteries. Am J Physiol Heart Circ Physiol 292, H1390–1397. [DOI] [PubMed] [Google Scholar]
- Matthews BD, Thodeti CK, Tytell JD, Mammoto A, Overby DR & Ingber DE (2010). Ultra‐rapid activation of TRPV4 ion channel by mechanical forces applied to cell surface β1 intergrins. Integr Bio (Camb) 2, 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza SA, Fang J, Gutterman DD, Wilcox DA, Bubolz AH, Li R, Suzuki M & Zhang DX (2010). TRPV4‐mediated endothelial Ca2+ influx and vasodilation in response to shear stress. Am J Physiol Heart Circ Physiol 298, H466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto K, Khosrof S, Bursell S, Moromizato Y, Aiello LP, Ogura Y & Anthony P (2000). Vascular endothelial growth factor (VEGF)‐induced retinal vascular permeability is mediated by intercellular adhesion molecule‐1 (ICAM‐1). Am J Pathol 156, 1733–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnar T, Yarishkin O, Iuso A, Barabas P, Jones B, Marc RE, Phuong TT & Križaj D (2016). Store‐operated calcium entry in Muller glia is controlled by synergistic activation of TRPC and Orai channels. J Neurosci 36, 3184–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan K, McNaughten J, McGahon MK, Kelly C, Kyle D, Yong PH, McGeown JG & Curtis TM (2015). Hyperglycemia and diabetes downregulate the functional expression of TRPV4 channels in retinal microvascular endothelium. PLoS One 10, e0128359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore TM, Chetham PM, Kelly JJ & Stevens T (1998). Signal transduction and regulation of lung endothelial cell permeability. Interaction between calcium and cAMP. Am J Physiol Lung Cell Mol Physiol 275, L203–222. [DOI] [PubMed] [Google Scholar]
- Narita K, Sasamoto S, Koizumi S, Okazaki S, Nakamura H, Inoue T & Takeda S (2015). TRPV4 regulates the integrity of the blood–cerebrospinal fluid barrier and modulates transepithelial protein transport. FASEB J 29, 2247–2259. [DOI] [PubMed] [Google Scholar]
- Navaratna D, McGuire PG, Menicucci G & Das A (2007). Proteolytic degradation of VE‐cadherin alters the blood retinal barrier in diabetes. Diabetes 56, 2380–2387. [DOI] [PubMed] [Google Scholar]
- Phuong TT, Yun YH, Kim SJ & Kang TM (2013). Positive feedback control between STIM1 and NFATc3 is required for C2C12 myoblast differentiation. Biochem Biophys Res Commun 430, 722–728. [DOI] [PubMed] [Google Scholar]
- Ratheesh A & Yap AS (2012). A bigger picture: classical cadherins and the dynamic actin cytoskeleton. Nat Rev Mol Cell Biol 13, 673–679. [DOI] [PubMed] [Google Scholar]
- Raviola G (1977). The structural basis of the blood–ocular barriers. Exp Eye Res 25 Suppl, 27–63. [DOI] [PubMed] [Google Scholar]
- Redmon SN, Shibasaki K & Križaj D (2017). Transient receptor potential cation channel subfamily V member 4 (TRPV4). Encyclopedia of Signaling Molecules, ed. Choi S, pp. 1–11. Springer Nature, New York. [Google Scholar]
- Rentería RC, Strehler EE, Copenhagen DR & Križaj D (2005). Ontogeny of plasma membrane Ca2+ ATPase isoforms in the neural retina of the postnatal rat. Vis Neurosci 22, 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryskamp DA, Jo AO, Frye AM, Vazquez‐Chona F, MacAulay N, Thoreson WB & Križaj D (2014). Swelling and eicosanoid metabolites differentially gate TRPV4 channels in retinal neurons and glia. J Neurosci 34, 15689–15700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryskamp DA, Witkovsky P, Barabas P, Huang W, Koehler C, Akimov NP, Lee SH, Chauhan S, Xing W, Renteria RC, Liedtke W & Križaj D (2011). The polymodal ion channel transient receptor potential vanilloid 4 modulates calcium flux, spiking rate, and apoptosis of mouse retinal ganglion cells. J Neurosci 31, 7089–7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryskamp DA, Frye AM, Phuong TT, Yarishkin O, Jo AO, Xu Y, Lakk M, Iuso A, Redmon SN, Ambati B, Hageman G, Prestwich GD, Torrejon KY & Križaj D (2016). TRPV4 regulates calcium homeostasis, cytoskeletal remodeling, conventional outflow and intraocular pressure in the mammalian eye. Sci Rep 6, 30583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliez J, Bouzin C, Rath G, Ghisdal P, Desjardins F, Rezzani R, Rodella LF, Vriens J, Nilius B, Feron O, Balligand JL & Dessy C (2008). Role of caveolar compartmentation in endothelium‐derived hyperpolarizing factor‐mediated relaxation: Ca2+ signals and gap junction function are regulated by caveolin in endothelial cells. Circulation 117, 1065–1074. [DOI] [PubMed] [Google Scholar]
- Schierling W, Troidl K, Apfelbeck H, Troidl C, Kasprzak PM, Schaper W & Schmitz‐Rixen T (2011). Cerebral arteriogenesis is enhanced by pharmacological as well as fluid‐shear‐stress activation of the Trpv4 calcium channel. Eur J Vasc Endovasc Surg 41, 589–596. [DOI] [PubMed] [Google Scholar]
- Seebach J, Donnert G, Kronstein R, Werth S, Wojciak‐Stothard B, Falzarano D, Mrowietz C, Hell SW & Schnittler HJ (2007). Regulation of endothelial barrier function during flow‐induced conversion to an arterial phenotype. Cardiovasc Res 75, 596–607. [DOI] [PubMed] [Google Scholar]
- Shin SH, Lee EJ, Hyun S, Chun J, Kim Y & Kang SS (2012). Phosphorylation on the Ser 824 residue of TRPV4 prefers to bind with F‐actin than with microtubules to expand the cell surface area. Cell Signal 24, 641–651. [DOI] [PubMed] [Google Scholar]
- Sokabe T & Tominaga M (2010). The TRPV4 cation channel: a molecule linking skin temperature and barrier function. Commun Integr Biol 3, 619–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI & Heppner TJ (2012). Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 336, 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strotmann R, Harteneck C, Nunnenmacher K, Schultz G & Plant TD (2000). OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol 2, 695–702. [DOI] [PubMed] [Google Scholar]
- Sun M, Fu H, Cheng H, Cao Q, Zhao Y, Mou X, Zhang X, Liu X & Ke Y (2012). A dynamic real‐time method for monitoring epithelial barrier function in vitro. Anal Biochem 425, 96–103. [DOI] [PubMed] [Google Scholar]
- Suresh K, Servinsky L, Reyes J, Baksh S, Undem C, Caterina M, Pearse DB & Shimoda LA (2015). Hydrogen peroxide‐induced calcium influx in lung microvascular endothelial cells involves TRPV4. Am J Physiol Lung Cell Mol Physiol 309, L1467–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thodeti CK, Matthews B, Ravi A, Mammoto A, Ghosh K, Bracha AL & Ingber DE (2009). TRPV4 channels mediate cyclic strain‐induced endothelial cell reorientation through integrin‐to‐integrin signaling. Circ Res 104, 1123–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorneloe KS, Sulpizio AC, Lin Z, Figueroa DJ, Clouse AK, McCafferty GP, Chendrimada TP, Lashinger ES, Gordon E, Evans L, Misajet BA, Demarini DJ, Nation JH, Casillas LN, Marquis RW, Votta BJ, Sheardown SA, Xu X, Brooks DP, Laping NJ & Westfall TD (2008). N‐((1S)‐1‐{[4‐((2S)‐2‐{[(2,4‐dichlorophenyl)sulfonyl]amino}‐3‐hydroxypropanoyl)‐1‐piperazinyl]carbonyl}‐3‐methylbutyl)‐1‐benzothiophene‐2‐carboxamide (GSK1016790A), a novel and potent transient receptor potential vanilloid 4 channel agonist induces urinary bladder contraction and hyperactivity: Part I. J Pharmacol Exp Ther 326, 432–442. [DOI] [PubMed] [Google Scholar]
- Toft‐Bertelsen TL, Križaj D & MacAulay N (2017). When size matters: transient receptor potential vanilloid 4 channel as a volume‐sensor rather than an osmo‐sensor. J Physiol 595, 3287–3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troidl C, Troidl K, Schierling W, Cai WJ, Nef H, Mollmann H, Kostin S, Schimanski S, Hammer L, Elsasser A, Schmitz‐Rixen T & Schaper W (2009). Trpv4 induces collateral vessel growth during regeneration of the arterial circulation. J Cell Mol Med 13, 2613–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ts'o DY, Begum M, Phuong TTT & Križaj D (2017). Epoxyeicosatrienoic acid (EET)‐mediated retinal functional hyperemia enhanced with inactivation of TRPV4 channels. Soc Neurosci Abstr, Neuroscience Meeting Planner Program No. 684.03. Washington, DC: Society for Neuroscience. Online. [Google Scholar]
- Villalta PC, Rocic P & Townsley MI (2014). Role of MMP2 and MMP9 in TRPV4‐induced lung injury. Am J Physiol Lung Cell Mol Physiol 307, L652–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriens J, Owsianik G, Fisslthaler B, Suzuki M, Janssens A, Voets T, Morisseau C, Hammock BD, Fleming I, Busse R & Nilius B (2005). Modulation of the Ca2+ permeable cation channel TRPV4 by cytochrome P450 epoxygenases in vascular endothelium. Circ Res 97, 908–915. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T & Nilius B (2003). Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature 424, 434–438. [DOI] [PubMed] [Google Scholar]
- Wheelock MJ & Johnson KR (2003). Cadherin‐mediated cellular signaling. Curr Opin Cell Biol 15, 509–514. [DOI] [PubMed] [Google Scholar]
- White JP, Cibelli M, Urban L, Nilius B, McGeown JG & Nagy I (2016). TRPV4: molecular conductor of a diverse orchestra. Physiol Rev 96, 911–973. [DOI] [PubMed] [Google Scholar]
- Willette RN, Bao W, Nerurkar S, Yue TL, Doe CP, Stankus G, Turner GH, Ju H, Thomas H, Fishman CE, Sulpizio A, Behm DJ, Hoffman S, Lin Z, Lozinskaya I, Casillas LN, Lin M, Trout RE, Votta BJ, Thorneloe K, Lashinger ES, Figueroa DJ, Marquis R, Xu X (2008). Systemic activation of the transient receptor potential vanilloid subtype 4 channel causes endothelial failure and circulatory collapse: Part 2. J Pharmacol Exp Ther 326, 443–452. [DOI] [PubMed] [Google Scholar]
- Xu Q, Qaum T & Adamis AP (2001). Sensitive blood‐retinal barrier breakdown quantitation using Evans blue. Invest Ophthalmol Vis Sci 42, 789–794. [PubMed] [Google Scholar]
- Yang D, Luo Z, Ma S, Wong WT, Ma L, Zhong J, He H, Zhao Z, Cao T, Yan Z, Liu D, Arendshorst WJ, Huang Y, Tepel M & Zhu Z (2010). Activation of TRPV1 by dietary capsaicin improves endothelium‐dependent vasorelaxation. Cell Metab 12, 130–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeiller C, Mebarek S, Jaafar R, Pirola L, Lagarde M, Prigent AF & Nemoz G (2009). Phospholipase D2 regulates endothelial permeability through cytoskeleton reorganization and occludin downregulation. Biochim Biophys Acta 1793, 1236–1249. [DOI] [PubMed] [Google Scholar]
- Zhang DX, Mendoza SA, Bubolz AH, Mizuno A, Ge ZD, Li R, Warltier DC, Suzuki M & Gutterman DD (2009). Transient receptor potential vanilloid type 4‐deficient mice exhibit impaired endothelium‐dependent relaxation induced by acetylcholine in vitro and in vivo . Hypertension 53, 532–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler N, Awwad K, Fisslthaler B, Reis M, Devraj K, Corada M, Minardi SP, Dejana E, Plate KH, Fleming I & Liebner S (2016). β‐Catenin is required for endothelial Cyp1b1 regulation influencing metabolic barrier function. J Neurosci 36, 8921–8935. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supporting information has been peer‐reviewed but not copyedited.
Video S1. MVEC transfected with the actin:mApple construct. Exposure to GSK101 reduces membrane ruffling, reduces the transcellular expression of actin stress fibres and induces redistribution of actin from cell cortex towards the nucleus.