

Abstract
Key points
The evoked cardiac response to bipolar cervical vagus nerve stimulation (VNS) reflects a dynamic interaction between afferent mediated decreases in central parasympathetic drive and suppressive effects evoked by direct stimulation of parasympathetic efferent axons to the heart.
The neural fulcrum is defined as the operating point, based on frequency–amplitude–pulse width, where a null heart rate response is reproducibly evoked during the on‐phase of VNS.
Cardiac control, based on the principal of the neural fulcrum, can be elicited from either vagus.
Beta‐receptor blockade does not alter the tachycardia phase to low intensity VNS, but can increase the bradycardia to higher intensity VNS.
While muscarinic cholinergic blockade prevented the VNS‐induced bradycardia, clinically relevant doses of ACE inhibitors, beta‐blockade and the funny channel blocker ivabradine did not alter the VNS chronotropic response.
While there are qualitative differences in VNS heart control between awake and anaesthetized states, the physiological expression of the neural fulcrum is maintained.
Abstract
Vagus nerve stimulation (VNS) is an emerging therapy for treatment of chronic heart failure and remains a standard of therapy in patients with treatment‐resistant epilepsy. The objective of this work was to characterize heart rate (HR) responses (HRRs) during the active phase of chronic VNS over a wide range of stimulation parameters in order to define optimal protocols for bidirectional bioelectronic control of the heart. In normal canines, bipolar electrodes were chronically implanted on the cervical vagosympathetic trunk bilaterally with anode cephalad to cathode (n = 8, ‘cardiac’ configuration) or with electrode positions reversed (n = 8, ‘epilepsy’ configuration). In awake state, HRRs were determined for each combination of pulse frequency (2–20 Hz), intensity (0–3.5 mA) and pulse widths (130–750 μs) over 14 months. At low intensities and higher frequency VNS, HR increased during the VNS active phase owing to afferent modulation of parasympathetic central drive. When functional effects of afferent and efferent fibre activation were balanced, a null HRR was evoked (defined as ‘neural fulcrum’) during which HRR ≈ 0. As intensity increased further, HR was reduced during the active phase of VNS. While qualitatively similar, VNS delivered in the epilepsy configuration resulted in more pronounced HR acceleration and reduced HR deceleration during VNS. At termination, under anaesthesia, transection of the vagi rostral to the stimulation site eliminated the augmenting response to VNS and enhanced the parasympathetic efferent‐mediated suppressing effect on electrical and mechanical function of the heart. In conclusion, VNS activates central then peripheral aspects of the cardiac nervous system. VNS control over cardiac function is maintained during chronic therapy.
Keywords: vagus nerve stimulation, neural fulcrum, neurocardiology, parasympathetic, cardiac afferent
Key points
The evoked cardiac response to bipolar cervical vagus nerve stimulation (VNS) reflects a dynamic interaction between afferent mediated decreases in central parasympathetic drive and suppressive effects evoked by direct stimulation of parasympathetic efferent axons to the heart.
The neural fulcrum is defined as the operating point, based on frequency–amplitude–pulse width, where a null heart rate response is reproducibly evoked during the on‐phase of VNS.
Cardiac control, based on the principal of the neural fulcrum, can be elicited from either vagus.
Beta‐receptor blockade does not alter the tachycardia phase to low intensity VNS, but can increase the bradycardia to higher intensity VNS.
While muscarinic cholinergic blockade prevented the VNS‐induced bradycardia, clinically relevant doses of ACE inhibitors, beta‐blockade and the funny channel blocker ivabradine did not alter the VNS chronotropic response.
While there are qualitative differences in VNS heart control between awake and anaesthetized states, the physiological expression of the neural fulcrum is maintained.
Introduction
Neural control of the heart involves multiple ongoing interactions among central and peripheral components of the cardiac neuroaxis (Ardell et al. 2015; Shivkumar et al. 2016). It is now recognized that during the evolution of heart disease, remodelling occurs not only at the level of the cardiomyocyte, but also throughout the cardiac neuraxis that impacts cardiac electrical and mechanical function (Zucker et al. 2012; Florea & Cohn, 2014; Fukuda et al. 2015). For instance, enhancement of sensory inputs derived from cardiac afferent neurons transducing the ischaemic myocardium elicit not only (i) central reflex‐induced cardiac sympatho‐excitation, but also (ii) central reflex reductions in parasympathetic efferent neuronal drive (Billman, 2006; Florea & Cohn, 2014; Wang et al. 2014). Since the cardiac neuraxis is integral to the evolution of cardiac pathology, it can also be considered as a highly relevant target for neuromodulation therapy to treat select cardiac pathologies (Ardell et al. 2016).
Vagus nerve stimulation (VNS) is one implementation in the emerging field of bioelectronics medicine. It has potential applications in treatment of arrhythmias (Stavrakis et al. 2015; Salavatian et al. 2016) and in preservation of cardiac function in reduced (HFrEF) and preserved (HFpEF) heart failure (Sabbah et al. 2011; Shinlapawittayatorn et al. 2013; De Ferrari, 2014; Beaumont et al. 2016). Pre‐clinical studies have recently been translated to the clinical setting, but with mixed results for HFrEF. Two studies reported neutral effects to chronic VNS (Zannad et al. 2015; Gold et al. 2016) and one demonstrated moderate clinical benefit to the patients (Premchand et al. 2014, 2016). Each of these clinical studies used different interfaces and stimulation protocols; such differences were likely a primary determinant of the different clinical outcomes. In some cases, therapeutic targets were not achieved due to off‐target adverse effects that were evoked during VNS (Zannad et al. 2015). These off‐target effects include cough, voice alteration and gastrointestinal (GI) discomfort (Bonaz et al. 2013). A major unmet need in the field of bioelectronics medicine is a structure–function understanding of the central and peripheral neural circuits that are being impacted by focal electrical neural stimulation and the dynamics of those network interactions as fundamental aspects of the stimulation protocols are altered – stimulation frequency, intensity, pulse width and duty cycle among them.
The cervical vagosympathetic nerve trunk is a mixed nerve containing afferent (∼80%) and efferent axonal projections with fibre types ranging from unmyelinated c‐fibres to larger myelinated axons (Paintal, 1963, 1973; Bonaz et al. 2013). VNS recruits (action potential forms and propagates) these axons based on proximity to the electrode and inversely related to size – with A‐fibres recruited first and c‐fibres last or most often never owing to their very high stimulation threshold (Yoo et al. 2013). In the anaesthetized state, at low VNS intensities, afferent axonal projections are activated before those of efferent neurons leading to a decrease in central parasympathetic drive (Ardell et al. 2015; Yamakawa et al. 2016). At higher VNS intensities, parasympathetic efferent axonal projections are recruited leading to the expected bradycardia (Ardell et al. 2015). When afferent‐driven decreases in central parasympathetic outflow are equivalently counteracted by direct activation of the cardiac parasympathetic efferent projections to the intrinsic cardiac nervous system (ICNS) and heart, the net result is a null HR response (Kember et al. 2014; Ardell et al. 2015). We have defined this point as the neural fulcrum (Ardell et al. 2015) and this point represents a dynamic equilibrium where neural circuits for cardiac control are engaged by electrical stimulation but where the endogenous neural networks remain capable of robust reflex control. It remains to be determined if the neural fulcrum functions in the intact state and if it is altered during prolonged periods of chronic vagus nerve stimulation.
The primary objective for this paper was to establish the stimulation protocol constraints that define the neural fulcrum for chronic VNS for cardiac control. This includes evaluating the effects of bipole electrode orientation, stimulation frequency, pulse width and intensity. Since VNS is currently deployed as an adjunct to standard of care for HFrEF (Premchand et al. 2014; Zannad et al. 2015; Gold et al. 2016; Premchand et al. 2016), we also determined the effects of beta blockers, angiotensin converting enzyme (ACE) inhibitors and funny channel blockers on VNS‐related cardiac control. Finally, we evaluated if chronic VNS altered central–peripheral neural network interactions for integrated control of the heart. The data presented herein indicate that VNS can be delivered chronically to either vagus nerve in a mechanistic manner to modulate cardiac function without compromising endogenous neural networks for control of regional cardiac function.
Methods
Cross‐breed dogs (n = 16, 11 female and 5 male, weighing 22.6 ± 0.7 kg) were used in this study. All experiments were performed in accordance with the guidelines set forth by the National Institutes of Health Guide for the Care and Use of Laboratory Animals and in compliance with the animal ethics checklist as defined by The Journal of Physiology (Grundy, 2015), and were approved by the East Tennessee State University Institutional Animal Care and Use Committee.
Chronic VNS implant procedures
Normal adult canines were pre‐medicated with buprenorphine (0.01 mg kg−1) and sedated with propofol (3–8 mg kg−1, i.v.), followed by endotracheal intubation and mechanical ventilation. General anaesthesia was maintained with isoflurane (1–2% inhalation therapy). The depth of anaesthesia was assessed throughout surgery by monitoring corneal reflexes, jaw tone, and haemodynamic indices. Body temperature was maintained via a circulating water heating pad (Gaymar T/Pump, Gaymar Industries Inc., Orchard Park, NY, USA).
Using aseptic techniques both cervical vagosympathetic trunks were isolated from a midline incision. Cervical vagosympathetic trunks were mobilized and bipolar electrodes (PerenniaFlex, Model 304, Cyberonics, Inc., Houston, TX, USA) were placed around each nerve with either the anode cephalad to cathode (n = 8, ‘cardiac’ configuration) or cathode cephalad to anode (n = 8, ‘epilepsy’ configuration). The leads were secured in place and tunnelled subcutaneously to subcutaneous pockets created over the dorsal aspect of the neck using separate incisions. Each lead was then attached to individual implantable programmable generators (IPG, Demipulse 103, Cyberonics, Inc.) and placed together within the subcutaneous pocket. Incisions were closed in layers with 2‐0 absorbable suture for the underlying muscle and 3‐0 non‐absorbable suture for the skin. Postoperative medications were given to ensure comfort (buprenorphine for 24 h post‐operative and carprofen 2× daily for 6 days thereafter) and reduce the risk of infection (cefazolin/cephalexin).
VNS titration protocol in awake states
All animals were trained to a Pavlov stand and each evaluated for chronotropic and off‐target effects to VNS. A lead II ECG was recorded continuously from each animal via a differential amplifier (Grass P511, Astro‐Med, West Warwick, RI, USA) and input to a Cambridge Electronics Design (Cambridge, UK, model 1401) data acquisition system for continuous monitoring of chronotopic function. VNS titration was initiated with parameters programmed starting from 10 Hz, 130 μs, 0.25 mA, with a duty cycle of 14 s on–1.1 min off (1080 cycles day−1). Currents were then increased to the tolerance boarder zone or a maximum of 3.0 mA at 130 μs. The tolerance boarder zone was defined as the point where the step up in current evoked cough and/or GI discomfort on three consecutive duty cycle stimulations. If the tolerance boarder zone was exceeded, current was stepped back 0.25 mA and left there until the next evaluation session 7 days later. If the 3.0 mA level was achieved, pulse width was increased to 250 μs, current reduced to 0.5 mA and increased during that session until the tolerance boarder zone was exceeded. At that point, current was stepped back 0.25 mA and left there until the next evaluation session 7 days later. This same up‐titration in current was continued at 250 μs pulse width to 3.0 mA and then with 500 μs pulse width. Frequency was then stepped to 20 Hz, 500 μs pulse width and current up‐titrated a final time. The dynamic titration protocol was completed within 30 days. Final current levels, maintained during the experimental phase, were set to intensities for each animal where during the active phase of VNS there was minimal change in heart rate (the neural fulcrum) but that with one additional step up in intensity (0.25 mA) bradycardia was reproducibly evoked.
VNS – chronotropic response protocol
Chronotropic responses to short‐duration VNS were evaluated in conscious animals trained to a Pavlov stand. A lead II ECG was recorded continuously and stored to computer as described above. Each specific combination of frequency, pulse width and intensity was delivered at a 17.5% duty cycle (14 s on‐phase). Five frequencies were evaluated: 2, 5, 10, 15 and 20 Hz. Four pulse widths were evaluated: 130, 250, 500 and 750 μs. Fourteen different intensities were evaluated, ranging from 0.25 to 3.50 mA in 0.25 mA increments. For a given recording session, stimulations were restricted to one nerve, two frequencies and the 14 current levels for a given frequency. Those current levels were randomized. Each recording session was completed in ∼90 min and no more than three sessions were done per week.
VNS – Holter monitoring protocol
Twenty‐four to forty‐eight hour Holter monitoring was done in a subset of animals during the course of chronic VNS. This was done primarily during right‐sided VNS and with the electrode in the anode cephalad to cathode ‘cardiac’ configuration (n = 8). Control Holter recordings were periodically obtained from the same animals whilst VNS was not active. Holter recording was done using a five‐lead DR200 recorder and data files analysed using Holter LX Analysis software (both from Northeast Monitoring, Maynard, MA, USA). Indices of heart rate variability (HRV) are presented in the time domain and include mean heart rate, pNN50% (number of time per hour in which change in consecutive normal sinus intervals exceeds 50 ms divided by total number of beats), pNN50 (NN50 count/total count), SDNN (standard deviation of NN intervals) and RMSSD (root mean squared difference of successive NN intervals). To evaluate the effects of VNS on circadian chronotropic rhythms, animals were instrumented at ∼17.00 h and the temporal data were subdivided into evening (18.00–00.00 h), overnight (00.00–06.00 h), morning (06.00–12.00 h) and afternoon (12.00–18.00 h) segments.
Pharmacological blockade – effects on VNS control of chronotropic function
Effects of selective autonomic blockade on the evoked chronotropic response to VNS were evaluated in both an acute and a sustained treatment phase. In all cases, animals were evaluated in the conscious state with animals standing quietly in a Pavlov stand. Chronotropic responses were evaluated in response to right‐sided VNS (10 Hz, 500 μs pulse width). For acute pharmacological treatment, VNS chronotropic responses were compared before and after metoprolol (1 mg kg−1, i.v.) and cholinergic blockade (glycopyrrolate, 0.4 mg kg−1, i.v.). For chronic pharmacological treatment, during successive 2‐week phases, animals were administered twice daily (PO) with (i) a beta blocker (metoprolol, 12.25 mg), (ii) an ACE inhibitor (enalapril, 2.5 mg), (iii) a funny channel blocker (ivabradine, 5.0 mg), (iv) metoprolol + enalapril, or (v) metoprolol + enalapril + ivabradine. In conscious animals, chronotropic responses to recurring cycles of 10 Hz RCV VNS (14 s on time–66 s off time) were measured at the end of each 2‐week therapy phase and compared to baseline.
Terminal study
Animals were sedated with propofol (3–8 mg kg−1, i.v.), followed by endotracheal intubation and mechanical ventilation. General anaesthesia was maintained with isoflurane (1–2%, inhalation). Depth of anaesthesia was assessed by monitoring corneal reflexes, jaw tone, and haemodynamic indices. Left femoral venous access was obtained for maintenance fluid and drug administration. Right femoral arterial access was obtained for monitoring aortic pressure. A pressure transducer catheter (Mikro‐Tip, Millar Instruments, Houston, TX, USA) was inserted into the left ventricle (LV) chamber via the left femoral artery. A lead II ECG was recorded via needle electrodes and amplified by a pre‐amplifier (P511, Grass Technologies). Haemodynamic data were acquired with a data acquisition system (Power1401, Cambridge Electronic Design) and analysed off‐line with Spike2 (Cambridge Electronic Design). At the completion of the surgery, general anaesthesia was switched to α‐chloralose (50 mg kg−1 i.v. bolus followed by 8–12 mg kg−1 h−1 continuous i.v. infusion). Acid–base status was evaluated hourly (Irma TruePoint, ITC, Edison, NJ, USA); respiratory rate and tidal volume were adjusted and/or sodium bicarbonate was infused as necessary to maintain blood gas homeostasis.
Vagus nerve stimulation – terminal study
The effects of VNS (right and left individually) on chronotropic, LV inotropic and LV lusitropic function were evaluated over a range of currents (0.25–3.5 mA), with 10 Hz frequency and 500 μs pulse width. VNS was performed for 14 s followed by a 66 s off‐phase. This time period was sufficient for cardiac indices to return to baseline values with no degradation in the responses to VNS over the duration of the experiments. Following stimulations in the intact state, the vagi were transected central to the stimulating electrode, allowing for the stimulation of only efferent fibres in subsequent parts of the protocol. At the completion of the experiments, animals were humanely euthanized under deep anaesthesia by inducing ventricular fibrillation via direct current and removing the heart.
Statistics
Off‐line analysis, using Spike2, was used to determine the average response for each of the parameters at baseline and during the 14 s VNS on‐phase. Derived indices included HR, aortic blood pressure (BP), LV end‐systolic pressure (LVSP), maximum rate of change in LV pressure (LV dP/dt maximum) and minimum rate of change in LV pressure (LV dP/dt minimum). Data are presented as mean ± standard error of the mean. A repeated measure mixed analysis of variance model was used for comparisons of mean current or mean frequency curves generated in the different states. HRV (Holter) data were assessed by two‐way repeated measures ANOVA using VNS and time of day as factors. P < 0.05 was considered to be statistically significant. Statistical analyses were performed with JMP Pro v13 (SAS Institute Inc., Cary, NC, USA).
Results
Identification of VNS tolerance
VNS tolerance was defined at any given frequency, pulse width and intensity as a level where the on‐phase of stimulation did not evoke off target effects of cough and/or GI discomfort. Default VNS protocols for epilepsy utilize 30 Hz 500 μs pulse width at a 9% duty cycle (30 s on, 5 min off). Utilizing this protocol, the maximal tolerated current achieved by the end of 60 days’ titration was 0.63 ± 0.16 mA (n = 4, data not shown); above that current amplitude off target effects were evoked. Using the accelerated protocol, using lower frequencies (10 Hz), shorter pulse widths and more robust duty cycle (17.5%; 14 s on, 66 s off), a tolerance threshold of 2.63 ± 0.13 mA was achieved within 4 weeks of titration onset and exceeded the current maximum (3.5 mA) by week 6. In the maintenance phase, VNS was programmed to 20 Hz, pulse width of 500 μs, duty cycles of 22.5% (14 s on‐phase) and average intensities of 1.91 ± 0.09 mA for right‐sided VNS or 2.80 ± 0.15 mA for left‐sided VNS. These parameter sets were chosen such that for each animal there was minimal change in heart rate during on‐phase VNS but that with one additional step up in current intensity (0.25 mA), bradycardia was reproducibly evoked. Unilateral VNS therapy was applied over a 14 ± 2 month duration (range 6–24 months) and operating point for VNS control of the heart chronotropic function remained stable over that time.
Chronotropic response to graded VNS
The evoked chronotropic response to VNS was dependent upon a number of factors including frequency, pulse width, intensity, the electrode–nerve interface and the site of stimulation. Figure 1 illustrates a representative response to bipolar stimulation of the right cervical vagosympathetic (RCV) nerve trunk. VNS was delivered with a 17.5% duty cycle (14 s on‐phase) at 10 Hz and 500 μs pulse width. Note the presence of inherent sinus arrhythmia in the animal standing awake and quietly in the Pavlov stand. Low intensity VNS (0.25 mA) evoked no change in basal chronotropic function. Increasing VNS intensity to 1.00 mA resulted in tachycardia during the on‐phase of VNS. Stepping the current to 1.75 mA dampened the sinus bradycardia with no change in average heart rate. Increasing the current to 3.00 mA induced the expected bradycardia during the on‐phase of VNS.
Figure 1. Chronotropic responses evoked during right cervical VNS in a conscious animal.
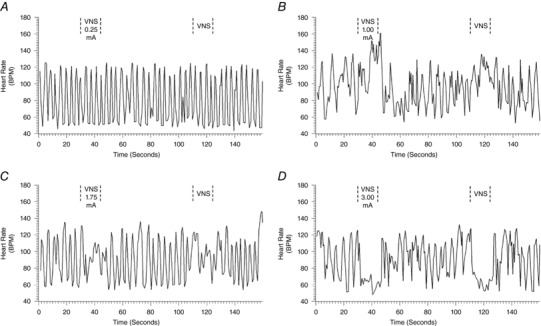
Frequency (10 Hz) and pulse width (500 μs) were held constant; duty cycle was 17.5% with a 14 s on‐phase. Current intensities were 0.25 mA (A), 1.00 mA (B), 1.75 mA (C) and 3.00 mA (D).
Average chronotropic responses (percentage change from baseline) in response to right‐sided (RCV) or left‐sided (LCV) bipolar VNS are summarized in Figs 2 and 3. For these animals (n = 8), the anode was cephalad to the cathode. The VNS stimulating protocol evaluated frequencies from 2 to 20 Hz at four different pulse widths (130 μs, Figs 2 A and 3 A; 250 μs, Figs 2 B and 3 B; 500 μs, Figs 2 C and 3 C; and 750 μs, Figs 2 D and 3 D) and with amplitude ranging from 0.25 to 3.50 mA. Across the entire response surface there were some commonalities: (i) the tachycardia responses during on‐phase VNS occurred preferentially at higher frequencies and lower amplitude; (ii) the response surfaces shifted dramatically going from 130 μs (Figs 2 A and 3 A) to 250 μs (Figs 2 B and 3B) pulse widths with little additional change going to 500 μs (Figs 2 C and 3C) or 750 μs (Figs 2 C and 3D) pulse widths; and (iii) there was a dynamic transition point (yellow shaded area) where the potential for evoked tachycardias changed to bradycardia. This transition zone we refer to as the neural fulcrum. Finally, while the same overall topographic characteristics of the VNS evoked chronotropic response surface was present to both right (Fig. 2) and left (Fig. 3) VNS, right‐sided VNS afforded a greater range of control with increased sensitivity to small changes in frequency or amplitude. Figure 4 A further details the concept of frequency–pulse width–amplitude interactions for RCV VNS where the bradycardic threshold (in mA) is plotted as a function of frequency at each of the four evaluated pulse widths. Bradycardia threshold was defined as a 5% decrease in heart rate during three consecutive VNS on‐phase stimulations. While the response curves to pulse widths of 130 μs and 250 μs were different from all others, there was no significant difference between the curves generated at 500 or 750 μs. It should also be noted that these response surfaces and thresholds were stable over time.
Figure 2. Chronotropic topographic response surface as a function of RCV VNS intensity (mA), frequency (Hz) and pulse width.
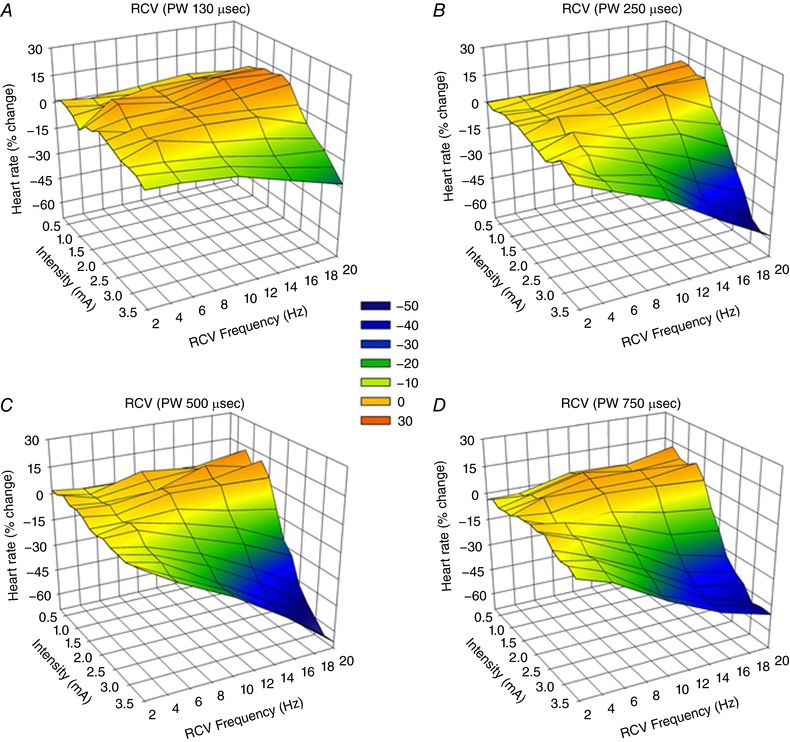
Animals were awake and standing quietly in Pavlov stand. VNS delivered at 17.5% duty cycle (14 s on‐phase). Bipolar electrodes were chronically implanted anode cephalad to cathode (n = 8). Pulse widths were 130 μs (A), 250 μs (B), 500 μs (C) and 750 μs (D). Heart rate depicted as percentage change from baseline during the on‐phase of VNS with magnitude indicated by colour scale.
Figure 3. Chronotropic topographic response surface as a function of LCV VNS intensity (mA), frequency (Hz) and pulse width.
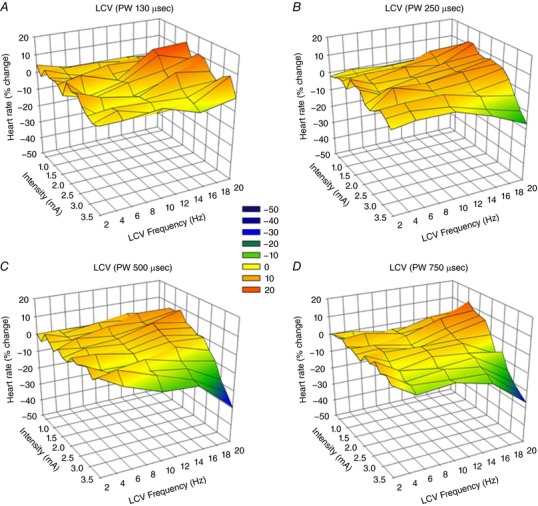
Animals were standing quietly in Pavlov stand. VNS delivered at 17.5% duty cycle. Bipolar electrodes were chronically implanted anode cephalad to cathode (n = 8). Pulse widths were 130 μs (A), 250 μs (B), 500 μs (C) and 750 μs (D).
Figure 4. Bradycardia threshold to RCV VNS as function of frequency, pulse width and electrode orientation.
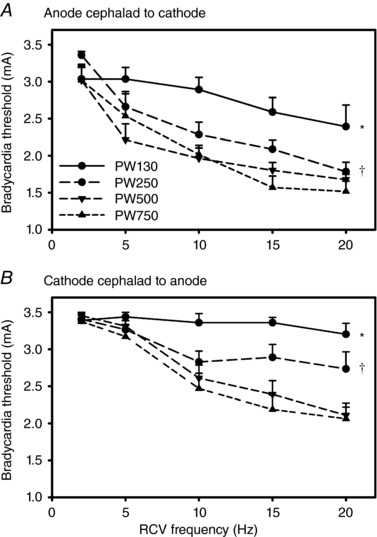
A, electrodes chronically implanted with anode cephalad to cathode (n = 8). B, electrodes chronically implanted with cathode cephalad to anode (n = 8). Pulse widths were 130 μs (PW130), 250 μs (PW250), 500 μs (PW500) or 750 μs (PW750). * P < 0.0005 PW130 vs. all other pulse widths; † P < 0.003 PW250 vs. PW500 or PW750.
Autonomic control of regional cardiac function demonstrates periodicities ranging from milliseconds to hours, rhythms that can be impacted by cardiac disease (Nolan et al. 1998; Zucker et al. 2012; Ardell et al. 2016). Figure 5 summarizes the circadian rhythm in HRV in animals with and without RCV VNS. Chronic VNS was delivered with a 22.5% duty cycle with a 14 s on‐time (n = 8). Four animals had RCV delivered at 10 Hz, 250 μs pulse width and with an intensity of 2.3 ± 0.2 mA. The remaining four animals had RCV delivered at 20 Hz, 500 μs pulse width and at an intensity of 1.9 ± 0.3 mA. These parameters were set as defined by achieving the null heart rate response during the on‐phase of VNS (see Fig. 1) while the animals were standing quietly in a Pavlov stand. Figure 5 shows the subsequent time domain profile as obtained by Holter monitor recording over 24–48 h time periods. Data are subdivided into consecutive 6 h segments starting at 18.00 h. Note the decrease in heart rate (Fig. 5 A) and increase in time domain indices of HRV (Fig. 5 B–D) as the animal's transition from evening to overnight. This pattern was reversed during the morning and afternoon time segments. RCV VNS, delivered within the constraints of the neural fulcrum, did not functionally disrupt the autonomic mechanisms underlying the circadian rhythm for any of the HRV parameters evaluated.
Figure 5. Time domain analysis of HRV in animals (n = 8) in response to cyclic RCV delivered at the neural fulcrum (grey bars) vs. the same animals with sham VNS (black bars).
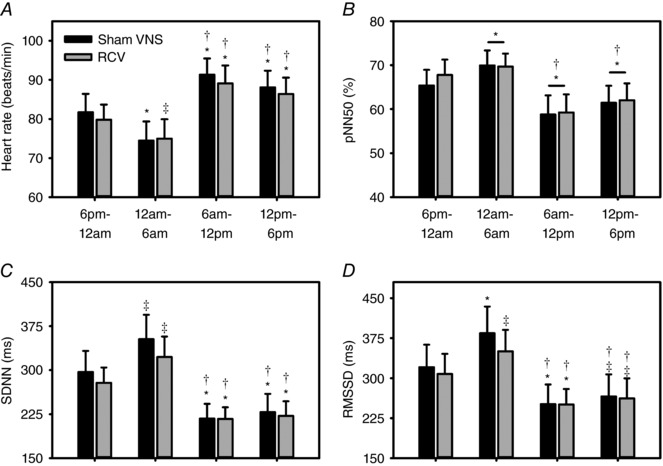
Data are subdivided into 6 h time segments starting at 18.00 h. Shown are heart rate (A), and indices of heart rate variability including pNN50 (%) (B), SDNN (C) and RMSSD (D). ‡ P ≤ 0.040 vs. 18.00–00.00 h; * P ≤ 0.010 vs. 18.00–00.00 h; and † P ≤ 0.001 vs. 00.00–06.00 h.
For bipolar stimulation of the vagi, the orientation of the cathode relative to the anode impacts the recruitment of the underlying fibres. It should also be considered that the cervical vagosympathetic truck contains nerve tracks running in opposite directions: afferent axons projecting cranial to the brainstem and efferent axons projecting caudal to the thorax and viscera. Figure 6 illustrates the response surfaces analogous to Figs 2 and 3 for cervical vagosympathetic stimulation, but in a separate group of animals (n = 8) where the cathode is cephalad to the anode. While the same general characteristics were maintained in this case, there was a more pronounced change in response surface going from 250 μs (Fig. 6 A and C) to 500 μs (Fig. 6 B and D) and the response surfaces were shifted upwards on the z‐axis (percentage change in heart rate). These same characteristics are evident in Fig. 4 B where the bradycardia threshold is plotted against RCV frequency, but with the bipole in the cathode cephalad to anode orientation. Note the increase in bradycardia threshold, especially at the shorter pulse widths (130 and 250 μs) and the significant shift in evoked response surface between pulse widths of 250 and 500 μs. Response surfaces and thresholds were stable over time.
Figure 6. Chronotropic topographic response surface as a function of RCV (A and B) or LCV VNS (C and D) intensity (mA), frequency (Hz) and pulse width.
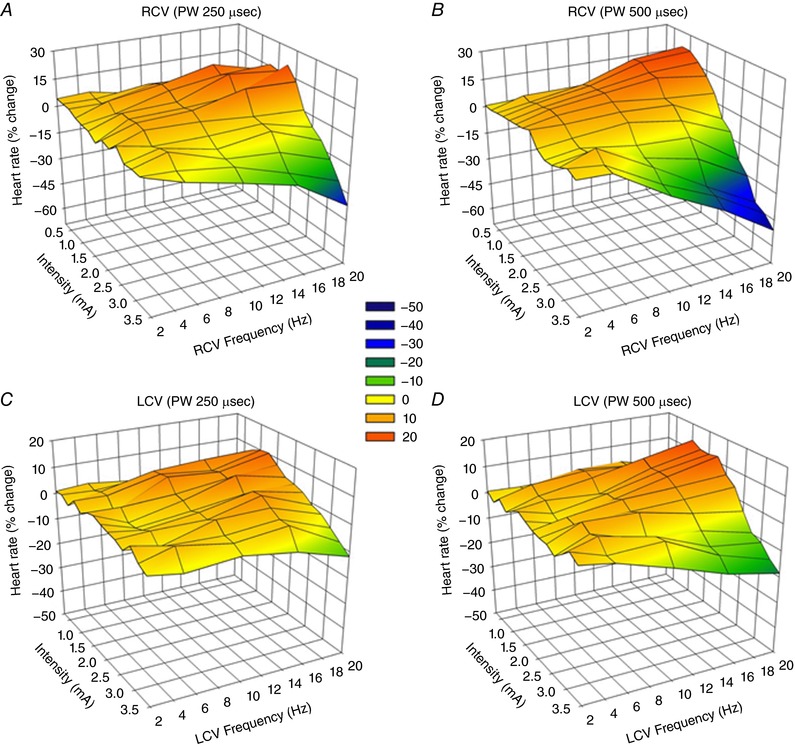
Bipolar electrodes were chronically implanted cathode cephalad to anode (n = 8). Other details as described in Fig. 2.
Pharmacological blockade – effects on VNS control of chronotropic function
Figure 7 summarizes the effects of acute and chronic treatments with cardiovascular relevant antagonists on the evoked chronotropic responses to graded right‐sided VNS. As discussed above and shown here, low intensity VNS was associated with tachycardia that transitioned to bradycardia at higher stimulus intensities. Figure 7 A shows the effects of metoprolol (1 mg kg−1, i.v.) on the response surface. Note that even with the high dose β‐adrenergic blockade the tachycardia phase to low amplitude VNS was maintained while the bradycardia to higher intensities of VNS was enhanced. That the bradycardia is mediated by muscarinic cholinergic receptors is indicated by the complete inhibition of bradycardia to VNS following glycopyrrolate (0.4 mg kg−1; Fig. 7 B). To evaluate the potential for longer term interactions between VNS and CHF relevant blockade, animals were treated twice daily for 2 weeks with a beta blocker (metoprolol, 12.25 mg), an ACE inhibitor (enalapril, 2.5 mg), a funny channel blocker (ivabradine, 5.0 mg) and combinations of them (metoprolol + enalapril or metoprolol + enalapril + ivabradine). Figure 7 C shows the average response curves for untreated animals (control), sole beta blockade (metoprolol) and all three blockers combined (metoprolol + enalapril + ivabradine). Overall, none of these agents interfered with the evoked chronotropic response to VNS.
Figure 7. Chronotropic response to RCV VNS (n = 8) prior to and 30 min following either β‐adrenergic blockade (metoprolol, 1 mg kg−1; A) or β‐adrenergic and muscarinic blockade (glycopyrrolate, 0.4 mg; B).
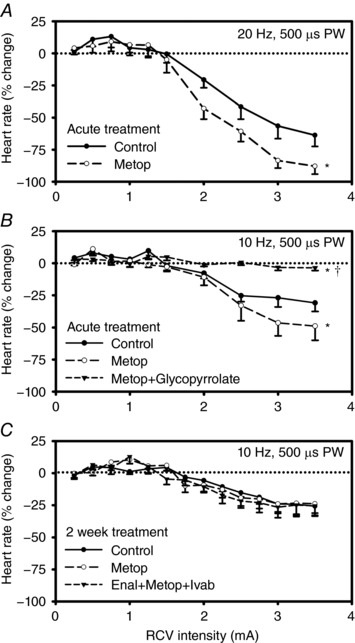
For chronic pharmacological treatment (n = 8), animals were administered for 2 weeks twice daily (PO) metoprolol (12.25 mg) or combination therapy with metoprolol, enalapril (2.5 mg) and ivabradine (5 mg). C, average data for the untreated condition (control), then following either metoprolol or combination therapy treatments with enalapril, metoprolol and ivabradine. * P < 0.002 vs. control; † P < 0.0001 metoprolol vs. metoprolol+glycopyrrolate.
Effects of anaesthesia on VNS chronotropic response surface
Figure 8 demonstrates the effects of graded VNS delivered in the awake state with the animals standing quietly in a Pavlov stand vs. the chronotropic response when VNS is delivered in the fully anaesthetized state, in this case with α‐chloralose. Shown are data from animals with the bipolar electrodes in the cathode cephalad to anode orientation (RCV, Fig. 8 A; LCV, Fig. 8 C; n = 8) and anode cephalad to cathode orientation (RCV, Fig. 8 B; LCV, Fig. 8 D; n = 8). Anaesthesia has the potential to alter neural control of the heart by acting at multiple levels of the cardiac neuraxis; this overall shift in response surface between awake and anaesthetized is evident for both nerves and for each bipole electrode orientation. The primary overall effect of α chloralose anaesthesia was to augment the bradycardias to higher intensity VNS.
Figure 8. Anaesthesia alters the chronotropic response to cervical VNS.
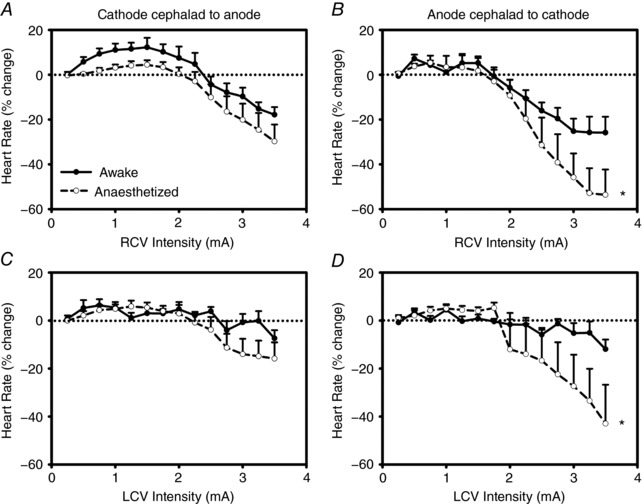
A and C reflect chronotropic responses to VNS with electrode bipole in cathode cephalad to anode orientation (n = 8); B and D reflect response surfaces with anode cephalad to cathode orientation (n = 8). While the response surfaces are similar including a tachycardia phase at low level intensity transitioning to bradycardia at higher intensities, there are quantitative differences including an enhanced bradycardia to higher intensity stimulations with the bipole in the anode cephalad to cathode orientation. Autonomic nerves intact in both states. * P < 0.0004 awake vs. anaesthetized.
Central–peripheral interactions during VNS for control of cardiac function
We have previously determined in preclinical studies that cervical vagal afferents inhibit centrally mediated parasympathetic efferent outflow (Ardell et al. 2015) and that chronic VNS can impact synaptic efficacy within the intrinsic cardiac nervous system (Beaumont et al. 2016). As we noted previously in acute animals (Ardell et al. 2015), Fig. 9 demonstrates that transection of the cervical vagosympathetic trunk, rostral to the stimulation site, eliminates the augmenting effects of VNS at lower stimulus intensities and shifts the entire VNS chronotropic response surface to the left (Fig. 9 A and B). The same findings apply to VNS control of inotropic (Fig. 9 C and D) and lusitropic (Fig. 9 E and F) function.
Figure 9. Evoked changes in chronotropic (A and B), left ventricular inotropic (C and D) and lusitropic (E and F) function in response right (left panels) and left (right panels) cervical VNS prior to and following cervical vagus transections rostral to stimulating electrode.
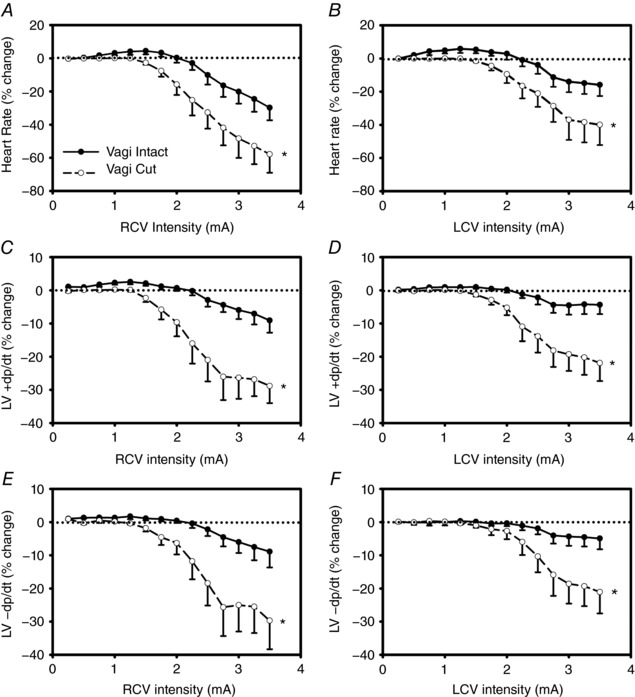
Animals were anaesthetized throughout. VNS delivered at 500 μs pulse width and a 17.5% duty cycle with a 14 s on‐phase. Responses reflect percentage change from baseline during VNS as a function of stimulus frequency. * P < 0.001 vs. intact.
Discussion
Vagus nerve stimulation is one of the primary tools in the evolving area of bioelectronic medicine (Bonaz et al. 2013). This study defines many of the critical structure–function factors that ultimately determine the efficacy of VNS to effectively modulate the neural network–heart interface and thereby impact disease progression linked to autonomic dysfunction. The primary findings of the study are the following: (i) the vagus and its projections can be ‘trained’ to VNS thereby reducing the potential for off‐target cough and GI discomfort and thereby maximizing the range of stimulation parameters that can be adjusted to effect cardiac control; (ii) the evoked cardiac response to bipolar cervical VNS reflects a dynamic interaction between afferent mediated decreases in central parasympathetic drive and suppressive effects evoked by direct stimulation of parasympathetic efferent axons to the heart; (iii) while cardiac control can be elicited from either vagus or bipole electrode configuration, lower intensities are required from the right‐sided cervical vagosympathetic nerve trunk with the bipole in the anode cephalad to cathode orientation; (iv) beta‐receptor blockade does not alter the tachycardia phase to low intensity VNS, but can increase the bradycardia to higher intensity VNS; (v) while muscarinic cholinergic blockade prevented the VNS‐induced bradycardia, clinically relevant doses of ACE inhibitors, beta‐blockade (Yancy et al. 2013, 2016) and the funny channel blocker ivabradine (Swedberg et al. 2010; Borer et al. 2016) did not alter the VNS chronotropic response; and (vi) while there are some qualitative difference in the VNS heart control between awake and anaesthetized states, the physiological expression of the neural fulcrum is maintained.
Structure–function considerations for VNS
Integrated cardiac control involves the dynamic interaction between intrathoracic and central reflexes (Ardell et al. 2016; Shivkumar et al. 2016). Intrathoracic reflexes include extracardiac (stellate/middle cervical) and intrinsic cardiac neural networks; central components include spinal cord, brainstem and higher centres (Ardell & Armour, 2016; Ardell et al. 2016). The cervical vagosympathetic truck is a major route for intrathoracic and visceral afferent projections to the medulla (Paintal, 1963, 1973; Williams et al. 2016) as well as parasympathetic preganglionic axons to the heart, lungs and visceral organs (Ardell & Armour, 2016). This region likewise contains a small number of sympathetic efferent projections and interspersed ganglia (Randall & Armour, 1974; Seki et al. 2014). Taken together, bioelectronic stimulation of cervical vagosympathetic trunk has the potential to alter intrathoracic and visceral organ function by multiple neural pathways, both directly by efferent activation and indirectly via afferent mediated changes in efferent outflows (Ardell et al. 2015; Yamakawa et al. 2015). If the control systems are ‘pushed’ in one direction by exogenous inputs (e.g. VNS), the endogenous reflexes ‘push back’ to maintain homeostasis (Kember et al. 2014).
Functional cardiac response to graded VNS
Autonomic control of regional cardiac function ultimately depends on the levels of sympathetic and parasympathetic activity and the interactions between them (Levy & Martin, 1979; Ardell & Armour, 2016). These interactions can take place within neural networks and at the end‐effectors themselves, including peripheral ganglia and at the nerve–myocyte interface (Ardell et al. 2016; Habecker et al. 2016). The fibre types being activated by cervical vagosympathetic trunk stimulation at the intensities evaluated here include myelinated afferents and parasympathetic efferents (Jewett, 1964; Yoo et al. 2013); unmyelinated fibres require stimulus intensities of ∼17 mA, perhaps 30 times higher than the stimulation intensity required to activate myelinated A or B fibres in close proximity to the cathodic electrode (Yoo et al. 2013). Confirming work from anaesthetized preparations (Ardell et al. 2015; Yamakawa et al. 2015), in conscious animals tachycardias are evoked at lower VNS intensities, especially at higher frequencies of stimulation, and bradycardias are manifest at higher VNS intensities. Higher level bradycardias during on‐phase VNS were followed by rebound tachycardias during the off‐phase of episodic VNS (data not shown). Rebound tachycardias likely reflect the endogenous autonomic control systems (e.g. baroreflex) responding to the hypotension accompanying the on‐phase bradycardias and corresponding decreases in blood pressure. As detailed in our previous publication in acute anaesthetized canine models (Ardell et al. 2015) and confirmed here, the tachycardia evident during the low intensity VNS and frequency ranges evaluated (2–20 Hz) likely reflect withdrawal of basal parasympathetic tone and not direct activation of sympathetic fibres contained within the cervical vagosympathetic trunk. Besides frequency and intensity, pulse width is a major determinant of axonal recruitment (Yoo et al. 2013; Arle et al. 2016). While only four different pulse widths were evaluated in this study, the data indicate that major changes in the evoked chronotropic response surface accompany increasing pulse widths from 130 to 250 μs. Increasing pulse widths above 250 μs did not fundamentally change the evoked cardiac response to incremental VNS. Bipole orientation is the last stimulation factor evaluated in this study. The cathode rostral to anode is the dipole configuration typically used in treatment of epilepsy (Morris & Mueller, 1999; Arle et al. 2016); anode rostral to cathode is the dipole configuration typically used in VNS for cardiac control (De Ferrari et al. 2014; Premchand et al. 2014). While cardiac control can be elicited from either vagus or bipole configuration, lower currents are required from the right cervical vagosympathetic nerve trunk with the bipole electrodes in the anode cranial to cathode orientation. The topography of the response surface also indicates that the anode cephalad orientation and right‐sided stimulation site affords the greatest flexibility in frequency–intensity control of evoked cardiac responses to VNS.
Pharmacological interactions with VNS
Standard of care pharmacological therapies for heart failure (Yancy et al. 2013, 2016) can impact autonomic control of the heart at multiple levels, including neural networks, the nerve–myocyte interface and at the end‐terminus on cardiac myocytes (e.g. second messenger interactions) (Armour, 2008; Ardell et al. 2016; Habecker et al. 2016). As expected, muscarinic blockade blocked the cardiac suppressor effects elicited by stimulation of parasympathetic preganglionic efferent axonal projections. In agreement with our previous results in the acute anaesthetized state (Ardell et al. 2015), beta adrenergic blockade at doses sufficient to block augmenting cardiac responses to direct activation of sympathetic post‐ganglionic nerves was ineffectual in preventing the tachycardia phase associated with higher frequencies and lower intensities of VNS delivered in the conscious state. Beta blockade did increase the bradycardia elicited by higher intensity stimulation, a response expected due to the mitigation of sympathetic–parasympathetic interactions at the heart (Levy & Martin, 1979; McGuirt et al. 1997). Chronic treatment (2 weeks) with clinically relevant doses of an ACE inhibitor (enalapril), beta blocker (metoprolol; Yancy et al. 2013; Yancy et al. 2016) and the funny channel blocker (ivabradine; Swedberg et al. 2010; Borer et al. 2016) did not alter the overall response to VNS, either alone or in any combination. Such is not the case for other relevant forms of bioelectronic therapies such as spinal cord stimulation where autonomic blockade can mitigate cardioprotection (Southerland et al. 2007, 2012). The minimal impact of drug therapy on VNS demonstrates that this therapy can be deployed as an adjunct to standard of care for heart failure over time and confirms safety.
Clinical relevance – VNS tolerance
Adverse effects of VNS often limit the therapeutic range that can be utilized for various indications including cardiac disease, epilepsy and depression (Bonaz et al. 2013). These off‐target effects include hoarseness, cough, GI discomfort, neck twitch and shortness of breath (Morris & Mueller, 1999; Rush & Siefert, 2009; Bonaz et al. 2013). We present data that demonstrate that the vagosympathetic trunk and its projections can be trained to VNS with the net result that the prevalence of cough and GI discomfort extinguish rapidly during the 4–6 week titration phase. While the precise neural mechanisms underlying this accommodation are unclear at this time, it is likely related to fibre selectivity resulting from the shorter pulse width stimuli. Once accommodated to the 130 μs pulse width VNS, step changes to the clinically relevant durations of greater than 250 μs were accomplished within 2–4 weeks and with intensities capable of exceeding the neural fulcrum at VNS frequencies from 2 to 20 Hz. It remains to be determined if this same protocol reduces other off‐target effects. In this regard, while off‐target adverse events are still evident in patients titrated with the accelerated VNS protocol (e.g. dysphonia, implant site pain, shoulder pain) the prevalence is reduced with time after implant (Premchand et al. 2014, 2016).
Clinical relevance – neural fulcrum
The neural fulcrum is defined as the operating point, based on frequency–amplitude–pulse width, where a null heart rate response is evoked during the on‐phase of VNS. The neural fulcrum represents a dynamic equilibrium between neural circuits rostral and caudal to the site of stimulation. As demonstrated here, and in agreement with previous work in the anaesthetized state (Ardell et al. 2015; Yamakawa et al. 2015), the potential for tachycardia induced by afferent activation and reflective of decreased central parasympathetic drive is counteracted by suppressing effects elicited by parasympathetic efferent projections to the heart. As is demonstrated in Fig. 5, delivery of cyclic VNS within the constraints of the neural fulcrum does not interfere with neural circuits underlying circadian variations in cardiac control. As demonstrated in Fig. 1, the fulcrum can be identified against a backdrop of sinus arrhythmia by damping in heart rate variability during the on‐phase VNS. The fulcrum point was stable over the average 14 months of investigation. To minimize environmental interference with establishing the neural fulcrum, titrations should take place in a quiet setting with the subject stationary. In the case of heart failure, against the backdrop of reduced central parasympathetic drive, the fulcrum can be identified by the stimulation protocol that induces minor (5% or less) bradycardias during the on‐phase of episodic VNS stimulation.
Multiple clinical trials have recently focused on the therapeutic efficacy of VNS in the setting of HFrEF, but with mixed results. Two of the studies failed to meet their clinical endpoints: INOVATE‐HF (Gold et al. 2016) and NECTAR‐HF (Zannad et al. 2015). Another study demonstrated efficacy: ANTHEM‐HF (Premchand et al. 2014, 2016). The three studies utilized different stimulation protocols and primarily relied on the right vagosympathetic nerve trunk as the target. Figure 10 is a replot of Fig. 2 C, but with the addition of the regions of the VNS chronotropic response surface that were utilized during the clinical studies referred to above. With the caveat that the magnitude of the response surface will likely vary between canine and man and the fact that the canine data are derived from a normal state as opposed to the altered autonomic state of heart failure, it is highly likely that the efficacy of VNS therapy was impacted by the constraints imposed by the choice of stimulation protocol. For the NECTAR‐HF design the goal was for VNS to be delivered at a 16.6% duty cycle (10 s on) at 20 Hz, pulse width of 300 μs and at an intensity to exceed 4 mA chronically (De Ferrari et al. 2014). However, at the end of the VNS titration, patients had a mean intensity of stimulation of 1.24 ± 0.74 mA (Zannad et al. 2015), a region corresponding to the tachycardia zone of the response surface. INOVATE‐HF utilized an intracardiac sensing lead with stimulation of the right cervical vagus synchronized to the cardiac cycle (Hauptman et al. 2012), with technology purported to produce block of ascending afferent input to limit off‐target effects (Anholt et al. 2011) and with an average intensity of 3.9 ± 1.0 mA (Gold et al. 2016) at the end of the 6 month follow‐up. As evident from Fig. 10, the topography of the VNS heart rate response is relatively flat over this range and with the potential complication of changes in effective gain function as the result of variable block of afferent inputs (see Fig. 9 for shift in response curve with interruption of afferent fibre projections). ANTHEM‐HF utilized the principal of the neural fulcrum at a frequency that corresponds to the synaptic efficacy for intrinsic cardiac neurons (Hardwick et al. 2008) and in the range of basal activity of cardiac‐related parasympathetic efferent projections to the heart (Jewett, 1964). Taken together, these data indicate that the characteristics of the electrode–nerve interface, the stimulation protocol utilized, and the disease process it is deployed against, are all relevant factors in determining the ultimate efficacy of VNS.
Figure 10. Clinical relevance of the VNS neural fulcrum.
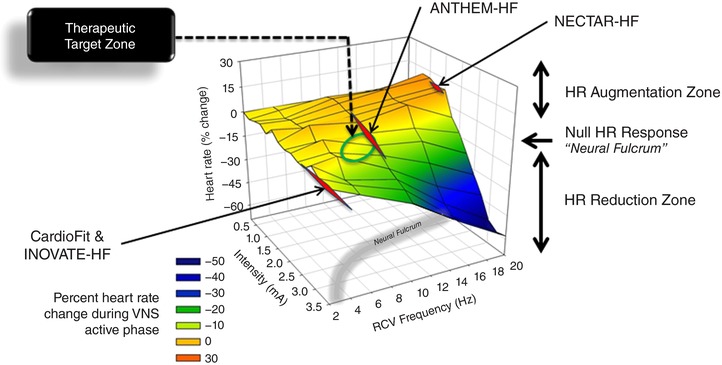
The chronotropic response surface is replotted from Fig. 2 C, but with the addition of specific points operationally used in three recent clinical trials for HFrEF: NECTAR‐HF (Zannad et al. 2015), INOVATE‐HF (Gold et al. 2016) and ANTHEM‐HF (Premchand et al. 2014, 2016). Yellow‐shaded region on response surface is approximately the neural fulcrum.
Study limitations
While this study evaluated many of the critical aspects that define the bioelectronic control of cardiac function via VNS, it should be recognized that cardiac pathology reorganizes autonomic control (Zucker et al. 2012; Ardell et al. 2016; Habecker et al. 2016) and thus will likely impact the response characteristics. At a given point in time, maintenance VNS was delivered unilaterally during this study and as a result there are qualitative differences in control between right and left sides of the heart (Randall et al. 1985). However, there are substantial overlaps in control of both right and left sides of the heart from either vagus (Yamakawa et al. 2014), such that a coordination resulting from neural network interactions within the intrinsic cardiac nervous system likely occurs (Ardell & Armour, 2016). The neural fulcrum was defined in a stable environment with the canine awake and standing quietly in a Pavlov stand with minimal changes in environment. It is likely that changes in arousal and behavioural stresses will alter the dynamics of bioelectronic control and should be evaluated in future studies. VNS was also delivered in an open loop configuration. Evolving designs for implantable programmable generators should consider adding cardiac event detection for dynamic closed‐loop of cardiac function using the principles of operating within the neural fulcrum. Finally, it is contingent on future study designs for VNS that structure–function of the cardiac nervous system be mechanistically leveraged to optimize protocols.
Additional information
Competing interests
None declared.
Author contributions
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines. J.L.A., B.K. and J.L.A. contributed to the conception and design of the experiments. H.N., E.B., E.M.S. and C.L.A. performed the experiments. H.N., J.L.A. and M.H. analysed the data. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
JLA has received grant support from Cyberonics. JLA and JAA served as scientific advisors to Cyberonics. BHK is an employee of Cyberonics Inc (LivaNova). The remaining authors have no conflicts of interest.
Acknowledgements
The authors acknowledge the technical assistance of Robin King, Shannon Ryan and Heather Johnson in animal husbandry and Dr Imad Libbus and Badri Amurthur in study design.
Linked articles This article is highlighted by a Perspective by Hsieh & Lewis. To read this Perspective, visit https://doi.org/10.1113/JP275204.
References
- Anholt TA, Ayal S & Goldberg JA (2011). Recruitment and blocking properties of the CardioFit stimulation lead. J Neural Eng 8, 034004. [DOI] [PubMed] [Google Scholar]
- Ardell JL, Andresen MC, Armour JA, Billman GE, Chen PS, Foreman RD, Herring N, O'Leary DS, Sabbah HN, Schultz HD, Sunagawa K & Zucker IH (2016). Translational neurocardiology: preclinical models and cardioneural integrative aspects. J Physiol 594, 3877–3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardell JL & Armour JA (2016). Neurocardiology: structure‐based function. Compr Physiol 6, 1635–1653. [DOI] [PubMed] [Google Scholar]
- Ardell JL, Rajendran PS, Nier HA, KenKnight BH & Armour JA (2015). Central‐peripheral neural network interactions evoked by vagus nerve stimulation: functional consequences on control of cardiac function. Am J Physiol Heart Circ Physiol 309, H1740–H1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arle JE, Carlson KW & Mei L (2016). Investigation of mechanisms of vagus nerve stimulation for seizure using finite element modeling. Epilepsy Res 126, 109–118. [DOI] [PubMed] [Google Scholar]
- Armour JA (2008). Potential clinical relevance of the ‘little brain’ on the mammalian heart. Exp Physiol 93, 165–176. [DOI] [PubMed] [Google Scholar]
- Beaumont E, Wright GL, Southerland EM, Li Y, Chui R, KenKnight BH, Armour JA & Ardell JL (2016). Vagus nerve stimulation mitigates intrinsic cardiac neuronal remodeling and cardiac hypertrophy induced by chronic pressure overload in guinea pig. Am J Physiol Heart Circ Physiol 310, H1349–H1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billman GE (2006). A comprehensive review and analysis of 25 years of data from an in vivo canine model of sudden cardiac death: implications for future anti‐arrhythmic drug development. Pharmacol Ther 111, 808–835. [DOI] [PubMed] [Google Scholar]
- Bonaz B, Picq C, Sinniger V, Mayol JF & Clarencon D (2013). Vagus nerve stimulation: from epilepsy to the cholinergic anti‐inflammatory pathway. Neurogastroenterol Motil 25, 208–221. [DOI] [PubMed] [Google Scholar]
- Borer JS, Deedwania PC, Kim JB & Bohm M (2016). Benefits of heart rate slowing with ivabradine in patients with systolic heart failure and coronary artery disease. Am J Cardiol 118, 1948–1953. [DOI] [PubMed] [Google Scholar]
- De Ferrari GM (2014). Vagal stimulation in heart failure. J Cardiovasc Transl Res 7, 310–320. [DOI] [PubMed] [Google Scholar]
- De Ferrari GM, Tuinenburg AE, Ruble S, Brugada J, Klein H, Butter C, Wright DJ, Schubert B, Solomon S, Meyer S, Stein K, Ramuzat A & Zannad F (2014). Rationale and study design of the NEuroCardiac Therapy for Heart Failure Study: NECTAR‐HF. Eur J Heart Fail 16, 692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florea VG & Cohn JN (2014). The autonomic nervous system and heart failure. Circ Res 114, 1815–1826. [DOI] [PubMed] [Google Scholar]
- Fukuda K, Kanazawa H, Aizawa Y, Ardell JL & Shivkumar K (2015). Cardiac innervation and sudden cardiac death. Circ Res 116, 2005–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MR, Van Veldhuisen DJ, Hauptman PJ, Borggrefe M, Kubo SH, Lieberman RA, Milasinovic G, Berman BJ, Djordjevic S, Neelagaru S, Schwartz PJ, Starling RC & Mann DL (2016). Vagus nerve stimulation for the treatment of heart failure: the INOVATE‐HF trial. J Am Coll Cardiol 68, 149–158. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 12, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habecker BA, Anderson ME, Birren SJ, Fukuda K, Herring N, Hoover DB, Kanazawa H, Paterson DJ & Ripplinger CM (2016). Molecular and cellular neurocardiology: development, and cellular and molecular adaptations to heart disease. J Physiol 594, 3853–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick JC, Southerland EM & Ardell JL (2008). Chronic myocardial infarction induces phenotypic and functional remodeling in the guinea pig cardiac plexus. Am J Physiol Regul Integr Comp Physiol 295, R1926–R1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauptman PJ, Schwartz PJ, Gold MR, Borggrefe M, Van Veldhuisen DJ, Starling RC & Mann DL (2012). Rationale and study design of the increase of vagal tone in heart failure study: INOVATE‐HF. Am Heart J 163, 954–962.e951. [DOI] [PubMed] [Google Scholar]
- Jewett DL (1964). Activity of single efferent fibres in the cervical vagus nerve of the dog, with special reference to possible cardio‐inhibitory fibres. J Physiol 175, 321–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kember G, Ardell JL, Armour JA & Zamir M (2014). Vagal nerve stimulation therapy: what is being stimulated? PLoS One 9, e114498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy MN & Martin PJ (1979). Neural control of the heart In Handbook of Physiology, Section 2, The Cardiovascular System, Vol. 1, The Heart, ed. Berne RM, pp. 581–620. The American Physiological Society, Bethesda. [Google Scholar]
- McGuirt AS, Schmacht DC & Ardell JL (1997). Autonomic interactions for control of atrial rate are maintained after SA nodal parasympathectomy. Am J Physiol Heart Circ Physiol 272, H2525–H2533. [DOI] [PubMed] [Google Scholar]
- Morris GL 3rd & Mueller WM (1999). Long‐term treatment with vagus nerve stimulation in patients with refractory epilepsy. The Vagus Nerve Stimulation Study Group E01‐E05. Neurology 53, 1731–1735. [DOI] [PubMed] [Google Scholar]
- Nolan J, Batin PD, Andrews R, Lindsay SJ, Brooksby P, Mullen M, Baig W, Flapan AD, Cowley A, Prescott RJ, Neilson JM & Fox KA (1998). Prospective study of heart rate variability and mortality in chronic heart failure: results of the United Kingdom heart failure evaluation and assessment of risk trial (UK‐Heart). Circulation 98, 1510–1516. [DOI] [PubMed] [Google Scholar]
- Paintal AS (1963). Vagal afferent fibres. Ergeb Physiol 52, 74–156. [PubMed] [Google Scholar]
- Paintal AS (1973). Vagal sensory receptors and their reflex effects. Physiol Rev 53, 159–227. [DOI] [PubMed] [Google Scholar]
- Premchand RK, Sharma K, Mittal S, Monteiro R, Dixit S, Libbus I, DiCarlo LA, Ardell JL, Rector TS, Amurthur B, KenKnight BH & Anand IS (2014). Autonomic regulation therapy via left or right cervical vagus nerve stimulation in patients with chronic heart failure: results of the ANTHEM‐HF trial. J Card Fail 20, 808–816. [DOI] [PubMed] [Google Scholar]
- Premchand RK, Sharma K, Mittal S, Monteiro R, Dixit S, Libbus I, DiCarlo LA, Ardell JL, Rector TS, Amurthur B, KenKnight BH & Anand IS (2016). Extended follow‐up of patients with heart failure receiving autonomic regulation therapy in the ANTHEM‐HF study. J Card Fail 22, 639–642. [DOI] [PubMed] [Google Scholar]
- Randall WC, Ardell JL & Becker DM (1985). Differential responses accompanying sequential stimulation and ablation of vagal branches to dog heart. Am J Physiol Heart Circ Physiol 249, H133–H140. [DOI] [PubMed] [Google Scholar]
- Randall WC & Armour JA (1974). Regional vagosympathetic control of the heart. Am J Physiol 227, 444–452. [DOI] [PubMed] [Google Scholar]
- Rush AJ & Siefert SE (2009). Clinical issues in considering vagus nerve stimulation for treatment‐resistant depression. Exp Neurol 219, 36–43. [DOI] [PubMed] [Google Scholar]
- Sabbah HN, Ilsar I, Zaretsky A, Rastogi S, Wang M & Gupta RC (2011). Vagus nerve stimulation in experimental heart failure. Heart Fail Rev 16, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salavatian S, Beaumont E, Longpre JP, Armour JA, Vinet A, Jacquemet V, Shivkumar K & Ardell JL (2016). Vagal stimulation targets select populations of intrinsic cardiac neurons to control neurally induced atrial fibrillation. Am J Physiol Heart Circ Physiol 311, H1311–H1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki A, Green HR, Lee TD, Hong L, Tan J, Vinters HV, Chen PS & Fishbein MC (2014). Sympathetic nerve fibers in human cervical and thoracic vagus nerves. Heart Rhythm 11, 1411–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinlapawittayatorn K, Chinda K, Palee S, Surinkaew S, Thunsiri K, Weerateerangkul P, Chattipakorn S, KenKnight BH & Chattipakorn N (2013). Low‐amplitude, left vagus nerve stimulation significantly attenuates ventricular dysfunction and infarct size through prevention of mitochondrial dysfunction during acute ischemia‐reperfusion injury. Heart Rhythm 10, 1700–1707. [DOI] [PubMed] [Google Scholar]
- Shivkumar K, Ajijola OA, Anand I, Armour JA, Chen PS, Esler M, De Ferrari GM, Fishbein MC, Goldberger JJ, Harper RM, Joyner MJ, Khalsa SS, Kumar R, Lane R, Mahajan A, Po S, Schwartz PJ, Somers VK, Valderrabano M, Vaseghi M & Zipes DP (2016). Clinical neurocardiology defining the value of neuroscience‐based cardiovascular therapeutics. J Physiol 594, 3911–3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southerland EM, Gibbons DD, Smith SB, Sipe A, Williams CA, Beaumont E, Armour JA, Foreman RD & Ardell JL (2012). Activated cranial cervical cord neurons affect left ventricular infarct size and the potential for sudden cardiac death. Auton Neurosci 169, 34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southerland EM, Milhorn DM, Foreman RD, Linderoth B, DeJongste MJ, Armour JA, Subramanian V, Singh M, Singh K & Ardell JL (2007). Preemptive, but not reactive, spinal cord stimulation mitigates transient ischemia‐induced myocardial infarction via cardiac adrenergic neurons. Am J Physiol Heart Circ Physiol 292, H311–H317. [DOI] [PubMed] [Google Scholar]
- Stavrakis S, Humphrey MB, Scherlag BJ, Hu Y, Jackman WM, Nakagawa H, Lockwood D, Lazzara R & Po SS (2015). Low‐level transcutaneous electrical vagus nerve stimulation suppresses atrial fibrillation. J Am Coll Cardiol 65, 867–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swedberg K, Komajda M, Bohm M, Borer JS, Ford I, Dubost‐Brama A, Lerebours G, Tavazzi L & Investigators S (2010). Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo‐controlled study. Lancet 376, 875–885. [DOI] [PubMed] [Google Scholar]
- Wang HJ, Wang W, Cornish KG, Rozanski GJ & Zucker IH (2014). Cardiac sympathetic afferent denervation attenuates cardiac remodeling and improves cardiovascular dysfunction in rats with heart failure. Hypertension 64, 745–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams EK, Chang RB, Strochlic DE, Umans BD, Lowell BB & Liberles SD (2016). Sensory neurons that detect stretch and nutrients in the digestive system. Cell 166, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakawa K, Howard‐Quijano K, Zhou W, Rajendran P, Yagishita D, Vaseghi M, Ajijola OA, Armour JA, Shivkumar K, Ardell JL & Mahajan A (2016). Central vs. peripheral neuraxial sympathetic control of porcine ventricular electrophysiology. Am J Physiol Regul Integr Comp Physiol 310, R414–R421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakawa K, Rajendran PS, Takamiya T, Yagishita D, So EL, Mahajan A, Shivkumar K & Vaseghi M (2015). Vagal nerve stimulation activates vagal afferent fibers that reduce cardiac efferent parasympathetic effects. Am J Physiol Heart Circ Physiol 309, H1579–H1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakawa K, So EL, Rajendran PS, Hoang JD, Makkar N, Mahajan A, Shivkumar K & Vaseghi M (2014). Electrophysiological effects of right and left vagal nerve stimulation on the ventricular myocardium. Am J Physiol Heart Circ Physiol 307, H722–H731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Colvin MM, Drazner MH, Filippatos G, Fonarow GC, Givertz MM, Hollenberg SM, Lindenfeld J, Masoudi FA, McBride PE, Peterson PN, Stevenson LW & Westlake C (2016). 2016 ACC/AHA/HFSA focused update on new pharmacological therapy for heart failure: an update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. J Am Coll Cardiol 68, 1476–1488. [DOI] [PubMed] [Google Scholar]
- Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines (2013). 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 62, e147–e239. [DOI] [PubMed] [Google Scholar]
- Yoo PB, Lubock NB, Hincapie JG, Ruble SB, Hamann JJ & Grill WM (2013). High‐resolution measurement of electrically‐evoked vagus nerve activity in the anesthetized dog. J Neural Eng 10, 026003. [DOI] [PubMed] [Google Scholar]
- Zannad F, De Ferrari GM, Tuinenburg AE, Wright D, Brugada J, Butter C, Klein H, Stolen C, Meyer S, Stein KM, Ramuzat A, Schubert B, Daum D, Neuzil P, Botman C, Castel MA, D'Onofrio A, Solomon SD, Wold N & Ruble SB (2015). Chronic vagal stimulation for the treatment of low ejection fraction heart failure: results of the NEural Cardiac TherApy foR Heart Failure (NECTAR‐HF) randomized controlled trial. Eur Heart J 36, 425–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker IH, Patel KP & Schultz HD (2012). Neurohumoral stimulation. Heart Fail Clin 8, 87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]