

Abstract
Osteoclastogenesis is essential for bone remodeling and normal skeletal maintenance. RANKL promotes osteoclast differentiation and function but requires costimulation of immunoreceptor tyrosine-based activation motif (ITAM)-coupled immunoreceptors. Triggering receptor expressed on myeloid cells-2 (TREM2) coupled to ITAM-adaptor protein DNAX activation protein 12kDA (DAP12) provides costimulation of intracellular calcium signaling during osteoclastogenesis. Previously, we found that downstream of kinase-3 (DOK3) physically associates with DAP12 to inhibit toll-like receptor (TLR)-induced inflammatory signaling in macrophages. However, whether and how DOK3 modulates DAP12-dependent osteoclastogenesis is unknown and the focus of this study. Bone micro-architecture and histology of sex and age matched WT and DOK3-deficient (DOK3-/-) mice were evaluated. Male and female DOK3-/- mice have significantly reduced trabecular bone mass compared to WT mice with increased TRAP+ osteoclasts in vivo. In vitro, DOK3-/- BMMs have increased M-CSF induced proliferation and increased sensitivity to RANKL-induced osteoclastogenesis. Compared to WT, DOK3-/- osteoclasts are significantly larger with more nuclei and have increased resorptive capacity. Mechanistically, DOK3 limits osteoclastogenesis by inhibiting activation of Syk and ERK in response to RANKL and M-CSF. DOK3 is phosphorylated in a DAP12-dependent manner and associates with Grb2 and Cbl. Compared to DAP12-/- mice with high bone mass, DOK3- and DAP12- doubly deficient mice (DKO) have normalized bone mass, indicating that DOK3 also limits DAP12-independent osteoclastogenesis in vivo. In vitro osteoclasts derived from DKO mice are mononuclear with poor resorptive capacity similar to DAP12-/- osteoclasts. Histomorphometry reveals that DOK3-/- mice also have reduced osteoblast parameters. DOK3-/- osteoblasts have reduced in vitro osteoblastogenesis and increased OPG to RANKL expression ratio compared to WT osteoblasts. Co-culture of WT and DOK3-/- osteoblasts with pre-osteoclasts reveals a reduced capacity of DOK3-/- osteoblasts to support osteoclastogenesis. These data indicate that DOK3 regulates bone remodeling by negatively regulating M-CSF and RANKL mediated osteoclastogenesis and positively regulating osteoblastogenesis.
Keywords: DOK3, TREM2, DAP12, M-CSF, RANKL
Introduction
Osteoclasts are derived from hematopoietic myeloid precursors in response to stimulation with M-CSF and RANKL. ITAM-adaptor proteins, including DAP12 and Fc Receptor common γ (FcRγ), costimulate RANKL-induced osteoclastogenesis (1,2). DAP12 also participates in M-CSF stimulated expansion and survival of BMMs (3). TREM2, a pattern recognition receptor, couples to DAP12 and mediates osteoclast multi-nucleation, migration, survival, and resorption (4). DAP12 also participates in intracellular signaling with a variety of other cell surface receptors including integrins and TLRs (5,6). TREM2 engagement leads to DAP12 ITAM activation by Src kinases and induces the recruitment and activation of cytoplasmic Syk protein tyrosine kinase as well as PI3K (7). Syk subsequently activates downstream effectors, including PLC-γ, VAV guanine nucleotide exchange factor, PI3K, and ERK (5,7). Additionally, DAP12 associates with SH2-domain inositol phosphatase-1 (SHIP1) in osteoclasts to mediate inhibitory ITAM signals induced by TREM2 crosslinking or M-CSF (7). However, there are limited studies on other molecular mechanisms regulating DAP12 signaling in osteoclasts.
DOK proteins are a family of adaptors that provide docking platforms for the assembly of multi- molecular signaling complexes and function to limit tyrosine kinase-mediated signaling downstream of numerous receptors (8). These adaptors have motifs and domains capable of mediating protein-protein or protein-lipid interactions including an N-terminal pleckstrin homology (PH) domain, a phosphotyrosine-binding (PTB) domain, and four tyrosine-containing motifs that bind to Src homology 2 (SH2) domains within other proteins (8). DOK1, DOK2 and DOK3 triple-deficient mice develop lung cancer and histiocytic sarcomas (9). DOK1 and DOK2 double-deficient mice have osteopenia with increased numbers of osteoclasts due to enhanced sensitivity to M-CSF but the molecular mechanism is unknown (10). We recently determined that DOK3 is required for inhibition of TLR-induced cytokine production downstream of TREM2 and DAP 12 in macrophages (6); therefore we hypothesized that DOK3 negatively regulates DAP 12 ITAM signaling in osteoclasts. To test this hypothesis, we used mice deficient in DOK3 to evaluate bone remodeling in vivo and osteoclastogenesis in vitro. Identifying negative regulators of osteoclastogenesis may provide new treatment options for osteoporosis or inflammatory arthritis.
Materials and Methods
Mice
129S1/SvImJ mice were housed at the Oklahoma Medical Research Foundation (OMRF). DOK3-/-129S1/SvImJ mice were kindly provided by Dr. Pier Paolo Pandolfi (Departments of Medicine and Pathology, Harvard Medical School) (11). DAP12-deficient (DAP12-/-) mice on a C57BL/6J background were provided by R. McEver (OMRF). Age and sex matched mice were used for in vivo bone remodeling studies. DAP12-/- and DOK3-/- mice were crossed to generate homozygous DAP12-/-DOK3-/-. These mice were backcrossed five generations onto C57Bl/6. Heterozygous DAP12+/-DOK3+/- mice were bred to obtain littermate controls for osteoclast studies and for microCT studies. Mice were socially housed with standard mouse chow. Animals were maintained according to the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care at OMRF with approval from IACUC committees at OMRF and the University of Oklahoma Health Science Center (OUHSC).
Reagents
Antibodies against the following proteins were used in this study: DOK3, C-Cbl, and Grb2 were from Santa Cruz Biotechnology (Santa Cruz, CA, USA); Phospho-ERK (pThr202/pTry204), ERK (pan ERK), Phospho-p38 MAPK (Thr180/Tyr182), p38 MAPK (pan p38 MAPK), Phospho-Syk (Tyr525/526), Syk (panSyk), IκBα, cyclin D1 antibody were from Cell Signaling Technology (Danvers, MA, USA) ; Anti-phosphotyrosine monoclonal antibody G410 was purchased from EMD Millipore (Billerica, MA, USA). TRAP staining kit was purchased from Sigma (St. Louis, MO, USA). Phospho-Dap12 rabbit antibody was previously described (6). Murine M-CSF and RANKL were obtained from Peprotech (Rocky Hill, NJ, USA). The enhanced chemiluminescence (ECL) was purchased from Thermo Scientific (Rockford, IL, USA). Tissue culture media and fetal bovine serum (FBS) were obtained from Life Technologies (Grand Island, NY, USA). MTT (3-(4, 5-dimethylthiazole-2-yl)-2, 5-dipenyltetrazolium bromide) cell proliferation assay kit was obtained from ATCC (Manassas, VA, USA).
Macrophage cell culture
The RAW264.7 macrophage cell line was purchased from the American Type Culture Collection and maintained in α-minimum essential medium (αMEM). For primary bone marrow derived macrophage (BMM) cultures, bone marrow was flushed from mouse long bones as previously described (7). After red cell lysis, mononuclear cells were cultured in complete α-MEM supplemented with 10% embryonic stem cell quality fetal bovine serum, 1% glutamine, Pen-Strep, and 5% CMG (conditioned medium containing ∼100 ng/ml M-CSF). After 2 days, non-adherent BMMs were collected and transferred at a density of 5 × 106 cells/10cm to tissue culture dishes and were cultured for an additional 2 to 5 days. BMMs were maintained in αMEM supplemented with 10% FBS, 5% CMG, penicillin, streptomycin, and glutamine. Osteoclasts were derived from BMMs in complete α-MEM with RANKL and M-CSF for an additional 3 to 5 days.
Osteoclast assays
BMMs were plated at 50,000 cells/well in 96-well dishes and treated with RANKL (5∼50 ng/ml) and M-CSF (20 ng/ml) for 5 days with media changed every 2 days. After 5 days, cells were fixed, stained for tartrate resistant acid phosphatase (TRAP) (Sigma), and multinucleated (≥3 nuclei), TRAP positive cells were counted. For resorption, 1 × 105 macrophages were seeded on BD BioCoat Osteologic Discs (BD Biosciences) or dentine slices and treated with RANKL (50 ng/ml) and M-CSF (20 ng/ml) for 10 days. Media was changed every 3 days. Resorption pits were analyzed via light microscopy and digital images recorded. The resorption area was determined by Metamorph Analysis software (Molecular Devices). For osteoclastic actin assay, 5.0 × 104 BMMs were seeded on slide chamber and treated with RANKL (50 ng/ml) and M-CSF (20 ng/ml) for 5 days. Then, the cells were fixed and stained with Alexa Fluor® 488 Phalloidin. Cells were stained with DAPI to reveal the nuclei. Osteoclastic actin rings were analyzed via confocal immunofluorescence microscopy and digital images recorded.
BMM proliferation assay
MTT cell proliferation assay was performed according to the instruction of the kit. WT or DOK3-/- BMM cells were seeded onto 96 well plate and cell numbers quantified after 24 hours stimulation with 20ng/ml M-CSF.
In vitro osteoblast cultures and co-cultures
Whole bone marrow and minced pieces of long bones were plated on 100-mm tissue culture plates in αMEM containing 10% FBS, 1% penicillin/streptomycin. After 7 days, cells were trypsin digested and 1 × 104 /cm2 cells were plated in 6 well plates in the presence of osteogenic medium [αMEM containing 10% FBS, 1% penicillin/streptomycin, 10 mM β-glycerophosphate, and 50 μM L-ascorbic acid-2-phosphate]. Media was exchanged every 3 days for up to 28 days. Alizarin red staining was performed following manufacturer's instructions (Sigma). For co-culture, confluent osteoblasts were split and 8000 osteoblasts/ well of 96 well plates were plated in αMEM containing 10% FBS, 1% penicillin/streptomycin, 1μM prostaglandin E2, and 10 nM 1,25-(OH)2D3). When confluent, 15,000 day 2, M-CSF stimulated non-adherent BMM were added per well. 50% media was refreshed every 3 days for 8 days.
RNA Isolation and Real-Time PCR
Total RNA was isolated from cultured osteoblasts according the RNeasy Mini Kit (Qiagen). Quantitative PCR was performed with a Quantitect SYBR Green PCR Kit (Bio-Rad), according to the manufacturers' instructions, and with a RealTime PCR System with analytical software (Opticon Monitor 3). The following primers were used: Runx2, 5′-TACCAGCCACCGAGACCAA-3′ and 5′-AGAGGCTGTTTGACGCCATAG-3′; Osterix, 5′-GGTCCAGGCAACACACCTAC-3′ and 5′ -GGTAGGGAGCTGGGTTAAG -3′; Col1a, 5′ -CTGGCGGTTCAGGTCCAAT-3′ and 5′-TTCCAGGCAATCCACGAGC-3′; ALP, 5′-TCAGGGCAATGAGGTCACATC-3′ and 5′-TCACAATGCCCACGGACTT-3′; GAPDH, 5′-AACAGCAACTCCCACTCTTC-3′ and 5′-CCTGTTGCTGTAGCCGTATT-3′; RANKL, 5′-AGCCATTTGCACACCTCAC-3′ and 5′-CGTGGTACCAAGAGGACAGAGT-3′; Osteoprotegerin (OPG), 5′-GTTTCCCGAGGACCACAAT-3′ and 5′-CCATTCAATGATGTCCAGGAG-3′
DNA plasmids construction and transfection
The plasmid pCMV-FLAG-DOK3 was constructed by subcloning complementary DNA encoding mouse DOK3 into pCMV-FLAG (Sigma) (7). Transfection of BMM with plasmids pCMV-FLAG-DOK3 or pCMV-FLAG was performed by nucleofection following the appropriate Amaxa protocol.
Immunoprecipitation
Stimulated cells were washed with PBS, lysed with ice-cold radioimmunoprecipitation assay (RIPA) buffer for 10 minutes and centrifuged at 14,000 rpm for 15 minutes at 4°C (2). The supernatants were collected and pre-cleared with protein A/G agarose bead for 30 minutes. Immunoprecipitations were performed by incubating the pre-cleared cell lysates with primary antibody at 4°C with rotation overnight followed by protein A or G beads at 4°C for 1 h with rotation. The beads were washed extensively and the immunoprecipitation complexes were dissolved in sample buffer for SDS-PAGE. The proteins were transferred to PVDF membrane, immunoblotted with the appropriate antibodies, and visualized with the enhanced chemiluminescence (ECL) system (Pierce).
Micro-computed tomography (μCT) and Histology
The right proximal tibias were scanned in a Scanco vivaCT 40 μCT scanner (Scanco Medical, Bassersdorf, Switzerland) according to our published protocol following published guidelines (12,13). A total of 100 consecutive 10.5-μm-thick sections of cancellous bone were semi- automatically analyzed. The segmentation values were set at 0.8/1/220 for thresholding. Three-dimensional reconstruction and structural parameters quantification were calculated using Scanco Medical software, including Bone volume (BV; mm3), Bone volume expressed per unit of total volume (BV/TV; %), trabecular number (Tb.N; 1/mm), trabecular thickness (Tb.Th; mm), trabecular spacing (Tb.Sp; mm), connectivity density (Conn. D; 1/mm3), Structure model index (SMI). Following microCT, bones were decalcified for 10 days in 10% EDTA, paraffin embedded, sectioned and stained for TRAP. TRAP+ osteoclasts on the bone surface were counted from 5-7 slices per bone from n = 3 each genotype.
Histomorphometry
Mouse femurs were excised, fixed in 70% ethanol, and embedded in methylmethacrylate (MMA). Longitudinal sections (5 μm thick) were cut at the 50% plane using a Leica 2265 microtome. Sections were stained with Goldner's Trichrome for the static measurements, and additional sections were cut at 10 μm, and left unstained for dynamic (fluorescent) measurements. A region of interest was selected that is exactly 250 μm distal to the growth plate, and extending 1 mm downward (thereby avoiding the primary spongiosa) through the metaphysis of the femur. Standard bone histomorphometry was performed by the methods adopted by the ASBMR (16) using Bioquant Image Analysis software (R & M Biometrics, Nashville, TN).
Statistical analysis
All statistical analyses were analyzed using GraphPad Prism 7.00 (GraphPad software, CA). Statistical values were calculated using a Student's t-test or one way ANOVA as indicated, *p < 0.05, **p < 0.01, n.s: no significance. Error bars indicate the mean ± SD of triplicates.
Results
DOK3-deficient mice are osteoporotic
DOK3-/- mice have normal growth with similar body length and weight as sex and aged matched mice at 3 and 6 months (Fig. S1). To evaluate the in vivo functional role of DOK3, we analyzed the bone phenotype of 16-week-old male and female WT and DOK3-/- mice by microCT and histology. Global deletion of DOK3, led to significantly decreased bone mass (BV/TV) at both the proximal tibia and the distal femur in both sexes indicating osteoporosis (Fig. 1A-B). Significant decreases in trabecular number (Tb.N), thickness (Tb.Th), and connection density (Conn.Dens) were found. As expected with an osteoporotic phenotype, trabecular spacing (Tb.Sp) increased and the structural model index (SMI) changed from a plate-like to a rod-like structure consistent with loss of trabeculae in the DOK3 -/- bones. 3D reconstructions of microCT tibia data and 2D histological evaluation of mouse tibias confirmed osteoporosis with decreases in trabeculae and preservation of cortical bone (Fig. 1C). TRAP staining of tibias from WT and DOK3-/- mice revealed an increase in TRAP+ osteoclasts lining the bone (Fig. 1D). Histomorphometric measurements of WT and DOK3-/- female tibias confirmed osteoporosis with fewer trabeculae and increased spacing in the absence of DOK3. Histomorphometric measurements of female WT and DOK3-/- tibias confirmed osteoporosis (Fig. S1A). Histomorphometric cellular analysis revealed similar numbers of osteoclasts, osteoclast surface area, and osteoclast perimeter between WT and DOK3-/- but significantly reduced osteoblast numbers and osteoblast perimeter (Fig. S1B-C). These data indicate that DOK3 is required for normal bone homeostasis and in its absence mice develop osteoporosis.
Figure 1. DOK3-deficient mice are osteoporotic.
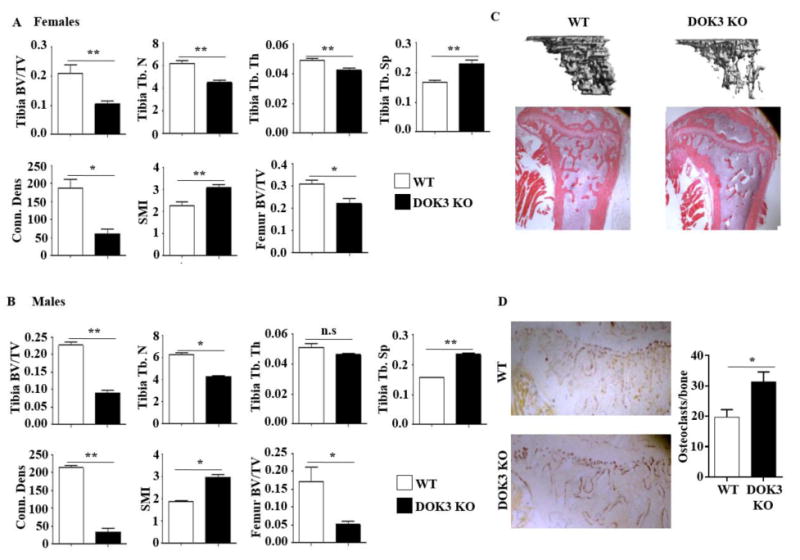
(A and B) Proximal tibias and distal femurs of 16-week-old female (A) or male (B) WT or DOK3-/- mice were scanned by microCT (n = 7∼9 each sex and genotype). Reconstructed tibias were analyzed for bone volume per tissue volume (BV/TV), trabecular number (Tb. N), trabecular thickness (Tb. Th), trabecular spacing (Tb. Sp), connection density (Conn. Dens), and structural model index (SMI). (C) Representative images of microCT reconstruction of proximal tibia (top) and H&E bone histology of 16-week-old male mice (bottom, original magnification: 20×). (D) TRAP stained tibia bones sections. TRAP+cells attached to bone were quantified. Data representative of >75 images quantified. n = 4 mice per group, original magnification: 40×. Statistical values were calculated using a Student's t-test unless otherwise indicated, *p < 0.05, **p < 0.01, n.s: no significance. Error bars indicate the mean ± SD.
DOK3 is required to limit osteoclast differentiation and resorption
The decreased bone mass in vivo suggests changes in both osteoclast and osteoblast differentiation and/or function. To further characterize the specific effect of DOK3-deletion on osteoclasts, we isolated BMM from WT and DOK3-/- mice and differentiated osteoclasts in vitro with increasing amounts of RANKL. Compared to WT, DOK3-/- BMM had increased sensitivity to RANKL with significantly increased osteoclastogenesis across RANKL concentrations (Fig. 2A-B). Even at low RANKL (5 ng/ml), DOK3-/- osteoclasts were much larger and complex compared to WT. Actin rings were comparable between WT and DOK3-/- osteoclasts and served as a visible indicator of the very large size and increased multinucleation of DOK3-/- osteoclasts (Fig. 2C). To confirm that the DOK3-/- osteoclast phenotype is due to loss of DOK3 and not off target effects, we reconstituted DOK3 in WT and DOK3-/- pre-osteoclasts. Pre-osteoclasts were transfected with either control or FLAG tagged -DOK3 expression plasmids and differentiated into osteoclasts with RANKL and M-CSF (Fig. 2D-E). Compared to WT, DOK3-/- pre-osteoclasts transfected with control plasmid maintained their enhanced osteoclastogenesis. However, overexpressing DOK3 in WT or DOK3-/- pre-osteoclasts significantly reduced osteoclastogenesis indicating that DOK3 inhibits osteoclastogenesis. To determine if DOK3 inhibits osteoclast function, DOK3-/- osteoclasts were generated on artificial calcium phosphate substrates and resorptive capacity evaluated. DOK3-/- osteoclasts had significantly increased resorptive capacity compared to WT cultures (Fig. S2). To quantify individual osteoclast resorptive capacity, day 3 osteoclasts were lifted and plated in equal numbers on an artificial calcium-phosphate substrate and dentine (Fig. 2F-H) Functionally, DOK3-/- osteoclasts had significantly increased percent resorption coupled with an overall increase in pit area per osteoclast indicating increased resorption capacity per osteoclast (Fig. 2F-H). Together, these data show DOK3 is essential for inhibiting osteoclast formation and function both in vitro.
Figure 2. DOK3 is required to limit osteoclast differentiation and function.
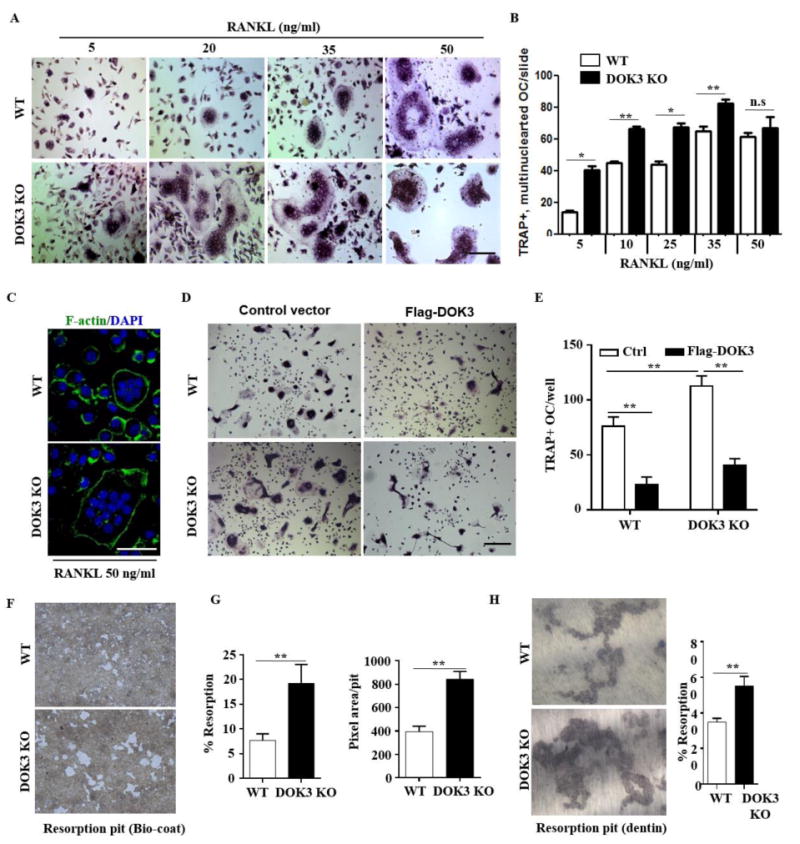
(A and B) BMMs were proliferated for 2 days 20 ng/ml M-CSF and equal numbers of non-adherent pre-osteoclasts were plated and differentiation was induced with 20 ng/ml M-CSF and increasing concentrations of RANKL 5-50 ng/ml for 5 days. Cells were fixed and stained for TRAP. Representative images were captured by light microscopy (A) and TRAP+ multinucleated (≥3 nuclei) osteoclasts quantified (B). (C) Osteoclasts derived in the presence of 20 ng/ml M-CSF and 50 ng/ml RANKL were fixed and permeabilized followed by staining with Alexa Fluor® 488 phalloidin to visualize actin rings. (D and E) WT or DOK3-/- BMMs were transfected with control or Flag-DOK3 expression plasmids and differentiated with 20 ng/ml M-CSF and 50 ng/ml RANKL on tissue culture plastic. Cells were fixed and stained for TRAP. Representative images were captured by light microscopy (D) and TRAP-positive stained cells (≥ 3 nuclei) were quantified as osteoclasts (E). (F-H) WT or DOK3-/- BMMs were plated and differentiated with RANKL 50 ng/ml and M-CSF 20 ng/ml for 3 days, lifted and plated in equal numbers onto BD Biocoat calcium-phosphate substrate and treated with RANKL 50 ng/ml and M-CSF 20 ng/ml for 5 days or 10 days for dentin slices. (F) Representative images of resorption pits (white areas) and (G) percent resorption (%) and pixel area per pit were quantified (n=∼500 pits per group). (H) Representative images of resorption pits were captured and percent resorption (%) was quantified. Scale bar: 100 μm (A and D) and 50 μm (C). Experiments was repeated three times. Statistical values were calculated using a Student's t-test unless otherwise indicated, *p < 0.05, **p < 0.01, n.s: no significance. Error bars indicate the mean ± SD of triplicates.
DOK3 negatively regulates RANKL signaling
To determine the molecular mechanism by which DOK3 negatively regulates osteoclast differentiation and function, we examined RANKL-induced signaling in the presence and absence of DOK3. Following serum starvation, WT and DOK3-/- BMMs were stimulated with 100 ng/mL RANKL and cell lysates were probed for activation of MAPK (phosphorylated forms of ERK and p38), phosphorylated Syk, and IκBα, as an indicator of NF-κB activation. RANKL induced increased ERK phosphorylation in DOK3-/- cells compared to WT cells (Fig. 3A). Basal Syk phosphorylation was increased in DOK3-/- BMMs (time 0) compared to WT cells, and RANKL induced increased Syk phosphorylation in DOK3-/- compared to WT BMMs (Fig. 3A). RANKL-induced NF-κB activation, as measured by degradation of IκBα, was also increased in DOK3-/- compared to WT with sustained degradation of IκBα out to 40 minutes (Fig. 3B). However, phosphorylation of MAP kinase p38 was relatively unaffected by DOK3-deficeincy (Fig. 3B). To examine if activation of NFATc1, a critical transcriptional factor, is affected in absence of DOK3. BMM were treated with RANKL 25 ng/ml for two days, serum starved for 4 hours and re-treated with RANKL 100 ng/ml for up to 2 hours. Confocal analyses of NFATc1 location was performed. WT osteoclasts had significant cytoplasmic NFATc1 at 0 and .5hour with nuclear translocation seen at 2 hours (Fig. 3C). However, DOK3-/- osteoclasts had increased nuclear NFATc1 at all time points. These data indicate that DOK3 negatively regulates RANKL-induced activation of ERK, Syk, NF-κB, and NFATc1.
Figure 3. DOK3 negatively regulates RANKL signaling.
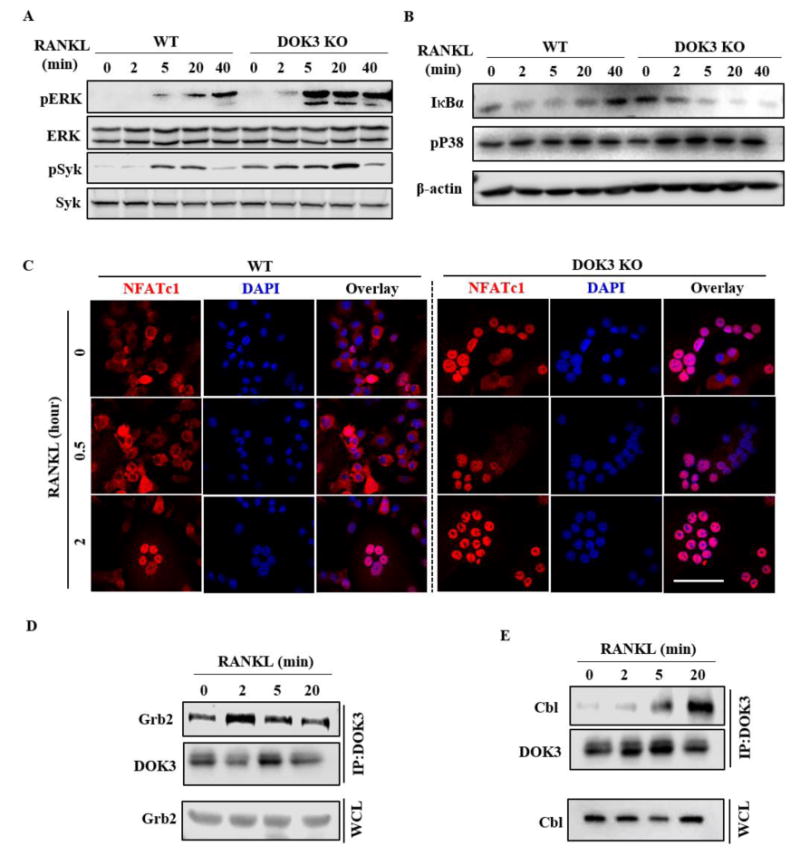
(A) WT and DOK3-/- BMMs were stimulated with RANKL (100 ng/ml) for the indicated times. Cell lysates were analyzed by Western blotting for phosphorylated and total Syk and ERK. (B) WT and DOK3-/- pre-osteoclasts were stimulated with RANKL (100 ng/ml) over time. Cell lysates were immunoblotted for IκBα, phosphorylated p38, and β-actin. (C) WT and DOK3-/- BMMs were stimulated with RANKL (20 ng/ml) for two days to induce NFATC1, serum starved for 4 hours, and stimulated with RANKL for indicated times to induce nuclear translocation of NFATc1. Cells were fixed, permeabilized, and NFATc1 localization detected by confocal microscopy. (D and E) BMMs were stimulated with RANKL (100 ng/ml) over time, cell lysates were immunoprecipitated with anti-DOK3 antibody and co-precipitated proteins analyzed by Western blot for Grb2 (D), Cbl (E) and DOK3. Scale bar: 200 μm (C). Experiments was repeated three times.
DOK3 recruits an inhibitory molecular complex in response to RANKL
We and others have shown that DOK3 forms a complex with growth factor receptor bound protein 2 (Grb2) and E3 ubiquitin ligase Cbl to limit LPS-induced TLR signaling and BCR activation (6,14,15). To determine whether DOK3 similarly regulates RANK signaling, we analyzed DOK3- associated proteins after RANKL stimulation. BMMs stimulated with RANKL were lysed, immunoprecipitated with anti-DOK3 antibody, and immunoprecipitated protein complexes were immunoblotted for Grb2 and Cbl. Grb2 and DOK3 were basally associated in adherent BMMs and RANKL further increased their association at 2 min (Fig. 3D). RANKL also induced DOK3 association with Cbl but this association occurred in the later stages of cellular signaling most notable at 5 and 20 minutes (Fig. 3E). Thus, DOK3 negatively regulates RANKL-induced osteoclastogenesis in part by forming a multimolecular complex with Grb2 and Cbl that limits RANKL-induced Syk, ERK, and NF-κB.
DOK3 regulates M-CSF-induced proliferation and signaling of BMMs
Since DOK proteins negatively regulate a variety of tyrosine kinase receptors, we explored the role of DOK3 as a negative regulator of the tyrosine kinase receptor, c-fms, that drives BMM proliferation. MTT cell proliferation assay confirmed the increased proliferation of DOK3 -/- BMM during 24h M-CSF stimulation (Fig. 4A). Thus, DOK3 negatively regulates BMM proliferation induced by M-CSF. To determine the mechanism allowing DOK3 to inhibit M-CSF-induced proliferation, we investigated M-CSF induced signaling in BMMs. Compared to WT, DOK3-/- BMM had significantly increased ERK and Syk activation in response to M-CSF (Fig. 4B). Activation of p38 and NF-κB was similar between WT and DOK3-/- M-CSF stimulated BMMs (Fig. 4B). M-CSF induced phosphorylation of DOK3, as indicated by the presence of a doublet and retarded migration of DOK3, and promoted the transient association of DOK3 with Grb2 at 2 and 5 minutes (Fig. 4C). M-CSF also significantly increased the transient association of DOK3 with Cbl at 2 and 5 minutes (Fig. 4C). These data suggest DOK3 negatively regulates M-CSF induced ERK and Syk activation via interactions with Grb2 and Cbl. Because M-CSF induces cyclin D1 downstream of ERK, we analyzed cyclin D1 expression induced by M-CSF stimulation of 0, 6, 18 and 24 hours. Compared to WT cells with increased cyclin D at 6 and 18 hours, DOK3-/- cells have sustained cyclin D out to 24 hours (Fig. 4D). These results indicate that in the presence of M-CSF, DOK3 inhibits osteoclast precursor proliferation by attenuating cyclin D1 expression.
Figure 4. DOK3 limits M-CSF induced signaling and proliferation of pre-osteoclasts.
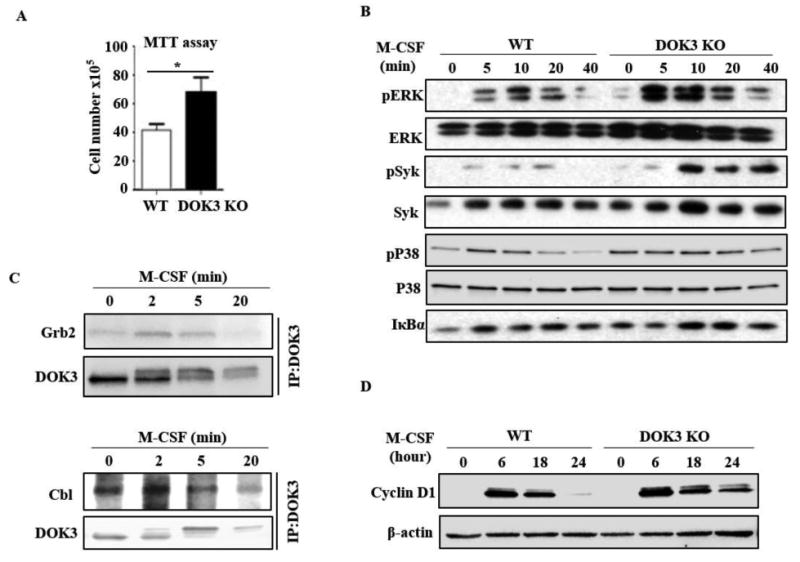
(A) WT and DOK3-/- BMMs were stimulated with M-CSF 100 ng/ml for 24 hours followed by MTT reduction assay. (B) WT and DOK3-/- BMMs were stimulated with M-CSF (100 ng/ml) for the indicated times and cell lysates were immunoblotted for phosphorylated and total ERK, Syk, and P38, and IκBα. (C) BMMs were stimulated with M-CSF (100 ng/ml), cell lysates were immunoprecipitated with anti-DOK3 antibody and co-precipitated proteins analyzed by Western blot for Grb2 (top) and Cbl (bottom) and DOK3. (D) WT and DOK3-/- BMMs were cultured with 20 ng/ml M-CSF for 3 days. Cell lysates was immunoblotted for cyclin D1 and β-actin. Statistical values were calculated using a Student's t-test unless otherwise indicated, *p < 0.05. Error bars indicate the mean ± SD of triplicates.
DOK3 associates with DAP12 in response to TREM2 ligation or M-CSF stimulation
Thus far, our data indicate that DOK3 is required to limit M-CSF and RANKL-induced signaling and osteoclastogenesis. Because DAP12 positively costimulates RANKL and M-CSF induced signaling, we asked whether DOK3 is downstream of DAP12 during osteoclastogenesis. RAW264.7 cells were stimulated with anti-TREM2 antibody to activate the TREM2-DAP12 complex and DAP12-associated proteins evaluated. TREM2 crosslinking induced the phosphorylation of DOK3 and SHIP1 at 2 and 5 min (Fig. 5A). Immunoprecipitation by antibodies to either DOK3 or phosphorylated DAP12 co-precipitated SHIP1 (Fig. 5A). Thus, TREM2 stimulation leads to the phosphorylation and association of DAP12, DOK3 and SHIP1. Similarly, M-CSF stimulation of WT BMM induced the phosphorylation of DOK3 and co-association of DOK3 with phosphorylated SHIP1 (Fig. 5B). These data suggest that DAP12 activation leads to DOK3 phosphorylation and the formation of a multimeric complex of DAP12, SHIP1 and DOK3.
Figure 5. DAP12 is required for M-CSF or RANKL-induced DOK3 phosphorylation.

(A) RAW264.7 cells were crosslinked with anti-TREM2 antibody for 2, 5, and 20 minutes. Cells were subjected to immunoprecipitation with antibodies against phosphorylated-DAP12 (pDAP12), DOK3, or phosphorylated-tyrosine (pTyr). Protein complexes were analyzed by Western blotting with antibodies specific for SHIP1, pDAP12, and DOK3. Whole cell lysates (WCL) were immunoblotted for phosphorylated-SHIP1 (pSHIP1), total SHIP1, and DOK3. (B) BMMs were stimulated with 100 ng/ml MCSF and lysates immunoprecipitated with anti-DOK3 and anti-pTyr antibodies. WCL or co-immunoprecipitated complexes were immunoblotted for SHIP1 and DOK3. (C and D) WT and DAP12-/- BMMs were stimulated with M-CSF (100 ng/ml). Cell lysates were immunoprecipitated with anti-DOK3 (C) and probed with pTyr antibody and DOK3 or anti-Grb2 (D) and immunoblotted for DOK3 and Grb2. Experiment was repeated three times.
DAP12 regulates M-CSF or RANKL-induced DOK3 phosphorylation
To determine if DAP12 was required for M-CSF-induced DOK3 phosphorylation, WT and DAP12-/- BMMs were stimulated with M-CSF or RANKL and the phosphorylation status of DOK3 determined. Basal phosphorylation of DOK3 was present in WT and DAP12-/- cells; while M-CSF- or RANKL induced phosphorylation of DOK3 at 2 and 5 minutes in WT but not DAP12-/- cells (Fig. 5C-D). To determine whether DOK3 phosphorylation was necessary for M-CSF-induced DOK3-Grb2 complex formation, WT and DAP12-/- BMMs were stimulated with M-CSF and cell lysates immunoprecipitated with anti-Grb2 antibody. M-CSF increased DOK3 co-precipitation with Grb2 at 2 and 5 min in WT but not in DAP12-/- cells (Fig. 5E). These data indicate that basal DOK3 phosphorylation and Grb2 association are independent of DAP12 but that DAP12 activation is required for M-CSF or RANKL-induced DOK3 phosphorylation and increased DOK3-Grb2 association.
DOK3 inhibits DAP12-dependent osteoclastogenesis
Having shown that DOK3 negatively regulates RANKL-induced osteoclastogenesis, M-CSF-induced pre-osteoclast proliferation, associates with TREM2 and DAP12, and is phosphorylated in a DAP12-dependent manner, we next determined whether the enhanced DOK3-/- osteoclastogenesis was dependent on DAP12. We generated global DOK3 and DAP12 double knock-out mice (designated as DKO) by crossing DOK3-/- and DAP12-/- mice. Heterozygous DOK3+/-AP12+/- mice were bred to generate littermates: DOK3+/+DAP12+/+ (WT), DOK3+/+DAP12-/- (DAP12 KO), DOK3-/-DAP12+/+ (DOK3 KO) and DOK3-/-DAP12-/- (DKO) mice. BMMs were derived from each genotype and treated with RANKL and M-CSF to induce osteoclastogenesis. We analyzed and quantified osteoclast differentiation, resorption, and actin ring formation. Similar to our prior publications (2,12), DAP12 KO osteoclasts were small with one or two nuclei and had significantly impaired resorption compared to WT or DOK3 KO (Fig. 6). On the mixed S129: C57Bl/6 background, DOK3 KO osteoclasts were enlarged and highly resorptive compared to WT or DAP12 KO; whereas DKO osteoclasts had the same phenotype as the DAP12 KO with very few multinucleated osteoclasts with poor resorptive potential (Fig. 6B-C). Actin ring formation counterstained with DAPI to reveal the nuclei confirmed the enlarged, highly multinucleated DOK3 KO osteoclasts and the predominantly mononuclear DAP12 KO and DKO osteoclasts (Fig. 6D). DKO pre-osteoclast proliferation was evaluated by MTT assay and revealed the enhanced proliferation of DOK3 KO was significantly suppressed to the reduced level of DAP12 KO (Fig. 6E). Collectively, these data indicate that DOK3 inhibits DAP12 positive co-stimulation of M-CSF and RANKL-induced osteoclastogenesis.
Figure 6. DOK3 inhibits DAP12-dependent osteoclastogenesis in vitro.
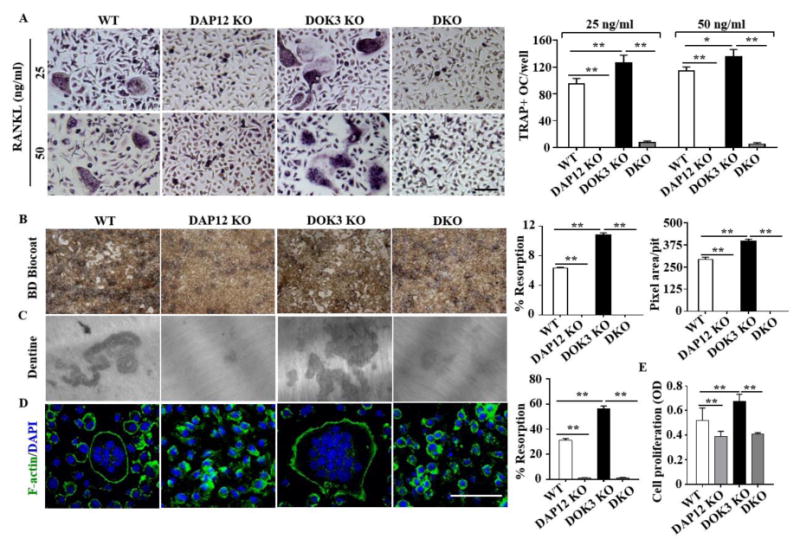
(A) BMMs from WT, DAP12 KO, DOK3 KO and DAP12/DOK3 DKO were cultured in the presence of 20 ng/ml M-CSF and treated with RANKL at concentration of 25 ng/ml and 50 ng/ml. Cells were fixed and stained for TRAP. Representative images were captured by light microscopy and TRAP-positive stained cells (≥ 3 nuclei) were quantified as osteoclasts. (B-D) BMMs were plated and differentiated with M-CSF (20 ng/ml) and RANKL (50 ng/ml). (B) BD Biocoat resorption pits at 7 days. Representative images of resorption pits (white areas) were captured (original magnification, 200×). Percent resorption (%) and pixel area per pit were quantified (n = 100 pits per group). (C) Representative images of dentin resorption pits and percent resorption (%) quantified. (D) Osteoclasts were fixed, permeabilized and stained with Alexa Fluor® 488 Phalloidin to visualize actin rings. (E) MTT assay of proliferation of BMMs from WT, DAP12 KO, DOK3 KO and DKO mice. BMMs were seeded and cultured with M-CSF (20 ng/ml) for 24 hours. Statistical values were calculated using a Student's t-test unless otherwise indicated Scale bar: 100 μm (A and D). Experiments were repeated three times. *p < 0.05, **p < 0.01. Error bars indicate the mean ± SD of triplicates.
DOK3 positively regulates osteoblast differentiation
Histomorphometry surprisingly revealed a significant decrease in osteoblast parameters and normal mineral apposition and bone formation rates (Fig. S1C-D). To address the role of DOK3 in osteoblasts, primary osteoblasts were derived from WT and DOK3-/- mice and differentiated in vitro. DOK3 was detected by immunblot in WT osteoblasts but not in DOK3-/- cultures (Fig. 7A). Alizarin red staining of osteoblast cultures at weeks of 2, 3 and 4 revealed significantly less staining in DOK3-/- cultures compared to WT cells (Fig. 7B). Real time PCR analyses revealed that expression levels of osteoblastogenesis-related genes, Runx2, Osterix, collagen α1 (Col1a) and alkaline phosphatase (ALP) were significantly reduced in in DOK3-/- cultures compared to WT (Fig. 7C). These data indicate that DOK3 is a positive regulator of osteoblastogenesis in vivo and in vitro.
Figure 7. DOK3 promotes osteoblastogenesis.
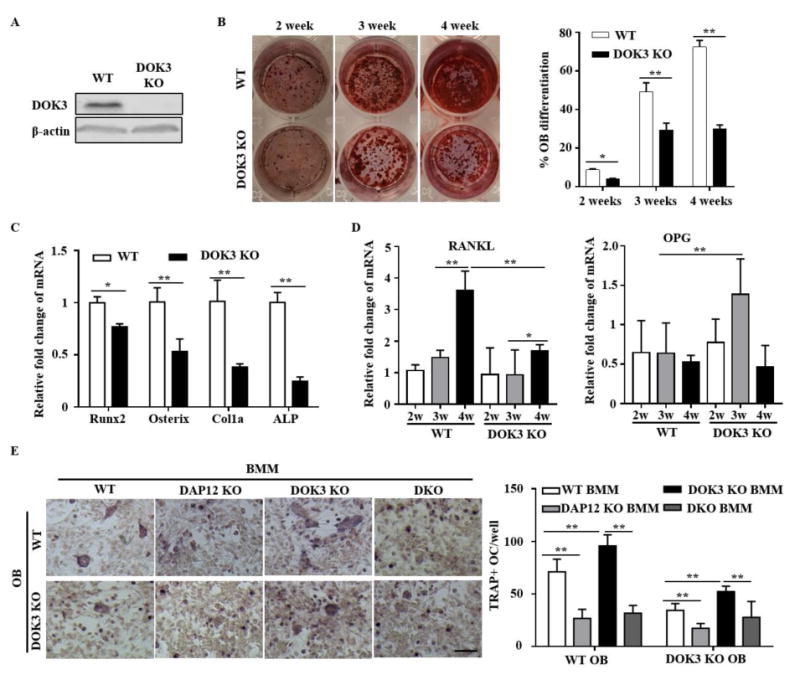
Osteoblasts derived from WT and DOK3-/- long bones were differentiated with β-glycerophosphate (BG) and ascorbic acid (AA) for 2, 3 and 4 weeks. (A) Whole cell lysates were analyzed by Western blot with anti-DOK3 and anti-β-actin. (B) Mineralized calcium nodules of WT and DOK3-/- osteoblasts were stained with Alizarin Red and nodule area quantified. (C) Real time PCR of osteoblastogenesis-related genes, Runx2, Osterix, collagen α1 (Col1a) and alkaline phosphatase (ALP) were measured at 4 weeks of WT and DOK3-/- osteoblasts differentiation. The results were normalized to GAPDH. (D) RANKL and Osteoprotegerin (OPG) real time PCR expression from WT and DOK3-/- osteoblasts differentiated for 4 weeks. The results were normalized to GAPDH. (E) Osteoblast osteoclast co-cultures experiments. BMMs from WT, DAP12 KO, DOK3 KO and DAP12/DOK3 DKO were overlain on confluent pre-osteoblasts derived from WT and DOK3-/- and co-cultured for an additional 8 days prior to fixation and TRAP staining. Representative images were captured by light microscopy and TRAP-positive stained cells (≥ 3 nuclei) were quantified as osteoclasts. Statistical values were calculated using a Student's t-test unless otherwise indicated, *p < 0.05, **p < 0.01. Error bars indicate the mean ± SD of triplicates.
To further investigate how osteoblasts interact with osteoclasts to modulate osteoclastogenesis, co-cultures of osteoblasts and osteoclasts were performed. WT and DOK3-/- primary osteoblasts were grown to confluency in the presence of vitamin D and prostaglandin E2 at which time, pre- osteoclasts were overlain and allowed to differentiate for 8 days. WT osteoblasts induced significantly more DOK3 KO multinucleated osteoclasts compared to WT osteoclasts (Fig. 7E). However, DOK3 KO osteoblasts had impaired ability to induce multinucleated WT and DOK3 KO osteoclasts. Osteoclasts derived from DAP12 KO and DKO formed small osteoclasts when co-cultured with WT or DOK3 KO osteoblasts. To further define why DOK3 KO osteoblasts fail to support robust osteoclastogenesis, expression of RANKL and osteoprotegrin (OPG) were examined by qPCR (Fig. 7D). RANKL expression was significantly reduced in DOK3 KO cultures compared to WT osteoblasts. OPG expression remained low in WT osteoblasts but was significantly elevated in DOK3 KO cells indicating an increase in the OPG/RANKL ratio that would be inhibitory to osteoclastogenesis. Collectively, these data have demonstrated DOK 3 promotes osteoblastogenesis, positively regulates osteoblast RANKL expression, and inhibits OPG expression.
DOK3 regulates DAP12-independent bone remodeling in vivo
To determine if DOK3 inhibition of osteoclastogenesis in vivo requires DAP12-dependent osteoclastogenesis, we generated cohorts of male and female 8 week old WT, DAP12 KO, DOK3 KO, and DKO mice for microCT analysis (Fig. S3). On this mixed S129: C57Bl/6 background (backcrossed 5 generations to C57Bl/6), female DAP12 KO mice maintained a high bone mass phenotype compared to DOK3 KO. However at this age, DOK3 KO mice in this mixed background did not have significant osteoporosis. DKO female mice had similar bone mass, trabecular number, thickness, spacing, and connectivity density as DOK3 KO and WT female mice. These data indicate that DOK3 deficiency normalizes the high bone mass DAP12 KO phenotype and suggests that DOK3 negatively regulates DAP12-dependent and independent osteoclastogenesis in vivo.
Discussion
We have identified novel roles of DOK3 in homeostatic bone remodeling using mice deficient in DOK3. This work has elucidated several novel findings modeled in Fig. S4. First, DOK3 -/- mice develop osteoporosis secondary to increased osteoclasts. Second, the molecular mechanism of increased osteoclastogenesis in the absence of DOK3 is due to enhanced M-CSF induced proliferation of osteoclast precursors and increased sensitivity to RANKL leading to faster differentiation and increased resorption capacity. Third, DOK3 activation occurs in a DAP12-dependent manner after M-CSF stimulation while basal DOK3 phosphorylation occurs in a DAP12-independent manner. Fourth, phosphorylation of DOK3 induces an inhibitory multimeric complex composed of DOK3, Grb2, and Cbl that limits Syk, NF-κB, and ERK activation inhibiting osteoclastogenesis. Fifth, DOK3 inhibition of osteoclastogenesis is particularly dependent on DAP12 in vitro but also limits DAP12-independent osteoclastogenesis in vivo. And finally, DOK3 has an unexpected positive role in osteoblastogenesis in vitro and in vivo. These findings have revealed a previously unknown role of DOK3 in osteoclastogenesis, osteoblastogenesis, and bone homeostasis.
Mechanistically, DOK3 has several inhibitory effects on cellular signaling downstream of M-CSF. These included DOK3 sequestration of Grb2 to limit M-CSF-induced ERK activation as well as inhibition of Syk phosphorylation. Based on the multimeric complex including DOK3, Grb2, Cbl, SHIP1 and DAP12, we speculate that DOK3 may bring these effector proteins into association with the ubiquitin ligase Cbl. We have previously shown that Cbl leads to ubiquitination of DOK3 and associated protein, SOS1, to limit LPS-induced ERK signaling in macrophages (15). ERK is a critical positive regulator of osteoclastogenesis and is required for proliferation, differentiation, bone resorptive activity, and migration (16). As shown in this study, DOK3 plays a critical role in limiting ERK activation in response to RANKL and M-CSF.
We have previously shown that SHIP1 inhibits TREM2-DAP12 induced osteoclastogenesis by competing with Syk for the DAP12 ITAM and limiting activation of ERK, AKT and VAV3 (7). Here we found that TREM2 receptor crosslinking and M-CSF stimulation also induce DOK3 association with DAP12 and SHIP. In macrophages, we have shown that DAP12 limits LPS induced cytokine responses in a DOK3-dependent manner and SHIP1 is not required for this negative regulation of TLR signaling (6). However, DOK3 and SHIP1 are both able to bind the closet immunoreceptor tyrosine-based inhibitory motif (ITIM) found within the ITAM of DAP12 (6,7). Additional studies are required to determine the interplay between DOK3 and SHIP1 as negative regulators of osteoclastogenesis.
In osteoclasts, M-CSF-induced cytoskeletal reorganization requires DAP12 and Syk (17). Here we have shown that DOK3 also participates in osteoclast cytoskeleton organization as assessed by actin ring formation and resorption. DOK3-/- osteoclasts are much larger and complex than WT, very similar to SHIP1-/- osteoclasts or WT osteoclasts treated with an activating anti-TREM2 antibody that stimulates phosphorylation of DAP12 and increases osteoclast multinucleation and size (4,7). Our findings that M-CSF induces DOK3 phosphorylation in a DAP12-dependent manner, indicates that DOK3 is negatively regulating M-CSF and DAP12-induced cytoskeletal changes likely via inhibition of ERK and Syk. Therefore DOK3-/- cells exhibit a lower M-CSF and RANKL threshold for cellular proliferation and differentiation because DAP12 costimulation proceeds without negative regulation (Fig. 7).
Although basal phosphorylation of DOK3 was still present in the absence of DAP12, M-CSF failed to induce increased DOK3 phosphorylation in DAP12-/- cells. This data suggest that basal DOK3 phosphorylation is independent of DAP12, likely occurring in response to other immunoreceptors, perhaps OSCAR coupled to FcRγ ITAM-adapter protein) or integrin-induced Src activators. Indeed, we have previously shown that inhibitors of Src, but not Syk, block basal and inducible DOK3 phosphorylation by LPS stimulation (6). Our data supports a model whereby M-CSF induces activation of Src that subsequently phosphorylates DAP12, Syk, and DOK3. DOK3 could limit Syk activation by competing for DAP12 ITAM binding or could bring Syk into close proximity to Cbl thereby allowing Syk to be targeted to the proteasome (18). Excessive Syk activation in response to M-CSF or RANKL in the absence of DOK3 is expected to lead to increased calcium signaling within the pre-osteoclast resulting in increased size and complexity of DOK3-/- osteoclasts (Fig. 2 and S4). Enhanced calcium signaling is also expected to lead to increased Pyk2 activation and β-catenin accumulation (3). Indeed, DOK3-/- osteoclasts have significantly increased expression of β-catenin target gene, cyclin D1, compared to WT cells (Fig. 4G). Functionally, this leads to increased proliferation of DOK3 -/- pre-osteoclasts compared to WT (Fig. 4E). β-Catenin and its target gene cyclin D1 are induced during the MCSF-mediated precursor proliferation, they are decreased during RANKL-mediated osteoclast differentiation.
We and others have previously shown that homeostatic bone remodeling requires either DAP12 or FcRγ- ITAM induced costimulation of RANKL in vivo (1,2). DAP12 KO mice have increased bone mass and here we observed normalization of the high bone mass in the DKO mice. This indicates that DOK3 is likely also negatively regulating FcRγ-induced osteoclastogenesis in vivo. Supporting this conclusion is the increased osteoclastogenesis seen in the DOK3 KO pre-osteoclast: WT osteoblast co-culture that relies heavily on the collagen receptor OSCAR coupled to FcRγ and less on DAP12 to drive osteoclastogenesis (19). Further studies are needed to determine the role of DOK3 downstream of FcRγ activation.
Based on our data showing a striking osteoclast phenotype in the absence of DOK3, we were surprised that histomorphometric analysis of DOK3-/- tibias revealed significantly decreased osteoblast parameters compared to WT (Fig. S1). Interestingly, despite decreases in these osteoblast parameters, the dynamic labeling of new bone formation revealed normal bone formation rate and mineral apposition rates in DOK3-/- (Fig. S1). In vitro DOK3-/- osteoblast precursors had impaired differentiation. Interestingly, this phenotype is similar to osteoblast-specific deletion of SHIP1 which leads to decreased mesenchymal stem cell osteolineage commitment leading to a low bone turnover osteoporosis with fewer osteoblasts and osteoclasts (20). Additional studies using cell-specific DOK3 knock outs are likely to delineate the contributions of DOK3 to osteoblasts and osteoclasts in vivo.
Unlike the inhibitory role of DOK3 in osteoclasts, our data supports a positive role for DOK3 in osteoblastogenesis. In the absence of DOK3, fewer osteoblasts differentiate to produce calcified nodules detected by alizarin red (Fig. 7B). Dok-1, Dok-2, and Dok-3 have been reported to be preferentially expressed in hematopoietic cells although they are also expressed in lung and colon adenocarcinomas (8,11). Unlike DOK3-deficiency, deficiency of either DOK1 or DOK2 failed to generate an overt bone phenotype; however, mice deficient in both DOK1 and DOK2 developed osteoporosis compared to WT mice (10). Histomorphometric analysis of those mice found increased osteoclast indices but normal osteoblast indices including BFR and MAR. Thus, DOK1-3 proteins regulate osteoclastogenesis while DOK3 is unique in its ability to promote osteoblastogenesis.
Limitations of our study include the use of globally deleted DOK3 and DAP12 mice instead of cell or tissue-specific deletions which are not available at this time. Additionally, the mixed background of the DKO mice limits our ability to fully interpret the results as there was significant variability between the littermates. Also the DOK3-deficient mice did not have a strong osteoporotic phenotype in this mixed background at 8 weeks of age. We completed our previous microCT analysis of the global DOK3 KO at the time of skeletal maturity at 16 weeks. Additional backcrossing and re-evaluation at 16 weeks of age may yield more informative results.
These studies may also inform Alzheimer's research as TREM2 and DAP12 are critical for normal microglial function and in their absence or when mutated in man, leads to a rapidly progressive dementia termed Nasu-Hakola (21). More recently, TREM2 mutations have been highly associated with Alzheimer's disease and may play a role in recognizing and limiting β-amyloid plaques (22). However, additional molecular mechanisms by which TREM2 and DAP12 contribute to neurodegenerative diseases is still limited. Our studies detailing important cellular signaling events downstream of TREM2 and DAP12 may shed new insights into microglial activation and proliferation.
In summary, our study demonstrates that DOK3 is a critical negative regulator of osteoclastogenesis and a positive regulator of osteoblastogenesis. DOK3 inhibits M-CSF induced macrophage proliferation and RANKL induced osteoclastogenesis in a DAP12-dependent manner by limiting ERK, Syk, NF-κB, and NFATc1 activation in vitro and DAP12-dependent and independent osteoclastogenesis in vivo. DOK3 positively regulates osteoblast expression of RANKL and represses OPG thereby providing additional osteoblast regulation of osteoclastogenesis. Further studies defining the independent roles DOK3 in osteoblasts and osteoclasts in homeostatic and pathological bone remodeling may provide new therapeutic targets for modulating the function of osteoclasts and osteoblasts for the treatment of osteoporosis.
Supplementary Material
Acknowledgments
The authors would like to thank the University of Alabama at Birmingham, Center for Metabolic Bone Disease Histomorphometry and Molecular analysis Core Laboratory for their technical services. Support was provided by NIH Grant P30-AR46031 (CLL), NIH R01-DE19398 (MBH), and VA PECASE Award (MBH).
Footnotes
This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. Please cite this article as doi: [10.1002/jbmr.3205]
Disclosure: All authors state that they have no conflicts of interest.
Authors' roles: Study design: XC, JX, CLL, QP, and MBH. Study conduct: XC, JX, QP and CLL. Data collection: XC, JX, QP, and CLL. Data analysis: XC, JX, CLL, QP and MBH. Data interpretation: XC, JX, CLL, QP, and MBH. Drafting manuscript: XC, JX, CLL, and MBH. Revising manuscript content: XC, JX, CLL, and MBH. Approving final version of manuscript: XC, JX, CLL, QP, and MBH. MBH takes responsibility for the integrity of the data analysis.
Additional Supporting Information may be found in the online version of this article.
References
- 1.Koga T, Inui M, Inoue K, Kim S, Suematsu A, Kobayashi E, Iwata T, Ohnishi H, Matozaki T, Kodama T, Taniguchi T, Takayanagi H, Takai T. Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature. 2004;428:758–763. doi: 10.1038/nature02444. [DOI] [PubMed] [Google Scholar]
- 2.Mocsai A, Humphrey MB, Van Ziffle JA, Hu Y, Burghardt A, Spusta SC, Majumdar S, Lanier LL, Lowell CA, Nakamura MC. The immunomodulatory adapter proteins DAP12 and Fc receptor gamma-chain (FcRgamma) regulate development of functional osteoclasts through the Syk tyrosine kinase. Proc Natl Acad Sci U S A. 2004;101:6158–6163. doi: 10.1073/pnas.0401602101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Otero K, Turnbull IR, Poliani PL, Vermi W, Cerutti E, Aoshi T, Tassi I, Takai T, Stanley SL, Miller M, Shaw AS, Colonna M. Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and beta-catenin. Nature immunology. 2009;10:734–743. doi: 10.1038/ni.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Humphrey MB, Daws MR, Spusta SC, Niemi EC, Torchia JA, Lanier LL, Seaman WE, Nakamura MC. TREM2, a DAP12-associated receptor, regulates osteoclast differentiation and function. J Bone Miner Res. 2006;21:237–245. doi: 10.1359/JBMR.051016. [DOI] [PubMed] [Google Scholar]
- 5.Long CL, Humphrey MB. Osteoimmunology: the expanding role of immunoreceptors in osteoclasts and bone remodeling. BoneKEy reports. 2012;1 doi: 10.1038/bonekey.2012.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peng Q, Long CL, Malhotra S, Humphrey MB. A physical interaction between the adaptor proteins DOK3 and DAP12 is required to inhibit lipopolysaccharide signaling in macrophages. Sci Signal. 2013;6:ra72. doi: 10.1126/scisignal.2003801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peng Q, Malhotra S, Torchia JA, Kerr WG, Coggeshall KM, Humphrey MB. TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci Signal. 2010;3:ra38. doi: 10.1126/scisignal.2000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mashima R, Hishida Y, Tezuka T, Yamanashi Y. The roles of Dok family adapters in immunoreceptor signaling. Immunol Rev. 2009;232:273–285. doi: 10.1111/j.1600-065X.2009.00844.x. [DOI] [PubMed] [Google Scholar]
- 9.Mashima R, Honda K, Yang Y, Morita Y, Inoue A, Arimura S, Nishina H, Ema H, Nakauchi H, Seed B, Oda H, Yamanashi Y. Mice lacking Dok-1, Dok-2, and Dok-3 succumb to aggressive histiocytic sarcoma. Lab Invest. 2010;90:1357–1364. doi: 10.1038/labinvest.2010.121. [DOI] [PubMed] [Google Scholar]
- 10.Kawamata A, Inoue A, Miyajima D, Hemmi H, Mashima R, Hayata T, Ezura Y, Amagasa T, Yamanashi Y, Noda M. Dok-1 and Dok-2 deficiency induces osteopenia via activation of osteoclasts. Journal of cellular physiology. 2011;226:3087–3093. doi: 10.1002/jcp.22909. [DOI] [PubMed] [Google Scholar]
- 11.Berger AH, Niki M, Morotti A, Taylor BS, Socci ND, Viale A, Brennan C, Szoke J, Motoi N, Rothman PB, Teruya-Feldstein J, Gerald WL, Ladanyi M, Pandolfi PP. Identification of DOK genes as lung tumor suppressors. Nat Genet. 2010;42:216–223. doi: 10.1038/ng.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Humphrey MB, Ogasawara K, Yao W, Spusta SC, Daws MR, Lane NE, Lanier LL, Nakamura MC. The signaling adapter protein DAP12 regulates multinucleation during osteoclast development. J Bone Miner Res. 2004;19:224–234. doi: 10.1359/JBMR.0301234. [DOI] [PubMed] [Google Scholar]
- 13.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res. 2010;25:1468–1486. doi: 10.1002/jbmr.141. [DOI] [PubMed] [Google Scholar]
- 14.Schnyder T, Castello A, Feest C, Harwood NE, Oellerich T, Urlaub H, Engelke M, Wienands J, Bruckbauer A, Batista FD. B cell receptor-mediated antigen gathering requires ubiquitin ligase Cbl and adaptors Grb2 and Dok-3 to recruit dynein to the signaling microcluster. Immunity. 2011;34:905–918. doi: 10.1016/j.immuni.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 15.Peng Q, O'Loughlin JL, Humphrey MB. DOK3 negatively regulates LPS responses and endotoxin tolerance. PLoS One. 2012;7:e39967. doi: 10.1371/journal.pone.0039967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He Y, Staser K, Rhodes SD, Liu Y, Wu X, Park SJ, Yuan J, Yang X, Li X, Jiang L, Chen S, Yang FC. Erk1 positively regulates osteoclast differentiation and bone resorptive activity. PLoS One. 2011;6:e24780. doi: 10.1371/journal.pone.0024780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zou W, Reeve JL, Liu Y, Teitelbaum SL, Ross FP. DAP12 couples c-Fms activation to the osteoclast cytoskeleton by recruitment of Syk. Molecular cell. 2008;31:422–431. doi: 10.1016/j.molcel.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peruzzi G, Molfetta R, Gasparrini F, Vian L, Morrone S, Piccoli M, Frati L, Santoni A, Paolini R. The adaptor molecule CIN85 regulates Syk tyrosine kinase level by activating the ubiquitin-proteasome degradation pathway. J Immunol. 2007;179:2089–2096. doi: 10.4049/jimmunol.179.4.2089. [DOI] [PubMed] [Google Scholar]
- 19.Barrow AD, Raynal N, Andersen TL, Slatter DA, Bihan D, Pugh N, Cella M, Kim T, Rho J, Negishi-Koga T, Delaisse JM, Takayanagi H, Lorenzo J, Colonna M, Farndale RW, Choi Y, Trowsdale J. OSCAR is a collagen receptor that costimulates osteoclastogenesis in DAP12-deficient humans and mice. J Clin Invest. 2011;121:3505–3516. doi: 10.1172/JCI45913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iyer S, Viernes DR, Chisholm JD, Margulies BS, Kerr WG. SHIP1 regulates MSC numbers and their osteolineage commitment by limiting induction of the PI3K/Akt/beta-catenin/Id2 axis. Stem cells and development. 2014;23:2336–2351. doi: 10.1089/scd.2014.0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xing J, Titus AR, Humphrey MB. The TREM2-DAP12 signaling pathway in Nasu-Hakola disease: a molecular genetics perspective. Res Rep Biochem. 2015;5:89–100. doi: 10.2147/RRBC.S58057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM, Cirrito JR, Colonna M. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell. 2015;160:1061–1071. doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.