

Developmental nail disorders are heterogeneous group of genodermatosis, with nonsyndromic congenital nail disorder (NDNC) being a rare subgroup inherited in autosomal dominant or autosomal recessive pattern. These are classified into ten different types (NDNC1-10), which are described in OMIM.1 The genes described for isolated nail disorders include PLCD1 (MIM 602142), RSPO4 (MIM 610573), FZD6 (MIM 603409), COL7A1 (MIM 120120), HPGD (MIM 601688) and SLCO2A1 (MIM 601460). In Addition, two other loci for NDNC have been mapped on chromosome 17p13 and 17q25.1–17q25.3.1, 2
In the present study we have investigated a five-generation consanguineous Pakistani family with multiple affected individuals, segregating fingernail dysplasia in autosomal recessive manner (Fig. 1a). Nail features were observed at birth. Age of affected members ranged from 15 to 27 years at the time of the study. All the affected members showed severe form of onychodystrophy. Digits had a bulbous appearance with mild erythema and swelling of the proximal and lateral nail folds. The nail bed was hyperkeratotic and the nail plate was thickened and highly dystrophic probably due to diffuse crumbling. Complete destruction of the nail plate was observed in the digits (Fig. 1b–f). Toe nails were normal. Severity of the disease phenotypes was varied among the affected individuals. No defect in other ectodermal structures was noted in affected individuals.
Fig 1.
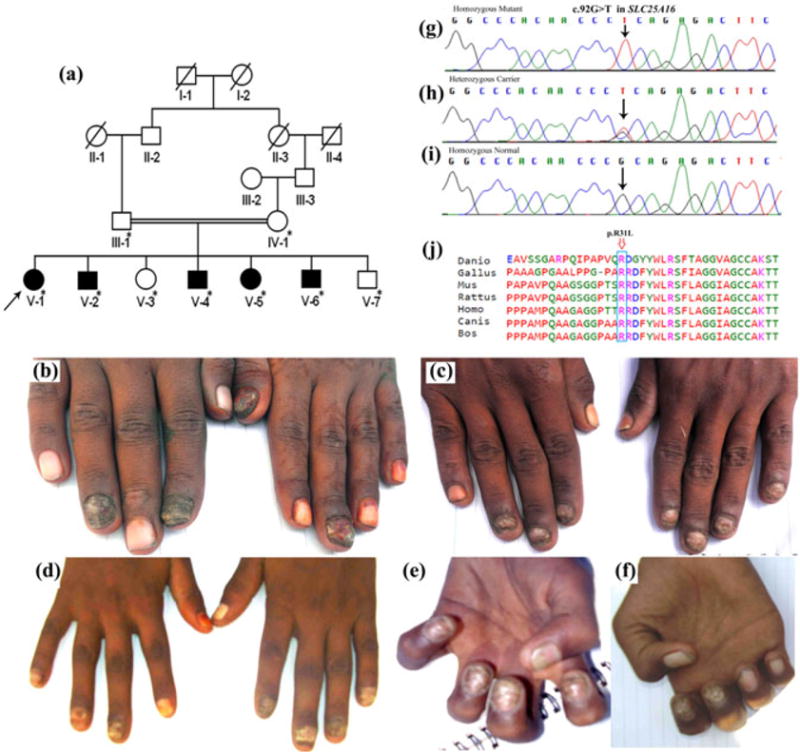
(a) Pedigree of consanguineous five-generation Pakistani family with autosomal recessive pure fingernail dysplasia. (b–f) Clinical features of patients with pure finger-nail dysplasia phenotypes. (b) Severe hyperkeratotic thickening of nail plate, nail bed and swelling of fingers is seen in individual V-1; (c) Keratotic lesions of the index, middle and ring fingers in the right hand and keratotic left fingernails and normal left thumb in individual V-2; (d) Severe onychodystrophy with hyperkeratosis, mild erythema and swelling in Patient V-4; (e) Patient V-5 with thickened dystrophic nail plate and hyperkeratotic nail bed; (f) Patient V-6 has severe destruction of nail plate and nail bed. (g–i) Sanger sequencing analysis of SLC25A16 gene in the study kindred. (g) The homozygous mutant allele in affected individual (V-1, V-2, V-4, V-5, V-6); (h) heterozygous SLC25A16 c.92G>T mutant and wild-type alleles in carriers (III-1, IV-1, V-3); (i) homozygous normal allele in normal individual (V-7). (j) Comparison of partial amino acid sequence of human SLC25A16 across different species. The missense mutation (p.Arg31Leu) affecting conserved arginine residue in human SLC25A16 is indicated by an arrow.
The study was approved by Institutional Review Boards of Quaid-i-Azam University (QAU) Islamabad, Pakistan and Baylor College of Medicine and Affiliated Hospitals, Houston, Texas, USA. Informed written consent for the study, including presentation of photographs for publication, was obtained from affected individuals and their legal guardian.
After exclusion of the known candidate genes (FZD6, PLCD1, RSPO4), genome wide autozygosity mapping was carried out using 580 short tandem repeat (STR) polymorphic microsatellite markers on DNA samples of four affected (V-1, V-2, V-4, V-5) and one unaffected (V-7) individual. Affected individuals showed homozygosity for five microsatellite markers including D5S2863 (5q35.2), D10S549 (10q21.1), D10S522 (10q21.3), D13S1493 (13q13.1–q13.2) and D18S454 (18q12.3). Upon genotyping all family members (III-1, IV-1, V-1, V-2, V-3, V-4, V-5, V-6, V-7) for these markers, only markers D10S549 and D10S522 co-segregated with the nail disorder. Additional genotyping within the region was performed using a total of 26 microsatellite markers. Twenty-three out of twenty-six markers were informative and considered for analysis. Markers analysis revealed an extended autozygous region of 22.34 Mb on chromosome 10q11.23–q22.1, with a maximum two point LOD score of 3.14 (θ=0) and maximum multiple LOD score of 4.09 (Supplementary Table 1). We then carried out whole exome sequencing using a DNA sample from affected individual V-1 (Fig. 1a). Exome sequencing was performed as described by Shah et al. (2016a).3 From the exome data of individual, we identified only nine homozygous exonic variants (Supplementary Table 2) that were pathogenic, and had minor allele frequency (MAF) <0.01 in South Asian alleles from the Exome Aggregation Consortium (ExAC) database. Of these, only the variant c.92G>T (p.Arg31Leu) in SLC25A16 gene lies within the linkage region. The variant is heterozygous in ExAC with South Asian MAF=0.00012. The variant is predicted to be deleterious by PolyPhen-2, CADD, SIFT, Mutationtaster2, and MutationAssessor. Sanger sequencing validated co-segregation of the variant (c.92G>T) with the disease phenotype in the entire family (Fig. 1g–i).
The SLC25A16 gene contains nine exons encoding 332 amino acids of the protein, which includes three tandem repeat mitochondrial transporter domains. Previously, mutations in other transporter genes have been associated with various forms of genodermatoses, including mutations in: (1) SLCO2A1 that cause primary hypertrophic osteoarthropathy with digital clubbing and pachydermoperiostitis; (2) SLC6A19 leading to Hartnup disorder which includes light-sensitive skin rash; (3) SLC2A10 that result in arterial tortuosity syndrome, including cutis laxa and skin telangiectasias/contractures; and (4) SLC17A5 causing infantile sialic acid storage disease which may include hypopigmented skin and fair hair.3–6
Solute carrier proteins comprise a large family of more than 300 membrane-bound proteins that enable the transport of a wide array of substrates across biological membranes. These proteins are involved in a wide range of physiological processes including uptake and absorbance of metabolites, nutrients, drugs and xenobiotics.7 The SLC25 family of transporters is the largest of solute carriers and consists of 53 members that are mostly associated with transport in mitochondria (mitochondrial carrier family) while few members of the family are located in other cell organelles such as peroxisomes, chloroplasts and mitosomes.8 SLC25A16 is localized in the inner mitochondrial membrane and mediates the carriage and exchange of molecular particles between cytosol and mitochondrial matrix. Yeast mitochondrial carrier protein LEU5 has 35% sequence homology with human SLC25A16. Studies on LEU5-deficient yeast strain have shown impaired mitochondrial coenzyme A (CoA level), while the cytosol containing CoA retains its normal activity. Moreover, the mutant strains were not able to produce alpha-isopropylmalate (IPM) due to entrapped IPM synthase inside mitochondria.9
In mitochondria Acetyl-CoA synthesis occurs either by beta-oxidation of fatty acids or direct linkage of CoA to acetate. Acetyl-CoA is transported to cytosol from mitochondria through citrate transport system. In cytosol acetyl-CoA is used in biosynthesis of lipids. Lipids are an essential part of nails in addition to keratins, water and minerals. Among the key lipids present in nails are ceramides, triglycerides, sterol and squalene; and free fatty acids.10 In nails lipids exist both in the intracellular and extracellular parts. The intracellular lipids provide pliability to nails while the extracellular lipids act as glue and hold the cells together in nail plate. It is highly likely that the variant, identified in the present study, might perturb CoA transport and alternately lipid biosynthesis resulting in nail phenotypes.
The substantial difference in the severity of the phenotypes, among affected individuals, suggest that other factors such as heterogeneity in genetic background, modifier genes or environmental factors might have played a role. Our findings and the evident extreme rarity of the SLC25A1 c.92G>T variant support causality of this variant for autosomal recessive form of isolated fingernail dysplasia. However, due to the non-availability of other families with similar phenotypes, further characterization of the gene to validate its contribution to the phenotypes was not possible.
Supplementary Material
Acknowledgments
The authors are highly obliged to affected and other members of the family for their invaluable cooperation and participation in the present study. We are also thankful to the “University of Washington Center for Mendelian Genomics” for carrying out exome sequencing.
Funding: The work presented here was supported by Higher Education Commission (HEC), Islamabad, Pakistan and National Institute of Health (NIH), USA.
Footnotes
This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. Please cite this article as doi: 10.1111/bjd.15661
PROFESSOR WASIM - AHMAD (Orcid ID : 0000-0003-1541-7258)
References
- 1.Khan S, Basit S, Habib R, Kamal A, Muhammad N, Ahmad W. Genetics of human isolated hereditary nail disorders. Br J Dermatol. 2015;173:922–29. doi: 10.1111/bjd.14023. [DOI] [PubMed] [Google Scholar]
- 2.Shah K, Ferrara TM, Jan A, Umair M, Khan S, Ahmad W, Spritz RA. Homozygous SLCO2A1 translation initiation codon mutation in a Pakistani family with recessive isolated congenital nail clubbing (ICNC) Br J Dermatol. 2016b doi: 10.1111/bjd.15094. [DOI] [PubMed] [Google Scholar]
- 3.Shah K, Ali RH, Ansar M, Lee K, Chishti MS, Abbe I, Li B, Smith JD, Nickerson DA, Shendure J, Coucke PJ. Mitral regurgitation as a phenotypic manifestation of nonphotosensitive trichothiodystrophy due to a splice variant in MPLKIP. BMC Med Genet. 2016a;17:13. doi: 10.1186/s12881-016-0275-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seow HF, Bröer S, Bröer A, Bailey CG, Potter SJ, Cavanaugh JA, Rasko JE. Hartnup disorder is caused by mutations in the gene encoding the neutral amino acid transporter SLC6A19. Nat Genet. 2004;36:1003–7. doi: 10.1038/ng1406. [DOI] [PubMed] [Google Scholar]
- 5.Callewaert BL, Willaert A, Kerstjens-Frederikse WS, De Backer J, Devriendt K, Albrecht B, Ramos-Arroyo MA, Doco-Fenzy M, Hennekam RC, Pyeritz RE, Krogmann ON. Arterial tortuosity syndrome: clinical and molecular findings in 12 newly identified families. Hum Mutat. 2008;29:150–58. doi: 10.1002/humu.20623. [DOI] [PubMed] [Google Scholar]
- 6.Lines MA, Rupar CA, Rip JW, Baskin B, Ray PN, Hegele RA, Grynspan D, Michaud J, Geraghty MT. Infantile sialic acid storage disease: two unrelated Inuit cases homozygous for a common novel SLC17A5 mutation. JIMD Reports. 2014;12:79–84. doi: 10.1007/8904_2013_247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin L, Yee SW, Kim RB, Giacomini KM. SLC transporters as therapeutic targets: emerging opportunities. Nat Rev Drug Discov. 2015;14:543–60. doi: 10.1038/nrd4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palmieri F. The mitochondrial transporter family SLC25: identification, properties and physiopathology. Mol Aspects Med. 2013;34:465–84. doi: 10.1016/j.mam.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 9.Prohl C, Pelzer W, Diekert K, Kmita H, Bedekovics T, Kispal G, Lill R. The yeast mitochondrial carrier Leu5p and its human homologue Graves’ disease protein are required for accumulation of coenzyme A in the matrix. Mol Cell Biol. 2001;21:1089–97. doi: 10.1128/MCB.21.4.1089-1097.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Helmdach M, Thielitz A, Röpke EM, Gollnick H. Age and sex variation in lipid composition of human fingernail plates. Skin Pharmacol Phys. 2000;13:111–9. doi: 10.1159/000029915. [DOI] [PubMed] [Google Scholar]
- 11.Matise TC, Chen F, Chen W, Francisco M, Hansen M, He C, Hyland FC, Kennedy GC, Kong X, Murray SS, Ziegle JS. A second-generation combined linkage physical map of the human genome. Genome Res. 2007;17:1783–6. doi: 10.1101/gr.7156307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.