

Abstract
The function of cystic fibrosis transmembrane conductance regulator (CFTR) channels is crucial in human airways. However unfortunately, chronic Pseudomonas aeruginosa infection has been shown to impair CFTR proteins in non-CF airway epithelial cells (AEC) and to alter the efficiency of new treatments with CFTR modulators designed to correct the basic CFTR default in AEC from cystic fibrosis (CF) patients carrying the F508del mutation. Our aim was first to compare the effect of laboratory strains, clinical isolates, engineered and natural mutants to determine the role of the LasR quorum sensing system in CFTR impairment, and second, to test the efficiency of a quorum sensing inhibitor to counteract the deleterious impact of P. aeruginosa both on wt-CFTR and on the rescue of F508del-CFTR by correctors. We first report that exoproducts from either the laboratory PAO1 strain or a clinical ≪Early≫ isolate (from an early stage of infection) altered CFTR expression, localization and function in AEC expressing wt-CFTR. Genetic inactivation of the quorum-sensing LasR in PAO1 (PAO1ΔlasR) or in a natural clinical mutant (≪Late≫ CF-adapted clinical isolate) abolished wt-CFTR impairment. PAO1 exoproducts also dampened F508del-CFTR rescue by VRT-325 or Vx-809 correctors in CF cells, whereas PAO1ΔlasR had no impact. Importantly, treatment of P. aeruginosa cultures with a quorum sensing inhibitor (HDMF) prevented the negative effect of P. aeruginosa exoproducts on wt-CFTR and preserved CFTR rescue by correctors in CF AEC. These findings indicate that LasR-interfering strategies could be of benefits to counteract the deleterious effect of P. aeruginosa in infected patients.
Keywords: cystic fibrosis, P. aeruginosa, infection, CFTR, Vx-809, correctors, LasR, furanone
Introduction
Anion secretion through cystic fibrosis transmembrane conductance regulator (CFTR) channels expressed at the apical membrane of airway epithelial cells (AEC) is essential to maintain an adequate airway periciliary liquid volume necessary for effective mucociliary clearance, a key mechanism to clear inhaled pathogens from the lungs. Bacterial colonization and infections are however frequent in patients with chronic obstructive pulmonary disease (COPD) for example (Matkovic and Miravitlles, 2013). In cystic fibrosis (CF) airways, mutations in the Cftr gene lead to dysfunctional Cl− and secretion associated with reduced periciliary liquid volume, impaired mucociliary clearance, accumulation of viscous mucus and airway surface liquid acidification. These phenomena, in turns, favor bacterial infections (Haq et al., 2015). The prevalence of respiratory pathogens varies among patients and over the course of the disease. However, ultimately most CF patients are chronically colonized by Pseudomonas aeruginosa (P. aeruginosa). This pathogen readily adapts to its environment and displays extensive genotypic and phenotypic changes over the course of chronic infection in the human lung (Folkesson et al., 2012). P. aeruginosa isolates from CF patients frequently exhibit host-adapted mutations, including in the lasR gene. LasR is the primary transcriptional regulator for quorum sensing, a bacterial communication system that allows for coordinated gene expression, including genes involved in virulence factor production (Feliziani et al., 2010; Jimenez et al., 2012; Qin et al., 2012; Rutherford and Bassler, 2012).
Exposure to P. aeruginosa bacteria and/or P. aeruginosa secreted exoproducts has been associated with loss of epithelial integrity in vitro and with progressive tissue damage and lung function decline in vivo (Coraux et al., 2004; Rejman et al., 2007; Folkesson et al., 2012). In addition, we recently showed that exoproducts under LasR control impair airway epithelial repair after injury (Ruffin et al., 2016). Interestingly, quorum sensing inhibitors have been identified within libraries of natural and chemical compounds and have been proposed as adjuvants to antibiotics based on their capability to restrain biofilm formation and bacterial virulence, without altering bacterial growth (Bhardwaj et al., 2013; Kalia, 2013). Our recent work provided the first proof of concept that a quorum sensing inhibitor, namely the 4-hydroxy-2, 5-dimethyl-3(2H)-furanone (HDMF; Choi et al., 2014; also known as furaneol or strawberry furanone) abrogates the deleterious effect of P. aeruginosa on the repair of CF and non-CF airway epithelia (Ruffin et al., 2016).
P. aeruginosa infections not only play a detrimental role on airway integrity but also impacts CFTR expression and function. Indeed, several studies, including from our group, showed that exposure to P. aeruginosa strains or exoproducts reduced the expression of wt-CFTR at the apical membrane and CFTR-mediated Cl− secretion through non-CF human AEC (Swiatecka-Urban et al., 2006; Rubino et al., 2014; Trinh et al., 2015). Such reduction of CFTR function in chronically infected airways may impair mucociliary clearance and consequently microbial clearance, even in non-CF airways, such as in COPD patients. The mechanisms by which P. aeruginosa alters CFTR is likely complex. Our data indicated that exposure to P. aeruginosa exoproducts reduced CFTR protein synthesis and enhanced CFTR protein degradation in AEC (Trinh et al., 2015). It has been recently reported that LasB elastase down-regulates wt-CFTR protein levels (Saint-Criq et al., 2017). Enhanced wt-CFTR ubiquitination and degradation, at least in part due to a CFTR inhibitory factor (Cif, present in P. aeruginosa outer membrane vesicles) has also been reported (Bomberger et al., 2011). The inhibition of CFTR endocytic recycling could also be mediated by a NHERF-dependent mechanism (Rubino et al., 2014).
During the last decade, the development of new mutation-specific CF therapeutics, with small molecules have given hope to rescue the basic CFTR defect (for review Bell et al., 2015). Specifically, a first CFTR potentiator (Vx-770, KalydecoMD) has been approved for patients with several class III (characterized by improper channel gating/regulation) and class IV (decreased channel conductance) mutations. In addition, several molecules called correctors (including VRT-325 and Vx-809) (Pedemonte et al., 2005; Goor et al., 2006; Van Goor et al., 2011) are directed against class II mutations, including the most frequent, F508del (characterized by improper CFTR protein folding, trafficking, membrane stability and function). Although CFTR correctors allow partial F508del-CFTR maturation and functional rescue in vitro, the first clinical trials with Vx-809 did not show significant changes in lung function (Brewington et al., 2015). Interestingly, a corrector/potentiator combined treatment (Vx-809+Vx-770) elicits more benefits (Wainwright et al., 2015) and this combination (Orkambi™) has recently been approved for homozygous F508del CF patients. However, clinical studies on CFTR modulators demonstrated that the response to treatments is variable among patients and the beneficial effects on lung function remained less than expected (Brewington et al., 2015). The cause of the limited and variable efficiency of CFTR modulators remains unclear and may be multiple, including modifier genes (Strug et al., 2016), disease severity, environmental factors, etc. In addition, work from our laboratory and others unveiled that P. aeruginosa infection restrains the functional rescue of F508del-CFTR by VRT-325 and Vx-809 correctors, alone or in combination with the Vx-770 potentiator (Stanton et al., 2015; Trinh et al., 2015). However, the bacterial products responsible for the deleterious effect of P. aeruginosa on CFTR rescue remain to be defined. Moreover, a strategy using quorum sensing inhibitors to decrease the production of virulence factors has, to the best of our knowledge, never been carry out in an attempt to counteract the harmful effect of P. aeruginosa on CFTR rescue impairment.
The aim of our study was first to define if P. aeruginosa exoproducts from clonally-related early and late-adapted clinical isolates elicit the same effect on wt-CFTR expression, localization and function. Then, using a wt-laboratory strain and an engineered lasR mutant, we tested the hypothesis that P. aeruginosa LasR quorum sensing play a role in wt-CFTR and F508del-CFTR rescue impairment. Finally, experiments were undertaken to evaluate the efficiency of a quorum sensing inhibitor to counteract the deleterious effect of P. aeruginosa on wt-CFTR and on the rescue of F508del-CFTR.
Materials and methods
Culture of CFBE-ΔF508 and CFBE-wt cell lines
CFBE-ΔF508 and CFBE-wt cell lines [CFBE41o- parental cells (Kunzelmann et al., 1993) stably transduced, respectively, with F508del-Cftr and wt-Cftr Bebok et al., 2005] were grown in EMEM medium (Wisent Inc., St-Bruno, QC, CA) supplemented with 10% FBS (Life technologies, Burlington, QC, CA), 2 mM L-glutamine (Life technologies) and 100U/ml of penicillin-streptomycin (Life technologies) on 35 mm petri dishes (Corning Inc., Corning, NY, USA), Lab-Tek 8 chamber slides (Thermo-Fisher Scientific Inc., Waltham, MA, USA) and permeant filters (4.67 cm2; Corning Inc.) coated with a LHC basal medium (Life technologies) solution containing 1 mg/ml BSA (Life technologies), 0.05 mg/ml bovine collagen I (Life technologies) and 1 mg/ml human fibronectin (VWR, Mont-Royal, QC, CA). Cells were cultured for 5 days on Lab-Tek chamber slides for immunofluorescence assays and 8 days on petri dishes before protein extraction, while cells on permeant filters were cultured for 3 weeks before electrophysiological measurements (Trinh et al., 2012, 2015; Bilodeau et al., 2016).
Culture of primary human airway epithelial cells
CF primary human AEC were isolated from bronchial tissues collected from CF patients (homozygous for the F508del mutation) who underwent a lung transplantation at CHUM hospital according to approved ethical protocols and with written informed consents, in accordance with the Declaration of Helsinki. After dissection, tissues were rinsed few times with sterile PBS and then incubated overnight on a rocking platform at 4°C with MEM medium (Life technologies) supplemented with 7.5% NaHCO3 (Sigma-Aldrich, Saint-Louis, MO, USA), 2 mM L-glutamine, 10mM HEPES (Thermo-Fisher Scientific Inc., #SH3023701), 0.05 mg/ml gentamycin, 50U/ml penicillin-streptomycin, 0.25 μg/ml Fungizone (Life technologies) and containing 0.1% protease (from Streptomyces griseus; Sigma-Aldrich) and 10 μg/ml DNAse (Deoxyribonuclease I from bovine pancreas; Sigma-Aldrich). The protease-DNAse activity was then neutralized with FBS, cells gently scraped off the remaining tissue and red blood cells removed by treatment with ACK lysis solution (0.1 mM NH4Cl, 10 μM KHCO3, 10 nM EDTA). After counting, cells were seeded on flasks coated with Purecol (Cedarlane Laboratory, Burlington, ON, CA) and cultured in CnT-17 medium (CellnTec Advanced Cell systems, Bern, CH) until confluency is reached. Cells were then detached with trypsin solution, seeded on permeant filters (1.1 cm2; Corning) coated with collagen IV (Sigma-Aldrich) and cultured in CnT-17 until confluency (~5 days). The apical medium was then removed to create an air-liquid interface and the basolateral medium was replaced by a differentiation medium (1:1 volume of BEGM (Lonza, Basel, CH) and DMEM (Life technologies) supplemented with 1.5 μg/ml BSA, 1 × 10−7 M retinoic acid and 100U/ml of penicillin-streptomycin) every 2 days for at least 35 days to obtain highly differentiated cultures (Trinh et al., 2012, 2015; Bilodeau et al., 2016; Ruffin et al., 2016).
P. aeruginosa strains, growth condition and preparation of diffusible material
Genotypic and phenotypic characteristics of P. aeruginosa strains used in this study are presented in Table 1. Two clonally related clinical isolates from the same CF patient recovered at 6 months of age (Early) and 8 years (Late) were used (Burns et al., 2001; Smith, 2006). Their whole genome have been sequenced and 68 mutations were identified in the Late isolate, including a nonsense mutation in the lasR gene (Burns et al., 2001; Smith, 2006). In addition, the common laboratory wild-type (wt) P. aeruginosa strains PAO1 (Nguyen et al., 2011) and PAO1-V [characterized by increased protease production compared with PAO1, Hobden, 2002], and the isogenic PAO1-V ΔlasR mutant (Hobden, 2002) were included in our study.
Table 1.
Genotypic and phenotypic characteristics of P. aeruginosa strains.
| Strain | Relevant genotypic and phenotypic characteristics | References |
|---|---|---|
| Early (E) | AMT0023-30 P. aeruginosa clinical isolate from a 6-month-old CF patient, with wild-type lasR allele | Burns et al., 2001; Smith, 2006 |
| Late (L) | AMT0023-34 P. aeruginosa clinical isolate clonally related to the Early strain, recovered from the same patient at age 8. 1-bp deletion in lasR at nucleotide 147 leading to a non-sense mutation | Burns et al., 2001; Smith, 2006 |
| PAO1 | P. aeruginosa wild-type laboratory strain | Nguyen et al., 2011 |
| PAO1-V | P. aeruginosa wild-type strain, variant of PAO1 with high basal expression of proteases | Hobden, 2002 |
| PAO1-VΔlasR | lasR::GmR mutation in lasR gene in PAO1-V parental P. aeruginosa strain | LaFayette et al., 2015 |
Bacterial strains were grown on Luria Bertani (LB, Difco, Montreal, QC, CA) 1.5% agar plates overnight (when needed, gentamicin (50 μg/ml) was used for bacterial selection), and single colonies were used to inoculate LB medium. Where indicated, LB medium was supplemented with 0.125 mg/ml HDMF (Sigma-Aldrich) and viable bacteria counts were determined by standard microdilution and colony forming units plate counting (Choi et al., 2014). To obtain the P. aeruginosa exoproducts, planktonic bacterial cultures, in the absence or presence of HDMF, were grown for 72 h with agitation at 250 rpm at 37°C, and then centrifuged at 7,200 g for 10 min at room temperature. Supernatants were filtered with low-protein binding 0.22 μm cellulose acetate filters (Corning) and aliquots of exoproducts were stored at −80°C, as previously done (Ruffin et al., 2016).
Immunoblotting
CFBE cells were scraped in ice-cold PBS, cell suspensions were centrifuged at 12,000 rpm for 5 min at 4°C and cell pellets were then solubilized in RIPA lysis buffer [150 mM NaCl, 20 mM Tris-HCl pH 8.0, 1% Triton X-100, 0.08% deoxycholic acid, 0.1% SDS, and protease inhibitor cocktail (≪complete mini EDTA free≫ Roche Diagnostic, Laval, QC, CA)] for 20 min on ice. After centrifugation (12,000 rpm, 15 min at 4°C), the supernatants were collected and protein concentration was quantified by Bradford assay. Cell lysates in 2X sample buffer [62.5 mM Tris-HCl pH 6.8, 2% SDS, 0.2% bromophenol blue, 10% glycerol and 40% β-mercapto-ethanol] were separated by SDS-PAGE (7.5%) and transferred onto nitrocellulose membranes. The membranes were first blocked with 10% dried fat-free milk in TBS-Tween (TBS-T) for 1 h at room temperature, then the upper part of the membrane was incubated for 18 h at 4°C with the polyclonal anti-CFTR 596 antibody (dilution 1:1,000, Cystic Fibrosis Foundation Therapeutics (CFFT), Bethesda, MA, USA) in TBS-T supplemented with 5% bovine serum albumin (BSA). The lower part of the membrane was incubated with purified rabbit anti-Pan-actin polyclonal antibody [Cell Signaling Danvers, MA, USA, dilution 1:1,000 in TBS-T supplemented with 5% bovine serum albumin (BSA, Sigma Aldrich)], to ensure equivalent loading and for signal normalization. Membranes were washed 3 × 15 min with TBS-T and then incubated for 1 h with goat anti-mouse HRP conjugated antibody (upper membrane; dilution 1:1,000; Millipore, Etobicoke, ON, CA) and goat anti-rabbit HRP conjugated antibody (lower membrane; dilution 1:1,000, Cell Signalling). Finally, the membranes were washed 3 × 15 min with TBS-T before chemiluminescent (ECL) detection (GE Healthcare Life Sciences, Baie d'Urfe, QC, CA). The anti-CFTR antibody recognized a 180 kDa (mature band C) and a 140–160 kDa (immature band B) protein and the intensity of bands was quantified with Multi Gauge software (Trinh et al., 2015). Each quantified CFTR band density was normalized (divided) by the value of actin density, for the same condition. Finally, the ratio (CFTR/actin) in each condition was presented as % of the control (LB) condition.
Immunofluorescence
CFBE-wt cells seeded on Lab-Tek 8 chamber slides (Thermo-Fisher Scientific) were fixed in 4% paraformaldehyde for 20 min and permeabilized with 0.1% triton X100 for 10 min at room temperature (Trinh et al., 2015). After blocking in PBS containing 10% fetal bovine serum (FBS) for 1 h, cells were first incubated with the polyclonal anti-CFTR 596 antibody (dilution 1:250, CFFT) overnight at 4°C, then with Alexa Fluor 488 conjugated anti-mouse antibody for 1 h at RT (dilution 1:200, Life Technologies Inc.). Slides were finally rinsed and counterstained with DAPI (dilution 1:1,000, Life Technologies). Fluorescence images were captured by an epifluorescent Olympus microscope. Absence of background signal and non-specific staining was verified in control experiments by omitting primary or secondary antibodies.
Electrophysiology
Primary CF human AEC and immortalized CFBE cells were cultured on permeable filters at the air-liquid interface. Where indicated, CFBE-ΔF508 and primary CF human AEC were treated (at the basolateral side) for 24 h before electrophysiological measurements with a CFTR corrector (VRT-325 (CFFT) or Vx-809 (Selleckchem, Houston, TX, USA), 5 μM). One hour prior to short-circuit current Isc measurements in an Ussing chamber, the apical side was submerged with their specific cell culture medium (see CFBE and primary cell culture sections). After washing, the filters were then mounted in a heated (37°C) Ussing chamber. To evaluate CFTR Cl− current through apical membranes, a Cl− gradient was established (low Cl− physiological solution at the apical side and high Cl− solution at the basolateral side) and the basolateral membranes were permeabilized with amphotericin B (7.5 μM). Transepithelial potential difference was clamped to zero by an external voltage clamp amplifier (VCCMC2, Physiological Instruments, San Diego, CA, USA) with KCl agar-calomel half-cells and Ag-AgCl electrodes, and the resulting Isc was recorded continuously on a computer with PowerLab system (ADInstruments, Toronto, ON, CA). Membrane resistance was verified with 1 mV pulses every 10 s (Trinh et al., 2015; Bilodeau et al., 2016).
Statistical analysis
Data are presented as mean ± SEM (Figures 1–6), except CFTR current measurements in primary airway epithelial cell cultures (Figure 7), which are presented individually for the 4 different patients tested. The number of repeated experiments is indicated in the figure legends. GraphPad Prism version 5.03 for Windows (GraphPad Software, San Diego, CA, USA, www.graphpad.com) was used to analyze all results. Paired t-tests were used to compare two groups as appropriate. One-way ANOVA were used for comparison of more than two groups and followed by Bonferroni's multiple post-hoc tests. P-values lower than 0.05 were considered to be significant. Non-significant (NS) and statistically significant differences with p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***) are indicated in the figures.
Figure 1.

Variable impact of P. aeruginosa exoproducts from Early and Late clinical isolates on CFTR expression, localization and function. CFBE-wt cells were challenged for 24 h with exoproducts from the Early or Late isolates, or LB medium (control condition). (A) Representative immunoblots (left) and densitometric analysis (right, reported as % of LB control) of mature (band C) CFTR protein expression levels (n = 6). (B) Representative immunofluorescence images (of n = 3 independent experiments) of CFBE-wt cells treated with LB, Early or Late. CFTR was detected with an anti-CFTR antibody (Ab) coupled to Alexa Fluor 488 conjugated anti-mouse antibody. Nuclei were stained with DAPI (blue) (n = 3). (C) Representative traces of short-circuit current (Isc) measurements in Ussing chamber and quantification of mean CFTRInh172 (20 μM)-sensitive currents (ΔICFTRInh−172) through CFBE-wt cells, in each condition. (n = 12). *p < 0.05, **p < 0.01, ***p < 0.001.
Results
The impact of P. aeruginosa on wt-CFTR varies as a function of bacterial strains
We recently demonstrated that exoproducts from P. aeruginosa modulate wt-CFTR expression and function in AEC (Trinh et al., 2015). However, the consequences of P. aeruginosa infection on CFTR may vary as a function of P. aeruginosa strains. Indeed, P. aeruginosa strains undergo adaptive genotypic and phenotypic changes during chronic infections in CF patients, including in the production of diffusible factors (Burns et al., 2001; Smith, 2006). We therefore tested the effect of the two clonally related isolates [Early and Late (Table 1) isolates Smith, 2006], which have been collected from the same CF patient at ages of 6 months (early stage of intermittent infection) and 8 years (chronic infection with CF-adapted isolates). CFBE-wt cells were stimulated for 24 h with LB medium (control condition) or P. aeruginosa exoproducts from the Early or Late isolates. As shown on Figure 1A, the level of mature CFTR (band C) expression was reduced by 79% after exposure to exoproducts from the Early isolate, whereas the level of band C CFTR expression measured after exposure to the Late exoproducts was similar to the one measured in LB condition.
The impact of Early and Late P. aeruginosa exoproducts on CFTR localization in CFBE-wt cells was then evaluated by immunofluorescence microscopy (Figure 1B). Results showed that both the intensity of intracellular CFTR staining and the presence of CFTR at the membrane (arrow heads), as observed in LB control conditions, were severely altered by exposure to exoproducts from the Early isolate but not from the Late isolate.
Finally, the effects of P. aeruginosa exoproducts on CFTR function were assessed in polarized CFBE-wt cells stimulated with Early or Late exoproducts, or LB controls, for 24 h before short-circuit (Isc) measurements in an Ussing chamber. To specifically measure apical CFTR Cl− currents, the basolateral membranes were permeabilized and a basolateral-to-apical Cl− gradient was applied before addition, in the apical compartment, of forskolin (Fsk) and 3-isobutyl-1-methylxanthine (IBMX), followed by the CFTRinh172 (see representative traces in Figure 1C, in LB, Early and Late conditions). Our data show that the CFTRinh172-sensitive currents (ΔICFTRinh172) are significantly reduced after treatments with exoproducts from the Early isolate (−22.38 ± 1.43 μA/cm2), compared to LB (−29.09 ± 1.79 μA/cm2). Cell cultures treated with exoproducts from the Late isolate exhibited significantly higher currents (−27.43 ± 1.36 μA/cm2) than cells treated with the Early isolate, reaching values similar to the control condition (LB).
Altogether, these data indicated that early intermittent infections with wt P. aeruginosa and chronic infections with Late CF-adapted strains may not elicit the same effects on CFTR expression, localization and function.
Role of LasR quorum sensing system in the deleterious effect of P. aeruginosa on wt-CFTR
To determine whether LasR quorum sensing system play a role in the deleterious effect of P. aeruginosa on CFTR, we compared the impact of exposure to exoproducts from a ΔlasR engineered mutant (PAO1-VΔlasR) to its isogenic wild-type parental strain PAO1-V. Whereas PAO1-V exoproducts severely reduced CFTR expression (significantly decreased level of mature band C, Figure 2A) and altered CFTR membrane localization (lack of CFTR membrane staining in immunofluorescence experiments, Figure 2B), this negative effect was no longer observed with the PAO1-VΔlasR mutant (Figures 2A,B). Similarly, CFBE-wt cells exposed to PAO1-VΔlasR exoproducts exhibited significantly higher CFTR Cl− currents than cells exposed to the PAO1-V strain (Figure 2C), indicating that LasR plays a key role in CFTR impairment.
Figure 2.

Role of LasR quorum sensing in the deleterious effect of P. aeruginosa on CFTR expression, localization and function. CFBE-wt cells were treated for 24 h with LB or exoproducts from PAO1-V or PAO1-V mutated in the lasR gene (PAO1-VΔlasR). (A) Representative immunoblots (left) and densitometric analysis (right, reported as % of LB control) of mature (band C) CFTR protein expression levels (n = 6). (B) Representative immunofluorescence images (of n = 4 independent experiments) of CFBE-wt cells treated with LB, PAO1-V or PAO1-VΔlasR exoproducts. CFTR was detected with an anti-CFTR antibody (Ab) coupled to Alexa Fluor 488 conjugated anti-mouse antibody. Nuclei were stained with DAPI (blue). (C) Representative traces of short-circuit current (Isc) measurements in Ussing chamber and quantification of mean CFTRInh172 (20 μM)-sensitive currents (ΔICFTRInh−172) through CFBE-wt cells, in each condition. (n = 9). NS, non-significant, *p < 0.05, **p < 0.01, ***p < 0.001.
Our data indicating that LasR-regulated exoproducts are key to the damaging action of P. aeruginosa on CFTR, prompted us to assess the efficiency of HDMF (4-hydroxy-2, 5-dimethyl-3(2H)-furanone), a quorum sensing inhibitor known to decrease P. aeruginosa exoproduct production and virulence (Choi et al., 2014; Ruffin et al., 2016). Accordingly, we previously showed that the elastase production, which is directly regulated by the LasR quorum sensing, by PAO1 grown in presence of HDMF (0.125 mg/ml) was strongly reduced. As expected from a quorum sensing inhibitor, we also verified that this dose of HDMF did not affect bacterial growth (Ruffin et al., 2016). We now compared the impact of exoproducts from PAO1 grown in the absence or presence of HDMF [harboring a 90% decrease in elastase activity, Supplementary Figure S1A)] on CFTR in CFBE-wt cells. We then found that quorum sensing inhibition of wild-type PAO1 with HDMF significantly mitigated the negative impact of P. aeruginosa on the level of mature CFTR expression (band C, Figure 3A), membrane localization (Figure 3B) and restored CFTR function (CFTRInh72-sensitive currents, Figure 3C), to levels similar to those observed in the LB condition. In control experiments, we also verified that HDMF itself does not exert any effect on CFTR protein expression (Supplementary Figure S1B). These data provide the first proof of concept that quorum sensing inhibitors may be useful tools to prevent the deleterious effect of P. aeruginosa on CFTR.
Figure 3.

Quorum sensing inhibition with HDMF counteracts the negative effect of P. aeruginosa on CFTR expression, localization and function. PAO1 bacteria were grown in LB with or without 0.125 mg/ml HDMF prior to collection of the bacterial filtrates. CFBE-wt cells were then treated for 24 h with exoproducts from PAO1 cultured with (PAO1+HDMF) and without HDMF (PAO1). LB medium was used as control. (A) Representative immunoblots (left) and densitometric analysis (right, reported as % of LB control) of mature (band C) CFTR protein expression levels (n = 6). (B) Representative immunofluorescence images (of n = 4 independent experiments) of CFBE-wt cells treated with LB, PAO1, or PAO1+HDMF exoproducts. CFTR was detected with an anti-CFTR antibody (Ab) coupled to Alexa Fluor 488 conjugated anti-mouse antibody. Nuclei were stained with DAPI (blue). (C) Representative traces of short-circuit current (Isc) measurements in Ussing chamber and quantification of mean CFTRInh172 (20 μM)-sensitive currents (ΔICFTRInh−172) through CFBE-wt cells, in each condition (n = 10). NS, non-significant, *p < 0.05, **p < 0.01, ***p < 0.001.
Quorum sensing inhibition abrogates the negative impact of P. aeruginosa on F508del-CFTR maturation and functional rescue by correctors
Our previous work revealed that the ability of the CFTR corrector VRT-325 to improve F508del-CFTR maturation (switch from immature band B to partial band C maturation) and partial functional rescue of CFTRInh172-sensitive CFTR currents were severely dampened by P. aeruginosa exoproducts from a clinical isolate (PACF508) (Trinh et al., 2015). Our data now confirmed that the maturation (restoration of a mature band C, Figure 4A) of F508del-CFTR in VRT-325 treated CFBE-ΔF508 cells (LB+VRT-325) are disrupted in the presence of PAO1-V exoproducts (PAO1-V+VRT-325). This lack of mature protein addressed to the membrane in the PAOI-V+VRT-325 condition, was also associated with impaired functional rescue. Indeed, the small rescue in CFTRInh172-sensitive currents by VRT-325 treatment (LB+VRT-325) was abolished by PAO1-V exoproducts (Figures 4B,C).
Figure 4.

Role of LasR quorum sensing in the deleterious effect of P. aeruginosa on F508del-CFTR maturation and functional rescue by VRT-325. CFBE-ΔF508 cells treated or not with the VRT-325 corrector (5 μM) were exposed to LB or exoproducts from PAO1-V or PAO1-V mutated in the lasR gene (PAO1-VΔlasR) for 24 h. (A) Representative immunoblots (left) and densitometric analysis (right, reported as % of LB control) of mature (band C) CFTR protein expression levels (n = 10). (B) Representative traces of short-circuit current (Isc) measurements in Ussing chamber and quantification of mean CFTRInh172 (20 μM)-sensitive currents (ΔICFTRInh−172) (C) through CFBE-ΔF508 cells, in each condition (n = 13). NS, non-significant, *p < 0.05, **p < 0.01, ***p < 0.001.
Importantly, the ΔlasR mutation abrogated the inhibitory effect of PAO1-V, leading to similar levels of F508del-CFTR maturation (Figure 4A) and functional rescue (Figures 4B,C) by VRT-325 in LB+VRT-325 and PAO1-VΔlasR +VRT-325 conditions. Similarly, quorum sensing inhibition of PAO1 cultures with HDMF prevented its deleterious action on CFTR maturation (Figure 5A) and currents (Figures 5B,C) in VRT-325-treated CFBE-ΔF508 cells.
Figure 5.

Quorum sensing inhibition abrogates the negative effect of P. aeruginosa on F508del-CFTR maturation and functional rescue with VRT-325. CFBE-ΔF508 cells were treated or not, for 24 h, with VRT-325 and LB or exoproducts from PAO1 bacteria cultured, or not, with 0.125 mg/ml of HDMF. (A) Representative immunoblots (left) and densitometric analysis (right, reported as % of LB control) of mature (band C) CFTR protein expression levels (n = 7). (B) Representative traces of short-circuit current (Isc) measurements in Ussing chamber and quantification of the mean CFTRInh172 (20 μM)-sensitive currents (ΔICFTRInh−172) (C) through CFBE-ΔF508 cells, in each condition (n = 9). NS, non-significant, **p < 0.01, ***p < 0.001.
The effect of HDMF-treated P. aeruginosa was also assessed on CFBE-ΔF508 cells rescued with the clinically used CFTR corrector Vx-809. As shown in Figure 6A, HDMF treatment restored the Vx-809 mediated CFTR maturation to levels similar to control condition (LB+Vx-809, without P. aeruginosa exoproducts). Moreover, HDMF treatment of PAO1 cultures (PAO1 HDMF+Vx-809 condition) counteracted the negative impact of P. aeruginosa (PAO1+Vx-809) on CFTRInh172-sensitive currents after activation with Vx-770 and cAMP (Figures 6B,C).
Figure 6.

Quorum sensing inhibition abrogates the effect of P. aeruginosa on F508del-CFTR maturation and functional rescue by Vx-809. CFBE-ΔF508 cells were treated or not, for 24 h, with the Vx-809 corrector (5 μM) and LB or exoproducts from PAO1 bacteria cultured, or not, in the presence of 0.125 mg/ml of HDMF. (A) Representative immunoblots (left) and densitometric analysis (right, reported as % of LB control) of mature (band C) CFTR protein expression levels (n = 11). (B) Representative traces of short-circuit current (Isc) measurements in Ussing chamber and quantification of mean CFTRInh172 (20 μM)-sensitive currents (ΔICFTRInh−172) (C) through CFBE-ΔF508 cells, in each condition (n = 5). NS, non-significant, *p < 0.05, **p < 0.01, ***p < 0.001.
Finally, we validated the beneficial effect of P. aeruginosa quorum sensing inhibition with HMDF on the CFTR functional rescue in differentiated primary airway epithelial cultures from 4 different CF patients homozygous for F508del mutations. Our data, presented individually for each tested patient, showed that despite variable levels of functional rescue of CFTR currents with Vx-809 among patients (LB+Vx-809 vs. LB), the inhibitory effect of PAO1 exoproducts (PAO1+Vx-809) was abrogated by HDMF (PAO1+HDMF+Vx-809) (Figure 7).
Figure 7.
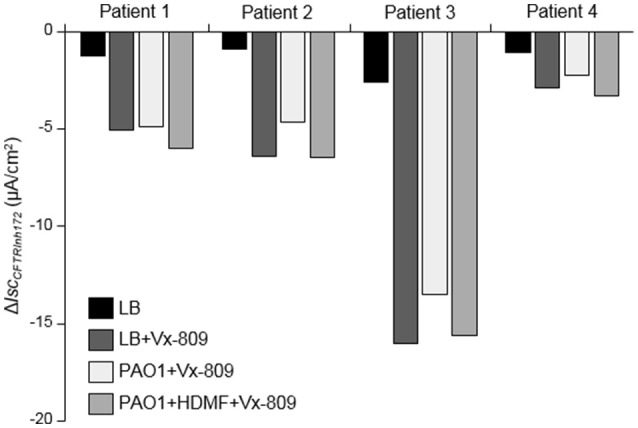
Improved CFTR rescue by Vx-809 in primary airway epithelial cells from CF patients with F508del mutations after P. aeruginosa quorum sensing inhibition. Short-circuit currents measurements were performed on human primary airway cells from 4 different CF patients homozygous for the F508del mutation cultured at the air-liquid interface for 35 days and then treated, or not, for 24 h with 5 μM Vx-809 and LB or exoproducts from PAO1 cultured with (PAO1+HDMF+Vx-809) and without (PAO1+Vx-809) 0.125 mg/ml HDMF. LB was used as control (LB and LB+Vx-809 conditions). CFTRInh172 (20 μM)-sensitive currents (ΔICFTRInh−172), in each condition, are presented individually, for each patient.
Discussion
Our study first showed that exoproducts from P. aeruginosa laboratory and clinical strains with functional LasR (wild-type lasR allele) impaired both expression and function of wt-CFTR, as well as the functional rescue of F508del-CFTR by VRT-325 and Vx-809 correctors. However, mutations in a CF-adapted isolate or loss of LasR function in an engineered lasR mutant, led to a loss of P. aeruginosa's inhibitory effect on CFTR. These data indicate that P. aeruginosa elicits different outcomes, as a function of their genotypic and phenotypic characteristics, on CFTR function in the airways of infected non-CF patients as well as on the efficacy of CFTR correctors in CF patients. Importantly, our data also unveiled that interfering with quorum sensing systems, and secondarily with virulence production, using a quorum sensing inhibitor, not only abrogated the negative impact of P. aeruginosa on wt-CFTR but also allowed to preserve the efficacy of CFTR correctors, despite the presence of P. aeruginosa infection.
Our data first demonstrated that CFBE-wt cells exposed for 24h to exoproducts from a clinical isolate (Early) or laboratory strains (PAO1 and PAO1-V) exhibited lower total protein expression of wt-CFTR and altered membrane localization, associated with reduced apical cAMP-activated CFTRInh172-sensitive Cl− currents. These observations are in agreement with our previous data on CFBE and primary AEC challenged with exoproducts from the P. aeruginosa CF clinical isolate PACF508 (Trinh et al., 2015). A dramatic decrease in CFTR total (Bomberger et al., 2011; Rubino et al., 2014; Saint-Criq et al., 2017) and membrane (Swiatecka-Urban et al., 2006; MacEachran et al., 2007; Bomberger et al., 2011) expression as well as Cl− currents (Swiatecka-Urban et al., 2006; Saint-Criq et al., 2017) have also been reported after exposure to live PAO1 (Rubino et al., 2014) and PA14 (Swiatecka-Urban et al., 2006; MacEachran et al., 2007; Bomberger et al., 2011) laboratory strains or bacteria-free PAO1/PA14 filtrates containing exoproducts (Swiatecka-Urban et al., 2006; MacEachran et al., 2007; Saint-Criq et al., 2017). However, the effect of P. aeruginosa on CFTR function may be time- and cell-type dependent. Indeed, a previous study showed increased CFTR function after acute (15 min) challenge with PA14 in 2WT2 epithelial cells expressing wt-CFTR (Haenisch et al., 2010). A CFTR-dependent increase in airway surface liquid secretion has also been observed in swine trachea submucosal glands after a 35 min exposure to P. aeruginosa (Luan et al., 2014). These observations could be interpreted as an early response to bacterial infection by the airway epithelium, in an attempt to favor the clearance of pathogens through improved CFTR-dependent Cl− and fluid secretion; whereas a longer exposure to P. aeruginosa elicits a negative effect on CFTR.
The mechanisms responsible for the observed decrease in CFTR expression and function may be multiple. Our previous study (Trinh et al., 2015) showed that exposure to PACF508 exoproducts decreased CFTR protein synthesis and enhanced protein degradation. The Stanton's group also reported a reduced number of CFTR channels at the apical membrane due to increased CFTR degradation in lysosomes and altered endocytic recycling in infectious conditions (Swiatecka-Urban et al., 2006; MacEachran et al., 2007; Bomberger et al., 2011). Several lines of evidence indicated that this effect may be mediated by the CFTR inhibitory factor (Cif, PA2394) from the outer membrane vesicles of P. aeruginosa, through a reduction of USP10-mediated deubiquitination of CFTR (MacEachran et al., 2007; Bomberger et al., 2011). Another study indicated that PAO1 inhibits CFTR endocytic recycling through a post-transcriptional modification of NHERF, by a mechanism dependent on bacterial pili and flagellin (Rubino et al., 2014). Furthermore, various secreted bacterial products may impair CFTR expression and function. A recent study (Saint-Criq et al., 2017) showed that LasB elastase in PAO1 secretome degrades CFTR and decreases CFTR activity. Our data also point toward a role for exoproducts under the control of quorum sensing, especially LasR. Indeed, P. aeruginosa strains carrying wild-type lasR alleles (PAO1, PAO1-V, Early) severely impaired CFTR, whereas an engineered (PAO1-V ΔlasR) mutant did not alter CFTR expression and function. Notably, CFTR impairment was also abolished with a spontaneous CF-adapted (Late) lasR mutant. This Late isolate harbors 68 different mutations compare to the Early, including in lasR and rhlR quorum sensing systems, as well as virulence factors and virulence regulators. Accordingly, we previously showed that the Late isolate exhibited reduced elastase/protease activities (Ruffin et al., 2016), which may explain why this isolate elicited less harmful impact on CFTR. Due to the number of mutations in the Late isolate, it is however not possible to ascertain whether the loss of deleterious effect on CFTR is due to the lasR mutation. LasR complementation in the Late strain would have been useful to test this hypothesis. Notably, the Late mutant exhibit reduced production of protease and elastase, as well as pyocyanine (Smith, 2006; Ruffin et al., 2016). The latter, a phenazine toxin, has been shown to down-regulate the vacuolar ATPase-dependent expression and apical membrane localization of CFTR (Kong et al., 2006). Our findings also provided the first proof of concept that a quorum sensing inhibitor (HDMF) decreased elastase production, which is directly regulated by LasR and abolished P. aeruginosa's negative effect on CFTR. We are aware however that HDMF does not elicit a specific effect on the LasR quorum sensing, since it has been shown to inhibit the production of virulence factors regulated by the las, rhl and pqs quorum sensing systems (Choi et al., 2014). Altogether, work from our group and others indicate that various bacterial factors and cellular mechanisms likely contribute to the regulation of CFTR expression and function by P. aeruginosa.
Our study showed that exposure to PAO1 and PAO1-V exoproducts reduced F508del-CFTR maturation as well as cAMP- and Vx-770 stimulated CFTR currents in CFBE-ΔF508 and primary CF AEC treated with either VRT-325 or Vx-809 correctors. These data are in agreement with previous reports (Stanton et al., 2015; Trinh et al., 2015), including from our group, revealing that certain P. aeruginosa strains and CF clinical isolates may have a deleterious impact on CFTR functional rescue. In clinical studies of CFTR modulators, including CF patients infected/colonized with P. aeruginosa, we speculate that the negative effect of P. aeruginosa on CFTR rescue contribute to the lesser clinical benefits of CFTR modulators on lung function (Wainwright et al., 2015), compared to their expected in vitro activity established in pathogen free conditions.
P. aeruginosa undergo genetic adaptation during CF infection, leading to mutations associated with phenotypes such as loss of LasR quorum sensing signaling function and mucoidy (alginate overproduction). Results from our group (Trinh et al., 2015) and others (Stanton et al., 2015) indicated that both mucoid and non-mucoid P. aeruginosa isolates altered CFTR expression and rescue. In this study, we now show for the first time that lasR loss-of-function mutations as well as mutations in genes coding for virulence factors and virulence factors regulators [as found in the Late isolate (Smith, 2006)] abolished the deleterious impact of P. aeruginosa on CFTR. This observation thus indicated that the effect of P. aeruginosa on CFTR varies as a function of the infecting bacteria and may evolve over the course of the disease. This may thus explain, at least in part, the marked variability in clinical efficacy of CFTR modulators among patients, even when they harbor the same CFTR mutation (Durmowicz et al., 2013; Wainwright et al., 2015; Ramsey et al., 2017). Moreover, the response of AEC to CFTR correctors/potentiators may be even more difficult to predict as genetically and phenotypically heterogeneous P. aeruginosa populations co-exist in different regions of the lungs within the same patients (Workentine et al., 2013). The impact of P. aeruginosa on CFTR thus adds to other factors that may modulate the response to treatments in CF patients, including drug pharmacokinetics, the presence of complex CFTR alleles and modifier genes (Strug et al., 2016), the level of residual CFTR function, the influence of environmental factors and the lung disease severity at the time of treatment initiation.
Finally, our results unveiled that the inhibitory effect of PAO1 on CFTR maturation and functional rescue in Vx-809 and Vx-770 treated CFBE-ΔF508 can be significantly reduced by a treatment of P. aeruginosa with HDMF. Individual analysis of CFTR currents in primary CF human airway epithelial cell cultures then indicated that HDMF treatment abolished the deleterious impact of P. aeruginosa for all the 4 tested patients. We are aware that the concentration of HDMF used in that study is high. However, we previously verified that bacterial growth was not affected by this dose of HDMF, indicating that the observed effect was not due to an antimicrobial activity of HDMF on P. aeruginosa (Ruffin et al., 2016). Moreover, we verified that HDMF did not elicit any effect per se on CFTR, but reduced P. aeruginosa virulence (Supplementary Figure S1). Quorum sensing inhibitors, such as HDMF, have indeed been shown to limit virulence factor production and biofilm formation, and have been proposed as adjuvant therapy to antibiotics (Hurley et al., 2012; Bhardwaj et al., 2013; Kalia, 2013; Choi et al., 2014). Importantly, our data demonstrated that quorum sensing inhibitors may also counteract the harmful action of P. aeruginosa infection on the rescue of F508del-CFTR in CF AEC. Although additional studies are required to further develop this approach, our work provide the first proof of evidence that interfering with quorum sensing systems, especially LasR, could improve the effectiveness of CFTR-directed treatments in CF patients.
Author contributions
EM, MR, DA, DN, and EB designed the study; EM, MR, DA, HM, SL, and GM performed the experiments; EM, MR, DA, HM, SL, DN, and EB analyzed and/or interpreted the data. EM and EB wrote the manuscript; MR, DA, and DN revised the manuscript for important intellectual content. All authors approved the final version of the manuscript and agree to be accountable for the content of the work.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thanks Jane L. Burns (Seattle Children's hospital, Seattle, WA, USA) (U.S. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases Grant P30 DK 089507) for providing the AMT0023-30 and AMT0023-34 isolates for this project. The PAO1-V was kindly provided by Jeffrey A. Hobden (Louisiana State University Health Sciences Center, New Orleans, LA, USA). We acknowledge R. J. Bridges (Rosalind Franklin University, North Chicago, IL, USA) and Cystic Fibrosis Foundation Therapeutic Inc. (CFFT) (Bethesda, MA, USA) for providing VRT-325 as well as J. Riordan (University of North Carolina, Chapel Hill, NC, USA) and CFFT for providing the CFTR Ab596.
Footnotes
Funding. The authors acknowledge the Respiratory Health Network of Fonds de Recherche du Québec – Santé (FRQS) for the support of our Respiratory Tissue and Cell Biobank of CRCHUM. This work was supported by Cystic Fibrosis Canada (CFC grant 2368 to EB), Canadian Institutes of Health Research (CIHR, grant PJT148593 to EB), Association Vaincre la Mucoviscidose (RF20160501674), les Fonds de Recherche du Québec en Santé (fellowships to MR and DA), Université de Montréal (COPSE summer award to HM); CRCHUM and Université de Montréal (scholarship to EB), CIHR and CFC (studentship to SL) and CIHR and FRQS (scholarship to DN). The funders had no role in study design, data collection and interpretation or on the decision to submit the work for publication.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2017.00470/full#supplementary-material
References
- Bebok Z., Collawn J. F., Wakefield J., Parker W., Li Y., Varga K., et al. (2005). Failure of cAMP agonists to activate rescued ΔF508 CFTR in CFBE41o - airway epithelial monolayers. J. Physiol. 569, 601–615. 10.1113/jphysiol.2005.096669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell S. C., De Boeck K., Amaral M. D. (2015). New pharmacological approaches for cystic fibrosis: promises, progress, pitfalls. Pharmacol. Ther. 145, 19–34. 10.1016/j.pharmthera.2014.06.005 [DOI] [PubMed] [Google Scholar]
- Bhardwaj A. K., Vinothkumar K., Rajpara N. (2013). Bacterial quorum sensing inhibitors: attractive alternatives for control of infectious pathogens showing multiple drug resistance. Recent Pat. Antiinfect. Drug Discov. 8, 68–83. 10.2174/1574891X11308010012 [DOI] [PubMed] [Google Scholar]
- Bilodeau C., Bardou O., Maillé É., Berthiaume Y., Brochiero E. (2016). Deleterious impact of hyperglycemia on cystic fibrosis airway ion transport and epithelial repair. J. Cyst. Fibros. 15, 43–51. 10.1016/j.jcf.2015.04.002 [DOI] [PubMed] [Google Scholar]
- Bomberger J. M., Ye S., MacEachran D. P., Koeppen K., Barnaby R. L., Oandapos Toole G. A., et al. (2011). A Pseudomonas aeruginosa toxin that hijacks the host ubiquitin proteolytic system. PLoS Pathog. 7:e1001325. 10.1371/journal.ppat.1001325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewington J. J., McPhail G. L., Clancy J. P. (2015). Lumacaftor alone and combined with ivacaftor: preclinical and clinical trial experience of F508del CFTR correction. Expert Rev. Respir. Med. 10, 5–17. 10.1586/17476348.2016.112252 [DOI] [PubMed] [Google Scholar]
- Burns J. L., Gibson R. L., McNamara S., Yim D., Emerson J., Rosenfeld M., et al. (2001). Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. J. Infect. Dis. 183, 444–452. 10.1086/318075 [DOI] [PubMed] [Google Scholar]
- Choi S. C., Zhang C., Moon S., Oh Y. S. (2014). Inhibitory effects of 4-hydroxy-2,5-dimethyl-3(2H)-furanone (HDMF) on acyl-homoserine lactone-mediated virulence factor production and biofilm formation in Pseudomonas aeruginosa PAO1. J. Microbiol. 52, 734–742. 10.1007/s12275-014-4060-x [DOI] [PubMed] [Google Scholar]
- Coraux C., Kileztky C., Polette M., Hinnrasky J., Zahm J.-M., Devillier P., et al. (2004). Airway epithelial integrity is protected by a long-acting beta2-adrenergic receptor agonist. Am. J. Respir. Cell Mol. Biol. 30, 605–612. 10.1165/rcmb.2003-0056OC [DOI] [PubMed] [Google Scholar]
- Durmowicz A. G., Witzmann K. A., Rosebraugh C. J., Chowdhury B. A. (2013). Change in sweat chloride as a clinical end point in cystic fibrosis clinical trials: the ivacaftor experience. Chest 143, 14–18. 10.1378/chest.12-1430 [DOI] [PubMed] [Google Scholar]
- Feliziani S., Luján A. M., Moyano A. J., Sola C., Bocco J. L., Montanaro P., et al. (2010). Mucoidy, quorum sensing, mismatch repair and antibiotic resistance in Pseudomonas aeruginosa from cystic fibrosis chronic airways infections. PLoS ONE 5:e12669. 10.1371/journal.pone.0012669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkesson A., Jelsbak L., Yang L., Johansen H. K., Ciofu O., Høiby N., et al. (2012). Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat. Rev. Microbiol. 10, 841–851. 10.1038/nrmicro2907 [DOI] [PubMed] [Google Scholar]
- Goor F., Van Straley K. S., Cao D., Hadida S., Hazlewood A., Joubran J., et al. (2006). Rescue of ΔF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Physiol. Lung Cell Mol. Physiol. 290, L1117–L1130. 10.1152/ajplung.00169.2005 [DOI] [PubMed] [Google Scholar]
- Haenisch M. D., Ciche T. A., Luckie D. B. (2010). Pseudomonas or LPS exposure alters CFTR iodide efflux in 2WT2 epithelial cells with time and dose dependence. Biochem. Biophys. Res. Commun. 394, 1087–1092. 10.1016/j.bbrc.2010.03.131 [DOI] [PubMed] [Google Scholar]
- Haq I. J., Gray M. A., Garnett J. P., Ward C., Brodlie M. (2015). Airway surface liquid homeostasis in cystic fibrosis: pathophysiology and therapeutic targets. Thorax 71, 284–287. 10.1136/thoraxjnl-2015-207588 [DOI] [PubMed] [Google Scholar]
- Hobden J. A. (2002). Pseudomonas aeruginosa proteases and corneal virulence. DNA Cell Biol. 21, 391–396. 10.1089/10445490260099674 [DOI] [PubMed] [Google Scholar]
- Hurley M. N., Camara M., Smyth A. R. (2012). Novel approaches to the treatment of Pseudomonas aeruginosa infections in cystic fibrosis. Eur. Respir. J. 40, 1014–1023. 10.1183/09031936.00042012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez P. N., Koch G., Thompson J. A., Xavier K. B., Cool R. H., Quax W. J. (2012). The multiple signaling systems regulating virulence in Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 76, 46–65. 10.1128/MMBR.05007-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia V. C. (2013). Quorum sensing inhibitors: an overview. Biotechnol. Adv. 31, 224–245. 10.1016/j.biotechadv.2012.10.004 [DOI] [PubMed] [Google Scholar]
- Kong F., Young L., Chen Y., Ran H., Meyers M., Joseph P., et al. (2006). Pseudomonas aeruginosa pyocyanin inactivates lung epithelial vacuolar ATPase-dependent cystic fibrosis transmembrane conductance regulator expression and localization. Cell Microbiol. 8, 1121–1133. 10.1111/j.1462-5822.2006.00696.x [DOI] [PubMed] [Google Scholar]
- Kunzelmann K., Schwiebert E. M., Zeitlin P. L., Kuo W. L., Stanton B. A., Gruenert D. C. (1993). An immortalized cystic fibrosis tracheal epithelial cell line homozygous for the delta F508 CFTR mutation. Am. J. Respir. Cell Mol. Biol. 8, 522–529. 10.1165/ajrcmb/8.5.522 [DOI] [PubMed] [Google Scholar]
- LaFayette S. L., Houle D., Beaudoin T., Wojewodka G., Radzioch D., Hoffman L. R., et al. (2015). Cystic fibrosis-adapted Pseudomonas aeruginosa quorum sensing lasR mutants cause hyperinflammatory responses. Sci. Adv. 1, 1–16. 10.1126/sciadv.1500199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan X., Campanucci V. A., Nair M., Yilmaz O., Belev G., Machen T. E., et al. (2014). Pseudomonas aeruginosa triggers CFTR-mediated airway surface liquid secretion in swine trachea. Proc. Natl. Acad. Sci. U.S.A. 111, 1–6. 10.1073/pnas.1406414111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacEachran D. P., Ye S., Bomberger J. M., Hogan D. A., Swiatecka-Urban A., Stanton B. A., et al. (2007). The Pseudomonas aeruginosa secreted protein PA2934 decreases apical membrane expression of the cystic fibrosis transmembrane conductance regulator. Infect. Immun. 75, 3902–3912. 10.1128/IAI.00338-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matkovic Z., Miravitlles M. (2013). Chronic bronchial infection in COPD. is there an infective phenotype? Respir. Med. 107, 10–22. 10.1016/j.rmed.2012.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D., Joshi-Datar A., Lepine F., Bauerle E., Olakanmi O., Beer K., et al. (2011). Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 334, 982–986. 10.1126/science.1211037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedemonte N., Lukacs G. L., Du K., Caci E., Zegarra-Moran O., Galietta L. J. V., et al. (2005). Small-molecule correctors of defective ΔF508-CFTR cellular processing identified by high-throughput screening. J. Clin. Invest. 115, 2564–2571. 10.1172/JCI24898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X., Zerr D. M., McNutt M. A., Berry J. E., Burns J. L., Kapur R. P. (2012). Pseudomonas aeruginosa syntrophy in chronically colonized airways of cystic fibrosis patients. Antimicrob. Agents Chemother. 56, 5971–5981. 10.1128/AAC.01371-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey B. W., Davies J., McElvaney G. N., Tullis E., Bell S. C., Dreninek P. D., et al. (2017). A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. Hum. Mol. Genet. 365, 1663–1672. 10.1056/NEJMoa1105185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rejman J., Di Gioia S., Bragonzi A., Conese M. (2007). Pseudomonas aeruginosa infection destroys the barrier function of lung epithelium and enhances polyplex-mediated transfection. Hum. Gene Ther. 18, 642–652. 10.1089/hum.2006.192 [DOI] [PubMed] [Google Scholar]
- Rubino R., Bezzerri V., Favia M., Facchini M., Tebon M., Singh A. K., et al. (2014). Pseudomonas aeruginosa reduces the expression of CFTR via post-translational modification of NHERF1. Pflugers Arch. Eur. J. Physiol. 466, 2269–2278. 10.1007/s00424-014-1474-6 [DOI] [PubMed] [Google Scholar]
- Ruffin M., Bilodeau C., Maille E., LaFayette S. L., McKay G. A., Trinh N. T. N., et al. (2016). Quorum-sensing inhibition abrogates the deleterious impact of Pseudomonas aeruginosa on airway epithelial repair. FASEB J. 30, 3011–3025. 10.1096/fj.201500166R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford S. T., Bassler B. L. (2012). Bacterial quorum sensing: its role in virulence and possibilities for its control. Cold Spring Harb. Perspect. Med. 2:a012427. 10.1101/cshperspect.a012427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-Criq V., Villeret B., Bastaert F., Kheir S., Hatton A., Cazes A., et al. (2017). Pseudomonas aeruginosa LasB protease impairs innate immunity in mice and humans by targeting a lung epithelial cystic fibrosis transmembrane regulator–IL-6–antimicrobial–repair pathway. Thorax. 10.1136/thoraxjnl-2017-210298. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E. E. (2006). Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl. Acad. Sci. U.S.A. 103, 8487–8492. 10.1073/pnas.0602138103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton B. A., Coutermarsh B., Barnaby R., Hogan D. (2015). Pseudomonas aeruginosa reduces VX-809 stimulated F508del-CFTR chloride secretion by airway epithelial cells. PLoS ONE 10:e0127742. 10.1371/journal.pone.0127742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strug L. J., Gonska T., He G., Keenan K., Ip W., Boëlle P.-Y., et al. (2016). Cystic fibrosis gene modifier SLC26A9 modulates airway response to CFTR-directed therapeutics HMG advance access. Hum. Mol. Genet. 25, 4590–4600. 10.1093/hmg/ddw290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiatecka-Urban A., Moreau-Marquis S., Maceachran D. P., Connolly J. P., Stanton C. R., Su J. R., et al. (2006). Pseudomonas aeruginosa inhibits endocytic recycling of CFTR in polarized human airway epithelial cells. Am. J. Physiol. Cell Physiol. 290, C862–C872. 10.1152/ajpcell.00108.2005 [DOI] [PubMed] [Google Scholar]
- Trinh N. T. N., Bardou O., Prive A., Maille E., Adam D., Lingee S., et al. (2012). Improvement of defective cystic fibrosis airway epithelial wound repair after CFTR rescue. Eur. Respir. J. 40, 1390–1400. 10.1183/09031936.00221711 [DOI] [PubMed] [Google Scholar]
- Trinh N. T. N., Bilodeau C., Maillé É., Ruffin M., Quintal M.-C., Desrosiers M.-Y., et al. (2015). Deleterious impact of Pseudomonas aeruginosa on cystic fibrosis transmembrane conductance regulator function and rescue in airway epithelial cells. Eur. Respir. J. 45, 1590–1602. 10.1183/09031936.00076214 [DOI] [PubMed] [Google Scholar]
- Van Goor F., Hadida S., Grootenhuis P. D. J., Burton B., Stack J. H., Straley K. S., et al. (2011). Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. U.S.A. 108, 18843–18848. 10.1073/pnas.1105787108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainwright C. E., Elborn J. S., Ramsey B. W., Marigowda G., Huang X., Cipolli M., et al. (2015). Lumacaftor-Ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N. Engl. J. Med. 373, 1–12. 10.1056/NEJMoa1409547 [DOI] [PubMed] [Google Scholar]
- Workentine M. L., Sibley C. D., Glezerson B., Purighalla S., Norgaard-Gron J. C., Parkins M. D., et al. (2013). Phenotypic heterogeneity of Pseudomonas aeruginosa populations in a cystic fibrosis patient. PLoS ONE 8:e60225. 10.1371/journal.pone.0060225 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.