

Abstract
Pain remains a challenging clinical condition and spinal GABAA receptors are crucial modulators of pain processing. α2/α3-subtype GABAA receptors mediate the analgesic actions of benzodiazepines. Positive allosteric modulators (PAMs) at α2/α3-subtype GABAA receptors may have analgesic potential. Here we report a new selective α2/α3-subtype GABAA receptor PAM in in vitro and in vivo pain assays. KRM-II-81 demonstrated similar efficacy at α1/α2/α3 GABAA receptors and negligible efficacy at α4/α5/α6 GABAA receptors, with α2 and α3- subtypes being 17- and 28-fold more potent than α1 subtypes in HEK-293T cells expressing GABAA receptors with different α subunits. In contrast, KRM-II-18B showed significant efficacy at α1/α2/α3/α5 subtypes, with similar potency at α1/α2/α3 subtypes. Both PAMs and morphine dose-dependently decreased 0.6% acetic acid- and 0.32% lactic acid-induced writhing. The effects of both PAMs were reversed by the benzodiazepine receptor antagonist flumazenil, confirming their action at the benzodiazepine binding site of GABAA receptors. Both PAMS and morphine all dose-dependently reversed 0.32% lactic acid (but not 0.6% acetic acid)-induced suppression of nesting behavior. Acetaminophen, but neither PAM, reversed acid-depressed locomotor activity. Combined, these findings suggest that KRM-II-81 is a selective α2/α3 subtype GABAA PAM with significant antinociceptive effects in chemical stimulation-induced pain in mice.
Keywords: GABAA, Positive allosteric modulator, Pain, Writhing, Nesting, Mice
Graphical abstract
INTRODUCTION
Over 100 million Americans suffer from chronic pain conditions and millions more suffer from acute pain1. Pain imposes not only physical and emotional discomfort on its sufferers but also a financial burden on the individual and society, with total costs estimated at $635 billion dollars annually2. Opioids and non-steroidal anti-inflammatory drugs (NSAIDs), while the current standards for pain management, have detrimental properties that limit their use. Opioids are associated with tolerance and abuse liability3–5 and NSAIDs are associated with increased risk of gastrointestinal and cardiovascular adverse effects6. Therefore, the development of novel analgesics remains a dire clinical need.
There have been no mechanistically novel analgesics developed in the past 50 years7. Some argue that the low success rate in the discovery of new analgesics could be partially due to the poor translation of existing animal models of pain8, 9. For example, most studies of pain only use pain-stimulated behaviors such as tail flick or paw withdrawal. Expanding the range of pain models used in preclinical studies to include pain-depressed behaviors may help to discover analgesics with better clinical success, and may also have higher face validity with respect to clinical pain conditions (e.g., chronic pain sufferers likely move less to avoid aggravating their pain as opposed to reacting more strongly to external noxious stimuli)8, 9.
According to the Gate Control Theory of pain, the transmission of pain signals from the periphery to the spinal cord is modulated by excitatory and inhibitory neurons emanating from the brain10. The primary inhibitory neurotransmitter in the spinal cord is gamma-aminobutyric acid (GABA). Chronic pain states are associated with a reduced GABA-mediated inhibitory function11. Therefore, it is theoretically plausible to reverse the GABAergic disinhibition by enhancing GABAergic function which should lead to analgesia. GABAA receptors are pentameric chloride channels usually comprising 2α, 2β and 1γ subunits (α2β2γ), in which there exists 6 different α subtypes (α1–α6). Benzodiazepines are a class of GABAA receptor positive allosteric modulators (PAMs) that are widely used for a number of clinical conditions. At the molecular level, benzodiazepines bind to and interact with four different α subtypes (α1–3, α5). However, there is little clinical evidence that benzodiazepines are analgesics, and we now know that the analgesic action of benzodiazepines is likely masked by their other effects such as sedation12. Point mutation studies and other studies employing pharmacological approaches have shown that different α subtypes mediate different pharmacological actions of benzodiazepines13. For example, the addiction-related effects of benzodiazepines are primarily mediated through α1/α2 subtypes, the analgesic effects through α2/α3 subtypes, and cognitive functions through α5 subtypes of GABAA receptors. Therefore, developing α2/α3 subtype- selective GABAA receptor PAMs may be a strategy to discover novel analgesics. Previous studies have identified a quite selective moderate-to-low efficacy α2/α3 subtype-selective GABAA receptor PAM NS16085, which demonstrated partial suppression of nociceptive behaviors in the formalin assay14.
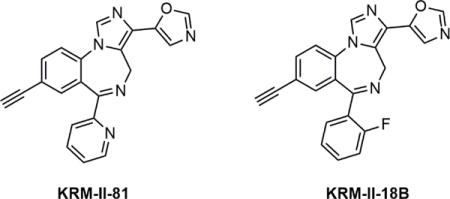
In an effort to develop novel α2/α3 subtype-selective GABAA receptor PAMs, here we report a new compound, KRM-II-81 (5-(8-ethynyl-6-(pyridin-2-yl)-4H-benzo[f]imidazo[1,5-a][1,4]diazepin-3-yl) oxazole) (Fig. 1), which demonstrated the profile as a highly selective α2/α3-specific GABAA receptor PAM. We first examined the α subtype specificity of KRM-II- 81 using electrophysiological recording in cells expressing different α subtype GABAA receptors. We then examined the antinociceptive effects of KRM-II-81 in several mice models of chemical stimulation induced visceral pain. Chemical-induced pain models were chosen because previous studies showed that they are sensitive to pharmacological modulation of GABAA receptors15–17. A structurally similar compound, KRM-II-18B (5-(8-ethynyl-6-(2-fluorophenyl)-4H-benzo[f]imidazo[1,5-a][1,4]diazepin-3-yl) oxazole) (a non-selective GABAA receptor PAM) was also studied in parallel for comparison.
Figure 1.
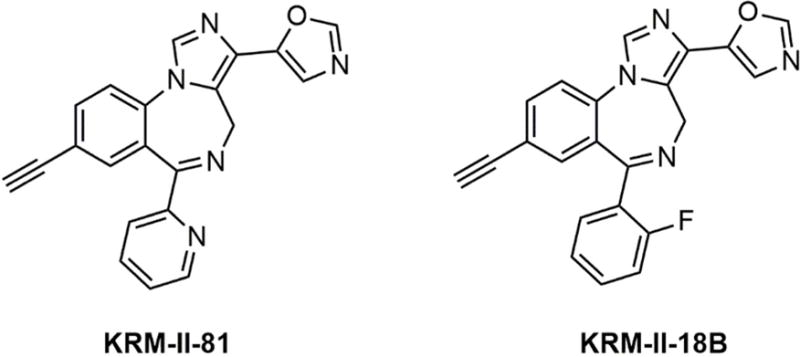
Chemical structures of KRM-II-81 and KRM-II-18B.
RESULTS AND DISCUSSION
This study examined the in vitro and in vivo effects of newly synthesized GABAA receptor PAMs, KRM-II-18B and KRM-II-81. Cellular electrophysiological tracing identified KRM-II-81 as a α2/α3 subtype-selective PAM while KRM-II-18B was non-selective. Two compounds were then evaluated in mice models of pain-stimulated (writhing) and pain-suppressed (acid-depressed nesting and locomotion) behaviors. Both PAMs significantly decreased acetic acid- and lactic acid-induced writhing, which was attenuated by the benzodiazepine receptor antagonist flumazenil. KRM-II-18B and KRM-II-81 restored lactic acid (but not acetic acid)-depressed nesting. In the assay of acid-depressed locomotion, acetaminophen but neither of the two GABAA receptor PAMs was effective in attenuating the decrease in locomotion. Combined, this study identified a novel high-efficacy α2/α3 subtype-selective PAM which showed significant antinociceptive effects in mouse assays of chemical stimulation-induced pain-like behaviors.
KRM-II-81 is a selective α2/α3-selective GABAA receptor PAM in in vitro characterization
Given the critical role of spinal modulation of pain processing by GABAergic neurons, one new strategy for novel analgesic discovery may involve the positive allosteric modulation of α2/α3 subtype-containing GABAA receptors. Previous research has identified GABAA receptor PAMs which has some selectivity on the individual α subtypes and shows some promising results with specific antinociceptive effects. For example, NS11394 has functional efficacy selectivity of α5 > α3 > α2 > α1 and produces significant antinociceptive effects in rat models of inflammatory and neuropathic pain but does not produce sedation at the same doses18. However, given the highest efficacy of NS11394 at α5 subtype GABAA receptors, its side effect on cognitive impairment could be serious13. A more selective compound NS16085 demonstrated moderate to low efficacy at α2 and α3 subtypes, no efficacy at α5 subtypes and a slight negative efficacy at α1 subtypes, which was partially effective in the formalin test14. In an effort to develop novel and high-efficacy selective α2/α3 subtype GABAA receptor PAMs, we discovered KRM-II-81.
HEK-293T cells were transiently transfected with one of the six different α subunit subtypes along with the same β (β3) and γ (γ2L) subunits. To determine the sensitivity to modulation, a submaximal concentration of GABA was co-applied with the modulator for 5 sec to cells voltage-clamped at −50 mV. The GABA concentration represented an EC<5 μM for each isoform19 and was 0.1 μM (α6), 0.3 μM (α4, α5), 1 μM (α1, α2) or 3 μM (α3). As expected, receptors containing α4 or α6 subunits were insensitive to a 1 μM concentration of any of these compounds (Fig. 2E). KRM-II-18B was an effective and potent modulator of all the benzodiazepine-sensitive receptor isoforms, enhancing the response to GABA to comparable maximum levels and with similar EC50’s (Fig. 2A, 2C). The average EC50 (and peak response) for potentiation of the response to GABA by KRM-II-18B was 738.8 ± 251.9 nM (410.6 ± 26.3%) for α1β3γ2L (n=7), 261.4 ± 115.8 nM (320.0 ± 14.3%) for α2β3γ2L (n=5) and 169.2 ± 39.8 nM (383.0 ± 33.2%) for α3β3γ2L (n=5). In contrast, the α1- and α5-containing receptors were much less sensitive to modulation by KRM-II-81, while the α2- and α3-containing receptors were robustly potentiated. From full concentration-response relationships, we found that the difference in sensitivity conferred by subunit subtype was largely in the relative potency of KRM-II-81, rather than its maximum efficacy (Fig. 2B, 2D). The average EC50 (and peak response) for potentiation of the response to GABA by KRM-II-81 was 1.73 ± 0.69 μM (316.9 ± 38.6%) for α1β3γ2L (n=5) compared to 101.9 ± 28.5 nM (350.1 ± 21.5%) for α2β3γ2L (n=5) and 60.9 ± 11.6 nM (352.2 ± 38.0%) for α3β3γ2L (n=5), which is equivalent to a 17-fold and 28-fold selectivity on α2 and α3 subtypes over α1 subtype GABAA receptors, respectively. As a comparison, KRM-II-18B showed no significant selectivity among the three subtypes. Therefore, KRM-II-81 represents a novel and the first high-efficacy selective α2/α3 subtype-selective GABAA receptor PAM.
Figure 2.
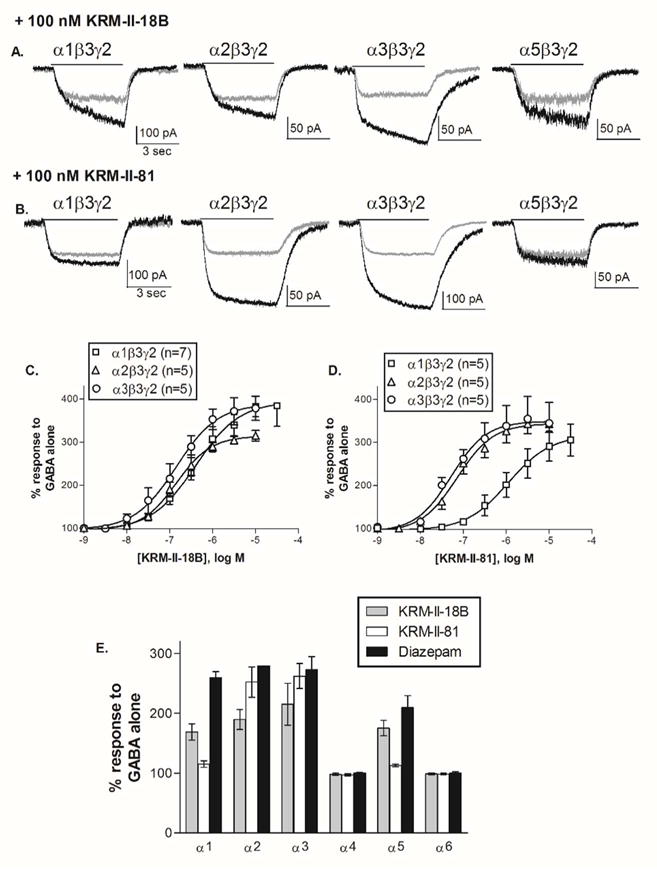
(A, B) Cells were transiently transfected with one of the α subtypes, as indicated, along with β3 and γ2L, and voltage clamped at −50 mV. Representative whole-cell currents are shown for 5 sec applications of GABA alone (gray) or GABA + 0.1 μM modulator (black). (C, D) Concentration-response relationships for the positive allosteric modulators at α1-, α2, and α3- containing receptors. The peak current amplitude was divided by the response to GABA alone for each cell. Symbols (±SEM) show the average response from 5–7 cells. (E) Average enhancement of the current evoked to GABA by 0.1 μM (α1, α2, α3, α5) or 1 μM (α4, α6) of the modulator indicated. The response was divided by the peak response to GABA alone for each cell. The dashed line at 100% indicates the response to GABA alone. Bars represent mean ± SEM (n=4–8).
KRM-II-81 and KRM-II-18B reduced acetic acid- and lactic acid-induced writhing
In order to examine the in vivo activity of KRM-II-81, we used an acid-induced writhing test in mice to evaluate its potential antinociceptive effects. Mice pre-treated with vehicle exhibited a mean of 46.29 ±3.859 writhes following treatment of 0.6% acetic acid. As a positive control, the opioid morphine dose-dependently attenuated acetic acid-induced writhing [F (3, 21) = 31.77, p < 0.001]. Post hoc analyses indicated that the effects of 1.0 and 3.2 mg/kg of morphine were significantly different from vehicle and 0.32 mg/kg morphine. KRM-II-18B and KRM-II-81 both dose-dependently decreased the acetic acid-induced writhes [KRM-II-18B: F (3, 22) = 11.16, p < 0.01, KRM-II-81: F (3, 23) = 7.05, p <0.05]. Post hoc analyses revealed that the effect of 3.2 and 10 mg/kg of KRM-II-18B were significantly different from those of vehicle and 1 mg/kg KRM-II-18B. The effects of of 5.6 and 10 mg/kg of KRM-II-81 were significantly different from those of vehicle and 3.2 mg/kg KRM-II-81 (Figure 3A). Larger doses of KRM-II-18B and KRM-II-81 were not studied because a pilot study found that a larger dose (32 mg/kg) produced marked sedation in mice, which may affect interpretation of the behavioral data.
Figure 3.
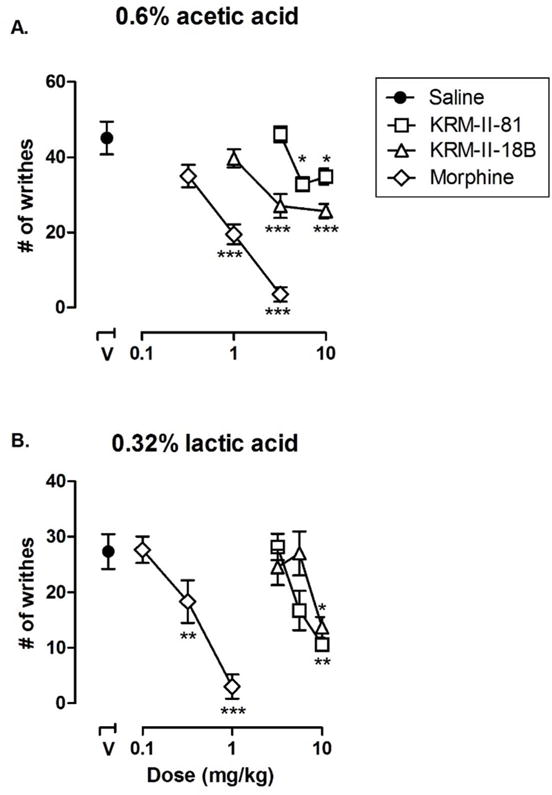
Effects of morphine, KRM-II-18B, and KRM-II81 on A) 0.6% acetic acid and B) 0.32% lactic acid-induced writhing. All points represent the mean and error bars show S.E.M. (n = 6–8 per group). Abscissa: dose of drug expressed as milligram per kilogram. Data point above “V” represents the number of writhes following administration of either A) acetic acid or B) lactic acid once pretreated with vehicle. Ordinate: number of writhes observed in the 25-minute observation period. Asterisks indicate data points that are significantly different from vehicle pretreatment (V) alone. *P < 0.05, ***p < 0.001.
Because both GABAA receptor PAMs only partially reduced 0.6% acetic acid-induced writhes, we hypothesized that such a chemical stimulus was too strong and these compounds might be more effective if a weaker painful stimulation were used. We then used 0.32% lactic acid-induced writhes to test the same doses of KRM-II-81 and KRM-II-18B. When 0.32% lactic acid was used, mice pre-treated with vehicle exhibited 27.33 ± 3.138 writhes. Under this condition, morphine also dose-dependently reduced lactic-acid induced writhing [F (3, 20) = 23.47, p < 0.001] and was more potent than in the study using 0.6% acetic acid. Post hoc analyses indicated that the doses of 0.32 and 1.0 mg/kg of morphine significantly decreased the writhes as compared to vehicle and 0.1 mg/kg morphine treatment conditions. KRM-II-18B and KRM-II-81 both decreased the lactic acid-induced writhes [KRM-II-18B: F (3, 20) = 4.23, p < 0.05, KRM-II-81: F (3, 22) = 9.61, p < 0.01]. Post hoc analysis revealed that 10 mg/kg KRM-II-18B significantly decreased the number of writhes as compared to vehicle- and 5.6 mg/kg-treated conditions; 10 mg/kg KRM-II-81 significantly decreased the number of writhes as compared to vehicle- and 3.2 mg/kg-treated conditions (Figure 3B). In order to confirm the receptor mechanisms mediating the effects of the GABAA receptor PAMs, a dose of 3.2 mg/kg flumazenil was used as a pretreatment which showed near complete blockade of the antinociceptive effects of KRM-II-18B (p < 0.05, Figure 4A) and KRM-II-81 (p < 0.001, Figure 4B). This dose of flumazenil was chosen according to published literature, which showed significant blockade of the discriminative stimulus effects of benzodiazepines in rats {Bai, 2011 #22}. This dose of flumazenil alone had no significant effect (data not shown). These results suggested that the antinociceptive effects of these GABAA receptor PAMs are primarily mediated through benzodiazepine binding site of GABAA receptors.
Figure 4.
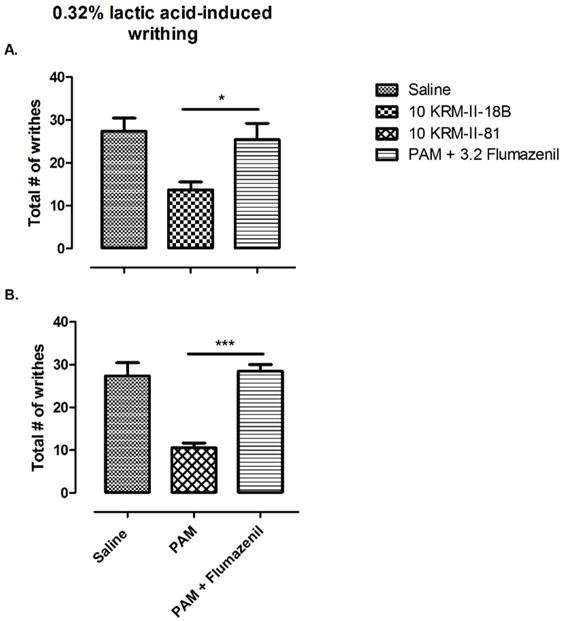
Effects of flumazenil on KRM-II-18B- and KRM-II-81-induced antinociception. Bars represent the mean and error bars show S.E.M. (n = 6–8 per group). Abscissa: pretreatment groups. Ordinate: total number of writhes induced by 0.32% lactic acid. A) KRM-II-18B pretreatment alone and with flumazenil. B) KRM-II-81 pretreatment alone and with flumazenil. Asterisks indicate significant difference in number of writhes in the presence and absence of flumazenil. *P < 0.05, ***p < 0.001.
KRM-II-81 and KRM-II-18B reverted lactic acid-but not acetic-acid-depressed nesting
Because pain not only stimulates nocifensive behaviors but also suppresses many adaptive behaviors, such as nesting or locomotion, measures of pain-depressed behaviors can provide new insights into the behavioral consequences of pain and the effects of candidate analgesics9, 20. We next examined the effects of the GABAA receptor PAMs on acid-depressed nesting behavior. When treated with vehicle, mice cleared 4.7 ± 0.2 out of 5 available zones by the end of the nesting period (data not shown). Acetic acid decreased the number of zones cleared to 1.4 ± 0.3. Morphine, KRM-II-18B, and KRM-II-81 all failed to significantly attenuate the decrease in cleared zones by acetic acid (Figure 5A). This lack of effect could be due to the strong pain stimulation induced by acetic acid, to which nesting behavior was particularly sensitive. When treated with lactic acid (Figure 5B), mice again displayed a decrease in the number of zones cleared to 1.6 ± 0.3. Morphine increased the number of zones cleared [F (3, 23) = 6.53, p < 0.01]. Post hoc analyses revealed that morphine significantly increased the number of zones that were cleared at 3.2 mg/kg as compared to vehicle (p < 0.01). KRM-II-18B and KRM-II-81 also both dose-dependently increased the number of zones cleared [KRM-II-18B: F (3, 26) = 5.97, p < 0.01, KRM-II-81: F (3, 27) = 3.55, p < 0.05]. Post hoc analysis revealed that doses of 10 mg/kg of KRM-II-18B and 10 mg/kg of KRM-II-81 significantly increased the number of zones cleared as compared to vehicle. While morphine, KRM-II-18B, and KRM-II-81 were effective in dose-dependently restoring nesting behavior in lactic acid-treated mice, no drug was effective in acetic acid-treated mice. When considered with the writhing data, these data support the notion that 0.6% acetic acid may produce a greater noxious stimulus than 0.32% lactic acid. In a previous study, morphine was partially effective in reversing nesting behavior in 0.32% lactic acid-treated mice20, and our results were consistent with that. More importantly, higher doses suppressed nesting behavior in non-acid treated (pain-free) mice20. It is conceivable that given the higher strength of pain induced by 0.6% acetic acid, morphine was able to reduce writhes but due to the nesting-suppressive effect of higher doses of morphine, the resultant effect was not statistically significant. Taken together, our results suggest that the ability of α2/α3 GABAA receptor PAMs to restore pain-depressed behavior may greatly depend on the degree of pain.
Figure 5.
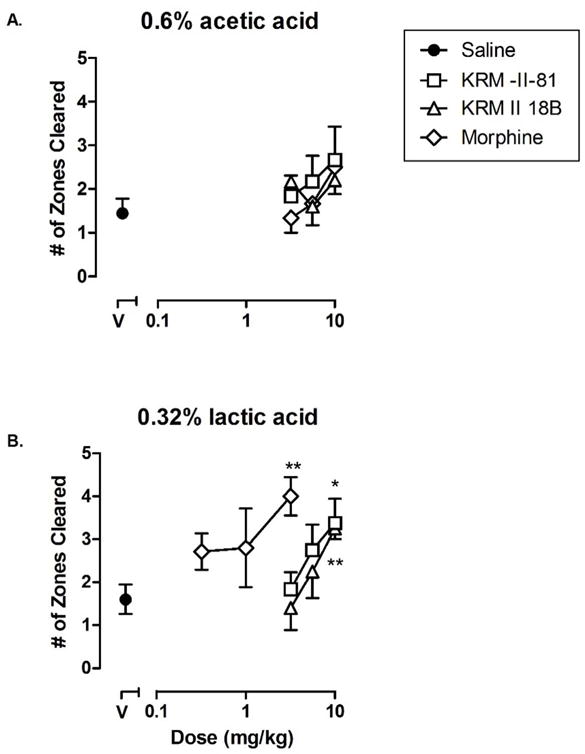
Effects of morphine, KRM-II-18B, and KRM-II-81 on A) acetic acid and B) lactic acid-depressed nesting (n=6–8). Data points show mean data and error bars show S.E.M. Abscissa: dose of drug expressed as milligram per kilogram. Data point above “V” (filled circle) represents the number of zones cleared following administration of either A) acetic acid or B) lactic acid once pretreated with vehicle. Ordinate: total number of zones cleared in the nesting procedure (60 min). Asterisks indicate that data points are significantly different from vehicle pretreatment (V) alone. *P < 0.05, ** p <0.001.
Acetaminophen but not KRM-II-18B nor KRM-II-81 restored acid depressed locomotion
Pain also reduces the spontaneous locomotion in mice21. Next we examined whether the GABAA receptor PAMs could restore acid-depressed locomotion. Because acetaminophen is among the most commonly used medicines to relieve pain and morphine has well-documented effect to increase locomotion22 which may complicate the interpretation of the data, we compared the novel PAMs with acetaminophen instead of morphine in this assay of pain-depressed behavior. When treated with vehicle, mice displayed a locomotor activity of 2323 ± 320.8 cm during the test period. Acetic acid (Figure 6A) and lactic acid (Figure 6B) decreased the locomotor activity to 577.1 ± 121.2 cm and 420.9 ± 62.44 cm, respectively, in agreement with previous reports21. Acetaminophen pretreatment, at a dose that did not significantly change the locomotor activity in naïve mice, dose-dependently increased the locomotor activity of mice treated with acetic acid (F (2, 16) = 3.39, p < 0.05) and lactic acid (F (2, 17) = 11.30, p < 0.001). Post hoc analyses revealed that acetaminophen at a dose of 100 mg/kg produced a significant increase in locomotion as compared to vehicle- and 56 mg/kg-treatment conditions in both the acetic acid- and lactic acid-treated mice. However, pretreatment with KRM-II-18B and KRM-II- 81 each failed to restore the locomotor activity of acetic acid- and lactic acid-treated mice. Larger doses of both PAMs were not studied because they produced effects (e.g., sedation) that are competing with behavioral measures of pain. The restoration of acid-depressed locomotion may require higher analgesic effectiveness than the alleviation of other pain-related behaviors since locomotion itself may intensify existing pain symptoms21, 23, 24. That is, in order to restore locomotion, an analgesic would need to not only reduce the basal noxious stimulus, but also the additional pain resulting from movement. This could be the reason why both GABAA receptor PAMs were not able to restore acid-depressed locomotion.
Figure 6.
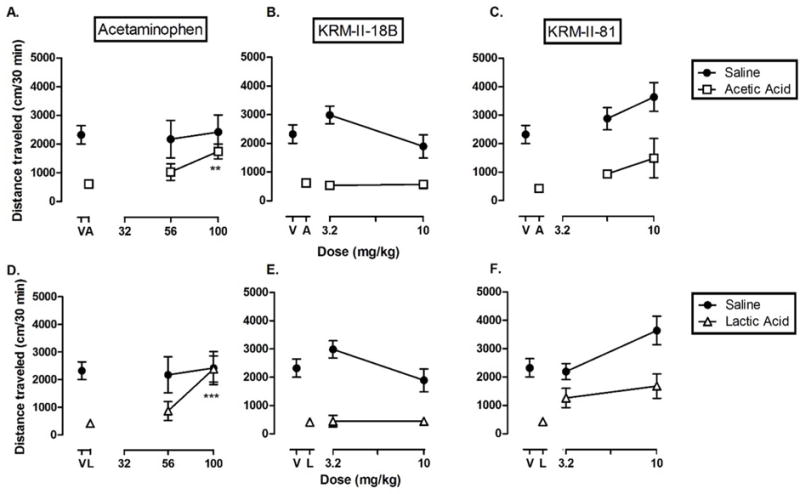
Effects of acetaminophen (A, D), KRM-II-18B (B, E) and KRM-II-81 (C.F) on acetic acid (top) and lactic acid (bottom)-depressed locomotor activity. Data points show mean±S.E.M. (n = 6–8 per group). Abscissa: dose of drug expressed as milligram per kilogram. Data point above “V” represents total distance traveled under control conditions (treated with vehicle). Data point above “A” represents total distance traveled following acid treatment. Ordinate: distance traveled in cm. Asterisks indicate data points significantly different from acetic acid (“A”) or lactic acid (“L”) alone. *P < 0.05, ** p < 0.001.
Although the nesting and locomotion assays used here were both assays of pain-depressed behavior, the effects of the PAMs differed, where the PAMs were effective in the nesting assay but not the locomotion assay. This finding suggests that the ability of the PAMs to alleviate pain-depressed behavior is dependent on the behavioral endpoint. This finding is consistent with previous reports. For example, the dose of analgesic required to restore acid-depressed locomotion was much higher than the dose required to restore acid-depressed feeding21, 25. Other studies have demonstrated disparities in behavioral endpoints such as pain-induced saccharin preference versus acid-depressed locomotor activity26 and pain-depressed wheel-running versus pain-depressed feeding27. Thus, the disparities in the findings of this study are not altogether surprising. Another somewhat surprising finding was that both PAMs showed similar effects in all the behavioral assays under this condition despite the fact that KRM-II-81 was selective for α2/α3 subtypes while KRM-II-18B was a non-selective GABAA receptor PAM. Benzodiazepines are non-selective GABAA receptor PAMs and systemic drug administration typically does not show analgesic effects due to significant sedation. However, intrathecal benzodiazepine administration can produce analgesia28, 29. In this study, the doses of both PAMs used for pain studies did not significantly alter the spontaneous activity, suggesting minimal sedation. Yet both compounds produced clear antinociceptive actions. This could be due to the differential efficacy demands of the behavioral endpoints (i.e., antinociception has lower efficacy demand than sedation), or differential target engagement of both compounds in the central nervous system underlying the behaviors. More work needs to be done to decipher this interesting finding.
In conclusion, this study reported a novel α2/α3-selective GABAA receptor PAM, which demonstrated good selectivity and significant antinociception without decreasing locomotion. Importantly, because the efficacy of GABAA receptor PAMs at α5-subtype is closely related to cognitive impairment and compounds with α5-subtype efficacy such as NS11821 impairs memory and cognition30, the lack of efficacy at α5 subtype GABAA receptors makes KRM-II-81 less likely to produce cognitive impairment. These results support the notion that developing subtype-selective GABAA receptor PAMs may be a viable strategy to discover novel analgesics. It should be noted that the GABAA receptor PAMs described here appear to be less effective than opioids for acid-induced visceral pain. This may not be surprising as acute pain condition may not involve marked spinal GABAergic disinhibition, a condition that GABAA receptor PAMs may be able to reverse. This is supported by studies using other GABAa PAMs. For example, the GABAa PAM NS11394 was only partially effective (33% reduction) in the formalin test18. Future studies should examine the antinociceptive effects of KRM-II81 in more persisting chronic pain conditions such as nerve injury induced neuropathic pain.
METHODS
Cellular studies
Transfection of mammalian cells and electrophysiological recordings
Full-length cDNAs for GABAA receptor subtypes (generously provided by Dr. Robert Macdonald, Vanderbilt University and Dr. David Weiss, University of Texas Health Sci. Center, San Antonio TX) in mammalian expression vectors were transfected into the human embryonic kidney cell line HEK-293T (GenHunter, Nashville, TN). All subtypes were rat clones except for α2, which was a human clone. Cells were maintained in Dulbecco’s modified Eagle medium (DMEM) plus 10% fetal bovine serum, 100 IU/ml penicillin and 100 μg/ml streptomycin.
HEK-293T cells were transiently transfected using calcium phosphate precipitation. Plasmids encoding GABAA receptor subtype cDNAs were added to the cells in 1:1:1 ratios (α:β:γ) of 2 μg each31. For identification of positively transfected cells, 1 μg of the plasmid pHook™-1 (Invitrogen Life Technologies, Grand Island NY) containing cDNA encoding the surface antibody sFv was also transfected into the cells11. Following a 4–6 hr. incubation at 3% CO2, the cells were treated with a 15% glycerol solution in BBS buffer (50 mM BES(N,N-bis[2-hydroxyethyl]-2-aminoethanesulfonic acid), 280 mM NaCl, 1.5 mM Na2HPO4) for 30 sec. The selection procedure for pHook expression was performed 18–52 hrs later. The cells were passaged and mixed for 30–60 min. with 3–5 μl of magnetic beads coated with antigen for the pHook antibody (approximately 6 × 105 beads)11. Bead-coated cells were isolated using a magnetic stand. The selected cells were resuspended into supplemented DMEM, plated onto glass coverslips treated with poly L-lysine and collagen, and used for recordings the next day.
Cells were patch-clamped at −50 mV in the whole-cell recording configuration. The bath solution consisted of (in mM): 142 NaCl, 8.1 KCl, 6 MgCl2, 1 CaCl2, and 10 HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) with pH = 7.4 and osmolarity adjusted to 295–305 mOsm. Recording electrodes were filled with a solution of (in mM); 153 KCl, 1 MgCl2, 5 K-EGTA (ethylene glycol-bis (β-aminoethyl ether N,N,N′N′-tetraacetate), and 10 HEPES with pH = 7.4 and osmolarity adjusted to 295–305 mOsm. GABA was diluted into the bath solution from freshly made or frozen stocks in water. Compounds were dissolved in DMSO and diluted into bath solution with the highest DMSO level applied to cells of 0.01%. Patch pipettes were pulled from borosilicate glass (World Precision Instruments, Sarasota, FL) on a two-stage puller (Narishige, Japan) to a resistance of 5–10 MΩ. Solutions containing GABA or GABA+ compounds were applied to cells for 5 sec. using a 3-barrelled solution delivery device controlled by a computer-driven stepper motor (SF-77B, Harvard Apparatus, Holliston, MA, open tip exchange time of <50 msec). There was a continuous flow of external solution through the chamber. Currents were recorded with an Axon 200B (Foster City, CA) patch clamp amplifier.
Whole-cell currents were analyzed using the programs Clampfit (pClamp9 suite, Axon Instruments, Foster City, CA) and Prism (Graphpad, San Diego, CA). Concentration-response data was fit with a four-parameter logistic equation (Current = [Minimum current + (Maximum current - Minimum Current)]/1+(10ˆ(log EC50 – log [modulator])*n) where n represents the Hill number. All fits were made to normalized data with current expressed as a percentage of the response to GABA alone for each cell.
Behavioral studies
Subjects
Adult male ICR mice (Envigo, Indianapolis, IN, USA) that were 8–12 weeks old and weighed 30–40g upon arrival were used in these studies. Mice were housed in pairs, except those used in the nesting procedure. Mice were on a 12/12 h reverse light/dark cycle (lights on at 6 PM and off at 6 AM) and had free access to water and food except during experimental sessions. All experiments were performed during the dark cycle. Animals (n = 6–8 per group) were maintained and experiments were conducted in accordance with guidelines of the International Association for the Study of Pain32 and were approved by the Institutional Animal Care and Use Committee, University at Buffalo, the State University of New York (Buffalo, NY), and with the 2011 Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources on Life Sciences, National Research Council, National Academy of Sciences, Washington, DC).
Writhing
Mice were habituated in a clean mouse cage with corn cob bedding for 20 min. Mice then received intraperitoneal (i.p.) injections of either 0.32% lactic acid or 0.6% acetic acid. The number of acid-induced writhes in a 25 min observation period was recorded, starting 5 min after the injection of acetic acid or 10 minutes after the injection of lactic acid. These starting times and observation period were decided according to a pilot study to ensure that the observation period included the majority of writhing responses. A writhe was defined as a contraction of the abdomen following a stretch of the hind limbs. Mice were randomly selected for treatment groups or vehicle groups. Mice in treatment groups received subcutaneous (s.c.) injections of either morphine (0.1–3.2 mg/kg, 10 min pretreatment), KRM-II-18B (1–10 mg/kg, 30 min pretreatment), or KRM-II-81 (3.2–10mg/kg, 30 min pretreatment). In flumazenil studies, mice received s.c. injections of flumazenil 15 minutes prior to injections of each PAM.
Nesting
Singly housed mice were tested in their home cages with familiar (≥ 2 days of habituation) corncob bedding (parameters established during pilot studies according to previous study18). Mice were randomly assigned to a particular dose of a treatment drug. Before the start of each test session, mice were acclimated to the procedure room for at least 30 min. Mice received pretreatments of vehicle, morphine, KRM-II-18B, or KRM-II-81 and were briefly transferred to a second cage while any existing nesting material (VWR Scientific, Randor, PA, USA) was removed from the home cage. Then, six 2 × 3 cm pieces of new nesting material were distributed around the home cage, each marking a designated zone within the cage18. Mice received i.p. injections of either acetic or lactic acid, then returned to the home cage for a 60 min nesting period, during which the cotton pad pieces were retrieved and used to build a nest. At the end of the nesting period, each zone from which a cotton pad piece was retrieved was considered a cleared zone.
Locomotion
Locomotor activity was measured using an infrared motion-sensor system (AccuScan Instruments, Inc., Columbus, OH) surrounding Plexiglas cages (40 × 40 × 30 cm). Versa Max software (Omnitech Electronics, Inc., Columbus, OH) was used to monitor the distance the animal travelled for a total of 65 min. Baseline sessions in which mice received s.c. injections of either acetaminophen, KRM-II-18B, or KRM-II-81 and were then placed in the locomotion chamber (n = 6–8 per group) were conducted before test sessions in order to verify that the doses used did not significantly decrease locomotor activity. After 30 minutes in the chamber, mice received i.p injections of vehicle. Test studies were identical except that mice received injections of either 0.6% acetic acid or 0.32% lactic acid instead of vehicle at the 30 min time point. In all sessions, the total distance traveled (cm) by each mouse was used to measure locomotor activity. The 5 minutes immediately following i.p. injection (either vehicle or acid) was excluded from the total distance due to increased locomotion after handling.
Drugs
KRM-II-18B and KRM-II-81 were obtained from Dr. James M. Cook (University of Wisconsin) according to published procedure33 and dissolved in a vehicle of 20% dimethyl sulfoxide (Amresco, Solon, OH), and 10% emulphor (Solvay, Cranbury, NJ), in 0.9% saline. Morphine sulfate was provided by Research Technology Branch, National Institute and Drug Abuse, National Institutes of Health (Rockville, MD, USA) and was dissolved in 0.9% saline. Acetaminophen was purchased from Sigma-Aldrich (St. Louis, MO, USA) and dissolved in a vehicle of 20% dimethyl sulfoxide in 0.9% saline. All drugs were administered subcutaneously. Lactic acid purchased from Sigma-Aldrich was diluted to 0.32% and acetic acid purchased from Macron Fine Chemicals (Center Valley, PA, USA) was diluted to 0.6% in 0.9% saline. Both lactic and acetic acid were administered intraperitoneally.
Data Analysis
Statistical analyses were performed with the GraphPad Prism 5.0 program (GraphPad Software, San Diego, CA, USA). Data are expressed as mean ± SEM. For studies of writhing, nesting, and locomotor activity, data were analyzed with one-way analysis of variance (ANOVA). Bonferroni’s multiple comparison post hoc test was used to determine statistical significance. For the study of the antagonism effects of flumazenil (writhing), data were analyzed with student’s t-test. P < 0.05 was considered statistically significant in all experiments.
Acknowledgments
This work was supported by the National Institutes of Health National Institute on Drug Abuse [Grant R01DA034806]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS
- cDNA
complementary deoxyribonucleic acid
- DMSO
Dimethyl sulfoxide
- GABA
gamma-aminobutyric acid
- NSAIDs
Opioids and non-steroidal anti-inflammatory drugs
- PAM
positive allosteric modulator
Footnotes
Notes
The authors declare no competing financial interest.
Author Contributions
LA Lewter, JL Fisher, JN Siemian and JX Li performed the studies and conducted the data analysis; KR Methuku, MM Poe and JM Cook synthesized KRM-II-81 and KRM-II-18B; LA Lewter, JL Fisher and JX Li prepared the manuscript; all authors approved the final version of the manuscript.
References
- 1.IOM. Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. National Academy of Sciences; Washington DC: 2011. [PubMed] [Google Scholar]
- 2.Gaskin DJ, Richard P. The Economic Costs of Pain in the United States. The Journal of Pain. 2012;13:715–724. doi: 10.1016/j.jpain.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 3.Reis DJ, Regunathan S. Is agmatine a novel neurotransmitter in brain? Trends Pharmacol Sci. 2000;21:187–193. doi: 10.1016/s0165-6147(00)01460-7. [DOI] [PubMed] [Google Scholar]
- 4.Dumas EO, Pollack GM. Opioid Tolerance Development: A Pharmacokinetic/Pharmacodynamic Perspective. The AAPS Journal. 2008;10:537. doi: 10.1208/s12248-008-9056-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ling W, Mooney L, Hillhouse M. Prescription opioid abuse, pain and addiction: clinical issues and implications. Drug Alcohol Rev. 2011;30:300–305. doi: 10.1111/j.1465-3362.2010.00271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sostres C, Gargallo CJ, Arroyo MT, Lanas A. Adverse effects of non-steroidal anti-inflammatory drugs (NSAIDs, aspirin and coxibs) on upper gastrointestinal tract. Best Pract Res Clin Gastroenterol. 2010;24:121–132. doi: 10.1016/j.bpg.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 7.Kissin I. The development of new analgesics over the past 50 years: a lack of real breakthrough drugs. Anesthesia and analgesia. 2010;110:780–789. doi: 10.1213/ANE.0b013e3181cde882. [DOI] [PubMed] [Google Scholar]
- 8.Burma NE, Leduc-Pessah H, Fan CY, Trang T. Animal models of chronic pain: Advances and challenges for clinical translation. J Neurosci Res. 2016 doi: 10.1002/jnr.23768. [DOI] [PubMed] [Google Scholar]
- 9.Negus SS, Vanderah TW, Brandt MR, Bilsky EJ, Becerra L, Borsook D. Preclinical assessment of candidate analgesic drugs: recent advances and future challenges. J Pharmacol Exp Ther. 2006;319:507–514. doi: 10.1124/jpet.106.106377. [DOI] [PubMed] [Google Scholar]
- 10.Melzack R, Wall PD. Pain mechanisms: a new theory. Science. 1965;150:971–979. doi: 10.1126/science.150.3699.971. [DOI] [PubMed] [Google Scholar]
- 11.Chesnut JD, Baytan AR, Russell M, Chang MP, Bernard A, Maxwell IH, Hoeffler JP. Selective isolation of transiently transfected cells from a mammalian cell population with vectors expressing a membrane anchored single-chain antibody. J Immunol Methods. 1996;193:17–27. doi: 10.1016/0022-1759(96)00032-4. [DOI] [PubMed] [Google Scholar]
- 12.Ralvenius WT, Benke D, Acuna MA, Rudolph U, Zeilhofer HU. Analgesia and unwanted benzodiazepine effects in point-mutated mice expressing only one benzodiazepine-sensitive GABAA receptor subtype. Nat Commun. 2015;6:6803. doi: 10.1038/ncomms7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crestani F, Rudolph U. Behavioral functions of GABAA receptor subtypes–the Zurich experience. Adv Pharmacol. 2015;72:37–51. doi: 10.1016/bs.apha.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 14.de Lucas AG, Ahring PK, Larsen JS, Rivera-Arconada I, Lopez-Garcia JA, Mirza NR, Munro G. GABAA alpha5 subunit-containing receptors do not contribute to reversal of inflammatory-induced spinal sensitization as indicated by the unique selectivity profile of the GABAA receptor allosteric modulator NS16085. Biochem Pharmacol. 2015;93:370–379. doi: 10.1016/j.bcp.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 15.Chiba S, Nishiyama T, Yoshikawa M, Yamada Y. The antinociceptive effects of midazolam on three different types of nociception in mice. J Pharmacol Sci. 2009;109:71–77. doi: 10.1254/jphs.08094fp. [DOI] [PubMed] [Google Scholar]
- 16.Green GM, Dickenson A. GABA-receptor control of the amplitude and duration of the neuronal responses to formalin in the rat spinal cord. Eur J Pain. 1997;1:95–104. doi: 10.1016/s1090-3801(97)90067-7. [DOI] [PubMed] [Google Scholar]
- 17.Knabl J, Witschi R, Hosl K, Reinold H, Zeilhofer UB, Ahmadi S, Brockhaus J, Sergejeva M, Hess A, Brune K, Fritschy JM, Rudolph U, Mohler H, Zeilhofer HU. Reversal of pathological pain through specific spinal GABAA receptor subtypes. Nature. 2008;451:330–334. doi: 10.1038/nature06493. [DOI] [PubMed] [Google Scholar]
- 18.Munro G, Lopez-Garcia JA, Rivera-Arconada I, Erichsen HK, Nielsen EO, Larsen JS, Ahring PK, Mirza NR. Comparison of the novel subtype-selective GABAA receptor-positive allosteric modulator NS11394 [3′-[5-(1-hydroxy-1-methyl-ethyl)-benzoimidazol-1-yl]-biphenyl-2-carbonitrile] with diazepam, zolpidem, bretazenil, and gaboxadol in rat models of inflammatory and neuropathic pain. J Pharmacol Exp Ther. 2008;327:969–981. doi: 10.1124/jpet.108.144568. [DOI] [PubMed] [Google Scholar]
- 19.Picton AJ, Fisher JL. Effect of the alpha subunit subtype on the macroscopic kinetic properties of recombinant GABA(A) receptors. Brain Res. 2007;1165:40–49. doi: 10.1016/j.brainres.2007.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Negus SS, Neddenriep B, Altarifi AA, Carroll FI, Leitl MD, Miller LL. Effects of ketoprofen, morphine, and kappa opioids on pain-related depression of nesting in mice. Pain. 2015;156:1153–1160. doi: 10.1097/j.pain.0000000000000171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stevenson GW, Cormier J, Mercer H, Adams C, Dunbar C, Negus SS, Bilsky EJ. Targeting pain-depressed behaviors in preclinical assays of pain and analgesia: drug effects on acetic acid-depressed locomotor activity in ICR mice. Life Sci. 2009;85:309–315. doi: 10.1016/j.lfs.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li JX, Shah AP, Patel SK, Rice KC, France CP. Modification of the behavioral effects of morphine in rats by serotonin 5-HT(1)A and 5-HT(2)A receptor agonists: antinociception, drug discrimination, and locomotor activity. Psychopharmacology (Berl) 2013;225:791–801. doi: 10.1007/s00213-012-2870-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moseley GL, Zalucki N, Birklein F, Marinus J, van Hilten JJ, Luomajoki H. Thinking about movement hurts: the effect of motor imagery on pain and swelling in people with chronic arm pain. Arthritis Rheum. 2008;59:623–631. doi: 10.1002/art.23580. [DOI] [PubMed] [Google Scholar]
- 24.van Weering M, Vollenbroek-Hutten MM, Kotte EM, Hermens HJ. Daily physical activities of patients with chronic pain or fatigue versus asymptomatic controls. A systematic review. Clin Rehabil. 2007;21:1007–1023. doi: 10.1177/0269215507078331. [DOI] [PubMed] [Google Scholar]
- 25.Stevenson GW, Bilsky EJ, Negus SS. Targeting Pain-Suppressed Behaviors in Preclinical Assays of Pain and Analgesia: Effects of Morphine on Acetic Acid-Suppressed Feeding in C57BL/6J Mice. The Journal of Pain. 2006;7:408–416. doi: 10.1016/j.jpain.2006.01.447. [DOI] [PubMed] [Google Scholar]
- 26.de la Puente B, Romero-Alejo E, Vela JM, Merlos M, Zamanillo D, Portillo-Salido E. Changes in saccharin preference behavior as a primary outcome to evaluate pain and analgesia in acetic acid-induced visceral pain in mice. J Pain Res. 2015;8:663–673. doi: 10.2147/JPR.S91230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller LL, Picker MJ, Schmidt KT, Dykstra LA. Effects of morphine on pain-elicited and pain-suppressed behavior in CB1 knockout and wildtype mice. Psychopharmacology (Berl) 2011;215:455–465. doi: 10.1007/s00213-011-2232-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tucker AP, Lai C, Nadeson R, Goodchild CS. Intrathecal midazolam I: a cohort study investigating safety. Anesth Analg. 2004;98:1512–1520. doi: 10.1213/01.ANE.0000087075.14589.F5. table of contents. [DOI] [PubMed] [Google Scholar]
- 29.Tucker AP, Mezzatesta J, Nadeson R, Goodchild CS. Intrathecal midazolam II: combination with intrathecal fentanyl for labor pain. Anesth Analg. 2004;98:1521–1527. doi: 10.1213/01.ANE.0000112434.68702.E4. table of contents. [DOI] [PubMed] [Google Scholar]
- 30.Zuiker RG, Chen X, Osterberg O, Mirza NR, Muglia P, de Kam M, Klaassen ES, van Gerven JM. NS11821, a partial subtype-selective GABAA agonist, elicits selective effects on the central nervous system in randomized controlled trial with healthy subjects. J Psychopharmacol. 2016;30:253–262. doi: 10.1177/0269881115620435. [DOI] [PubMed] [Google Scholar]
- 31.Fisher JL, Zhang J, Macdonald RL. The role of alpha1 and alpha6 subtype amino-terminal domains in allosteric regulation of gamma-aminobutyric acida receptors. Mol Pharmacol. 1997;52:714–724. doi: 10.1124/mol.52.4.714. [DOI] [PubMed] [Google Scholar]
- 32.Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. PAIN. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]
- 33.Poe MM, Methuku KR, Li G, Verma AR, Teske KA, Stafford DC, Arnold LA, Cramer JW, Jones TM, Cerne R, Krambis MJ, Witkin JM, Jambrina E, Rehman S, Ernst M, Cook JM, Schkeryantz JM. Synthesis and Characterization of a Novel gamma-Aminobutyric Acid Type A (GABAA) Receptor Ligand That Combines Outstanding Metabolic Stability, Pharmacokinetics, and Anxiolytic Efficacy. J Med Chem. 2016;59:10800–10806. doi: 10.1021/acs.jmedchem.6b01332. [DOI] [PMC free article] [PubMed] [Google Scholar]