

Abstract
There are twenty-five published structures of Mycobacterium tuberculosis anthranilate phosphoribosyltransferase (Mtb-AnPRT) that use the same crystallization protocol. The structures include protein complexed with natural and alternative substrates, protein:inhibitor complexes, and variants with mutations of substrate-binding residues. Amongst these are varying space groups (i.e. P21, C2, P21212, P212121). This article outlines experimental details for 3 additional Mtb-AnPRT:inhibitor structures. For one protein:inhibitor complex, two datasets are presented – one generated by crystallization of protein in the presence of the inhibitor and another where a protein crystal was soaked with the inhibitor. Automatic and manual processing of these datasets indicated the same space group for both datasets and thus indicate that the space group differences between structures of Mtb-AnPRT:ligand complexes are not related to the method used to introduce the ligand.
Keywords: Crystallography, Macromolecules, X-ray diffraction, Ligand binding, Structure-based inhibitor design
Specifications Table
| Subject area | Proteomics and Biochemistry |
| More specific subject area | Structural biology |
| Type of data | Tables, figures and X-ray diffraction images |
| How data was acquired | X-ray macromolecular crystallography; |
| Australian Synchrotron MX1 and MX2 beamlines | |
| Data format | Unprocessed, processed, deposited with crystal packing analyzed. |
| Experimental factors | Protein crystals soaked and co-crystallized with ligands |
| Experimental features | Contrast of the different protein crystal packing associated with different inhibitors and/or introduced by soaking or co-crystallization. |
| X-ray diffraction datasets for co-crystallization and soaked-in experiments with the same inhibitor indicate space group changes are independent of method used to introduce the inhibitor. | |
| Data source location | Data collected in Melbourne, Australia. |
| Data accessibility | The atomic coordinates and observed structure factors are available from Protein Data Bank with accession numbers:5BO2,5BO3and5BNE. |
| The X-ray diffraction image files corresponding to datasets from two experiments are hosted by Mendeley: | |
| http://dx.doi.org/10.17632/2zrfgv34nb.1 | |
| http://dx.doi.org/10.17632/xgn5z8jnr7.1 | |
| Related research article | Anthranilate phosphoribosyltransferase: Binding determinants for 5′-phospho-alpha-d-ribosyl-1′-pyrophosphate (PRPP) and the implications for inhibitor design “in press”. |
| https://doi.org/10.1016/j.bbapap.2017.08.018 |
Value of the data
-
•
Mtb-AnPRT is a target of interest in developing novel anti-tuberculosis agents. This protein's capacity to crystallize thus yield new protein:ligand complexes makes it of interest for structure-based inhibitor design.
-
•
Previously Mtb-AnPRT:ligand complexes have been found with different space groups (e.g. P21, C2, P21212, and P212121), generated by using the same crystallization protocol.
-
•
Mtb-AnPRT:inhibitor complex structures described herein have C2 and P21 space groups. These structures were solved using X-ray diffraction datasets from protein:ligand crystals generated by streak-seeded with wild-type P212121 crystal.
-
•
For one inhibitor, X-ray diffraction datasets are presented for both co-crystallization and soaked crystal experiments. Space group C2 occurred in both datasets and indicates the space group change between ligand-free and inhibitor-bound structures are independent of method used to introduce the inhibitor.
-
•
X-ray diffraction datasets utilizing different methods of ligand introduction and yielding equivalent protein:ligand structures are typically not made available. These publically available datasets in the context of multiple space groups observed for Mtb-AnPRT:ligand structures, and the analysis presented herein, demonstrate that space group changes can be independent of co-crystallization and soaking methods of ligand introduction. This has relevance to academic and industrial researchers who are pursuing structure-based inhibitor design.
1. Data
1.1. Overview
The experimental and data processing details for 3 new protein:ligand structures of Mycobacterium tuberculosis anthranilate phosphoribosyl transferase (Mtb-AnPRT) are described herein (Table 1). The protein structures are complexed with Mtb-AnPRT inhibitors characterized in [1] and annotated as:
-
•
8k (2-(2-carboxyphenylamino)-5-(5-phosphonopentyloxy)benzoic acid)
-
•
8j (2-(2-carboxyphenylamino)-5-(4-phosphonobutoxy)benzoic acid)
-
•
8i (2-(2-carboxyphenylamino)-5-(3-phosphonopropoxy)benzoic acid)
Table 1.
Summary of Mtb-AnPRT structures described herein.
| PDB ID | Ligand ID | Solvent content | Unit Cell |
Space group | Resolution (Å) | Chains | PDB DOI | |||
|---|---|---|---|---|---|---|---|---|---|---|
| β (°) | A (Å) | B (Å) | C (Å) | |||||||
| 5BO2 | 8i | 46% | 111° | 94 | 78 | 103 | C2 | 2.00 | A,B | 10.2210/pdb5bo2/pdb |
| 5BO3 | 8j | 46% | 111° | 95 | 78 | 103 | C2 | 1.75 | A,B | 10.2210/pdb5bo3/pdb |
| 5BNE | 8k | 46% | 91° | 77 | 78 | 117 | P21 | 2.15 | A,B,C D | 10.2210/pdb5bne/pdb |
These structures were determined with protein crystallized in presence of imidazole-malate and PEG4000. Crystallization drops seeded with crystal nuclei from a pre-existing Mtb-AnPRT crystal generate better crystal morphology for Mtb-AnPRT [2]. All new structures presented herein were generated by streak-seeding using wild-type P212121 crystals. Also presented are two X-ray diffraction datasets corresponding to protein crystals either soaked and co-crystallized with the same inhibitor (Table 2).
Table 2.
Crystallization of complexes, along with space group and unit cell from data processing.
| PDB ID | Ligand(s) bound | [Protein] mg mL−1 | Reservoir condition | Cryoprotectant/Soak | Notes | Space group | Unit cell |
|---|---|---|---|---|---|---|---|
| 5BO2 | 8i : | 3.0 | 0.2 M imidazole.malate, pH 7.0, 9% PEG4000 | 0.2 M imidazole.malate, pH 7.0, 15% PEG4000, 1 mM 8i | Streak seeded; soaked crystal for 4 h; | C2 | 94×78×103 Å |
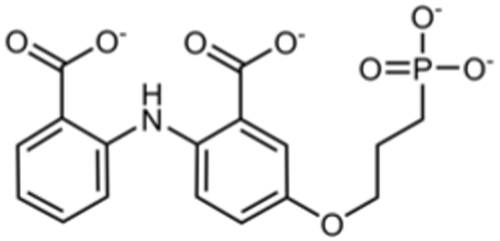 |
2 day old crystal | ||||||
| X-ray images available at:http://dx.doi.org/10.17632/xgn5z8jnr7.1 | ßβ=111° | ||||||
| Not applicable | 8i : | 3.0 | 0.2 M imidazole.malate, pH 7.0, 11% PEG4000, 1 mM 8i | 0.2 M imidazole.malate, pH 7.0, 15% PEG4000, 1 mM 8i | Streak seeded; protein co-crystallized with ligand; | C2 | 95×78×103 Å |
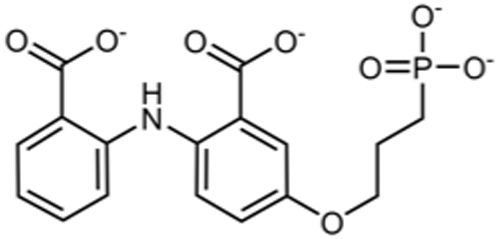 |
2 day old crystal | ||||||
| β=111° | |||||||
| X-ray images available at:http://dx.doi.org/10.17632/2zrfgv34nb.1 | |||||||
| 5BO3 | 8j : | 3.0 | 0.2 M imidazole.malate, pH 7.0, 11% PEG4000, | 0.2 M imidazole.malate, pH 7.0, 15% PEG4000, 1 mM 8j | Streak seeded; soaked crystal for 10 min; | C2 | 95×78×103 Å |
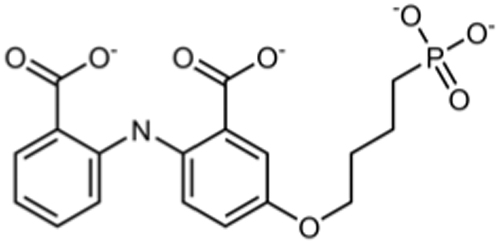 | |||||||
| 2 day old crystal | |||||||
| β=111° | |||||||
| 5BNE | 8k : | 3.1 | 0.2 M imidazole.malate, pH 7.0, 15% PEG4000, 1 mM 8k | No cryo used, because crystallization condition contained 15% PEG4000 | Streak seeded; protein co-crystallized with ligand; | P21 | 77×78×117 Å |
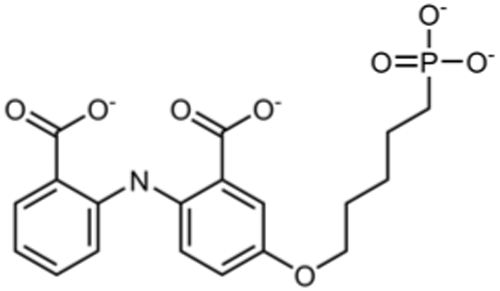 | |||||||
| 2 day old crystal | |||||||
| β=91° |
Of the previously published structures of Mtb-AnPRT, 25 were determined from protein crystallized using this protocol (Table 3). Most of these structures have a ligand bound (e.g. inhibitor or substrate) and/or are protein variants with mutations in substrate-binding residues (Table 3). Amongst both cohorts (Table 1, Table 3) several different space groups have been observed, including P21, C2, P21212, P212121. Amongst the 28 structures referred to in this article (Table 1, Table 3), similar unit cells correspond to each space group.
Table 3.
Space groups, unit cells and other information for previously published Mtb-AnPRT structures crystallized in imidazole-malate and PEG4000.
| PDB ID | Ref | Solvent content | Unit Cell |
Space Group | Resolution (Å) | Chains | Notes | PDB DOI | |||
| β (°) | A (Å) | B (Å) | C (Å) | ||||||||
| 3QS8 | [2] | 46% | 90° | 78 | 81 | 111 | P21 | 2.00 | A,B,C,D | Co-crystal | 10.2210/pdb3qs8/pdb |
| 3UU1 | [2] | 45% | 90° | 78 | 111 | 80 | P21 | 1.82 | A,B,C,D | Co-crystal | 10.2210/pdb3uu1/pdb |
| 3R6C | [2] | 44% | 110° | 94 | 78 | 100 | C2 | 1.83 | A,B | Co-crystal | 10.2210/pdb3r6c/pdb |
| 4IJ1 | [5] | 45% | 111 | 95 | 78 | 102 | C2 | 1.79 | A,B | Co-crystal | 10.2210/pdb4ij1/pdb |
| 4X58 | [6] | 50% | 111 | 95 | 78 | 101 | C2 | 1.75 | A,B | Mutant | 10.2210/pdb4x58/pdb |
| 4X59 | [6] | 50% | 112 | 95 | 78 | 102 | C2 | 1.80 | A,B | Mutant | 10.2210/pdb4x59/pdb |
| 4X5A | [6] | 49% | 112 | 94 | 78 | 102 | C2 | 1.93 | A,B | Mutant | 10.2210/pdb4x5a/pdb |
| 4X5B | [6] | 45% | 111 | 94 | 78 | 100 | C2 | 2.47 | A,B | Mutant | 10.2210/pdb4x5b/pdb |
| 4X5C | [6] | 49% | 111 | 94 | 78 | 101 | C2 | 2.33 | A,B | Mutant | 10.2210/pdb4x5c/pdb |
| 4X5E | [6] | 50% | 110 | 95 | 79 | 101 | C2 | 1.77 | A,B | Mutant | 10.2210/pdb4x5e/pdb |
| 4GIU | [5] | 46% | 90 | 111 | 81 | 79 | P21212 | 1.67 | A,B | Co-crystal | 10.2210/pdb4giu/pdb |
| 4GKM | [5] | 46% | 90 | 111 | 81 | 78 | P21212 | 1.67 | A,B | Co-crystal | 10.2210/pdb4gkm/pdb |
| 3QR9 | [2] | 57% | 90 | 79 | 92 | 120 | P212121 | 1.87 | A,B | Ligand-free | 10.2210/pdb3qr9/pdb |
| 4M0R | [5] | 56% | 90 | 79 | 92 | 120 | P212121 | 1.96 | A,B | Co-crystal | 10.2210/pdb4m0r/pdb |
| 4N5V | [7] | 57% | 90 | 80 | 93 | 121 | P212121 | 1.90 | A,B | Soak | 10.2210/pdb4n5v/pdb |
| 4N8Q | [7] | 56% | 90 | 80 | 91 | 120 | P212121 | 2.08 | A,B | Soak | 10.2210/pdb4n8q/pdb |
| 4N93 | [7] | 57% | 90 | 80 | 92 | 121 | P212121 | 2.03 | A,B | Soak | 10.2210/pdb4n93/pdb |
| 4OWM | [7] | 60% | 90 | 79 | 92 | 121 | P212121 | 1.99 | A,B | Soak | 10.2210/pdb4owm/pdb |
| 4OWN | [7] | 60% | 90 | 80 | 92 | 121 | P212121 | 2.11 | A,B | Soak | 10.2210/pdb4own/pdb |
| 4OWO | [7] | 60% | 90 | 79 | 92 | 121 | P212121 | 1.99 | A,B | Soak | 10.2210/pdb4owo/pdb |
| 4OWQ | [7] | 61% | 90 | 79 | 92 | 122 | P212121 | 1.89 | A,B | Soak | 10.2210/pdb4owq/pdb |
| 4OWS | [7] | 60% | 90 | 80 | 92 | 121 | P212121 | 2.43 | A,B | Soak | 10.2210/pdb4ows/pdb |
| 4OWU | [7] | 60% | 90 | 79 | 92 | 121 | P212121 | 1.89 | A,B | Soak | 10.2210/pdb4owu/pdb |
| 4OWV | [7] | 60% | 90 | 80 | 92 | 120 | P212121 | 1.90 | A,B | Soak | 10.2210/pdb4owv/pdb |
| 4X5D | [6] | 60% | 90 | 80 | 92 | 121 | P212121 | 2.30 | A,B | Mutant | 10.2210/pdb4x5d/pdb |
P212121 is the most common space group for macromolecular structures, and it has been proposed that this is due to its capacity to accommodate repositioning, i.e. rotations or translations, within the asymmetric unit, without loss of crystal contacts [3]. The structure of wild-type Mtb-AnPRT without ligands (PDB ID: 3QR9) has previously been solved from protein crystallized in imidazole-malate and PEG4000 in the space group P212121, with two monomers (A, B) in the asymmetric unit, a unit cell of 79×92×120 Å, and 57% solvent content [2].
Mtb-AnPRT is a homodimeric protein with an extended “S”-shape, with each subunit containing two domains [4]. In the ligand-free structure, a single dimer (the biological assembly) is found in the asymmetric unit [2]. The two subunits of the dimer are related by a non-crystallographic two-fold symmetry axis (NCS).
1.2. Data for protein complexes with inhibitors
The three protein:inhibitor complexes (PDB IDs: 5BO2, 5BO3, 5BNE) were solved in the absence of metals and substrate. For these structures, the solvent content has decreased by ~ 10%, the unit cell has changed (i.e. dimension(s) decreased by 10–20 Å), and the space group has changed to P21 or C2, compared to the ligand-free wild-type structure.
In structures of AnPRT from other prokaryote species, domain movement is observed within subunits due to substrate binding and results in compression of the homodimer by 10 Å (e.g. Dmax (maximum distance) changes from 110 to 100 Å [8]). However, superposition of the subunits in the new Mtb-AnPRT structures onto the subunits of the ligand-free structure indicates there are no large changes (Table 4; [9]). Additionally, the longest dimension of the Mtb-AnPRT dimer is relatively unchanged between the ligand-free protein structure and the protein:ligand complex structures (Table 4; [10]). Thus, the changes in space group are not driven by domain movements within each subunit.
Table 4.
Comparison Mtb-AnPRT subunits found in the 3 new structures to ligand-free structure.
| RMSDa to chain A-3QR9 (Å) | RMSDa to chain B-3QR9 (Å) | Longest dimension of dimer (Å) | |
|---|---|---|---|
| 3qr9.pdb:A | – | 0.89 | 109 |
| 3qr9.pdb:B | 0.89 | – | |
| 5bo2.pdb:A | 0.67 | 0.68 | 110 |
| 5bo2.pdb:B | 0.66 | 0.73 | |
| 5bo3.pdb:A | 0.63 | 0.53 | 110 |
| 5bo3.pdb:B | 0.62 | 0.73 | |
| 5bne.pdb:A | 0.57 | 0.53 | 109 |
| 5bne.pdb:B | 0.46 | 0.74 | |
| 5bne.pdb:C | 0.46 | 0.92 | 109 |
| 5bne.pdb:D | 0.55 | 0.60 |
Root mean standard difference (RMSD) between the Calpha atoms
The P21 structure (PDB ID: 5BNE) contains an inhibitor annotated as 8k, and is the third structure of Mtb-AnPRT with this space group determined for protein crystallized in the imidazole-malate condition. The increased components in this structure's asymmetric unit (chains A-D, vs. chains A and B; Fig. 1A) means that the lower symmetry described by P21 can generate equivalent protein content in a similarly-sized unit cell as is observed with structures defined by space groups P212121 or P21212. The ß angle of 91° could be taken to suggest that the space group should be orthorhombic (e.g. P21212 or P212121). Both POINTLESS [11] and ZANUDA [12] indicated that P21 was the correct space group for this dataset, however.
Fig. 1.

Understanding changes in crystal packing in the Mtb-AnPRT structures with inhibitors. (A) Superposition of the dimer (cartoon) from a Mtb-AnPRT:inhibitor structure defined by space group P21, PDB ID: 5BNE (chain A, B, C and D in green, cyan, pink and yellow, respectively), onto that of the ligand-free Mtb-AnPRT structure (PDB ID: 3QR9 [2]; P212121; chain A and B in dark and light grey, respectively). The figure includes adjacent dimers (ribbons) in equivalent crystal layers (c-b plane in 3QR9 [2] and c-a plane in 5BNE). In (B) the superposition is re-colored with the Mtb-AnPRT:inhibitor structure in yellow and the ligand-free Mtb-AnPRT structure in dark grey. Arrows highlight the reorientation of dimers relative to each other. (C) Superposition of the dimer (cartoon) from a Mtb-AnPRT:inhibitor structure defined by space group C2, PDB ID: 5BO2 (marine blue) onto that of the ligand-free Mtb-AnPRT structure (PDB ID: 3QR9 [2]; P212121; dark grey). In (D) superpositions in panels B-C are combined.
(A) Superposition of the dimer (cartoon) from a Mtb-AnPRT:inhibitor structure defined by space group P21, PDB ID: 5BNE (chain A, B, C and D in green, cyan, pink and yellow, respectively), onto that of the ligand-free Mtb-AnPRT structure (PDB ID: 3QR9 [2]; P212121; chain A and B in dark and light grey, respectively). The figure includes adjacent dimers (ribbons) in equivalent crystal layers (c-b plane in 3QR9 [2] and c-a plane in 5BNE). In (B) the superposition is re-colored with the Mtb-AnPRT:inhibitor structure in yellow and the ligand-free Mtb-AnPRT structure in dark grey. Arrows highlight the reorientation of dimers relative to each other. (C) Superposition of the dimer (cartoon) from a Mtb-AnPRT:inhibitor structure defined by space group C2, PDB ID: 5BO2 (marine blue) onto that of the ligand-free Mtb-AnPRT structure (PDB ID: 3QR9 [2]; P212121; dark grey). In (D) superpositions in panels B-C are combined.
Structural superposition of the dimers from Mtb-AnPRT:inhibitor structures (PDB ID: 5BNE and 5BO2) onto the dimer present in the P212121 ligand-free Mtb-AnPRT structure (PDB ID: 3QR9 [2]) revealed a reorientation of the protein dimers relative to each other (Fig. 1B and C). The combination of these superpositions (Fig. 1D) indicates that the lattice in the P21 structure corresponds to an intermediate position between the lattice observed for P212121 and C2 structures. We propose the subunits that are related by crystallographic symmetry elements in the P212121 structures are related by pseudosymmetry elements in the P21 structure. Pseudosymmetry occurs where a non-crystallographic symmetry element within the asymmetric unit is close to a crystallographic symmetry operators [13]. Thus, the P21 space group has been correctly assigned, even though the unit cell has a β of approximately 90 °.
The generation of the protein: inhibitor structures, involved experiments, with all three inhibitors, using both co-crystallization and soaking-in methods. The structures deposited on the PDB correspond to those where the clearest density was observed for the inhibitor (i.e. modelled with full occupancy; Fig. 2). In Table 5 there are data statistics corresponding to Mtb-AnPRT co-crystallized with inhibitor 8i. In Table 6 are data statistics corresponding to inhibitor 8i soaked into a wild-type ligand-free Mtb-APRT crystal. In both cases, with no manual intervention, the datasets processed in XDS [14] and AIMLESS [11] as space group C2. Thus, in this study, there no correlation was found between space group type and the method by which the ligand was introduced (i.e. soaking vs. co-crystallization).
Fig. 2.

Omit and fitted map for inhibitors bound in Mtb-AnPRT structures. The Fo-Fc map calculated (green, contoured at 3 σ) prior to the addition of ligands to the model for A) 8k, B) 8j and C) 8i (PDB entries 5BNE, 5BO3 and 5BO2, respectively). The 2Fo-Fc map calculated (blue, contoured at 1 σ) after final refinement, with model including the ligands for D) 8k (black), E) 8j (green) and F) 8i (pink).
Table 5.
Statistics for dataset of Mtb-AnPRT co-crystallized with inhibitor 8i.
| Data collection | |
| AnPRT complexed with | 8i |
| Space group | C2 |
| Cell dimensions | |
| a, b, c (Å) | 94.6, 78.1, 102.8 |
| β (deg) | 110.9 |
| Unique reflectionsa | 387946 (24179) |
| Resolution range (Å)a | 48-1.95 (2.00-1.95) |
| Rmergea | 0.138 (1.237) |
| Rp.i.m.a | 0.054 (0.488) |
| Mean I/σ(I)a | 9.9 (1.5) |
| CC1/2a | 0.997 (0.679) |
| Completeness (%)a | 99.6 (94.0) |
| Redundancya | 7.6 (7.1) |
| Wilson B factor | 20.5 |
b The average atomic temperature factor. cRwork=(|Fobs| - |Fcalc|)/|Fobs| and Rfree=∑T (|Fobs| - |Fcalc|)/∑T |Fobs|, where T is a test dataset of 5% of the total reflections randomly chosen and set aside before refinement. d RMSD from ideal geometry values from Engh and Huber (1991),[15].
Outer resolution shell is shown in parentheses.
Table 6.
Data and refinement statistics for AnPRT complexes with inhibitors.
| Data collection | |||
| AnPRT complexed with | 8i | 8j | 8k |
| PDB code | 5BO2 | 5BO3 | 5BNE |
| Space group | C2 | C2 | P21 |
| Cell dimensions | |||
| a, b, c (Å) | 94.5, 78.0, 102.9 | 95.0, 78.1, 102.6 | 77.3, 78.4, 117.2 |
| β (deg) | 111.0 | 111.1 | 90.7 |
| Unique reflectionsa | 47170 (3415) | 68993 (3669) | 75079 (4387) |
| Resolution range (Å)a | 47-2.00 (2.05-2.00) | 48-1.75 (1.78-1.75) | 47-2.15 (2.19-2.15) |
| Rmergea | 0.121 (0.856) | 0.121 (1.762) | 0.111 (0.711) |
| Rp.i.m.a | 0.076 (0.564) | 0.046 (0.688) | 0.073 (0.477) |
| Mean I/σ(I)a | 8.5 (1.5) | 13.3 (1.2) | 6.9 (1.5) |
| CC1/2a | 0.993 (0.564) | 0.998 (0.465) | 0.993 (0.502) |
| Completeness (%)a | 99.8 (97.7) | 97.8 (93.7) | 98.4 (77.3) |
| Redundancya | 3.4 (3.3) | 7.8 (7.4) | 3.0 (2.9) |
| Wilson B factor | 15.1 | 16.3 | 21.4 |
| Refinement | |||
| Atoms, B factor (Å2)b | |||
| Protein | 4788, 25.8 | 4746, 24.6 | 9280, 29.0 |
| Solvent | 352, 29.7 | 402, 29.2 | 353, 29.5 |
| Ligands | 79, 33.2 | 66, 26.6 | 107, 30.6 |
| Rwork/Rfree (%/%)a,c | 0.192/0.232 | 0.206/0.240 | 0.207/0.235 |
| (0.276/0.324) | (0.319/0.336) | (0.284/0.332) | |
| Ramachandran outliers (%) | 0.31 | 0.16 | 0.31 |
| R.m.s.d. of | |||
| Bond lengths (Å)d | 0.003 | 0.005 | 0.003 |
| Bond angles (°)d | 0.774 | 0.899 | 0.745 |
Outer resolution shell is shown in parentheses.
The average atomic temperature factor.
Rwork=(|Fobs| - |Fcalc|)/|Fobs| and Rfree=∑T (|Fobs| - |Fcalc|)/∑T |Fobs|, where T is a test dataset of 5% of the total reflections randomly chosen and set aside before refinement.
RMSD from ideal geometry values from Engh and Huber,[15].
2. Experimental design, materials and methods
2.1. Materials
Unless otherwise stated, all chemicals were obtained from Sigma-Aldrich, Scharlau, or Pure Science. The purification of Mtb-AnPRT, as well as the synthesis and biochemical characterization of its inhibitors annotated as 8i, 8j and 8k (Table 2), are outlined in the article entitled “Anthranilate phosphoribosyltransferase: Binding determinants for 5′-phospho-alpha-d-ribosyl-1′-pyrophosphate (PRPP) and the implications for inhibitor design” [1].
2.2. Crystallization
Crystals of Mtb-AnPRT were obtained in hanging drops of 1–2 μL protein solution (3.0–3.1 mg mL−1 in final storage buffer) with the equivalent amount of reservoir solution in a fine screen of 0.2 M imidazole-malate pH 7.0–8.5, 5–15% polyethylene glycol (PEG) 4000, as previously reported [2]. Complexes were prepared either by co-crystallization or by soaking with ligands (details in Table 2). The crystallization drops were seeded from previously formed crystals of Mtb-AnPRT, by streak seeding using a cat whisker. Data collection occurred within a few days after crystallization. In preparation for data collection, crystals were typically soaked in a reservoir solution containing cryoprotectant 15% PEG4000 and appropriate ligands (details in Table 2), before being flash-cooled in liquid nitrogen.
2.3. Data collection, structure solution and refinement
Data were collected at the Australian Synchrotron, Beamlines MX1 and MX2 at 110 K. The crystals of the Mtb-AnPRT complexes diffracted to a maximum resolution that varied between 1.75 and 2.15 Å. X-ray diffraction spots were indexed and integrated with XDS [14] and scaled with AIMLESS [11]. The high resolution cut-off was determined based on a correlation coefficient (CC1/2) [16] exceeding 0.5, with mean I/σ between 1 and 1.5, and a Rp.i.m. of 0.7 or less, as calculated by AIMLESS [11]. Rp.i.m. is defined as ∑hkl [1/(n−1)]×Rmerge, with Rmerge defined as ∑hkl|I(hkl)-<I(hkl)>|/∑hkl<I(hkl)>, where <I(hkl)> is the mean of symmetry-equivalent reflections of I(hkl) [17]. Data and refinement statistics are given in Table 5, Table 6.
Mtb-AnPRT (chain A of 3QR9 [2]), without solvent or ligands, and with loops I and II removed, was used as the search model for structure determination by molecular replacement using Phaser [18]. Refinement and model building was performed with COOT [19], Refmac5 [20], and PHENIX [21]. After positioning waters, omit maps were examined (Fig. 2) and ligands placed, with subsequent refinement including restraints for the ligands generated by phenix.elbow [21]. Restraints on protein bond lengths and angles were based on the ideal values of Engh and Huber [15] and model quality was assessed using MolProbity [22]. Figures illustrating structural details were prepared using PyMOL.
The Fo-Fc map calculated (green, contoured at 3 σ) prior to the addition of ligands to the model for A) 8k, B) 8j and C) 8i (PDB entries 5BNE, 5BO3 and 5BO2, respectively). The 2Fo-Fc map calculated (blue, contoured at 1 σ) after final refinement, with model including the ligands for D) 8k (black), E) 8j (green) and F) 8i (pink).
Acknowledgements
This work was funded by grants from FRST (Foundation for Research, Science and Technology grant number UOAX1005) and the Health Research Council of New Zealand (Programme award 12/1111). Coco the cat is acknowledged for her contribution of a whisker for protein crystallization. We also thank Dr Farah Javid-Majd and Prof. Jim Sacchettini from the Department of Biochemistry and Biophysics, Texas A&M University for the original gift of the Mtb-AnPRT expression plasmid. This work was performed as part of the M. tuberculosis Structural Genomics Consortium (http://www.webtb.org). The authors also wished to thank the anonymous reviewer who suggested examination/explanation of various space groups observed in Mtb-AnPRT structures.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.dib.2017.10.051.
Contributor Information
Genevieve L. Evans, Email: g.evans@auckland.ac.nz.
J. Shaun Lott, Email: s.lott@auckland.ac.nz.
Transparency document. Supplementary material
Supplementary material
References
- 1.Evans G.L., Furkert D.P., Abermil N., Kundu P., de Lange K.M., Parker E.J., Brimble M.A., Baker E.N., Lott J. Shaun. Anthranilate phosphoribosyltransferase: Binding determinants for 5'-phospho-alpha-D-ribosyl-1'-pyrophosphate (PRPP) and the implications for inhibitor design. Biochim. Biophys. Acta. 2017 doi: 10.1016/j.bbapap.2017.08.018. (In press) (PII: S1570-9639(17)(30204-2)) [DOI] [PubMed] [Google Scholar]
- 2.Castell A., Short F.L., Evans G.L., Cookson T.V., Bulloch E.M., Joseph D.D., Lee C.E., Parker E.J., Baker E.N., Lott J.S. The substrate capture mechanism of Mycobacterium tuberculosis anthranilate phosphoribosyltransferase provides a mode for inhibition. Biochemistry. 2013;52:1776–1787. doi: 10.1021/bi301387m. [DOI] [PubMed] [Google Scholar]
- 3.Wukovitz S.W., Yeates T.O. Why protein crystals favour some space-groups over others. Nat. Struct. Biol. 1995;2:1062–1067. doi: 10.1038/nsb1295-1062. [DOI] [PubMed] [Google Scholar]
- 4.Lee C.E., Goodfellow C., Javid-Majd F., Baker E.N., Shaun Lott J. The crystal structure of TrpD, a metabolic enzyme essential for lung colonization by Mycobacterium tuberculosis, in complex with its substrate phosphoribosylpyrophosphate. J. Mol. Biol. 2006;355:784–797. doi: 10.1016/j.jmb.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 5.Evans G.L., Gamage S.A., Bulloch E.M., Baker E.N., Denny W.A., Lott J.S. Repurposing the chemical scaffold of the anti-arthritic drug lobenzarit to target tryptophan biosynthesis in Mycobacterium tuberculosis. Chembiochem. 2014;15:852–864. doi: 10.1002/cbic.201300628. [DOI] [PubMed] [Google Scholar]
- 6.Cookson T.V., Evans G.L., Castell A., Baker E.N., Lott J.S., Parker E.J. Structures of Mycobacterium tuberculosis anthranilate phosphoribosyltransferase variants reveal the conformational changes that facilitate delivery of the substrate to the active site. Biochemistry. 2015;54:6082–6092. doi: 10.1021/acs.biochem.5b00612. [DOI] [PubMed] [Google Scholar]
- 7.Cookson T.V., Castell A., Bulloch E.M., Evans G.L., Short F.L., Baker E.N., Lott J.S., Parker E.J. Alternative substrates reveal catalytic cycle and key binding events in the reaction catalysed by anthranilate phosphoribosyltransferase from Mycobacterium tuberculosis. Biochem. J. 2014;461:87–98. doi: 10.1042/BJ20140209. [DOI] [PubMed] [Google Scholar]
- 8.Marino M., Deuss M., Svergun D.I., Konarev P.V., Sterner R., Mayans O. Structural and mutational analysis of substrate complexation by anthranilate phosphoribosyltransferase from Sulfolobus solfataricus. J. Biol. Chem. 2006;281:21410–21421. doi: 10.1074/jbc.M601403200. [DOI] [PubMed] [Google Scholar]
- 9.Krissinel E., Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 10.Ortega A., Amoros D., Garcia de la Torre J. Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys. J. 2011;101:892–898. doi: 10.1016/j.bpj.2011.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evans P.R. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 2011;67:282–292. doi: 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lebedev A.A., Isupov M.N. Space-group and origin ambiguity in macromolecular structures with pseudo-symmetry and its treatment with the program Zanuda. Acta Crystallogr. D Biol. Crystallogr. 2014;70:2430–2443. doi: 10.1107/S1399004714014795. [DOI] [PubMed] [Google Scholar]
- 13.Zwart P.H., Grosse-Kunstleve R.W., Lebedev A.A., Murshudov G.N., Adams P.D. Surprises and pitfalls arising from (pseudo)symmetry. Acta Crystallogr. D Biol. Crystallogr. 2008;64:99–107. doi: 10.1107/S090744490705531X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Engh R.A., Huber R. Accurate bond and angle parameters for X-ray protein-structure refinement. Acta Crystallogr. Sect. A. 1991;47:392–400. [Google Scholar]
- 14.Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karplus P.A., Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weiss M.S. Global indicators of X-ray data quality. J. Appl. Crystallogr. 2001;34:130–135. [Google Scholar]
- 18.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murshudov G.N., Vagin A.A., Dodson E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 21.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., McCoy A.J., Moriarty N.W., Oeffner R., Read R.J., Richardson D.C., Richardson J.S., Terwilliger T.C., Zwart P.H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen V.B., Arendall W.B., 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material