

Abstract
Accumulation of tumor‐associated macrophages (TAMs) associates with malignant progression in cancer. However, the mechanisms that drive the pro‐tumor functions of TAMs are not fully understood. ZEB1 is best known for driving an epithelial‐to‐mesenchymal transition (EMT) in cancer cells to promote tumor progression. However, a role for ZEB1 in macrophages and TAMs has not been studied. Here we describe that TAMs require ZEB1 for their tumor‐promoting and chemotherapy resistance functions in a mouse model of ovarian cancer. Only TAMs that expressed full levels of Zeb1 accelerated tumor growth. Mechanistically, ZEB1 expression in TAMs induced their polarization toward an F4/80low pro‐tumor phenotype, including direct activation of Ccr2. In turn, expression of ZEB1 by TAMs induced Ccl2, Cd74, and a mesenchymal/stem‐like phenotype in cancer cells. In human ovarian carcinomas, TAM infiltration and CCR2 expression correlated with ZEB1 in tumor cells, where along with CCL2 and CD74 determined poorer prognosis. Importantly, ZEB1 in TAMs was a factor of poorer survival in human ovarian carcinomas. These data establish ZEB1 as a key factor in the tumor microenvironment and for maintaining TAMs’ tumor‐promoting functions.
Keywords: EMT, macrophages, TAMs, tumor microenvironment, ZEB1
Subject Categories: Cancer, Molecular Biology of Disease, Signal Transduction
Introduction
Macrophages play central roles in tissue homeostasis, in pathophysiological responses as well as in a number of diseases, including cancer (Wynn et al, 2013; Franklin & Li, 2016). Extravasation of monocytes into inflamed tissues and the tumor microenvironment promote their differentiation into macrophages and subsequent activation along a continuum of functional phenotypes that, at the extremes, are referred to as pro‐inflammatory/M1 and anti‐inflammatory/M2 (Lawrence & Natoli, 2011; Xue et al, 2014). Based on their expression of the cell surface marker F4/80, mouse macrophages are ontogenetically divided into F4/80low (that differentiate from circulating monocytes) and F4/80high (that originate from embryonic precursors) (Schulz et al, 2012). The F4/80high subpopulation—that in the peritoneal cavity are referred as large peritoneal macrophages, LPMs—is the largest under basal conditions but are replaced by F4/80low macrophages—referred in the peritoneum as small peritoneal macrophages, SPMs—in the context of inflammation and cancer (Goshn et al, 2010; Okabe & Medzhitov, 2014; Rei et al, 2014).
Tumor‐associated macrophages (TAMs) are critical players in the crosstalk between cancer cells and their microenvironment (Franklin & Li, 2016). Secretion by cancer cells and non‐malignant cells in the microenvironment of chemokines (e.g., CCL2/MCP‐1), cytokines (e.g., IL‐4, IL‐13), and growth factors (e.g., VEGF, CSF1/M‐CSF, CSF2/GM‐CSF) triggers the recruitment into the tumor of pro‐inflammatory F4/80low circulating monocytes—expressing the CCL2 receptor CCR2—and their acquisition of a TAM phenotype (Qian et al, 2011; Ostuni et al, 2015; Franklin & Li, 2016). Although TAMs have some gene expression overlap with M2 macrophages, their transcriptomes are still distinct (Biswas et al, 2006). The capacity of carcinomas to recruit and activate TAMs largely depends on malignant cells having acquired an undifferentiated mesenchymal phenotype as part of the so‐called epithelial‐to‐mesenchymal transition (EMT) (Su et al, 2014; Nieto et al, 2016). In turn, TAMs release factors that promote cancer cell stemness, tumor angiogenesis and progression, and chemotherapy resistance, all cancer hallmarks associated with an EMT (Lu et al, 2014; Ostuni et al, 2015). These tumor‐promoting functions of TAMs are stronger in F4/80low TAMs (Hagemann et al, 2006; Cain et al, 2013; Rei et al, 2014).
A key inducer of EMT in cancer cells is the transcription factor ZEB1 (also known as δEF1), which is expressed by malignant cells at the invasive front of carcinomas (Wellner et al, 2009; Sánchez‐Tilló et al, 2011). ZEB1 endows cancer cells with a pro‐invasive and stem‐like phenotype and determines a worse clinical prognosis in most cancers (Sánchez‐Tilló et al, 2012; Hill et al, 2013). ZEB1 can directly activate or repress gene expression by binding to the regulatory regions of its target genes (Postigo & Dean, 1999; Postigo, 2003; Postigo et al, 2003).
Since TAMs are the most abundant immune cells in the tumor microenvironment—particularly in tumors that underwent an EMT—and ZEB1 is expressed by cancer cells at the leading edge of tumors—where cancer cells interact with their microenvironment—we sought to investigate whether ZEB1 plays a role in the crosstalk between cancer cells and TAMs. While ZEB1 has been characterized in normal and malignant lymphocytes (Genetta et al, 1994; Sánchez‐Tilló et al, 2014), its expression and function in macrophages remain to be determined.
Using Zeb1 (+/−) mice—null Zeb1 (−/−) mice are embryonic lethal (Takagi et al, 1998)—we characterized here the expression and function of ZEB1 in normal macrophages and TAMs and found that TAMs depend on ZEB1 for their tumor‐promoting role. ZEB1 was restricted to the F4/80low fraction of macrophages and TAMs where it activates SPM‐ and TAM‐associated genes. Macrophages required full levels of ZEB1 for their activation toward TAMs with an F4/80low tumor‐promoting phenotype, including expression of Ccr2 and Mmp9. In addition, ZEB1 expression by TAMs induced Ccl2, Cd74, and a mesenchymal/stem‐like phenotype in cancer cells. Thus, expression of ZEB1 by F4/80low TAMs activated a CCR2‐MMP9‐CCL2 loop between tumor cells and TAMs. Expression of ZEB1 by TAMs determined worse survival in patients with ovarian carcinoma. In turn, TAM infiltration correlated with increased ZEB1 in tumor cells, where concomitant high expression of CCL2 and/or CD74 determined a poorer prognosis.
TAMs constitute an important target in oncology and efforts are being made to regulate their recruitment and/or pro‐tumor functions (Mantovani et al, 2017). The unexpected role for ZEB1 established by these results offers additional approaches to target TAMs in cancer therapy.
Results
Zeb1 is restricted to F4/80low macrophages
Before investigating a potential role of ZEB1 in macrophages at the tumor microenvironment, we examined its expression and role during monocyte‐to‐macrophage differentiation. The intraperitoneal (i.p.) administration of CSF1 (M‐CSF) into mice triggers the mobilization of circulating monocytes (CD11b+F4/80−) into the peritoneal cavity, their differentiation into macrophages (CD11b+F4/80+), and a decrease in the F4/80high subpopulation.
We first assessed Zeb1 mRNA expression in bone marrow total cells and in peritoneal monocytes (CD11b+F4/80−), macrophages (CD11b+F4/80+), and macrophage subpopulations (SPM, LPM) isolated from wild‐type mice treated with CSF1 (Fig 1A). We found that Zeb1 was higher in peritoneal monocytes than in bone marrow precursors and that, interestingly, Zeb1 was almost restricted to the F4/80low SPM fraction (Fig 1A).
Figure 1. Zeb1 is restricted to F4/80low macrophages, activates the expression of SPM‐related genes, and inhibits maturation toward F4/80high macrophages.
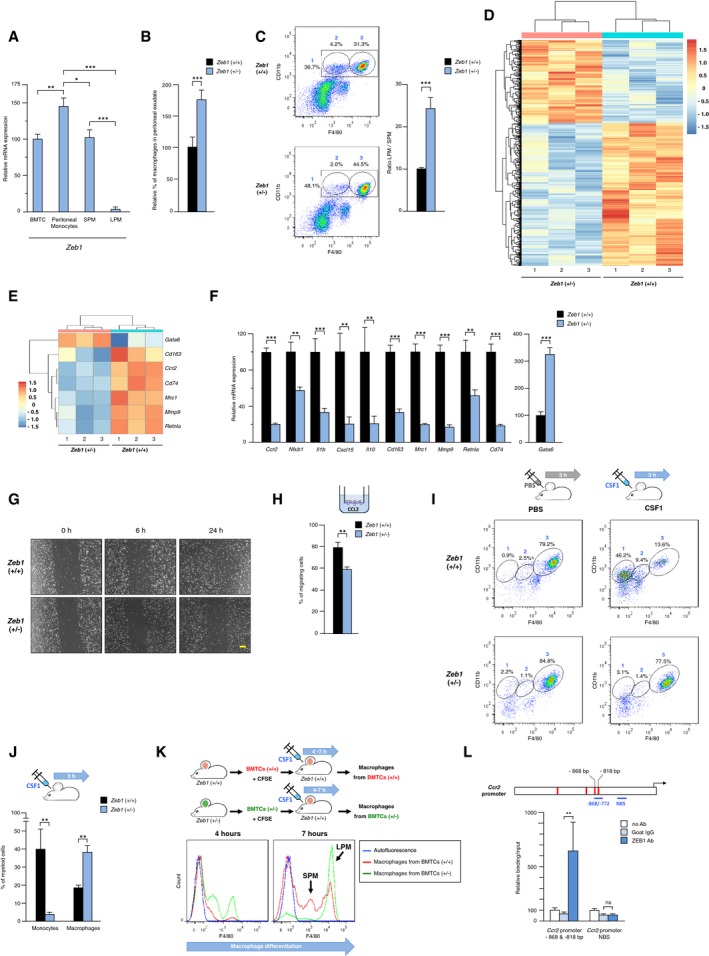
- Zeb1 expression in bone marrow total cells (BMTCs), peritoneal monocytes, and F4/80low (SPM) and F4/80high (LPM) peritoneal macrophages. Wild‐type mice were injected i.p. with rmCSF1, and 4 h later, BMTC and peritoneal cells were harvested and sorted for CD11b and F4/80. Expression of Zeb1 mRNA was assessed by qRT–PCR with respect to Gapdh. At least three mice for each genotype were analyzed.
- The share of macrophages as a percentage of total peritoneal exudate cells is higher in Zeb1 (+/−) mice than in wild‐type ones. At least six mice for each genotype were analyzed.
- Zeb1 downregulation decreases the SPM fraction and increases the F4/80high to F4/80low ratio. Peritoneal macrophages from wild‐type and Zeb1 (+/−) mice were examined by FACS for CD11b and F4/80 expression. Left panel: A representative plot for each genotype is shown. Gates numbering corresponds to (1) total macrophages, (2) SPM, and (3) LPM. Right panel: Values for the LPM to SPM ratio are the mean with standard errors of six mice for each genotype.
- Heatmap of genes differentially expressed in wild‐type versus Zeb1 (+/−) peritoneal macrophages for a false discovery rate (FDR) ≤ 0.1. Units in the color scale represent a z‐score.
- As in (D) but for selected SPM‐ and LPM‐associated genes. Units in the color scale represent a z‐score.
- Zeb1 activates the expression of F4/80low SPM‐related genes. Peritoneal macrophages from 6‐ to 8‐week‐old wild‐type and Zeb1 (+/−) mice, at least four mice for each genotype, were assessed for mRNA expression of the indicated genes by qRT–PCR.
- Wild‐type peritoneal macrophages migrated more than Zeb1 (+/−) counterparts in a wound‐healing assay. Images shown are representative of at least four different mice for each genotype. Scale bar: 100 μm.
- Transwell® chemotaxis assay of CFSE‐labeled peritoneal macrophages from wild‐type and Zeb1 (+/−) in the presence of rmCCL2. Values are the mean of relative fluorescence units (RFU) of five mice for each genotype.
- Decreased number of monocytes and increased number of F4/80high macrophages (LPM) in Zeb1 (+/−) mice in response to CSF1. At least four mice for each genotype were injected i.p. with either PBS or rmCSF1. Monocyte chemotaxis into the peritoneum and the distribution of the different peritoneal subpopulations were examined at 3 h. See also Fig EV2A for additional time periods. Gate numbers correspond to the following subpopulations: (1) monocytes, (2) F4/80low macrophages, and (3) F4/80high macrophages.
- Decreased number of monocytes and increased number of macrophages in Zeb1 (+/−) mice upon administration of CSF1. Mice were treated with rmCSF1 and the peritoneal myeloid subpopulations assessed by FACS as in (I).
- Adoptive transfer of Zeb1 (+/−) BMTCs into wild‐type mice yields increased number of F4/80high macrophages (LPM) than do wild‐type BMTCs. CFSE‐labeled BMTCs from mice of both genotypes were inoculated into wild‐type mice. Mice were then injected i.p. with rmCSF1, and myeloid cell mobilization and their distribution between SPM and LPM subpopulations were assessed at 4 h and 7 h.
- ZEB1 binds to the mouse Ccr2 promoter. Upper panel: Scheme of 2 kb of the Ccr2 promoter. At least four potential ZEB1 binding sequences (red boxes) were identified at −1,398 bp (CACCTG), −984 bp (CACGTA), −868 bp (CAGGTG), and −818 bp (CAGGTG). Lower panel: DNA from macrophages was immunoprecipitated with antibodies against ZEB1 (clone E‐20) or a matched IgG control (goat IgG) and amplified by qRT–PCR for two regions of the Ccr2 promoter (blue lines in the upper panel): a first region containing two ZEB1 consensus binding sites at −868 bp and −818 bp (−868/−772‐bp region) and a second region lacking consensus binding sites for ZEB1 (non‐binding site, NBS, −574/−422‐bp region). A condition without antibody is also shown. For each promoter fragment, the condition without antibody was set to 100. Values shown represent relative binding in relation to input and are the average of at least three independent experiments, each one performed in triplicate.
Next, we explored whether Zeb1 regulates macrophage homeostasis. It was found that: (i) the number and the share of macrophages as a percentage of total peritoneal cells, and (ii) the ratio LPM to SPM (F4/80high to F4/80low) were all larger in Zeb1 (+/−) mice than in wild‐type counterparts (Figs 1B and C, and EV1A). These data indicate that Zeb1 modulates the distribution of LPM and SPM subpopulations, increasing the latter.
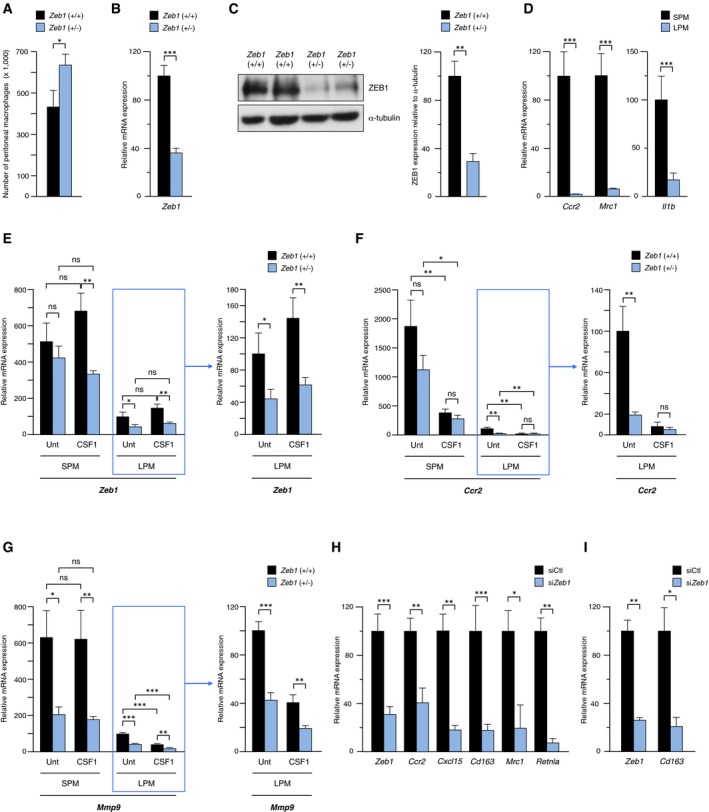
Figure EV1. Zeb1 upregulates the F4/80low SPM macrophage subpopulation where it activates the expression of SPM‐related genes.
-
ANumber of peritoneal macrophages in wild‐type and Zeb1 (+/−) mice. Macrophages (CD11b+F4/80+) were sorted and quantified by FACS. Data represent the mean with standard errors from at least eight mice for each genotype.
-
BmRNA expression of Zeb1 in wild‐type and Zeb1 (+/−) peritoneal macrophages. mRNA levels were assessed by qRT–PCR using Gapdh as reference gene. Data represent the mean with standard errors from at least five mice for each genotype.
-
CProtein expression of ZEB1 in wild‐type and Zeb1 (+/−) peritoneal macrophages. Left panel: Cell lysates were blotted for ZEB1 (clone HPA027524) along with α‐tubulin (clone B‐5‐1‐2) as loading control. A representative blot of three independent Western blots is shown. Full unedited blots for ZEB1 and α‐tubulin are shown in the Source Data file. Right panel: Quantification of ZEB1 protein expression relative to α‐tubulin. The average of three independent Western blots is shown.
-
DCcr2, Mrc1, and Il1b mRNA levels in SPM (F4/80low) and LPM (F4/80high) subpopulations isolated from wild‐type mice. Data represent the mean with standard errors from at least four wild‐type mice.
-
ELeft panel: Zeb1 expression in SPM and LPM subpopulations sorted from 6‐ to 8‐week‐old wild‐type and Zeb1 (+/−) mice in basal conditions (injected with PBS, referred as Unt) or treated for 3 h with 1 ml CM from L929/CSF1 cells. Right panel: Since Zeb1 expression in LPMs is low, the LPM fraction is represented again using a smaller scale. mRNA levels were assessed by qRT–PCR with respect to Gapdh. Data represent the mean with standard errors from at least three mice for each genotype.
-
F, GAs in (E) but for Ccr2 and Mmp9, respectively. Data represent the mean with standard errors from at least three mice for each genotype.
-
HExpression of selected SPM‐associated genes in wild‐type peritoneal macrophages interfered with a siRNA against mouse Zeb1 (siZeb1) or a siRNA control (siCtl). Data are the average of at least three independent experiments. Data represent the mean with standard error from at least three mice for each genotype.
-
IMacrophages derived from bone marrow cells treated with rmCSF1 were interfered with siCtl or siZeb1 and assessed for Zeb1 and Cd163 mRNA levels by qRT–PCR. Data represent the mean with standard error from at least three mice for each genotype.
Zeb1 induces SPM‐associated genes
To elucidate the gene expression profile regulated by Zeb1 in macrophages, we carried out RNA sequencing (RNA‐seq) using RNA from wild‐type and Zeb1 (+/−) macrophages (Fig 1D). Expression of ZEB1 mRNA and protein in Zeb1 (+/−) macrophages is around 40 and 30%, respectively, of the levels found in wild‐type macrophages (Fig EV1B and C). Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses of RNA‐seq data revealed that many genes differentially expressed (DE) between both genotypes corresponded to immune functions (Appendix Tables S1 and S2). Interestingly, DE genes included those associated with SPM and LPM phenotypes (Fig 1E).
We then validated the expression of selected SPM‐ and LPM‐associated genes (Okabe & Medzhitov, 2014; Rei et al, 2014; and Fig EV1D) in wild‐type and Zeb1 (+/−) macrophages by quantitative real‐time PCR (qRT–PCR). It was found that Zeb1 activated SPM‐related genes as Zeb1 (+/−) macrophages expressed lower levels of Ccr2, Nfκ1b, Il1b, Cxcl15 (Il8), Cd163, Mrc1 (Cd206), Mmp9, Retnla, and Cd74 (Fig 1F). Conversely, Gata6, whose expression is linked to LPMs, was higher in Zeb1 (+/−) macrophages (Fig 1F). Zeb1 did not induce an archetypal M1 or an M2 phenotype because, compared to wild‐type counterparts, Zeb1 (+/−) macrophages expressed lower levels of both pro‐ and anti‐inflammatory genes (Fig 1F). Overall, these data indicate that Zeb1 increases the F4/80low SPM population by activating SPM‐associated genes. Among these genes was Ccr2, the high‐affinity receptor for the chemokine Ccl2 that is produced at sites of inflammation and in tumors.
The expression of Zeb1, Ccr2, and Mmp9 was also examined in sorted SPM and LPM fractions from mice of both genotypes and both in basal conditions and upon CSF1 stimulation. In line with the above results, these genes were predominantly expressed in F4/80low macrophages (Fig EV1E–G). The expression of SPM‐associated genes was also examined in wild‐type peritoneal macrophages transfected with either a siRNA control (siCtl) or a specific siRNA against mouse Zeb1 (siZeb1). Knockdown of Zeb1 downregulated the expression of Ccr2, Cxcl15, Cd163, Mrc1, and Retnla (Fig EV1H). Cd163 was also downregulated in bone marrow‐derived macrophages upon Zeb1 knockdown (Fig EV1I).
Zeb1 inhibits the maturation of macrophages toward F4/80high LPMs
Macrophage chemotaxis and migration are essential steps during the immune response, inflammation, and cancer, and we investigated whether Zeb1 regulates these functions. First, we analyzed the basal motility of wild‐type and Zeb1 (+/−) peritoneal macrophages in an in vitro wound‐healing assay. In the absence of a chemotactic stimulus, Zeb1 (+/−) macrophages showed a slower closure of the scratch wound than wild‐type ones (Fig 1G). We then examined the chemotaxis of macrophages from both genotypes to recombinant mouse CCL2 (rmCCL2). CCL2 is required for the mobilization of pro‐inflammatory CCR2+ monocytes into inflammatory and infection foci, and tumors (Serbina & Pamer, 2006; Ostuni et al, 2015). Again, Zeb1 (+/−) macrophages migrated less efficiently than wild‐type counterparts (Fig 1H). These data indicate that Zeb1 promotes macrophage motility in vitro under both basal and chemotactic conditions.
Next, we explored whether Zeb1 regulates the in vivo bone marrow mobilization of monocytes induced by CSF1 and CCL2. The i.p. injection of recombinant mouse CSF1 (rmCSF1) into Zeb1 (+/−) mice failed to elicit both the mobilization and peritoneal infiltration of monocytes as well as the reduction in F4/80high macrophages that occurred in wild‐type mice after only 3 h (Figs 1I and J, and EV2A). After 7 h, SPM and LPM subpopulations almost recovered to basal conditions (Fig EV2A). Likewise, the i.p. injection of rmCCL2 also failed to mobilize monocytes into the peritoneal cavity of Zeb1 (+/−) mice (Fig EV2B).
Figure EV2. Decreased number of monocytes and increased number of F4/80high macrophages in Zeb1 (+/−) mice in response to CSF1 and CCL2.
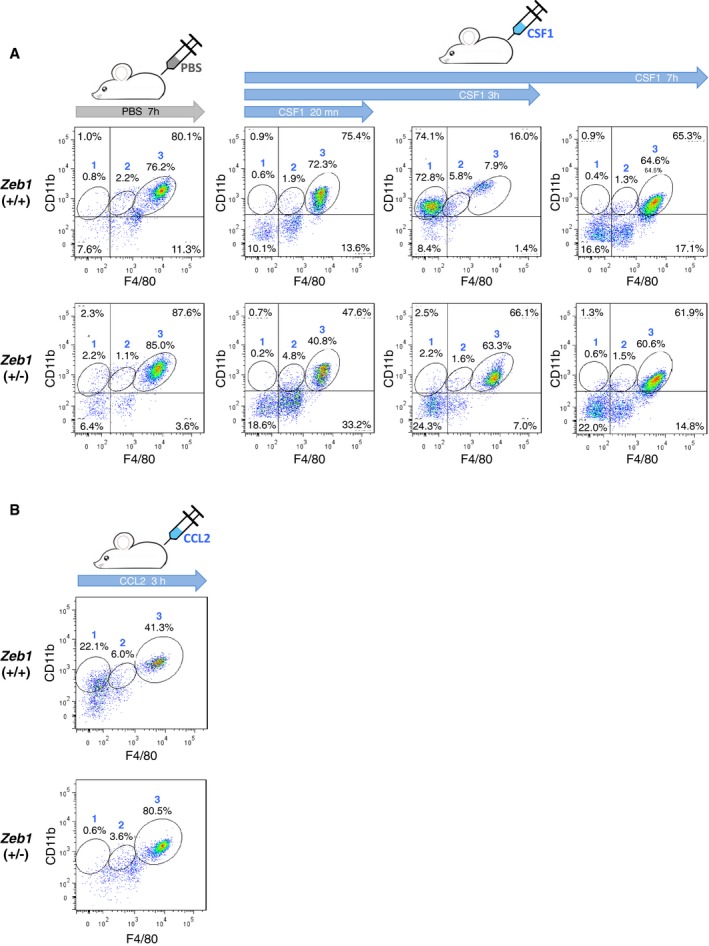
- As in Fig 1I, mice from both genotypes were injected i.p. with rmCSF1 and peritoneal subpopulations were assessed at different time points by FACS. Gate numbers correspond to the following subpopulations: (1) monocytes, (2) F4/80low macrophages, and (3) F4/80high macrophages. A representative dot plot from at least three mice for each genotype is shown.
- As in (A) but using rmCCL2 and with peritoneal cells assessed at 3 h.
We also studied whether a wild‐type or Zeb1 (+/−) microenvironment background influences the mobilization of myeloid precursors and their subsequent maturation into macrophages. Bone marrow total cells (BMTCs) from both genotypes were labeled with the dye 5(6)‐carboxyfluorescein N‐hydroxysuccinimidyl ester (CFSE) and injected intravenously into wild‐type mice. After only 4 h, a larger share of Zeb1 (+/−) BMTCs had differentiated toward macrophages (F4/80+) than in the case of wild‐type BMTCs (Fig 1K). At later times (7 h), virtually all macrophages that originated from Zeb1 (+/−) BMTCs were F4/80high LPMs (Fig 1K). In contrast, only about half of those derived from wild‐type BMTCs were F4/80high LPMs (Fig 1K).
ZEB1 binds to a subset of E‐box sequences in the regulatory regions of its target genes (Sekido et al, 1994). Analysis of the first 2 kb of the mouse Ccr2 promoter revealed the existence of at least four potential ZEB1 high‐affinity binding sequences (Fig 1L). The ability of endogenous ZEB1 to directly bind to two of them (−868 bp and −818 bp) was tested by a chromatin immunoprecipitation (ChIP) assay. An antibody against ZEB1, but not its respective host‐matched control IgG, immunoprecipitated a region of the Ccr2 promoter containing both sites but not a region lacking consensus binding sites for ZEB1 (Fig 1L).
Altogether, these data indicate that ZEB1 blocks monocyte‐to‐macrophage differentiation and that a partial downregulation of Zeb1 was sufficient to unleash differentiation. It can be also concluded that the failure of Zeb1 (+/−) BMTCs to generate F4/80low SPMs does not depend on the genotype of the microenvironment (all recipient hosts were wild type); instead, this failure was intrinsic to the genotype of the BMTCs.
Zeb1 is required for the activation of macrophages toward pro‐tumor F4/80low TAMs
We then examined whether the expression of Zeb1 in macrophages is modulated by the presence of cancer cells. We used a well‐established syngeneic mouse cancer model to study the interaction between malignant cells and their microenvironment in the context of fully immunocompetent C57BL/6 mice. Intraperitoneal injection of ID8 ovarian carcinoma cells into female recipient mice results in their rapid growth and the development of ascites with abundant by TAMs (Hagemann et al, 2006, 2008; Yin et al, 2016). Tumor growth in the ID8 model is dependent on the accumulation of TAMs (Hagemann et al, 2008; Rei et al, 2014; Yin et al, 2016). In addition, the peritoneal microenvironment also stably enhances ID8 cells’ tumorigenic potential compared to that of parental ID8 cells that had been maintained in culture medium (Cai et al, 2015). Thus, when ID8 cells isolated from a mouse are inoculated into new into a new recipient mouse, these transferred ID8 cells—that will be referred hereafter as ID8RI, standing for “ID8 Re‐Injected” cells—display accelerated tumor onset and progression and generate more ascites (Cai et al, 2015).
Notably, we found that, for both genotypes, Zeb1 expression increased when macrophages were exposed to cancer cells. Zeb1 was higher in TAMs isolated from mice injected with ID8 cells during 13 weeks than in basal macrophages (Fig 2A). These results indicate that cancer cells induce ZEB1 in TAMs and support a role for ZEB1 in the regulation of a TAM phenotype.
Figure 2. Macrophage activation toward TAMs requires Zeb1 expression, which increases the F4/80low TAM subpopulation.
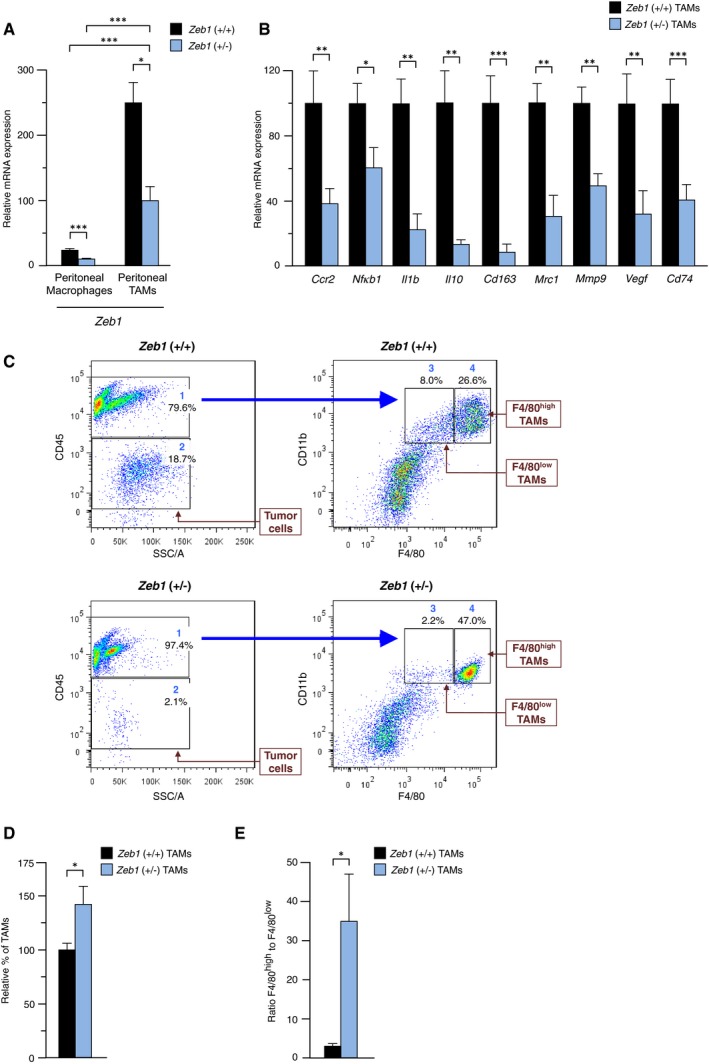
- Zeb1 increases during activation of peritoneal macrophages into TAMs. Zeb1 mRNA levels were determined by qRT–PCR in sorted peritoneal macrophages from wild‐type and Zeb1 (+/−) mice either under homeostasis conditions (peritoneal macrophages) or 13 weeks after injection of ID8 cells (peritoneal TAMs). Values represent the means of at least three mice for each genotype. The last bar was set to 100.
- Zeb1 promotes the in vivo acquisition of a TAM phenotype. Mice from both genotypes were injected with ID8 cells, and 6 weeks later, peritoneal TAMs were isolated and assessed for gene expression by qRT–PCR. Data represent the mean with standard errors from at least three for each genotype.
- TAMs from Zeb1 (+/−) mice failed to acquire a pro‐tumoral F4/80low TAM phenotype. Left panels: Mice were injected with ID8 cells and the distribution of tumor (CD45−) and immune (CD45+) cells in peritoneal exudates was examined 6 weeks later. Right panels: CD45+ cells were then analyzed for CD11b and F4/80 expression. Gate numbers correspond to the following subpopulations: (1) total leukocytes, (2) ID8 tumor cells, (3) F4/80low TAMs, and (4) F4/80high TAMs. Shares in (3) and (4) are calculated out of (1).
- The relative share of TAMs, as a percentage of total peritoneal cells, was higher in Zeb1 (+/−) mice than in wild‐type counterparts. Data represent the mean with standard errors from at least eight mice for each genotype.
- Zeb1 (+/−) peritoneal TAMs displayed a higher F4/80high to F4/80low ratio. As in (C), expression of F4/80 in TAMs was assessed 6 weeks after injection of ID8 cells. Data represent the mean with standard errors from at least four mice for each genotype.
Next, we explored whether macrophage activation toward TAMs requires full levels of ZEB1 expression. Wild‐type and Zeb1 (+/−) mice were injected with ID8 cells during 8 weeks and peritoneal TAMs were then isolated and examined for the expression of TAM‐associated genes. Expression levels of Ccr2, Nfkb1/p50, Il1b, Il10, Cd163, Mrc1, Mmp9, Vegf, and Cd74 were lower in Zeb1 (+/−) TAMs (Fig 2B), indicating that Zeb1 drives in vivo activation of a tumor‐promoting gene expression profile in TAMs.
In the ID8 model, F4/80low TAMs display a stronger pro‐tumor phenotype than F4/80high TAMs (Hagemann et al, 2006; Cain et al, 2013; Rei et al, 2014). The overall TAM population and its distribution between F4/80low and F4/80high fractions were examined in the peritoneal exudate of mice from both genotypes injected with ID8 cells. In wild‐type mice, the number of TAMs increased with respect to the number of peritoneal macrophages in homeostatic conditions (Fig EV3A). As the tumor load after 6 weeks was higher in wild‐type mice than in Zeb1 (+/−) counterparts (Fig 2C and see below), the share of TAMs as a percentage of total peritoneal cells was higher in the latter (Fig 2C and D). In addition, the pro‐tumor F4/80low TAM subpopulation did not expand in Zeb1 (+/−) mice and the ratio F4/80high TAMs to F4/80low TAMs was higher in these mice than in wild‐type ones (Fig 2C and E).
Figure EV3. Number of peritoneal cells in wild‐type and Zeb1 (+/−) mice under basal conditions and in the ID8 cancer model.
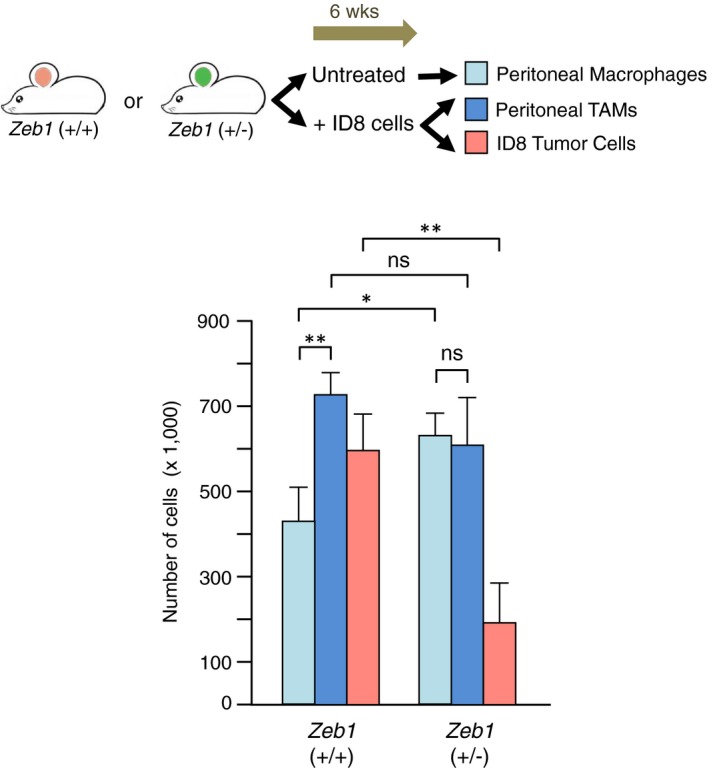
In wild‐type mice, higher tumor load corresponds to an increase in the number of TAMs. Wild‐type and Zeb1 (+/−) mice were either left untreated or injected with ID8 cells. After 6 weeks, mice were euthanized and analyzed for the number of (i) peritoneal macrophages under basal conditions in those mice left untreated, or (ii) peritoneal TAMs, and (iii) peritoneal ID8 cells in those mice injected with ID8 cells. Cell counts were determined by FACS. At least eight mice of each genotype were examined, of which half were left untreated and half injected with ID8 cells. Statistical significance was assessed with a non‐parametric Mann–Whitney U‐test: **P ≤ 0.01; *P ≤ 0.05; ns, P > 0.05. Error bars represent standard error of the mean.
TAMs require expression of full levels of Zeb1 to promote tumor progression
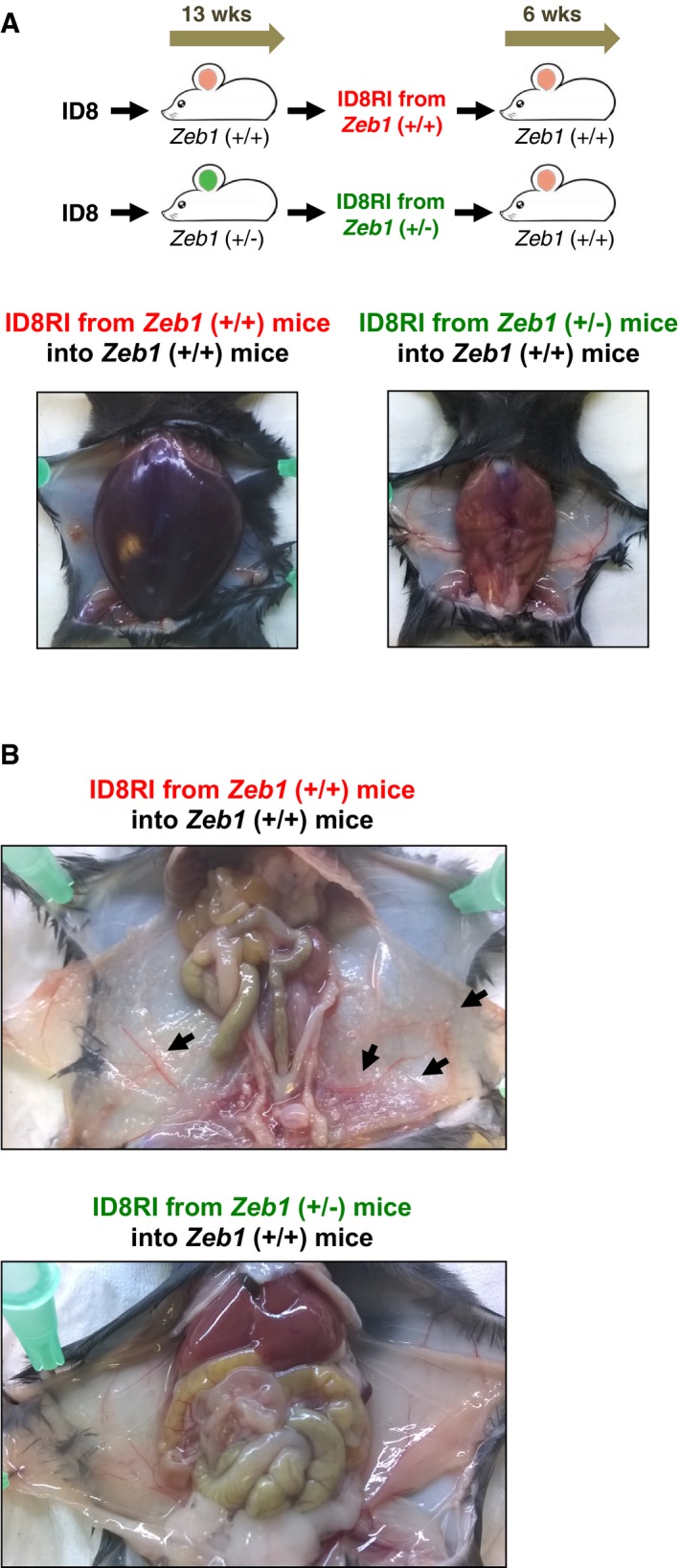
In Fig 2C, we have showed that when mice from both genotypes were injected with ID8 cells and sacrificed after 6 weeks, the pro‐tumor F4/80low TAM subpopulation was reduced in Zeb1 (+/−) mice. In addition, both the number of ID8 tumor cells (CD45− cells) and their share out of total peritoneal cells were also greatly diminished (left panel in Fig 2C, and Figs 3A and EV3A). In addition, at the end point, there were fewer tumor deposits in the peritoneal lining of Zeb1 (+/−) mice than in wild‐type counterparts (Fig 3B). The tumor load and the progression of the ID8 cancer model in both genotypes were assessed over time by bioluminescence imaging upon injection of ID8 cells carrying the luciferase‐2 gene (ID8‐luc) (Fig 3C and D). In contrast to wild‐type mice, where the tumor load progressively increased up to the end point, tumor cells barely grew in Zeb1 (+/−) mice (Fig 3C).
Figure 3. TAMs require full levels of Zeb1 expression to promote tumor progression and to induce an undifferentiated phenotype in cancer cells.
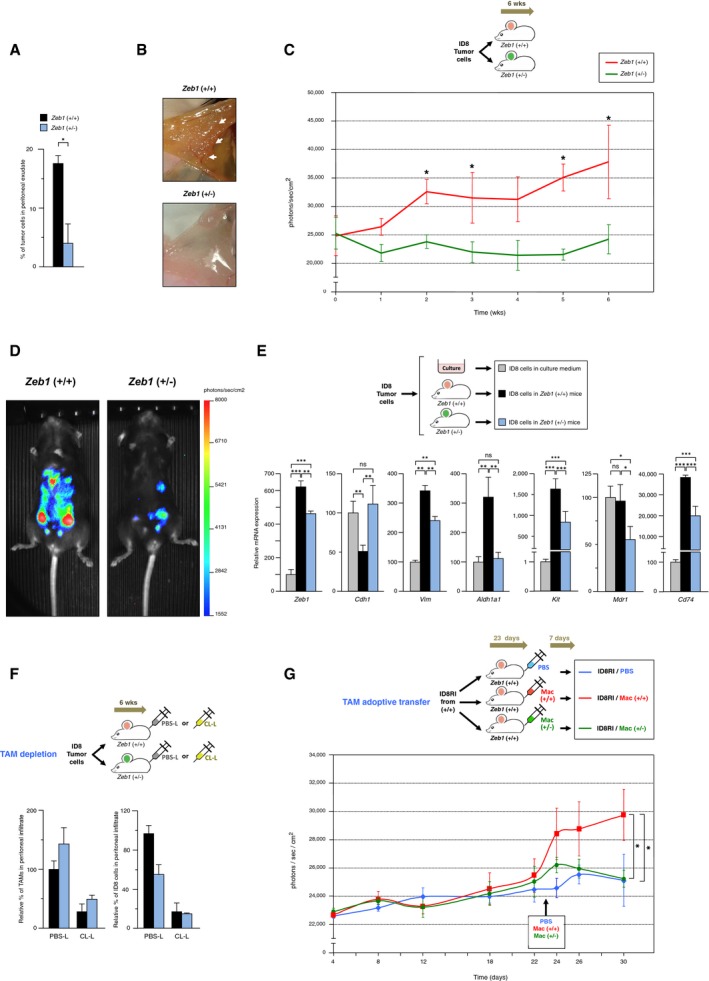
- Reduced tumor growth in Zeb1 (+/−) mice. The percentage of tumor (CD45−) cells out of the total number peritoneal cells was assessed in mice from both genotypes 6 weeks after injection of ID8 cells as in Fig 2C. Data represent the mean with standard errors from at least four mice for each genotype.
- Injection of ID8 cells yields fewer peritoneal tumor deposits in Zeb1 (+/−) mice. Representative images of the peritoneal lining of mice 13 weeks after i.p. injection with ID8 cells. White arrows mark selected tumor deposits.
- The progression of the ID8 cancer model is inhibited in Zeb1 (+/−) mice. Mice were injected with ID8‐luc cells, and the progression of the tumor load was followed up to 6 weeks by bioluminescence imaging. At least five mice of each genotype were included. The statistical significance (*P ≤ 0.05) between the tumor load in wild‐type and Zeb1 (+/−) mice is displayed on top of the standard error bar of wild‐type mice.
- As in (C), bioluminescence signal rendered by a representative mouse for each genotype at week 6.
- Zeb1 expression in TAMs induces an undifferentiated phenotype in cancer cells. ID8 cells were either cultured in vitro in complete medium or injected i.p. during 13 weeks into mice and then sorted out by FACS. Gene expression in ID8 cells was assessed by qRT–PCR. Data represent the mean with standard errors from at least three mice for each genotype.
- Depletion of TAMs in ID8 tumor‐bearing wild‐type mice mimicked the effect of downregulation of Zeb1 in TAMs. Mice from both genotypes were injected with ID8 cells at the start of the experiments and every 4 days also with liposomes containing either PBS (PBS‐L) or clodronate (CL‐L). The distribution of subpopulations was examined 6 weeks later. A representative experiment from at least four mice, half of each genotype, is shown.
- TAMs require full levels of Zeb1 to promote tumor progression upon adoptive transfer in the ID8 cancer model. First, ID8 cells that had previously grown in wild‐type mice (ID8RI cells) were re‐injected into new wild‐type mouse recipients that were then randomly divided into three cohorts. At day 23, each cohort was inoculated i.p. with either PBS, wild‐type macrophages, or Zeb1 (+/−) macrophages and euthanized 7 days later. Tumor load was assessed by bioluminescence. For each time point, data represent the mean with standard errors from at least three mice for each genotype.
Expression of a mesenchymal phenotype in carcinomas is associated with an aggressive behavior (Nieto et al, 2016). The phenotype of ID8 cells that had grown during 13 weeks in wild‐type and Zeb1 (+/−) mice was examined for several cancer biomarkers, namely Zeb1 itself, whose expression determines worse prognosis in most cancers (Sánchez‐Tilló et al, 2012); E‐cadherin (Cdh1) and vimentin (Vim), the archetypal epithelial and mesenchymal markers, respectively; Aldh1a1 and Kit, two cancer stem cell markers whose expression is associated with tumor progression in ovarian and other cancers (Chau et al, 2011; Silva et al, 2011); Mdr1 (Abcb1), an efflux drug transporter associated with chemotherapy resistance (Gottesman et al, 2002); and Cd74, which is expressed by both F4/80low macrophages and ovarian cancer cells and that activates CCL2 transcription and promotes tumor progression (Wilkinson et al, 2015; Gil‐Yarom et al, 2017). Interestingly, Zeb1 was higher in ID8 cells isolated from wild‐type mice than in those from Zeb1 (+/−) mice (Fig 3E). Expression of Vim, Aldh1a1, Kit, Mdr1, and Cd74 was also higher in ID8 cells recovered from wild‐type mice while that of Cdh1 was lower (Fig 3E). From all the above data, it could be concluded TAMs expressing full levels of Zeb1 promote both a more de‐differentiated cancer cell phenotype and greater tumor growth than Zeb1 (+/−) TAMs.
We then explore whether the reported essential role of TAMs in the progression of the ID8 cancer model (Yin et al, 2016) was affected by the expression of Zeb1 in TAMs. To that effect, macrophages were either depleted or transplanted in tumor‐bearing mice. First, mice from both genotypes were inoculated with ID8 cells at the beginning of the experiment and during 6 weeks injected regularly with liposomes carrying either PBS (PBS‐L, as control) or clodronate (CL‐L), a bisphosphonate that causes macrophage apoptosis and is widely used for in vivo specific depletion of macrophages (van Rooijen & Hendrikx, 2010; Yin et al, 2016). As expected, in the control PBS‐L group, the number of ID8 cells was lower in Zeb1 (+/−) mice than in wild‐type ones (Fig 3F). Treatment of tumor‐bearing mice with CL‐L reduced the number of TAMs and tumor cells in both genotypes, to the point that the number of cancer cells in wild‐type mice leveled to that in Zeb1 (+/−) mice (Fig 3F). Regarding the growth of the ID8 tumor model, depletion of TAMs in wild‐type mice mimicked the effect of Zeb1 downregulation in TAMs.
If macrophage depletion blocks tumor growth, adoptive transfer of macrophages into ID8 tumor‐bearing mice accelerates tumor progression (Hagemann et al, 2008). To confirm that the reduced progression of the ID8 tumor model in Zeb1 (+/−) mice was related to the deficient pro‐tumor function of Zeb1 (+/−) TAMs, we conducted the adoptive transfer of macrophages from both genotypes into wild‐type tumor‐bearing mice (see scheme in Fig 3G). Wild‐type mice were first injected with ID8 cells that had previously grown in recipient mice (ID8RI cells), and 23 days later, individuals were randomly divided into three cohorts. One of the cohorts was injected i.p. with PBS while the other two cohorts were injected i.p. with either wild‐type or Zeb1 (+/−) macrophages. Tumor progression was followed by bioluminescence imaging for up to 30 days (Fig 3G). Notably, tumor‐bearing mice injected with wild‐type macrophages displayed an accelerated tumor growth compared to those injected with PBS or with Zeb1 (+/−) macrophages (Fig 3G). These results indicate that the reduced progression of the ID8 model in Zeb1 (+/−) mice is due to the partial downregulation of Zeb1 in TAMs. It can be also concluded that for TAMs to promote tumor growth, they have to express full levels of Zeb1 and that a partial downregulation of Zeb1 was sufficient to reduce tumor progression.
Zeb1 expression in TAMs drives a Ccr2‐Mmp9‐Ccl2 loop between TAMs and cancer cells
We then turned to investigate the molecular mechanisms by which the expression of ZEB1 in TAMs promotes an enhanced tumor progression in the ID8 cancer model. The CCL2‐CCR2 axis helps to recruit monocytes to the tumor microenvironment and promotes tumor growth and metastasis (Qian et al, 2011). We found that, compared to ID8 cells that had been maintained in culture medium (referred hereafter as “DMEM” condition), ID8RI cells that were isolated upon injection in wild‐type mice during 13 weeks expressed higher levels of Zeb1 and Ccl2 (Fig 4A). We also compared Ccl2 expression in ID8RI cells grown in wild‐type versus Zeb1 (+/−) mice. Notably, ID8RI cells isolated from the peritoneal cavity of wild‐type mice expressed higher levels of Ccl2 than those recovered from Zeb1 (+/−) counterparts (Fig 4B, left panel). In fact, ID8RI cells that had grown in Zeb1 (+/−) mice displayed similar levels of Ccl2 than ID8 cells that had never been injected into mice but rather maintained in DMEM.
Figure 4. Expression of Zeb1 by TAMs activates a Ccr2‐Mmp9‐Ccl2 loop between TAMs and cancer cells and increases the resistance of cancer cells to chemotherapy drugs.
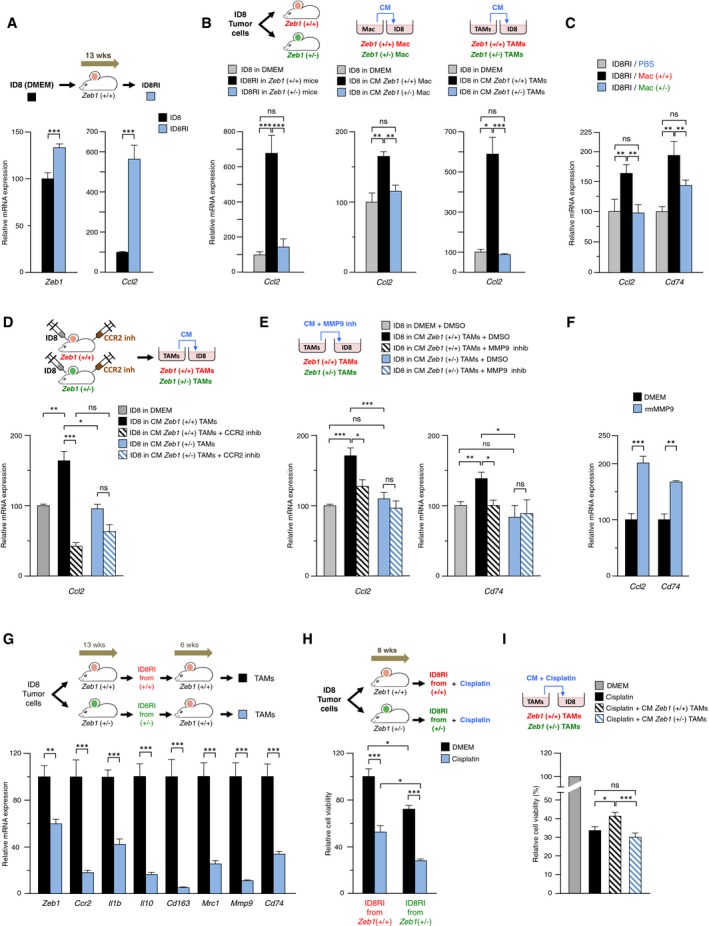
- ID8RI cells express higher levels of Zeb1 and Ccl2 than parental ID8 cells. ID8 cells maintained in complete medium (referred here as DMEM) and ID8RI cells isolated from 13 weeks wild‐type ID8 tumor‐bearing mice were assessed for gene expression by qRT–PCR. Data represent the mean with standard errors from at least three replicates for each condition.
- Macrophages require full levels of Zeb1 expression to induce Ccl2 in cancer cells. Ccl2 was examined by qRT–PCR in ID8RI cells isolated from the peritoneal cavity of wild‐type and Zeb1 (+/−) mice 13 weeks after injection (left panel), ID8 cells cultured in vitro (DMEM) or co‐cultured with conditioned medium (CM) from either peritoneal macrophages (central panel), or TAMs (right panel) isolated from mice of both genotypes. Data represent the mean with standard errors from at least three replicates for each condition.
- Adoptive transferred TAMs require full levels of Zeb1 expression to induce Ccl2 and Cd74 in cancer cells. ID8RI cells isolated from the experiment in Fig 3G were assessed for Ccl2 and Cd74 by qRT–PCR. Data represent the mean with standard errors from at least three replicates for each condition.
- Blocking of CCL2‐CCR2 signaling in TAMs inhibited Ccl2 in cancer cells. ID8 tumor‐bearing wild‐type and Zeb1 (+/−) mice were injected with a CCR2 antagonist. The CM produced by their respective TAMs was added to ID8‐luc cells and their expression of Ccl2 assessed by qRT–PCR. Data represent the mean with standard errors from at least six mice for each genotype.
- MMP9 from wild‐type TAMs, but not from Zeb1 (+/−) TAMs, upregulated Ccl2 and Cd74 in cancer cells. ID8 cells were incubated with CM from TAMs of both genotypes in the presence or absence of a MMP9 inhibitor. Data represent the mean with standard errors from at least three replicates for each condition.
- Recombinant mouse MMP9 (rmMMP9) upregulated Ccl2 and Cd74 in ID8‐luc cells. Data represent the mean with standard errors from at least three replicates for each condition.
- The undifferentiated phenotype in cancer cells induced by wild‐type TAM, but not by Zeb1 (+/−) TAMs, (Fig 3E) promotes, in turn, a pro‐tumor phenotype in TAMs. First, mice from both genotypes were injected with ID8‐luc cells. After 13 weeks, mice were sacrificed and sorted tumor cells were re‐injected (ID8RI cells) into wild‐type mouse recipients. After 6 weeks, mice were euthanized and TAMs were isolated and analyzed for gene expression by qRT–PCR. Six wild‐type were randomly divided in two cohorts, half were injected with ID8RI cells from wild‐type mice and half from ID8RI cells from Zeb1 (+/−) mice.
- Expression of Zeb1 in TAMs determines increased resistance of cancer cells to chemotherapy. Wild‐type and Zeb1 (+/−) mice were injected with ID8‐luc cells during 8 weeks. Mice were then euthanized and their ID8RI cells cultured in the absence (referred as DMEM) or presence of 50 μg/ml of cisplatin. Cell viability was assessed by an MTT assay. Data represent the mean with standard errors from at three replicates.
- ID8 cells were cultured in the absence (DMEM) or presence of 50 μg/ml of cisplatin and incubated with CM derived from wild‐type and Zeb1 (+/−) TAMs. Data represent the mean with standard errors from at three replicates.
In order to explore whether Zeb1 expression in macrophages modulates Ccl2 expression in cancer cells, ID8 cells were cultured with conditioned medium (CM) collected from either normal peritoneal macrophages or TAMs from both genotypes. Ccl2 expression was higher in ID8 cells incubated with CM from wild‐type macrophages or TAMs than in ID8 cells that had been cultured with CM from Zeb1 (+/−) counterparts (Fig 4B, central and right panels, respectively). The CM from wild‐type TAMs, but not that from Zeb1 (+/−) TAMs, also induced higher expression of Cd74 in ID8 cells (Fig EV4A). These results indicate that Zeb1 expression in TAMs not only upregulates Ccr2 in TAMs (Fig 2B) but also induces Ccl2 in cancer cells, thus triggering a positive Ccr2‐Ccl2 loop between TAMs and cancer cells.
Figure EV4. Expression of Zeb1 by TAMs induces a Ccr2‐Mmp9‐Ccl2 loop between TAMs and cancer cells.
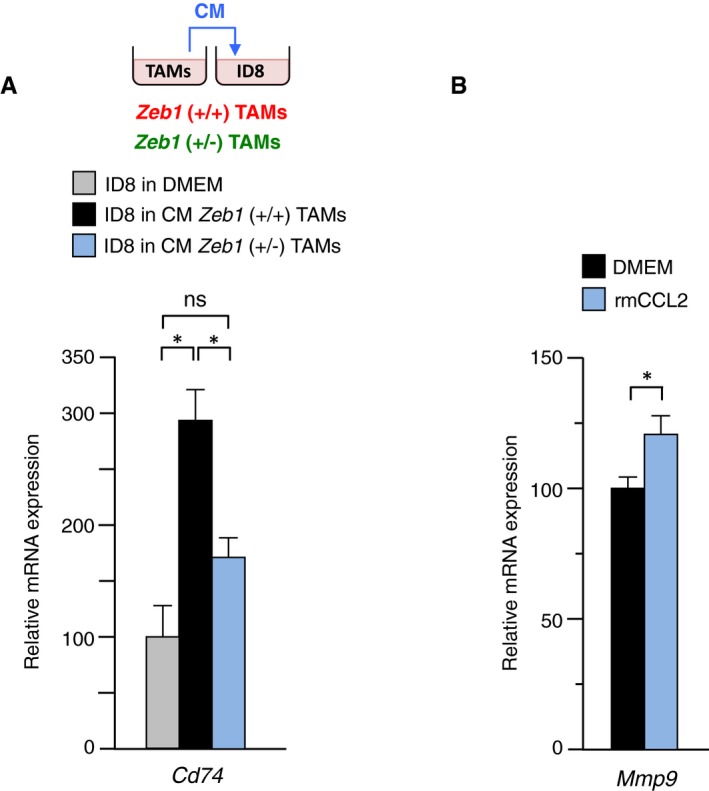
- Compared to wild‐type counterparts, Zeb1 (+/−) TAMs induce lower Cd74 expression in tumor cells. Cd74 mRNA levels were examined by qRT–PCR in ID8 cells cultured in vitro in complete medium (referred here as DMEM) or co‐cultured with conditioned medium (CM) from either wild‐type and Zeb1 (+/−) peritoneal TAMs.
- rmCCL2 induces Mmp9 in peritoneal macrophages. Wild‐type peritoneal macrophages were cultured in complete medium in the absence (referred as DMEM) or presence of rmCCL2. Mmp9 mRNA levels were examined by qRT–PCR.
ID8RI tumor cells isolated at day 30 in the experimental setting of Fig 3G were also assessed for Ccl2 and Cd74 expression. ID8RI cells in the cohort that received wild‐type macrophages displayed higher expression of both genes than ID8RI cells sorted from mice that were injected with Zeb1 (+/−) macrophages (Fig 4C). These results confirm that Zeb1 (+/−) macrophages are unable to induce Ccl2 and Cd74 in tumor cells and contribute to explaining the differential effect of macrophages from both genotypes in Fig 3G.
Next, we investigated the mechanism by which Zeb1 expression in TAMs upregulates Ccl2 in cancer cells. We tested the effect of in vivo blocking CCL2‐CCR2 signaling in TAMs on the expression of Ccl2 by ID8 cells. ID8 tumor‐bearing mice from both genotypes were injected with a CCR2 small molecule antagonist and the CM produced by their respective TAMs was then used to culture ID8 cells (see scheme in Fig 4D). In contrast to the lack of effect of the CM derived from Zeb1 (+/−) TAMs, the CM from wild‐type TAMs induced Ccl2 expression in ID8 cells and this upregulation was inhibited by the CCR2 antagonist (Fig 4D).
MMP9 mediates plasminogen‐induced migration of macrophages in the context of inflammation, but plasminogen fails to increase the chemotaxis of Mmp9‐deficient macrophages toward CCL2 (Gong et al, 2008). In addition, MMP9 expression by TAMs promotes cancer cell invasiveness and associates with clinical aggressiveness in ovarian cancer (Huang et al, 2002). Thus, among the genes upregulated by Zeb1 in TAMs (Fig 2B), we focused our attention on Mmp9 and explored whether the lower Mmp9 expression in Zeb1 (+/−) TAMs participated in the reduced Ccl2 levels displayed by ID8RI cells that had grown in Zeb1 (+/−) mice. ID8 cells were incubated with the CM from wild‐type and Zeb1 (+/−) TAMs and in either the presence or absence of an MMP9 inhibitor. We found that the upregulation of Ccl2 in ID8 cells induced by their incubation with the CM derived from wild‐type TAMs was reduced by the MMP9 inhibitor (Fig 4E, left panel). In contrast, Ccl2 expression was neither altered by the CM from Zeb1 (+/−) TAMs or by the treatment with the MMP9 inhibitor. Similar results were found for Cd74: The upregulation of Cd74 induced by the CM from wild‐type TAMs, but not by the CM from Zeb1 (+/−) TAMs, was reduced by the MMP9 inhibitor (Fig 4E, right panel). In turn, recombinant mouse MMP9 (rmMMP9) increased the expression of Ccl2 and Cd74 in ID8 cells (Fig 4F). These results indicated that the MMP9 produced by TAMs expressing full levels of Zeb1, but not from Zeb1 (+/−) TAMs, induces Ccl2 and Cd74 in cancer cells. Altogether, the above data indicate that Zeb1 expression in TAMs drives a CCR2‐MMP9‐CCL2 loop between TAMs and cancer cells.
TAM‐mediated induction of Ccl2 in tumor cells triggers a pro‐tumor phenotype in TAMs
Since tumor cells also modulate TAMs’ phenotype, we examined whether a more or less differentiated phenotype of ID8 cells elicits a different TAM response. ID8 cells were injected into wild‐type and Zeb1 (+/−) mice. Thirteen weeks later, tumor cells were isolated from mice of both genotypes (therefore, ID8RI cells) and re‐injected again but in both cases into wild‐type mice during 6 weeks (see scheme in Fig 4G). In line with the higher tumorigenic capacity of ID8RI cells (Cai et al, 2015), ID8RI cells isolated from wild‐type mice developed similar levels of tumor deposits and ascites after only 6 weeks than those observed upon 13 weeks with parental ID8 cells. Interestingly, the ID8RI cells isolated from Zeb1 (+/−) mice yielded lower ascites and produced fewer tumor deposits than those rendered by the ID8RI cells sorted from wild‐type mice (Fig EV5A and B). These results indicate that the slower tumor progression of ID8RI cells isolated from Zeb1 (+/−) tumor‐bearing mice is transferable into wild‐type mice.
Figure EV5. ID8RI cells from Zeb1 (+/−) mice yield lower ascites and tumor deposits than those rendered by the ID8RI cells from wild‐type mice.
- As in Fig 4G, ID8RI cells isolated after 13 weeks from ID8 tumor‐bearing wild‐type or Zeb1 (+/−) mice were re‐injected into wild‐type mice. ID8RI cells that originated from Zeb1 (+/−) mice induced lower volume of ascites than ID8RI cells isolated from wild‐type mice.
- As in (A), but mice were examined for the formation of tumor deposits on the peritoneal lining. Black arrows mark selected tumor deposits.
TAMs from both experimental conditions in Fig 4G were then characterized by their gene expression profile. ID8RI cells isolated from wild‐type mice, which were less differentiated and expressed higher levels of Ccl2 and Cd74 (Figs 3E and 4B), triggered a stronger macrophage activation toward TAMs (e.g., with higher levels of Ccr2, Il1b, Il10, Cd163, Mrc1, Mmp9, and Cd74) than did the ID8RI cells isolated from Zeb1 (+/−) mice (Fig 4G).
TAM infiltration associates with higher tumor cell resistance to chemotherapy (Mitchem et al, 2013; Mantovani et al, 2017). We found that the ID8RI cells isolated from Zeb1 (+/−) mice displayed higher sensitivity to cisplatin than those sorted out from wild‐type counterparts (Fig 4H). Likewise, incubation of ID8 cells with CM derived from wild‐type TAMs, but not with CM from Zeb1 (+/−) TAMs, rendered cancer cells more resistant to cisplatin (Fig 4I). These results indicate that for TAMs to enhance resistance to cisplatin, they have to express full levels of Zeb1.
ZEB1 expression correlates with TAM infiltration in human ovarian carcinomas where ZEB1 expression in TAMs and ZEB1/CCL2/CD74 in cancer cells determine poorer survival
Expression of ZEB1 and CCL2 in tumor cells determines poorer prognosis in a wide range of carcinomas, including ovarian (Balkwill, 2012; Sánchez‐Tilló et al, 2012). Using survival data for 395 epithelial ovarian carcinoma patients, we found that patients displaying concomitant high expression of both ZEB1 and CCL2 (left panel in Fig 5A) had a lower progression‐free survival rate than those with discordant levels of these genes. In turn, patients with low expression of both ZEB1 and CCL2 displayed the best survival probability. Likewise, concomitant high expression of both ZEB1 and CD74 had a worse prognosis than those with lower levels of ZEB1 and/or CD74 (central panel in Fig 5A). Of note, when combining all three genes, the survival prediction was superior to either of the previous combinations (right panel in Fig 5A). In other words, the maximum effect of ZEB1 as a predictor of reduced survival in ovarian carcinoma requires high levels of CCL2 and CD74, thus suggesting that, at least for some of its tumor‐promoting functions, ZEB1 depends on CCL2 and CD74 expression.
Figure 5. ZEB1 expression correlates with TAM infiltration in human ovarian carcinomas where ZEB1 expression in TAMs and ZEB1/CCL2/CD74 in cancer cells determine poorer survival.
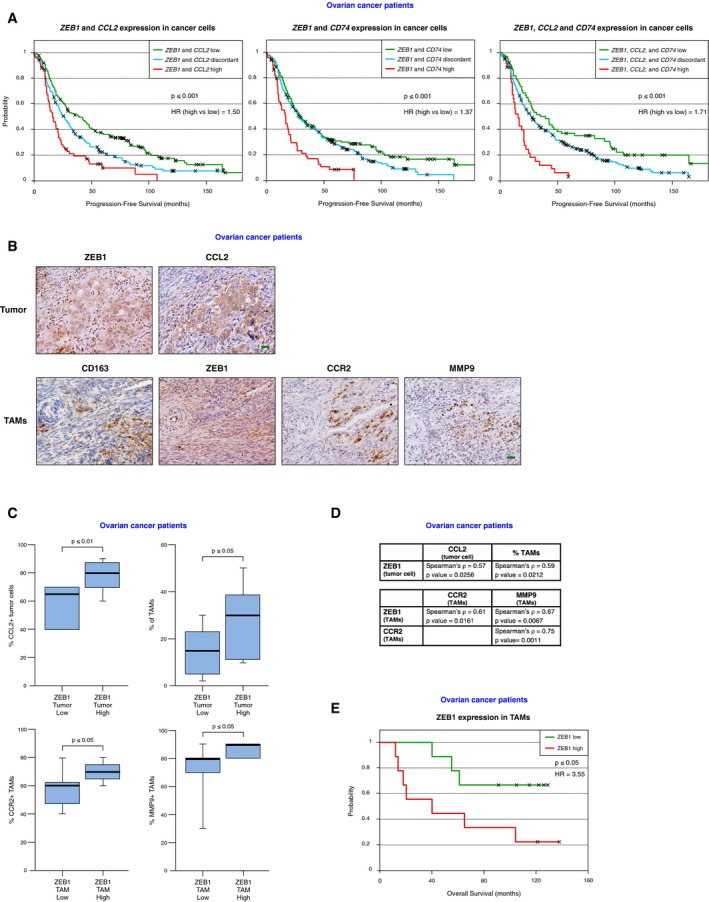
- Ovarian cancer patients with concomitant high expression of ZEB1 and CCL2 and/or CD74 (red line) have shorter progression‐free survival than those that display discordant (blue) or concomitant low (green) expression of these genes. See Materials and Methods for details. HR: hazard ratio.
- Expression of ZEB1 (clone HPA027524), CCL2 (clone 2D8), CD163 (clone 10D6), CCR2 (clone 48607), and MMP9 (clone E11) in human serous ovarian carcinomas. Representative pictures of tumor cells and TAMs are shown. Scale bar: 20 μm.
- High expression of ZEB1 in tumor cells correlates with higher expression of CCL2 in tumor cells and higher infiltration of TAMs. Higher expression of ZEB1 in TAMs correlates with higher expression of CCR2 and MMP9 in TAMs. See Materials and Methods for details. The percentages of tumor cells and TAMs expressing each protein were determined for each patient, and cases were then segregated by their expression of ZEB1—in tumor cells or in TAMs—below or above the median. The box spans the first quartile to the third quartile, the horizontal line corresponds to the median and the whiskers above and below the box show the locations of the minimum and maximum values.
- Correlations between relevant protein expression pairs in tumor cells and/or TAMs in ovarian carcinomas were assessed by a Spearman correlation test (ρ coefficient). Statistical significance was assessed by their P‐value.
- High expression of ZEB1 in TAMs in human serous ovarian carcinomas determined poorer overall survival. Expression of ZEB1 (clone HPA027524) in TAMs (CD163, clone 10D6) was assessed by immunostaining. Overall survival was calculated as described in Materials and Methods. HR: hazard ratio.
In most epithelial tissues, ZEB1 is not present in the normal mucosa but is expressed in most human carcinomas. We investigated ZEB1 expression in TAMs and its association with TAM‐associated markers in human epithelial ovarian carcinomas. A series of serous ovarian carcinomas was stained for ZEB1, CCL2, CD163, CCR2, and MMP9. It was found that infiltration of the tumor by TAMs correlated with higher expression of ZEB1 by tumor cells (Fig 5B–D). Higher expression of ZEB1 in TAMs associated with simultaneous higher expression of CCR2 and MMP9 (Fig 5B–D). Lastly, in ovarian tumor cells, expression of ZEB1 and CCL2 was positively correlated (Fig 5C and D). These data further support that ZEB1 is expressed by both TAMs and tumor cells and that ZEB1 expression in TAMs induces the expression of TAM‐associated markers.
Since the expression of Zeb1 by TAMs in the ID8 mouse model promoted tumor progression, we studied whether ZEB1 expression in TAMs affected the prognosis of ovarian carcinoma patients. Importantly, we found that higher expression of ZEB1 in TAMs was associated with poorer survival in these patients (Fig 5E).
Discussion
ZEB1 expression in cancer cells promotes tumor progression (reviewed in Sánchez‐Tilló et al, 2012; Hill et al, 2013). We show here that ZEB1 is also induced in TAMs where it is required for maintaining a pro‐tumor F4/80low TAM phenotype and for sustaining their tumor‐promoting functions. Mechanistically, ZEB1 activates a CCR2‐MMP9‐CCL2 loop between TAMs and tumor cells to promote tumor growth (see a summary model in Fig 6).
Figure 6. Summary model.
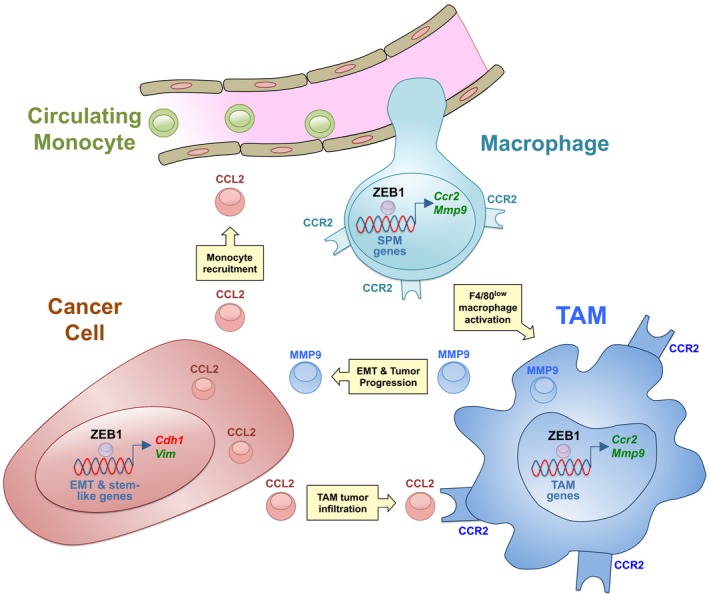
The expression of ZEB1 in TAMs induces tumor progression by the activation of a Ccr2‐Mmp9‐Ccl2 positive loop between TAMs and cancer cells.
ZEB1 is expressed in lymphoid cells where it represses key hematopoietic transcription factors (Genetta et al, 1994; Postigo et al, 1997; Sánchez‐Tilló et al, 2014), but there was no evidence for a role of ZEB1 in the regulation of lymphoid or myeloid differentiation. We showed here that Zeb1 inhibits the differentiation of bone marrow precursors toward F4/80high LPM macrophages. ZEB1 inhibits cell differentiation in other cell types (e.g., epithelial cells, myoblasts, chondroblasts) (e.g., Postigo & Dean, 1997; Aigner et al, 2007; Siles et al, 2013) and plays important roles during embryogenesis—ablation of both alleles of Zeb1 in mice results in embryonic lethality (Takagi et al, 1998)—but the molecular mechanisms involved are still not fully understood.
ZEB1 has been extensively characterized in cancer cells where it promotes their stemness, survival, and invasiveness, thus determining a poorer survival in a wide range of cancers (reviewed in Sánchez‐Tilló et al, 2012; Hill et al, 2013). Within tumors, ZEB1 is restricted to a subpopulation of stem‐like malignant cells at the invasive front (Wellner et al, 2009; Sánchez‐Tilló et al, 2011), where cancer cells interact with their microenvironment. Although ZEB1 expression among stromal cells has been previously noted (e.g., Gemmill et al, 2011; Isella et al, 2015), the identity of the cell types expressing ZEB1, let alone its function in these cells, have not been established. We found here that Zeb1 is expressed in F4/80low TAMs—known to display a stronger pro‐tumor phenotype (Rei et al, 2014)—whose share was expanded by ZEB1. We also showed that ZEB1 in TAMs determined a poorer prognosis in ovarian carcinoma patients. ZEB1 modulated the interplay between cancer cells and TAMs and this crosstalk, in turn, also regulated ZEB1 expression itself. Thus, Zeb1 increased both in macrophages that have interacted with cancer cells and in cancer cells that have interacted with TAMs. The tumor‐promoting functions of ZEB1 are therefore supported by a positive feedback of its expression between TAMs and malignant cells.
The role of the CCR2‐CCL2 signaling axis in tumor progression is well established (Qian et al, 2011; Balkwill, 2012). The data presented here show that Zeb1 is an important inducer of this loop at both sides of the tumor cell–TAM crosstalk. In the ID8 mouse cancer model, Zeb1 directly activated Ccr2 in macrophages and, in turn, only TAMs expressing full levels of Zeb1 induced Ccl2 in cancer cells. In the same line, infiltration of CCR2+ TAMs in human ovarian carcinomas correlated with ZEB1 and CCL2 in cancer cells. Furthermore, concomitant expression of ZEB1 and CCL2 determined poorer prognosis in ovarian carcinoma patients. These results indicate that at least some of ZEB1's pro‐tumor functions require and are mediated by CCR2 in TAMs and CCL2 in cancer cells.
We found that TAMs expressing full levels of Zeb1 induce Ccl2 in cancer cells via upregulation of Mmp9. MMP9 promotes tumor growth in ovarian carcinomas, but the molecular mechanism involved was unclear (Huang et al, 2002). We also showed that Zeb1 expression in TAMs induces Cd74 in cancer cells. Although CD74 is best known as an MHC class II chaperon and a co‐receptor for MIF, recent evidence indicates that CD74 also acts as a transcription factor activating the expression of CCL2 and survival genes (Wilkinson et al, 2015; Gil‐Yarom et al, 2017). We also found that rmMMP9 activated both Cd74 and Ccl2 in cancer cells. Thus, in the model proposed here, MMP9 released by wild‐type TAMs—but not by Zeb1 (+/−) TAMs—activates CCL2 in cancer cells through upregulation of CD74. Concomitant high expression of ZEB1, CCL2, and CD74 increased the survival prediction value of ZEB1 in human ovarian carcinomas. Of note, CCL2 and CD74 signaling induces MMP9 in a myelomonocytic cell line and osteoblasts, respectively (Onodera et al, 2002; Robinson et al, 2002) as we also found here in macrophages (Fig EV4B). Therefore, it appears to exist a self‐reinforcing feedback loop between MMP9 in TAMs and CD74/CCL2 in cancer cells. As there are ongoing clinical trials to inhibit MMP9 and CD74 expression and/or function, our results here offer a potentially new approach to target them.
It is highly relevant to note that while Zeb1 expression in TAMs promotes their acquisition of an F4/80low pro‐tumor phenotype and induces TAMs’ tumor‐promoting roles, these were inhibited by just a partial downregulation of Zeb1 in TAMs. In a mouse model of lung carcinoma, a partial downregulation of ZEB1 in cancer cells—from the deletion of one Zeb1 allele—is sufficient to block the transition from adenoma to adenocarcinoma, indicating that ZEB1's pro‐tumor role in cancer cells depends on a fine threshold of its expression (Liu et al, 2014). Our data here showed that the pro‐tumor function of ZEB1 in TAMs also depends on a similarly narrow threshold of expression. Zeb1 (+/−) TAMs still express about 30–40% of ZEB1 levels of wild‐type counterparts but, importantly, this downregulation was enough to render Zeb1 (+/−) TAMs unable to promote tumor growth.
Expression of ZEB1 in cancer cells determines increased chemotherapy resistance (Sánchez‐Tilló et al, 2014). Here, we found that expression of ZEB1 in TAMs also increased cancer cell resistance to chemotherapy. In that line, Zeb1 in TAMs increased the expression of Il1b, Il10, Mmp9, and Mdr1 in TAM themselves and/or in cancer cells (Figs 2B and 3E), genes that have been shown to have suppressor effects on chemotherapy response (Mitchem et al, 2013; Ostuni et al, 2015). The role of ZEB1 promoting tumor progression, chemotherapy resistance, and poorer survival through its dual expression in cancer cells and TAMs—albeit acting through different mechanisms—has important translational implications. Targeting ZEB1 in cancer cells is being considered in clinical trials, but data here suggest that inhibiting tumor growth and improving chemotherapy response would also require targeting of ZEB1 in TAMs. The fact that just a partial downregulation of ZEB1 in TAMs was sufficient to abolish TAMs pro‐tumor roles is also important in therapy approaches aiming at blocking ZEB1 expression and/or function.
In sum, this study has uncovered a new role for ZEB1 as a key factor in the activation of a TAM pro‐tumor phenotype and in the maintenance of TAMs’ tumor‐promoting functions.
Materials and Methods
Mouse models and human samples
The use of mice in this work—CL57BL/6 wild‐type and Zeb1 (+/−) mice (Takagi et al, 1998)—followed the guidelines established by the Animal Experimental Committee of the University of Barcelona School of Medicine (Barcelona, Spain) and was approved as protocol 535/16. Human samples in this work were obtained from the Hospital Clínico San Carlos (Madrid, Spain), and their use was approved by the local Clinical Ethics Research Committees, had the informed consent of patients, and conformed with the principles of the Helsinki Declaration.
Antibodies
The primary antibodies (conjugated and unconjugated) used in this article originated as follows: anti‐mouse F4/80 conjugated to APC (clone BM8, reference 17‐4801; Bioscience Inc., San Diego, CA, USA), anti‐mouse CD11b conjugated to PE (clone M1/70.15, reference 22159114; Immuno Tools GmbH, Friesoythe, Germany), anti‐mouse CD45 conjugated to PerCP/Cy5.5 (clone 30‐F11, reference 103131; BioLegend, San Diego, CA, USA), anti‐human/mouse ZEB1 (HPA027524, Sigma‐Aldrich, St. Louis, MO, USA; and E‐20, sc‐10572, Santa Cruz Biotechnologies, Dallas, TX, USA), anti‐human CCL2 (clone 2D8, Invitrogen, Thermo Fisher, Carlsbad, CA, USA), anti‐human CD163 (clone 10D6, reference PA0090, Leica Biosystems, Newcastle Upon Tyne, UK), anti‐human CCR2 (Clone 48607, R&D Systems, Minneapolis, MN, USA), anti‐human MMP9 (clone E‐11, sc‐393859, Santa Cruz Biotechnologies), and anti‐human/mouse α‐tubulin (clone B‐5‐1‐2, Sigma‐Aldrich). The secondary antibodies used in this article originated as follows: Peroxidase‐AffiniPure donkey anti‐mouse IgG (H+L) (reference 715‐035‐151), peroxidase‐AffiniPure goat anti‐rabbit IgG (H+L) (reference 111‐035‐144), and mouse gamma globulin (reference 015‐000‐002) were purchased from Jackson ImmunoResearch Europe (Newmarket, United Kingdom).
Isolation and culture of mouse primary cells
Bone marrow total cells (BMTCs) were obtained from 6‐ to 8‐week‐old CL57BL/6 wild‐type and Zeb1 (+/−) mice and were differentiated into macrophages as described (Zhang et al, 2008). Briefly, femur and tibia bone marrows were flushed with PBS and BMTCs collected were centrifuged and resuspended in Dulbecco's modified Eagle medium (DMEM) (Lonza, Basel, Switzerland) supplemented with 10% FBS (Sigma‐Aldrich) and 1% penicillin–streptomycin (Pen/Strep) (Lonza), medium hereafter referred as complete medium. To generate bone marrow‐derived macrophages (BMDM), BMTCs were cultivated with 20 ng/ml of recombinant mouse CSF1 (Immuno Tools GmbH, Friesoythe, Germany) during 6 days. Every 2 days, half of the medium was replaced with fresh medium supplemented with CSF1. Identical results were obtained when BMTCs were cultivated with 40% of conditioned medium (CM) from L929 culture cells (ATCC, Manassas, VA, USA) stably transfected with CSF1 expression vectors. Peritoneal macrophages were isolated from 6‐ to 8‐week‐old wild‐type and Zeb1 (+/−) mice as per standard protocols (Zhang et al, 2008). Briefly, mice were euthanized and the peritoneal cavity was washed twice with 6 ml of ice‐cold PBS supplemented with 3% FBS. Cells from the peritoneal lavage were centrifuged at 400 × g for 10 min at 4°C, and erythrocytes were osmotically lysed with red blood cell lysis buffer (Sigma‐Aldrich), followed by a wash with PBS and resuspension in PBS or complete medium. Peritoneal cells were then sorted by flow cytometry for F4/80 and CD11b cell surface expression (see below), and isolated cells were then processed for analysis of mRNA or cell surface markers expression or tested in functional assays. To generate CM from peritoneal macrophages and TAMs, both cell types were sorted by flow cytometry and 5 × 105 cells cultured on ultra‐low‐attachment 6‐well plates (Costar, Corning) during 24 h in 2 ml of DMEM supplemented with 2% FBS. The CM was then collected and dialyzed overnight against PBS. Wherever indicated (see below in Culture of ID8 cells), the CM was concentrated in a high retention dialysis tubing (Sigma‐Aldrich) with poly‐ethylene glycol 20,000 MW (Thermo Fisher Scientific, Waltham, MA, USA) as per standard protocols. For RNA interference of mouse macrophages, these were transfected with 200 nM of either siRNA control or a siRNA against mouse Zeb1 with Lipofectamine® RNAimax (Thermo Fisher, Waltham, MA, USA) as per manufacturer's instructions. siRNA oligonucleotide duplexes were purchased from Sigma‐Aldrich, and their sequences are detailed in Appendix Table S3.
Cell surface protein expression analysis and cell sorting by flow cytometry (FACS)
Cells were first blocked for Fc receptors with mouse gamma globulin (Jackson ImmunoResearch Europe). Cells were then incubated in PBS with 2% FBS during 45 min at 4°C with the corresponding fluorochrome‐labeled antibodies. Expression of cell surface proteins was assessed in a BD FACSCanto™ II analyzer (BD Biosciences, San Jose, CA, USA). Wherever indicated, cells were sorted for specific subpopulations in a FACS Aria™ II cell sorter (BD Biosciences) for subsequent experimentation. The acquired data were analyzed using FlowJo for Windows, version 7.6.1 (FlowJo®, Ashland, OR, USA).
Protein expression analysis by Western blot
Peritoneal macrophages were plated on 12‐well plates during 30 min, washed with ice‐cold PBS, and directly resuspended in RIPA lysis buffer (150 mM NaCl, 50 mM Tris pH 8, 1% NP‐40, 0.5% SDS, 2 mM EDTA) containing protease inhibitors as previously described (Sánchez‐Tilló et al, 2011). Lysates were clarified several times by centrifugation and quantified by Bradford assay. Lysates were then boiled and loaded onto 10% polyacrylamide gels and transferred to a PVDF membrane (Immobilon‐P, Millipore, Bedford, MA, USA). Membranes were blocked with 5% non‐fat milk and blotted with ZEB1 (HPA027524, 1:300 dilution) or α‐tubulin (B‐5‐1‐2, 1:400 dilution) antibodies for 1 h at 37°C. After washing with TBS‐T (137 mM NaCl, 20 mM Tris, 0.1% Tween‐20, pH 7.6), membranes were incubated for 2 h at room temperature with anti‐rabbit (1:10,000) and anti‐mouse (1:5,000) HRP‐conjugated secondary antibodies, respectively. The chemiluminescent reaction was detected with Clarity™ ECL Western Blotting Substrate (Bio‐Rad, Hercules, CA, USA). Figure EV1C is representative of at least three independent experiments. Full unedited gel images are shown as a supplemental Source Data File with the area displayed in Fig EV1C boxed. Quantification of ZEB1 expression with respect to α‐tubulin in three independent Western blots was assessed with ImageJ software (https://imagej.nih.gov/) (NIH, Bethesda, MD, USA).
In vitro macrophage migration
In vitro migration of macrophages was assessed through two approaches: first, using a wound‐healing assay. Briefly, 1 × 106 macrophages in complete medium were seeded in 6‐well plates and incubated overnight. A defined wound field was then created with a pipette tip, and cell migration across the gap was monitored by light microscopy (Olympus BX41, Olympus, Hicksville, NY, USA) for up to 24 h. Second, macrophage migration was also examined in a Transwell® migration/chemotaxis assay. Briefly, 0.5 × 106 peritoneal macrophages were labeled with 5 μM CFSE. After washing once with complete medium and thrice with PBS, cells were resuspended in 200 μl of DMEM supplemented with 2.5% FBS and added on top of 6.5‐mm‐diameter/8‐μm pore polycarbonate Transwell® inserts (Corning Inc., Corning, NY, USA), which in turn were placed over a 24‐well plate. The lower chamber was filled with 0.6 ml of complete medium containing 80 ng/ml recombinant mouse CCL2 (rmCCL2) (Immuno Tools GmbH). After 2 hrs, the medium in the lower chamber was collected and macrophage migration was assessed by the CFSE fluorescence signal measured in a Modulus II GloMax®‐Multi‐Detection System microplate reader (Promega Corp., Madison, WI, USA). Macrophage migration was then expressed as relative fluorescence units (RFU) with respect to a 100% value represented by the fluorescence of the cell suspension initially loaded on top of the Transwell® insert. RFU values are expressed as the mean with their standard errors of five mice for each genotype.
In vivo myeloid cell differentiation
In vivo monocyte maturation was carried out as described (Tsou et al, 2007). Briefly, bone marrow total cells (BMTCs) from wild‐type and Zeb1 (+/−) mice were first labeled with CFSE (Sigma‐Aldrich). 2–3 × 107 CSFE‐labeled BMTCs from both genotypes were then inoculated i.v. into wild‐type recipients. For in vivo mobilization and differentiation of myeloid precursors into macrophages, either 1 ml of conditioned medium from L929/M‐CSF cells or 1 μg of rmCCL2 (Peprotech) was inoculated i.p. into 6‐ to 8‐week‐old wild‐type and Zeb1 (+/−) mice. The mobilization of monocytes into the peritoneal cavity and their maturation into macrophages were followed up to 7 h by FACS analysis.
RNA extraction and quantitative real‐time PCR
Total RNA was extracted with RNAzol® RT reagent (Sigma‐Aldrich) or TRIzol® (Life Technologies, Thermo Fisher Scientific, Carlsbad, CA, USA) and reverse‐transcribed with oligo‐dT using High‐Capacity cDNA Reverse Transcription Kit (Life Technologies). mRNA levels were then determined by quantitative real‐time PCR (qRT–PCR) at 60°C using GoTaq® qPCR Master Sybr Green Mix (Promega). Results were analyzed using Opticon Monitor 3.1.32 software (Bio‐Rad) by ΔΔCt method and normalizing values with respect to mouse Gapdh. qRT–PCR data shown are the average of a minimum of three mice of each genotype run in triplicate. DNA primers used in qRT–PCR were purchased from Sigma‐Aldrich, and their sequences are described in Appendix Table S4. The nomenclature for mouse genes adheres to MGI (Mouse Genome Informatics, http://www.informatics.jax.org/).
Chromatin immunoprecipitation assays
Chromatin immunoprecipitation (ChIP) assays were performed using EpiQuick ChIP kit (Epigentek, Farmingdale, NY, USA) as per manufacturer's instructions. Briefly, 1.5 × 107 bone marrow‐derived macrophages were incubated for 10 min with 1% formaldehyde solution (Electron Microscopy Sciences, Hatfield, PA, USA) at room temperature followed by incubation with 1.25 mM glycine. Lysates were sonicated as described elsewhere (Sánchez‐Tilló et al, 2011). Goat anti‐ZEB1 (E‐20) and its corresponding normal goat IgG (Jackson ImmunoResearch) were used. Identification of DNA binding sequences for ZEB1 and design of primers for qRT–PCR amplification were conducted using MacVector software (MacVector Inc, Apex, NC, USA). DNA fragments were quantified by qRT–PCR as detailed above using the primers detailed in Appendix Table S5. In all qRT–PCRs, values shown represent relative binding in relation to input and are the average of at least three independent ChIP experiments, each one performed in triplicate.
RNA sequencing and data analysis
Peritoneal macrophages (CD45+, CD11b+, F4/80+) from 6‐ to 8‐week‐old wild‐type and Zeb1 (+/−) female mice—6 for each genotype—were isolated and their RNA was extracted using RNAzol® RT reagent (Sigma‐Aldrich). RNA was quantified and its quality (RNA integrity numbers ≥ 8.5) assessed on an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA). Part of the RNA samples was reverse‐transcribed as described above to examine Zeb1 expression. To obtain at least 1 mg of RNA required for the preparation of libraries in triplicate, two samples from each genotype were pooled. The libraries from the mouse total RNA were prepared using the TruSeq® Stranded mRNA LT Sample Prep Kit (Rev.E, October 2013) (Illumina Inc., San Diego, CA, USA) according to the manufacturer's protocol. Briefly, 0.5 μg of total RNA was used for poly‐A‐based mRNA enrichment with oligo‐dT magnetic beads. The mRNA was fragmented (fragment size: 80–250 nt, with the major peak at 130 nt) and the first‐strand cDNA synthesis was conducted by random hexamers and reverse transcriptase. The second‐strand cDNA synthesis was performed in the presence of dUTP instead of dTTP, to achieve strand specificity. The blunt‐ended double‐stranded cDNA was 3′adenylated and Illumina indexed adapters were ligated. The ligation product was then enriched with 15 PCR cycles, and the final library was validated on an Agilent 2100 Bioanalyzer with the DNA 7500 assay. Libraries were sequenced on HiSeq 2000 (Illumina, Inc) in a paired‐end mode with a read length of 2 × 76 bp using TruSeq SBS Kit v3‐HS. Over 20 million paired‐end reads were generated for each sample in a fraction of a sequencing flow cell lane, following the manufacturer's protocol. Image analysis, base calling, and quality scoring of the run were processed using the manufacturer's software Real Time Analysis (RTA 1.13.48) and followed by generation of FASTQ sequence files by Illumina's CASAVA. Reads were mapped against the mouse reference genome (GRCm38) with STAR (Dobin et al, 2013) using the ENCODE parameters for long RNA. Gene quantifications were performed with RSEM (Li & Dewey, 2011) using default options and mouse GENCODE annotation version 11. Normalization and differential expression analyses were done with edgeR software (Bioconductor) (Robinson et al, 2010) using default options. GO and KEGG enrichment analyses were performed with the beta version of DAVID database (http://david.ncifcrf.gov/). The RNA‐seq data have been submitted to the GEO database and assigned accession number GSE103830.
Culture of ID8 cells with conditioned medium, soluble factors, inhibitors, and chemotherapy drugs
The C57BL/6 mouse ovarian carcinoma cell line ID8 was obtained from K. Roby, (University of Kansas, Kansas City, KS, USA) (Roby et al, 2000) and cultured in complete medium. ID8‐luc cells, harboring the luciferase gene, have been previously described (Hagemann et al, 2008). In selected experiments, 0.5 × 106 ID8‐luc cells were plated on 6‐well plates with 2 ml of complete medium and supplemented with 100–200 μl of concentrated CM from peritoneal macrophages or TAMs during 16–24 h. Inhibition of MMP9 in the CM from TAMs was conducted as follows. First, the CM from 5 × 105 TAMs isolated from 13‐week ID8 tumor‐bearing wild‐type or Zeb1 (+/−) mice was collected and concentrated as detailed above. Then, 30 μl of concentrated CM in 1 ml of complete medium incubated for 45 min with 20 μM of MMP9 inhibitor dissolved in DMSO (MMP9 PEX inhibitor 444293, Calbiochem, Millipore, USA) or the corresponding volume of DMSO (1 μl of MMP9 inhibitor or DMSO per 1 ml of diluted CM). Lastly, 0.3 × 106 ID8‐luc cells were plated on 12‐well plates and incubated for 16 h with 1 ml diluted CM (with or without MMP9 inhibitor) before cells were processed for gene expression by qRT–PCR. In selected experiments using rmMMP9, 0.3 × 106 ID8‐luc cells were plated on 12‐well plates containing 1 ml complete medium supplemented with 1 mg/ml of rmMMP9 (590502, BioLegend) during 36 h. Co‐culture of ID8‐luc cells with CM from TAMs where CCR2 signaling was blocked was carried out as follows. First, six wild‐type and six Zeb1 (+/−) mice were inoculated i.p. with 5 × 106 ID8RI cells previously grown in wild‐type mice. After 72 h, mice for each genotype were divided into two cohorts and injected i.p. every 12 h during 3 days with either PBS or 2.5 mg/kg of a small molecule CCR2 inhibitor (RS 504393, Tocris‐Biotechne, Bristol, UK) before being euthanized. The CM from TAMs for each mice and cohort was collected as detailed above. The CM was diluted twice in complete medium and 1 ml of the diluted CM was added onto ID8‐luc cells for an additional 72 h before being assessed for Ccl2 expression by qRT–PCR. The effect of cisplatin on ID8 cells was examined through two different approaches. First, 5 × 106 ID8‐luc were injected in wild‐type and Zeb1 (+/−) mice and allowed to grow in these mice for 8 weeks. At this point, ID8RI cells were sorted out by FACS and 5 × 104 ID8RI cells were plated onto 96‐well plates and incubated for 24 h in the absence (only complete medium) or presence of 50 μg/ml of cisplatin (Pharmacia Nostrum, Madrid, Spain). In the second set of experiments, 5 × 106 ID8‐luc were injected in wild‐type and Zeb1 (+/−) mice and TAMs were then sorted out 96 h after. TAMs were then cultured for 24 h as described above to generate CM. 5 × 104 ID8‐luc cells were then plated on 96‐well plate and cultured for 24 h in the absence (only complete medium) or presence of 50 μg/ml of cisplatin and the CM from wild‐type and Zeb1 (+/−) TAMs. In both cases, the response of ID8 cancer cells to cisplatin was examined for cell viability in an MTT assay. Briefly, 15 μl of a 5 mg/ml MTT solution (Sigma‐Aldrich) was added to each well for 3 h at 37°C. Then, 100 μl of DMSO was added and incubated for 15 min under dark and the absorbance at 590 nm was read with a reference filter 750 in Glomax microplate reader (Promega).
ID8 ovarian cancer model and bioluminescence imaging
5 × 106 ID8 cells (either ID8‐luc or ID8RI) were resuspended in 500 μl of PBS and injected i.p. into 8‐week‐old wild‐type and Zeb1 (+/−) female mice. At the indicated periods, mice were euthanized and ID8 cells and macrophages were sorted and processed for additional analyses. At the end of each protocol, the abdominal perimeter, ascites volume, and tumor deposits on the peritoneal lining were assessed. In selected experiments, tumor progression was followed by bioluminescent imaging (Evans et al, 2014). Briefly, mice were injected i.p. with 1.5 mM of CycLuc1 substrate (Calbiochem®, EMD Millipore, Billerica, MA, USA) in 100 μl of PBS. Ten min later, mice were anesthetized with 2.5% isoflurane and the photon flux signal was collected in a charge‐coupled ORCA‐2BT imaging system (Hamamatsu Photonics, Hamamatsu City, Japan). Bioluminescence data were analyzed with Wasabi! Imaging Software (Hamamatsu Photonics) and represented as the total photon flux/sec/cm2 signal emitted from the abdominal cavity. In experiments with ID8RI cells, ID8‐luc cells that had been previously injected in wild‐type or Zeb1 (+/−) mice were isolated from the ascites after 13 weeks and re‐injected into new recipients. As ID8RI cells accelerate tumor progression (Cai et al, 2015), mice were euthanized 6 weeks post‐inoculation and peritoneal cells were isolated for subsequent analyses. For macrophage depletion in ID8 tumor‐bearing mice, 6‐ to 8‐week‐old wild‐type and Zeb1 (+/−) female mice were inoculated with 5 × 106 ID8‐luc cells. Four days later, they were injected i.p. with 200 μl of liposomes containing either PBS (PBS liposomes, PBS‐L) or clodronate (clodronate liposomes, CL‐L) (ClodronateLiposomes.org, Haarlem, the Netherlands). Liposome injections were repeated every 4 days for 6 weeks at which point mice were euthanized and peritoneal cells were isolated for subsequent analyses. Adoptive transfer of macrophages into ID8 tumor‐bearing mice was performed as in Hagemann et al (2008). Female wild‐type mice were injected with 1 × 106 ID8RI cells that had been previously inoculated in wild‐type female mice during 13 weeks. Twenty‐three days after re‐injection of ID8RI cells, mice were inoculated with PBS or 3–4 × 106 bone marrow‐derived macrophages from either wild‐type or Zeb1 (+/−) mice. Tumor progression was monitored over time by bioluminescence imaging. Mice were euthanized 7 days after macrophage inoculation, and peritoneal cells were isolated for subsequent analyses.
Survival plots of ovarian carcinoma patients
Correlation between the expression of ZEB1/CCL2, ZEB1/CD74, and ZEB1/CCL2/CD74 and progression‐free survival was examined in published array databases of ovarian carcinoma. Only datasets publishing survival data with a mean follow‐up of at least 2 years and using the Affymetrix Human Genome chips were considered to avoid platform differences when using different gene arrays. The following probes were used: 212758_s_at for ZEB1, 216598_s_at for CCL2, and 209619_at for CD74. Four datasets were included in the study, namely GSE15622 (Ahmed et al, 2007), GSE26193 (Mateescu et al, 2011), GSE30161 (Ferriss et al, 2012), and GSE26712 (Bonome et al, 2008). Datasets were processed separately and assembled into a single final database. Cox proportional hazard regression analysis was calculated and a univariate Kaplan–Meier analysis was calculated for each gene separately as described (Mihály et al, 2013). Then, genes were combined to have three cohorts: samples with a concordant low expression of the two genes, samples with a discordant expression of the two genes (e.g., one high and the other low), and those where both genes have concordant high expression. The final Kaplan–Meier plots display the survival using these three cohorts. Correlation between ZEB1 expression in TAMs and overall survival in ovarian carcinomas patients was assessed as follows. A series of 18 cases of human serous ovarian carcinomas grade III and IV that had undergone complete surgery was stained for ZEB1 (clone HPA027524) and CD163 (clone 10D6) (see below for the immunochemistry protocol). ZEB1 expression in TAMs was then correlated with overall survival probability with a Cox proportional hazards regression and a univariate Kaplan–Meier analysis using WinStat software v.2016 (R. Fitch Software, Bad Krozingen, Germany).
Immunohistochemistry of human ovarian carcinomas
Immunohistochemistry of formalin‐fixed, paraffin‐embedded human samples of a series of 35 cases of human serous epithelial ovarian carcinomas was carried out as follows. Slides were deparaffinized and rehydrated before being subjected to antigen retrieval with 10 mM sodium citrate pH 6.0 for 5 min. Slides were then incubated with a non‐specific binding blocking solution (5% donkey/goat normal serum plus 4% BSA in PBS, 0.5% Tween‐20) followed by the corresponding primary and HRP‐conjugated secondary antibodies. Staining was performed manually except for CD163 that was performed automatically in a Bond‐Max automatic stainer (Leica Biosystems). The primary antibodies were used as follows: ZEB1: 1/250 dilution overnight at 4°C; CCL2: 1/50 dilution overnight at 4°C; CCR2: 1/100 dilution overnight at 37°C; MMP9: 1/50 dilution overnight at room temperature; and CD163: 1/3,000 dilution 30 min at room temperature. Secondary antibodies were incubated at 1/100 during 1 h at 37°C, except for ZEB1 (1/200, 1 h at 37°C) and CD163 (1/200, 30–60 min at room temperature). For CD163, automated staining was conducted as follows. Antigen retrieval was carried out with low pH Bond ER2 Buffer Solution® (Vision Biosystems, Leica, Wetzlar, Germany) for 30 min, followed by incubations with CD163 antibody (clone 10D6) and Bond Refine Polymer (Vision Biosystems, Leica) (20 min, room temperature). The immunohistochemistry reaction was developed with a DAB substrate kit (Vector Labs, Burlingame, CA, USA) before slides were counterstained with hematoxylin and mounted in Di‐N‐butylphthalate in xylene solution (DPX, Sigma‐Aldrich). The number of positive cells was scored by microscopic analysis and statistical analyses were performed as follows. After the percentage of tumor cells and TAMs expressing each protein had been determined, cases were segregated by their expression of ZEB1—in tumor cells or in TAMs—below or above the median. The means for CCL2 expression in tumor cells, the percentage of TAM infiltration—determined by CD163 expression—and the expression of CCR2 and MMP9 in TAMs were calculated for the low and high ZEB1 cohorts.
Statistical analysis
Statistical analysis of data shown was performed using Prism for Mac version 5.0a (GraphPad Software Inc., La Jolla, CA). Statistical significance was assessed with a non‐parametric Mann–Whitney U‐test except in Kaplan–Meier survival plots (see above). In histograms, deviations represent standard errors of means. In the immunohistochemistry of human ovarian carcinomas, correlations between relevant protein expression pairs in tumor cells and/or TAMs were assessed by a Spearman's correlation coefficient (ρ). In all Figures and Expanded View (EV) Figures, relevant comparisons were labeled as either significant at the P ≤ 0.001 (***), P ≤ 0.01 (**), or P ≤ 0.05 (*) levels, or non‐significant (ns) for values of P > 0.05.
Author contributions
MC performed most of the experimental work in the study. LS‐M and OdB carried out the immunohistochemistry and ChIP experiments, respectively. MCMC assisted in some experiments. MJF‐A facilitated the collection of human samples and conducted the pathological analysis of their immunostaining. AE‐C was responsible for the bioinformatics analysis of RNA‐seq data. BG conducted the bioinformatics analysis of arrays for survival and gene expression. DSD, DCD, and TL provided important materials and insights to the study. MC and AP designed and interpreted experiments. AP conceived and supervised the study and wrote the manuscript. All authors critically reviewed the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Acknowledgements
We regret that due to space limitations some articles were only cited indirectly through reviews. We greatly appreciate insightful comments to an earlier version of the manuscript by Dr. A Corbí (CSIC‐CIB, Madrid, Spain) and help with pathological analysis by Dr. M Cuatrecasas (Hospital Clínic, Barcelona, Spain). We are also grateful to the FACS Unit at IDIBAPS for technical help. The IDIBAPS Institute is partly funded by the CERCA Program of the Government of Catalonia. The different parts of this work were independently funded by grants to the corresponding author (AP) from Ministry of Economy and Competitiveness (MINECO, SAF2014‐52874‐R, 2013‐2016 National Scientific and Technical Research and Innovation Plan, co‐financed by the European Union's Regional Development Fund), Fundació La Marató de TV3 (201330.10), Catalan Agency for Management of University and Research Grants (AGAUR, 2014‐SGR‐475), Leukemia Research Foundation (Hollis Brownstein Grants 2014), and Avon Breast Cancer Campaign (ACSAU). MC was funded by a PhD scholarship from the Chilean Ministry of Education (Comisión Nacional de Investigación Científica y Tecnológica, CONICYT) and from grants to AP (SAF2014‐52874‐R and 201330.10). LSM is recipient of a PhD scholarship from the Spanish Ministry of Education, Culture and Sports (FPU Program, FPU14/06217). OdB was subsequently supported by an FPU Program PhD scholarship (AP2010‐5489) and from grants to AP (2014‐SGR‐475 and 201330.10). MCMC is recipient of a predoctoral of a PhD scholarship from the Spanish Ministry of Economy and Competitiveness (FPI Program, BES‐2015‐075757).
The EMBO Journal (2017) 36: 3336–3355
References
- Ahmed AA, Mills AD, Ibrahim AE, Temple J, Blenkiron C, Vias M, Massie CE, Iyer NG, McGeoch A, Crawford R, Nicke B, Downward J, Swanton C, Bell SD, Earl HM, Laskey RA, Caldas C, Brenton JD (2007) The extracellular matrix protein TGFBI induces microtubule stabilization and sensitizes ovarian cancers to paclitaxel. Cancer Cell 12: 514–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aigner K, Dampier B, Descovich L, Mikula M, Sultan A, Schreiber M, Mikulits W, Brabletz T, Strand D, Obrist P, Sommergruber W, Schweifer N, Wernitznig A, Beug H, Foisner R, Eger A (2007) The transcription factor ZEB1 (δEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 26: 6979–6988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill FR (2012) The chemokine system and cancer. J Pathol 226: 148–157 [DOI] [PubMed] [Google Scholar]
- Biswas SK, Gangi L, Paul S, Schioppa T, Saccani A, Sironi M, Bottazzi B, Doni A, Vincenzo B, Pasqualini F, Vago L, Nebuloni M, Mantovani A, Sica A (2006) A distinct and unique transcriptional program expressed by tumor‐associated macrophages (defective NF‐κB and enhanced IRF‐3/STAT1 activation). Blood 107: 2112–2122 [DOI] [PubMed] [Google Scholar]
- Bonome T, Levine DA, Shih J, Randonovich M, Pise‐Masison CA, Bogomolniy F, Ozbun L, Brady J, Barrett JC, Boyd J, Birrer MJ (2008) A gene signature predicting for survival in suboptimally debulked patients with ovarian cancer. Cancer Res 68: 5478–5486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Yan L, Xu Y (2015) Anoikis resistance is a critical feature of highly aggressive ovarian cancer cells. Oncogene 34: 3315–3324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain DW, O'Koren EG, Kan MJ, Womble M, Sempowski GD, Hopper K, Gunn MD, Kelsoe G (2013) Identification of a tissue‐specific, C/EBPβ‐dependent pathway of differentiation for murine peritoneal macrophages. J Immunol 191: 4665–4675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau WK, Ip CK, Mak AS, Lai HC, Wong AS (2011) c‐Kit mediates chemoresistance and tumor‐initiating capacity of ovarian cancer cells through activation of Wnt/β‐catenin–ATP‐binding cassette G2 signaling. Oncogene 32: 2767–2781 [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis C, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MS, Chaurette JP, Adams ST Jr, Reddy GR, Paley MA, Aronin N, Prescher JA, Miller SC (2014) A synthetic luciferin improves bioluminescence imaging in live mice. Nat Methods 11: 393–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferriss JS, Kim Y, Duska L, Birrer M, Levine DA, Moskaluk C, Theodorescu D, Lee JK (2012) Multi‐gene expression predictors of single drug responses to adjuvant chemotherapy in ovarian carcinoma: predicting platinum resistance. PLoS ONE 7: e30550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RA, Li MO (2016) Ontogeny of tumor‐associated macrophages and its implication in cancer regulation. Trends Cancer 2: 20–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemmill RM, Roche J, Potiron VA, Nasarre P, Mitas M, Coldren CD, Helfrich BA, Garrett‐Mayer E, Bunn PA, Drabkin HA (2011) ZEB1‐responsive genes in non‐small cell lung cancer. Cancer Lett 300: 66–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genetta T, Ruezinsky D, Kadesch T (1994) Displacement of an E‐box‐binding repressor by basic helix‐loop‐helix proteins: implications for B‐cell specificity of the immunoglobulin heavy‐chain enhancer. Mol Cell Biol 14: 6153–6163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goshn EE, Cassado AA, Govoni GR, Fukuhara T, Yang Y, Monack DM, Bortoluci KR, Almeida SR, Herzenberg LA, Herzenberg LA (2010) Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proc Acad Natl Sci USA 107: 2568–2573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil‐Yarom N, Radomir L, Sever L, Kramer MP, Lewinsky H, Bornstein C, Blecher‐Gonen R, Barnett‐Itzhaki Z, Mirkin V, Friedlander G, Shvidel L, Herishanud Y, Lolise EJ, Becker‐Hermana S, Amita I, Shachara I (2017) CD74 is a novel transcription regulator. Proc Natl Acad Sci USA 114: 562–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Hart E, Shchurin A, Hoover‐Plow J (2008) Inflammatory macrophage migration requires MMP‐9 activation by plasminogen in mice. J Clin Invest 118: 3012–3024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman MM, Fojo T, Bates SE (2002) Multidrug resistance in cancer: role of ATP–dependent transporters. Nat Rev Cancer 2: 48–58 [DOI] [PubMed] [Google Scholar]
- Hagemann T, Wilson J, Burke F, Kulbe H, Li NF, Plüddemann A, Charles K, Gordon S, Balkwill FR (2006) Ovarian cancer cells polarize macrophages toward a tumor‐associated phenotype. J Immunol 176: 5023–5032 [DOI] [PubMed] [Google Scholar]
- Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, Robinson SC, Balkwill FR (2008) “Re‐educating” tumor‐associated macrophages by targeting NF‐κB. J Exp Med 205: 1261–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill L, Browne G, Tulchinsky E (2013) ZEB/miR‐200 feedback loop: at the crossroads of signal transduction in cancer. Int J Cancer 132: 745–754 [DOI] [PubMed] [Google Scholar]
- Huang S, Van Arsdall M, Tedjarati S, McCarty M, Wu W, Langley R, Fidler IJ (2002) Contributions of stromal metalloproteinase‐9 to angiogenesis and growth of human ovarian carcinoma in mice. J Natl Cancer Inst 94: 1134–1142 [DOI] [PubMed] [Google Scholar]
- Isella C, Terrasi A, Bellomo SE, Petti C, Galatola G, Muratore A, Mellano A, Senetta R, Cassenti A, Sonetto C, Inghirami G, Trusolino L, Fekete Z, De Ridder M, Cassoni P, Storme G, Bertotti A, Medico E (2015) Stromal contribution to the colorectal cancer transcriptome. Nat Genet 47: 312–319 [DOI] [PubMed] [Google Scholar]
- Lawrence T, Natoli G (2011) Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol 11: 750–761 [DOI] [PubMed] [Google Scholar]
- Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA‐Seq data with or without a reference genome. BMC Bioinformatics 12: 323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Lu X, Huang L, Wang W, Jiang G, Dean KC, Clem B, Telang S, Jenson AB, Cuatrecasas M, Chesney J, Darling DS, Postigo A, Dean DC (2014) Different thresholds of ZEB1 are required for Ras‐mediated tumour initiation and metastasis. Nat Commun 5: 5660 [DOI] [PubMed] [Google Scholar]
- Lu H, Clauser KR, Tam WL, Fröse J, Ye X, Eaton EN, Reinhardt F, Donnenberg VS, Bhargava R, Carr SA, Weinberg RA (2014) A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat Cell Biol 16: 1105–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P (2017) Tumour‐associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol 14: 399–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateescu B, Batista L, Cardon M, Gruosso T, de Feraudy Y, Mariani O, Nicolas A, Meyniel JP, Cottu P, Sastre‐Garau X, Mechta‐Grigoriou F (2011) miR‐141 and miR‐200a act on ovarian tumorigenesis by controlling oxidative stress response. Nat Med 17: 1627–1635 [DOI] [PubMed] [Google Scholar]
- Mihály Z, Kormos M, Lánczky A, Dank M, Budczies J, Szász MA, Győrffy B (2013) A meta‐analysis of gene expression‐based biomarkers predicting outcome after tamoxifen treatment in breast cancer. Breast Cancer Res Treat 140: 219–232 [DOI] [PubMed] [Google Scholar]
- Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica‐Worms D, Hewitt S, Udupi GM, Gallagher WM, Wegner C, West BL, Wang‐Gillam A, Goedegebuure P, Linehan DC, DeNardo DG (2013) Targeting tumor‐infiltrating macrophages decreases tumor‐initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res 73: 1128–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA, Huang RYJ, Jackson RA, Thiery JP (2016) EMT:2016. Cell 166: 21–45 [DOI] [PubMed] [Google Scholar]
- Okabe Y, Medzhitov R (2014) Tissue‐specific signals control reversible program of localization and functional polarization of macrophages. Cell 157: 832–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onodera S, Nishihira J, Iwabuchi K, Koyama Y, Yoshida K, Tanaka S, Minami A (2002) Macrophage migration inhibitory factor up‐regulates matrix metalloproteinase‐9 and‐13 in rat osteoblasts relevance to intracellular signaling pathways. J Biol Chem 277: 7865–7874 [DOI] [PubMed] [Google Scholar]
- Ostuni R, Kratochvill F, Murray PJ, Natoli G (2015) Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol 36: 229–239 [DOI] [PubMed] [Google Scholar]
- Postigo A, Dean DC (1997) ZEB, a vertebrate homolog of Drosophila Zfh‐1, is a negative regulator of muscle differentiation. EMBO J 16: 3935–3943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postigo A, Sheppard AM, Mucenski ML, Dean DC (1997) c‐myb and ets proteins synergize to overcome transcriptional repression by ZEB. EMBO J 16: 3924–3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postigo A, Dean DC (1999) ZEB represses transcription through interaction with the corepressor CtBP. Proc Natl Acad Sci USA 96: 6683–6688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postigo A (2003) Opposing functions of ZEB proteins in the regulation of the TGFβ/BMP signaling pathway. EMBO J 22: 2443–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postigo AA, Depp JL, Taylor JJ, Kroll K (2003) Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins. EMBO J 22: 2453–2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW (2011) CCL2 recruits inflammatory monocytes to facilitate breast‐tumour metastasis. Nature 475: 222–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rei M, Gonçalves‐Sousa N, Lança T, Thompson RG, Mensurado S, Balkwill FR, Kulbe H, Pennington DJ, Silva‐Santos B (2014) Murine CD27(−) Vγ6(+) γδ T cells producing IL‐17A promote ovarian cancer growth via mobilization of protumor small peritoneal macrophages. Proc Natl Acad Sci USA 111: E3562–E3570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson SC, Scott KA, Balkwill FR (2002) Chemokine stimulation of monocyte matrix metalloproteinase‐9 requires endogenous TNF−α. Eur J Immunol 32: 404–412 [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O, Persons DL, Smith PG, Terranova PF (2000) Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis 21: 585–591 [DOI] [PubMed] [Google Scholar]
- van Rooijen N, Hendrikx E (2010) Liposomes for specific depletion of macrophages from organs and tissues. Liposomes: Methods Protoc 1: 189–203 [DOI] [PubMed] [Google Scholar]
- Sánchez‐Tilló E, de Barrios O, Siles L, Cuatrecasas M, Castells A, Postigo A (2011) β‐catenin/TCF4 complex induces the epithelial‐to‐mesenchymal transition (EMT)‐activator ZEB1 to regulate tumor invasiveness. Proc Natl Acad Sci USA 108: 19204–19209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez‐Tilló E, Liu Y, de Barrios O, Siles L, Fanlo L, Cuatrecasas M, Darling DS, Dean DC, Castells A, Postigo A (2012) EMT‐activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell Mol Life Sci 69: 3429–3456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez‐Tilló E, Fanlo L, Siles L, Montes‐Moreno S, Moros A, Chiva‐Blanch G, Estruch R, Martinez A, Colomer D, Győrffy B, Roué G, Postigo A (2014) The EMT activator ZEB1 promotes tumor growth and determines differential response to chemotherapy in mantle cell lymphoma. Cell Death Differ 21: 247–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz C, Perdiguero EG, Chorro L, Szabo‐Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, Frampton J, Liu KJ, Geissmann F (2012) A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336: 86–90 [DOI] [PubMed] [Google Scholar]
- Sekido R, Murai K, Funahashi JI, Kamachi Y, Fujisawa‐Sehara A, Nabeshima YL, Kondoh H (1994) The delta‐crystallin enhancer‐binding protein delta EF1 is a repressor of E2‐box‐mediated gene activation. Mol Cell Biol 14: 5692–5700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serbina NV, Pamer EG (2006) Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol 7: 311–317 [DOI] [PubMed] [Google Scholar]
- Siles L, Sánchez‐Tilló E, Lim JW, Darling DS, Kroll KL, Postigo A (2013) ZEB1 imposes a temporary stage‐dependent inhibition of muscle gene expression and differentiation via CtBP‐mediated transcriptional repression. Mol Cell Biol 33: 1368–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva IA, Bai S, McLean K, Yang K, Griffith K, Thomas D, Ginestier C, Johnston C, Kueck A, Reynolds RK, Wicha MS, Buckanovich RJ (2011) Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer Res 71: 3991–4001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S, Liu Q, Chen J, Chen J, Chen F, He C, Huang D, Wu W, Lin L, Huang W, Zhang J, Cui X, Zheng F, Li H, Yao H, Su F, Song E (2014) A positive feedback loop between mesenchymal‐like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 25: 605–620 [DOI] [PubMed] [Google Scholar]
- Takagi T, Moribe H, Kondoh H, Higashi Y (1998) δEF1, a zinc finger and homeodomain transcription factor, is required for skeleton patterning in multiple lineages. Development 125: 21–31 [DOI] [PubMed] [Google Scholar]
- Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF (2007) Critical roles for CCR2 and MCP‐3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest 117: 902–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, Zur Hausen A, Brunton VG, Morton J, Sansom O, Schüler J, Stemmler MP, Herzberger C, Hopt U, Keck T, Brabletz S, Brabletz T (2009) The EMT‐activator ZEB1 promotes tumorigenicity by repressing stemness‐inhibiting microRNAs. Nat Cell Biol 11: 1487–1495 [DOI] [PubMed] [Google Scholar]
- Wilkinson RD, Magorrian SM, Williams R, Young A, Small DM, Scott CJ, Burden RE (2015) CCL2 is transcriptionally controlled by the lysosomal protease cathepsin S in a CD74‐dependent manner. Oncotarget 6: 29725–29739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Chawla A, Pollard JW (2013) Macrophage biology in development, homeostasis and disease. Nature 496: 445–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, Ganesan H, Nino‐Castro A, Mallmann MR, Labzin L, Theis H, Kraut M, Beyer M, Latz E, Freeman TC, Ulas T et al (2014) Transcriptome‐based network analysis reveals a spectrum model of human macrophage activation. Immunity 40: 274–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin M, Li X, Tan S, Zhou HJ, Ji W, Bellone S, Xu X, Zhang H, Santin AD, Lou G, Min W (2016) Tumor‐associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J Clin Invest 126: 4157–4173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Goncalves R, Mosser DM (2008) The isolation and characterization of murine macrophages. Curr Protoc Immunol 83: 14.1.1–14.1.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File