

Abstract
Low molecular weight gels are formed by the self-assembly of a suitable small molecule gelator into a three-dimensional network of fibrous structures. The gel properties are determined by the fiber structures, the number and type of cross-links and the distribution of the fibers and cross-links in space. Probing these structures and cross-links is difficult. Many reports rely on microscopy of dried gels (xerogels), where the solvent is removed prior to imaging. The assumption is made that this has little effect on the structures, but it is not clear that this assumption is always (or ever) valid. Here, we use small angle neutron scattering (SANS) to probe low molecular weight hydrogels formed by the self-assembly of dipeptides. We compare scattering data for wet and dried gels, as well as following the drying process. We show that the assumption that drying does not affect the network is not always correct.
Introduction
Low molecular weight gels (LMWG) are receiving a lot of attention.1−9 Unlike covalently cross-linked polymer gels, LMWG are formed when small molecules self-assemble into one-dimensional structures such as fibrils, fibers, or tubes. At a sufficiently high concentration (the so-called minimum gelation concentration (mgc)), these structures entangle and branch to a sufficient degree that a sample spanning network is formed. This immobilizes the solvent, resulting in a gel. Typically, the mgc will be less than 1 wt%. Such gels are reversible, for example reverting to a solution on heating.7 For peptide-based LMWG, the main driving forces of gel formation are noncovalent interactions. Changes in temperature or pH and the addition of salts can all lead to changes in the interactions between LMWG molecules that drive self-assembly into a kinetically trapped state. The kinetics and thermodynamics of dipeptide gelation, specifically focusing on diphenylalanine, has been reviewed recently,10 although the thermodynamic aspects of gelation remain less well understood. Drying could lead to changes in the kinetically trapped structures to a thermodynamic energy minimum such as crystallization or the fibers could be maintained.
There is significant interest in such gels for applications in cell culturing,4,11 controlled release,12 optoelectronics,5 drug therapies,13 and oil recovery.14 For these applications, key properties include the absolute mechanical strengths, the recoverability after shear (for example, in drug delivery where the gel would be passed through a needle),15 or the thermal reversibility.16,17 All of these properties depend on the fiber network, which means that characterizing and understanding this network is absolutely vital.
To characterize such gels, a range of methods have been used. Rheological methods inform as to the mechanical properties, but the network type has to then be inferred.18,19 Techniques such as infrared spectroscopy or circular dichroism can provide information as to the molecular packing, but cannot provide detail about the network. It is most common for a range of microscopy techniques to be used.
Moving down in microscopy length scales, optical microscopy simply cannot provide the necessary resolution to image the self-assembled fibers. Confocal microscopy can provide information at a relatively large length scale (although these techniques are improving constantly, and there are some extremely effective high resolution methods that are just coming on line).20,21 However, for confocal microscopy, either the molecule has to be synthetically adjusted such that it is fluorescent, which is likely to change its self-assembly behavior, or a fluorescent stain has to be added, which may affect the system. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) have been widely used.22 Both of these methods usually require that the gel is dried. Further, for SEM it is common to sputter a metal onto the structures, and for TEM it is common to stain the structures, for example, with a heavy metal salt. The structures are then imaged under high vacuum. Although it is commonly assumed that the structures are related to those in the native, wet gel, there is often little evidence that this is the case. Cryo-SEM can be used, but there can easily be artifacts arising from freeze-concentration effects. In some cases, cryo-TEM is used.23,24 Here, the sample is imaged in a vitrified hydrated state, which is presumably closer to the native structure. However, the sample preparation requires a thin film, which is difficult to access for a gel sample. Most experimental procedures involve placing a TEM grid on a gel or dipping into the gel. As such, the network may be disrupted and it might be that truly entangled fibers are not removed, but rather only free fibers are adsorbed. Certainly, the requirement of a maximum thickness means that analyzing the gel state is difficult. Finally, it should also be noted that such microscopy can only possibly access a tiny fraction of the structures in the gel due to the magnification used.
Scattering methods, however, allow the bulk sample to be analyzed. For gels, small-angle X-ray and neutron scattering experiments are widely used.25−30 While requiring access to a facility, the data is relatively easy to collect. Bulk samples can be analyzed and, critically while in the wet, solvated state at ambient temperatures. This means that there is no need to dry or stain the sample (although for small angle neutron scattering, it is necessary to use a deuterated solvent for contrast). Having collected the data, these are then fitted with a mathematical model to access information about the fibers, and the network if a sufficiently wide Q-range can be accessed (Q is the scattering variable, an inverse length scale defined as Q = (4π/λ)sin(θ/2), where λ is the neutron wavelength and θ is the scattering angle).
As stated, it is critical to understand the fiber network. There are few examples where multiple forms of analysis are used to probe the network, including examples showing a combination of scattering and microscopy.17,29,31−33 In some cases, there is a discrepancy between the microscopy data and the scattering. Since the scattering is collected in the wet state, it is tempting to assume that the microscopy suffers from drying artifacts, especially since the structures imaged have a higher apparent radius than that suggested by the scattering data.34−39
Here, we use small angle neutron scattering (SANS) to probe a number of hydrogels formed from dipeptide gelators. We compare the wet gels and the corresponding xerogels. We also use SANS to allow the influence of remaining water within the structures to be better understood through the contrast difference between hydrogen and deuterium. We compare these data with microscopy on the gels. These data allow us to discuss the effect of drying on these LMWGs.
Experimental Methods
Materials
The dipeptide LMWG were prepared as we have described previously.40,41 The deuterated analogues were prepared from deuterated 2-naphthol or deuterated amino acids following the same synthetic procedures. Full experimental and characterization data are provided in the Supporting Information. D2O, GdL, and NaOD were purchased from Sigma Aldrich and used as received. The NaOD was purchased as a 40 wt% solution and diluted with D2O to 0.1 M.
Gel Preparation and Drying Procedures
Solutions of each LMWG were prepared at 10 mg/mL in H2O (or D2O where required), including 1 mol equiv of 0.1 M NaOH (or NaOD). The pH values of the solutions were between 10 and 11 at this point (pD was converted to pH using a standard approach).42 The gels were formed by adding 1 mL of solution of a gelator to GdL (16 mg) in a vial; depending on the technique, the sample was transferred to a cuvette (for wet SANS), or transferred to a mold (all dried samples) with aluminum foil, providing the base layer onto which the gel is eventually dried (or a glass coverslip for the AFM/SEM samples). The samples were then left to gel sealed overnight. Air-dried samples were released from the mold, loosely covered to reduce dust or other sources of contamination or damage while drying in air on the bench at room temperature. A small control set of samples were instead dried inside an incubator at 25 °C to ensure temperature fluctuations did not affect the drying process. Freeze-dried samples were kept in the mold and placed into a lab freezer at −20 °C for approximately 7 h before being placed into a Labconco freezone 4.5 freeze-dryer with a condenser temperature of −50 °C and a shelf temperature of 20 °C overnight. A small number of samples were placed in liquid nitrogen instead of the lab freezer prior to freeze-drying.
Analytical Techniques
Small Angle Neutron Scattering (SANS)
Solutions were prepared as described for other techniques, with D2O and NaOD (unless otherwise stated). Gels were prepared as above using GdL. UV spectrophotometer grade, quartz cuvettes (Starna) with a 5 mm path length were filled with the solution immediately after the addition of GdL, allowed to gel sealed overnight and housed in a temperature-controlled sample rack at 25 °C during the measurements. For the dried film samples, the films were mounted over apertures in a cadmium plate, which was then placed in the same sample rack. For the in situ drying experiment, the gel was released from the mold after overnight gelation and mounted on the same cadmium plate as the dried films. SANS measurements were performed using the SANS2D instrument (ISIS pulsed neutron source, Oxfordshire, U.K.). A neutron beam allowed measurements over a large range in Q [Q = 4π sin(θ/2)/λ] of 0.005–0.7 Å–1 using incident wavelengths (λ) from 1.75 to 16.5 Å and employing a sample-to-detector distance of 4 m, with the 1 m2 detector offset vertically 60 mm and sideways 100 mm. The measuring times were 1–4 h dependent on the contrast.
Each raw scattering data set was corrected for the detector efficiencies, sample transmission and background scattering and converted to scattering cross-section data (∂Σ/∂Ω vs Q) using the instrument-specific software.43 These data were placed on an absolute scale (cm–1) using the scattering from a standard sample (a solid blend of hydrogenous and perdeuterated polystyrene) in accordance with established procedures.44 The scattering from D2O (the solvent) was also measured and subtracted from the wet data. For data from dried samples, the empty beam was subtracted.
The instrument-independent data were then fitted to customized models in the SasView software package;45 these combined an absolute power law with a (Kratky-Porod) flexible cylinder, as described previously.46 The Q-dependent power law (Q–N) accounts for the mass fractal contribution to the scattering intensity, which is superimposed on that from the cylindrical structures, that is, the fibers. The fibers of the gel are represented as a flexible worm-like chain of cylindrical Kuhn segments.
Atomic Force Microscopy
AFM imaging was performed in ambient conditions using the ScanAsyst mode using a Bruker Multimode 8 Nanoscope instrument with a J-scanner (Bruker Nano Inc., Santa Barbara, CA). The samples were imaged with a Bruker RTESPA-150 probe from with a nominal spring constant of 5 N/m. Images with size 5 μm × 5 and 1 μm × 1 μm were collected at a probe modulation frequency of 2 kHz. The topography images were analyzed offline using Bruker NanoScope Analysis v 1.5 software for topography measurements. The Section tool was used for measuring fiber diameter. Fiber diameters were measured using the “Section tool” within the software.
Scanning Electron Microscopy
SEM images were collected using a Hitachi S-4800 FE-SEM at 3 keV in deceleration mode at a height between 2 and 3 mm. For air-dried samples the gel was dried onto a glass coverslip and attached to a 15 mm screw in aluminum stub attached via a sticky carbon tab (Agar Scientific). For freeze-dried samples a small amount of the freeze-dried samples was stuck directly onto the sticky carbon tab. The freeze-dried samples were very fluffy and so had to be gently pressed flat using a glass coverslip to make them easier to focus on for imaging. Loose freeze-dried gel was then removed using compressed air. These images were still difficult to collect due to the nonflat surface the freeze-drying creates making it difficult to focus the beam properly. None of the samples were sputter coated with metal as the fibers were very small, this ensured that measuring the fiber widths was accurate. Images were collected in different places on the sample chosen at random to ensure that the images were representative.
Cryo-TEM
Cryogenic TEM imaging was performed on the FEI Tecnai 12 TWIN Transmission Electron Microscope, operating at 100 kV. Gels were immediately diluted five times with water to reduce their viscosity and 6 μL of sample solution was placed on a holey carbon film supported on a TEM copper grid (Electron Microscopy Services, Hatfield, PA). All the TEM grids used for cryo-TEM imaging were treated with plasma air to render the lacey carbon film hydrophilic. A thin film of the sample solution was produced using the Vitrobot with a controlled humidity chamber (FEI). After loading of the sample solution, the lacey carbon grid was blotted using preset parameters and plunged instantly into a liquid ethane reservoir precooled by liquid nitrogen. The vitrified samples were then transferred to a cryo-holder and cryo-transfer stage, which was cooled by liquid nitrogen. To prevent sublimation of vitreous water, the cryo-holder temperature was maintained below −170 °C during the imaging process. All images were recorded by a SIS Megaview III wide-angle CCD camera.
Thermal Gravimetric Analysis
TGA measurements were carried out on a TA Instruments SDT Q600 TGA machine using a constant air flow of 100 mL/min. Samples were heated at a rate of 10.00 °C/min to 120.00 °C and held there for 20 min before further heating at 10.00 °C/min to 200.00 °C. The sample mass used in all cases was between 3.5 and 7.5 mg. There was no difference in sample preparation or storage from that of the SANS samples.
Fiber Width Measurements
For cryo-TEM and SEM, fiber width measurements were collected using ImageJ. The scale bar was used to set the scale for the width measurement. A total of 70 measurements of fibers were used to create the histograms. These were done on several images of the same gels to ensure the widths were representative. Only objects that were clear single fibers (rather than aggregates or undetermined fibers) were measured to ensure primary fibers were being measured.
Results and Discussion
The gels examined here are all formed using dipeptide gelators.6 Initially, solutions at high pH (or pD) are prepared in water (or D2O) at a concentration of 1.0 wt%. The pH is then lowered by the addition of glucono-δ-lactone (GdL). GdL quickly dissolves, and then slowly hydrolyses to gluconic acid, resulting in a slow pH change and the formation of very reproducible gels.47,48 For this work, we have used a subset of our expanded library (Figure 1).
Figure 1.

Structures of the gelators used here (synthesis and characterization details in Supporting Information). Note, for 3, the alpha substituent is a deuteron rather than a proton, as it is for 1, 2, 4, 5, and 6.
The in situ hydrated primary self-assembled structures of LMWG that lead to the gel can be probed using SANS across a wide length scale from a few nanometers to a couple of hundred nanometres.25,26 SANS is particularly suited to aqueous systems such as those described here, as the water component is easily replaced by D2O to provide scattering length density (ρ) contrast; we refer to this as a H-gel in D2O since the gelator is fully hydrogenous (Figure 2). It is also possible to change the contrast by preparing an analogous deuterated gelator, a so-called D-gel (Figure 2). The scattering length densities (SLDs) for all gelators are shown in Table S5 (Supporting Information).
Figure 2.

Schematic to represent the contrast differences when (a) wet gels in D2O of hydrogenous LMWGs (H-gel) and partially deuterated LMWGs (D-gel) are dried. The darker the fibers and background the higher the scattering length density (ρ). (b) The ideal scenario where all remaining water is removed and therefore in the matrix ρ = 0 Å–2. (c) The more realistic scenario where a small amount of D2O remains within the sample, as observed by thermal gravimetric analysis (Figures S1–3 and Table S1 in Supporting Information), and therefore, ρ for the matrix could be slightly higher than 0. This illustrates how deuteration enhances the contrast of the xerogel.
The results from SANS of the wet H-gels (Figure 3 and Figure S4, Supporting Information) investigated here are consistent with our previous work, which have included the gelators 1, 4, and 6.38,46,49 Model fitting to the data using a modified Kratky-Porod flexible cylinder model provides information on the radius and Kuhn length of the primary fibers.38 An indication of the network segregation is given by the power law exponent, which fits the decay of the data in the low-Q region, attributed to mass-fractal type behavior. When wet, the H-gels of 1, 4, and 6 have fiber radii in the region of 3–4 nm (Tables S2–4 in Supporting Information).
Figure 3.
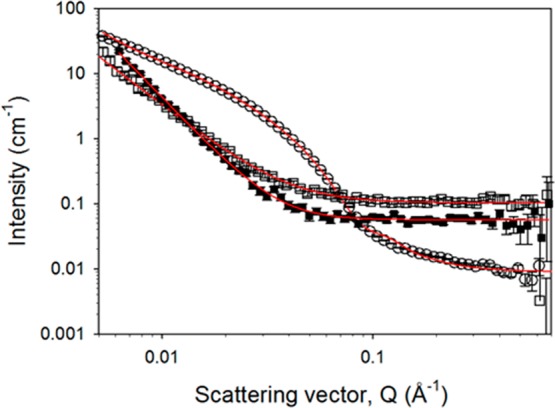
Fitted SANS profiles for the hydrogenous gelator 4. Open circles represent the wet gel, open squares the air-dried xerogel, and filled squares the freeze-dried xerogel. The solid lines depict the model fits to the data as parametrized in the Supporting Information (Table S3). Data for the other systems is shown in the Supporting Information.
Applying the same scattering approach to dried material requires careful experimental design (Figure 2) as the removal of D2O from the gel significantly changes the scattering length density difference (Δρ) between the gelator fibers (ρ in the region of 1–2 × 10–6 Å–2; Table S5, Supporting Information) and the matrix by which they are surrounded (D2O, ρ = 6.3 × 10–6 Å–2 or air, ρ = 0 Å–2). Hence, there would be a significant decrease in contrast from a Δρ of >4 × 10–6 Å–2 to <2 × 10–6 Å–2 on drying. Thus, the overall scattering intensity and the intensity of the features arising from the form factor are reduced, as illustrated by the general scattering equation (eq 1). The scattering intensity I(Q) is also determined from the volume fraction, defined as ϕ, the particle volume Vp and the form and structure factors of the scattering, P(Q) and S(Q), respectively. Simultaneously, the incoherent background, B, from the increased proportion of hydrogenous material overshadows any features at high Q.
| 1 |
Scattering from Hydrogenous Xerogels
Initially, to examine the effect of drying, we used our hydrogenous gelators. Gels were prepared in molds (Figure S5a, Supporting Information). After complete gelation, the samples were either air-dried on the benchtop or frozen and dried using a freeze-dryer. Both of these are typical sample preparation methods reported in the literature for preparation of samples for imaging. On air-drying, the networks collapse to form a thin film (hence, aerogels are not formed). Using the freeze-drier, a more expanded aerogel-type material was formed (Figure S5, Supporting Information). It should be noted that it was not possible to scale the SANS data to gain absolute intensities for the xerogel samples. This is because the thickness of the samples was not as uniform as would normally be achieved for wet samples in cuvettes. Therefore, one measured thickness would not provide an accurate representation of the sample, particularly for the freeze-dried xerogels containing a high proportion of air. A nominal thickness of 0.1 mm was used to reduce all the xerogel data, providing an estimated scaling. The data have been left as reduced on the same axes as the absolutely scaled wet gel data for ease of comparison. The shape of the data, however, is not affected by the scaling and in the fitting the change in scale is absorbed by the free fitting scale factors and background parameter.
For the xerogels (Figures 3 and S4, Supporting Information), the scattering intensity in the region between 0.01 and 0.1 Å–1 decreases compared to the wet gels, reducing any features that can be associated with fiber structures. This is most clearly seen for the air-dried and freeze-dried sample of 4, as shown in Figure 3. The fit to this data shows that only the power-law component and incoherent background remain in the scattering data. This suggests that ρ of the matrix is close to contrast matching the fibers, despite only 6–10% water remaining in the samples (as measured using thermal gravimetric analysis (TGA), Figures S1–3 and Table S1, Supporting Information), which would result in ρ = 0.4 to 0.6 × 10–6 Å–2. However, the features associated with fibers, in particular the inflection between 0.01 and 0.1 Å–1, are present in other xerogels but are difficult to define by eye, for example in the case of 1 (Figure S4, Supporting Information). The reproducibility of SANS from the xerogel films, the benchtop air-drying method, and film stability were confirmed (Figures S6–8, Supporting Information).
All of these data show that it is difficult to determine whether the fibers remain in a form that can be detected by SANS upon drying. Where there are features, they are less well defined, reducing the confidence in the sizes determined for the H-gels due to a lack of contrast. Hence, following the drying process in situ using the H-gels would not be possible. Therefore, deuteration of the LMWG itself was used to increase the scattering length density of the material and subsequently both increase Δρ within the xerogels and lower the incoherent background.
In Situ Drying
Initially, we examined the effect of drying in situ using a partially deuterated gelator to maximize the contrast in its more dehydrated states. A gel of 2 was prepared and placed in the neutron beam while still wet (Figures 4 and S5c,d, Supporting Information). The sample was exposed to air, allowed to dry in a temperature-controlled environment at 25 °C. As the sample dried, the overall scattering intensity first decreased with the flexible cylinder features disappearing after 8 h (Figures 4 and S9, Supporting Information). The intensity then increased again until the sample had been drying for 24 h. In the latter stages of drying, between 18 and 24 h, the features attributable to fibers had returned to the original scattering pattern. Close to absolute scaling was achieved here by using extrapolated thicknesses assuming a linear rate of D2O loss based on multiple thickness measurements at three time points during the experiment. We highlight here that it is not possible to discriminate during the drying process with SANS between water that interacted strongly with the dipeptides compared with the water that was weakly trapped and, therefore, would be lost first from the gel.
Figure 4.

Time-dependent SANS profiles of 2 while drying. The data have been split into plot (a) showing 0 h represented by filled circles, 4.2 h by open circles, and 8.3 h by filled triangles, when the overall scattering intensity was decreasing (with time, as shown by the arrow) and (b) showing 11.9 h represented by filled circles, 13.6 h by open circles, 15.2 h by filled triangles, 15.6 by open triangles, 18.3 h by filled squares, 24.0 h by open squares, 29.3 h by filled diamonds, and 33.2 h by open diamonds, when the scattering intensity began to increase again (with time, as shown by the arrow). The data for 24 h and beyond overlap each other completely. The data were normalized and background subtracted based on an extrapolated thickness assuming a linear rate of D2O loss from measurements at three time points.
There are a few contributing factors to this changing SANS intensity and increased incoherent background. As the sample is open to the air, there could be a small effect of exchange between the D2O in the gel and the H2O (ρ = −0.56 × 10–6 Å–2) in the atmosphere, which would reduce ρ for the water surrounding the fibers. Any interface between the evaporating water and air could add to the fractal contribution to the scattering, exhibited by a Q-dependent power law, but as the slope in the lowest Q region below 0.01 Å–1 does not vary, this is unlikely to be a significant factor. However, the larger contribution is likely to come from the contrast change caused by the evaporation of the D2O and the partial replacement of the solvent with air inside the fiber network, in addition to the collapse of the network on formation of a thin film. As the ratio of hydrated and dried parts of the network changes, the average ρ of the matrix decreases toward that of air. As the chosen gelator, 2, is partially deuterated, the contrast match point where the SANS intensity is lowest is relatively early in the drying process (between 8 and 12 h). Another factor that could potentially contribute to the reduced intensity is the rearrangement of the structures during drying. However, when the flexible cylinder features return in the scattering pattern (most clearly after 18 h), their position and shape are relatively similar to the initial state. This indicates that there has not been a dramatic change in structure. This assertion is also supported by a second in situ sample of 2 examined just for the first 13 h prepared in air contrast matched water (H2O and D2O mixed to ρ = 0 Å–2). The larger incoherent background makes the fiber scattering features less clearly defined than in the pure D2O sample but as the background decreases and the scattering intensity at low Q increases, the fiber-matrix contrast increases (Figure S10, Supporting Information). However, the inflection between 0.01 and 0.1 Å–1 is approaching the same shape and position as in the D2O data, indicating that the structure sizes have not changed with the contrast of the water matrix. This highlights the importance of contrast in the changes to the overall scattering intensity and confirms that the solvent contrast change is the main reason for the scattering intensity changes.
Along with the data for the hydrogenous gelators above, these results show that the reduction in contrast that arises from either dried H-gels or D-gels part way through the drying process (when the contrast match point is found) makes the size of the fibers impossible to determine under those conditions. As it would be impractical to improve the dried gel contrast of the H-gel samples by changing the air (e.g., drying them in a D2 atmosphere), more contrast needs to be introduced by modifying the hydrogen/deuterium content of the gelator itself.
Scattering from Deuterated Xerogels
The partially deuterated compounds, used to enhance contrast, show much clearer scattering patterns in their xerogel forms (Figure 5 and Tables S6–8, Supporting Information). Comparisons of how the best fit from the wet (D2O matrix) gel would look with the background matrix replaced by air or an air/D2O mix of ρ = 0.5 × 10–6 Å–2 are shown to illustrate the effects of contrast change on the scattering pattern (Figure S11, Supporting Information).
Figure 5.

Fitted SANS profiles for the deuterated LMWGs (a) 2, (b) 3, and (c) 5. Open circles represent the wet gel, open squares the air-dried xerogel, and filled squares the freeze-dried xerogel. The solid lines depict the model fits to the data as parametrized in the Supporting Information (Tables S6–8).
The gelators that were partially deuterated show similar flexible cylinder features to the wet hydrogenous gels. Subtle changes in the dimensions of the cylinders are to be expected, owing to the slightly differently defined boundaries to the fibers, arising from the packing of the molecules within the fibers, and the location of any remaining water. There will also be some expected shifts in the scattering pattern owing to the changes in contrast between the systems (Figure S11, Supporting Information). However, the deuterated versions 2 and 3 show a slight decrease in radius upon drying compared to a slight increase in the radius for 1. The very small contribution of the flexible cylinder features to the xerogel scattering patterns of 1 mean that the data from 2 and 3 seem more reliable. LMWG 5 appears to retain relatively similar dimensions on drying. This could not be understood from its hydrogenous equivalent 4, which suffered from apparent contrast matching upon drying.
We hypothesized that the freeze-drying approach would provide clearer scattering patterns owing to a more open network, with the boundaries between the fibers and the air matrix more clearly defined. Hence, ρ of the matrix would be much closer to 0 Å–2, despite TGA indicating that some water is either retained or reabsorbed once processed and stored under atmospheric conditions. In general, we observed that the freeze-dried xerogels were more likely to retain features attributable to the fibers in the SANS data. The only exceptions to that hypothesis were gelators 1 and 4. When the data was fitted in some cases, such as for 6, the fitted radii of the freeze-dried xerogel were the same as for the wet gels, within the uncertainty of the fitting (Table S4, Supporting Information).
Cryogenic freeze-drying of one sample of LMWG 2 was also used in order to see whether the vitrification of the water made a difference to the fiber network as seen by SANS (Figure S12, Supporting Information). The power law contribution was seen to dominate with the simple power law model providing the best fit to the data with an exponent of 3.5 ± 0.1. This indicates that the network has moved from the mass fractal into the surface fractal regime, which arises from the rough surface scattering from dense clusters where there is minimal internal contrast.
Comparison between SANS and Microscopy
As LMWG 5, the deuterated version of 4, was shown to retain similar fiber dimensions upon drying when measured using scattering, SEM and AFM images were collected for xerogels of 4 and 5 (Figures 6a and S13–16, Supporting Information) and compared to the values obtained from scattering. The diameters, D, of the fibers recorded in the AFM and SEM images are generally between 6 and 12 times larger than the values obtained from model fits to the scattering. A comparison of these with SANS parameters is given in Table 1 for LMWGs 4 and 5. Model fits for both 4 and 5 using fiber radii approximately equivalent to those observed in the SEM (at 28.5 nm radius, polydispersity of 0.2) are shown in Figure 6c,d. While the fringe features in these model fits are smoothed out by a polydispersity in the radius, the shapes remain different from the experimental data recorded. These model fits highlight how the xerogel data from 4 is dependent only on the power law as asserted earlier and therefore the size of the fibers cannot be deduced from that data. However, for both the wet gel of 4 and all gels of 5, the fits are clearly not as good when the larger radius is used. This also confirms that the SANS technique is capable of measuring features in this size range, if they were the primary fiber size over the whole sample area. The difference in fiber radius between the SANS and microscopy also appears larger than we might expect simply from the different ways in which the edges of the fibers will be defined by the three techniques.
Figure 6.

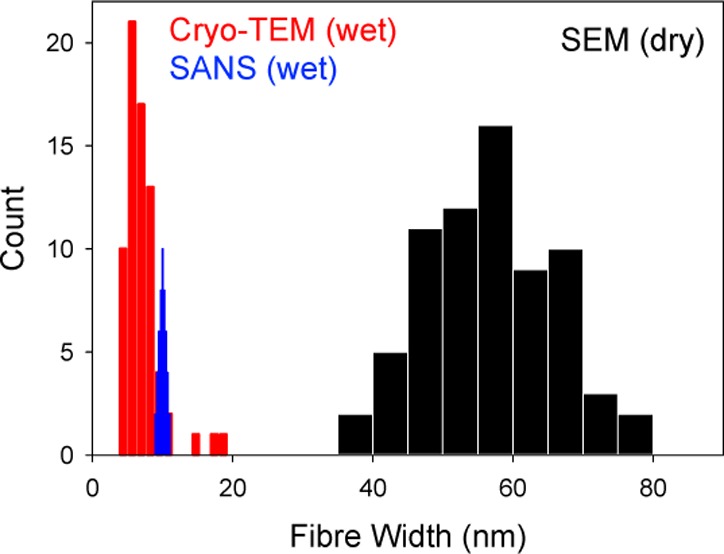
(a) SEM images of the air-dried xerogel of 5, where the scale bar represents 2 μm. Widths averaged over at least 70 measurements were 57 ± 23 nm. Other images are provided in the SI (Figures S13–S15). (b) Histogram of the widths of fibers measured from the SEM images (black) and cryo-TEM (red) of 5, along with the distribution expected from a Gaussian distribution (generated from SigmaPlot with a standard deviation of 0.4) around the mean diameter determined by SANS (blue). (c) SANS data for 5 with the model fits with radius (R = 28.5 nm), Kuhn length (50 nm), and length (2 μm) equivalent to the sizes determined from the AFM and SEM images both with and without the power law exponent in order to highlight the differences with the best fits to the data. Other parameters were kept the same as in the best fits. The long dash line is without both polydispersity and the power law model, the medium dash line is with the polydispersity of 0.2 and without the power law model, the dotted line is without the polydispersity but with the power law model (N = 2.5) and the short dash line is with both a polydispersity of 0.2 and a power law model (N = 2.5). Open circles represent the wet gel, open squares the air-dried xerogel, and filled squares the freeze-dried xerogel. The solid red lines depict the model fits to the data as parametrized in Supporting Information (Tables S8). (d) Example of the cryo-TEM image for 5, with a scale bar of 200 nm. The white arrow highlights where two primary fibers seem to wrap around each other. Other images are shown in Figures S18 and S19, Supporting Information.
Table 1. Comparison of Diameters Measured Using SANS and Microscopy for 4 and 5a.
| diameters measured (in nm) by: | SANS |
AFM | SEM | cryo-TEM | ||
|---|---|---|---|---|---|---|
| wet | air-dried | freeze-dried | ||||
| 4 | 8.0 ± 0.8 | - | - | 52 ± 13 | 57 ± 28 | 7.6 ± 2.0 |
| 5 | 10.0 ± 1.0 | 12.0 ± 1.0 | 9.2 ± 1.0 | - | 57 ± 23 | 7.1 ± 3.0 |
“-” indicates that the value was not determined.
Since the SANS data shows that the scattering before and after drying is very similar, there remains a discrepancy between the SANS and microscopy. As mentioned above, there are relatively few comparisons between small angle scattering data and microscopy for such gels. In some cases, a close match between the radii measured using both methods are found. Examples include Pochan and Schneider’s β-hairpin based LMWG,31,50 some examples of Stupp’s peptide amphiphiles,51,52 and some ionic peptides.53,54 In other examples, there are significant discrepancies between the data. These include some of our work,38,46,49 as well as related work from Thordarson.39,55 In these cases, the microscopy implies that the radii are significantly greater than that measured by small angle scattering.
Hence, there are two questions that need answering. First, why do the data for microscopy and scattering differ for our systems and not for others? We hypothesize that the apparent differences between samples can be explained by the degree of charge on the self-assembled fibers. For the examples where the data from the microscopy and scattering are in agreement, there is significant charge left on the self-assembled fibers.29,53 However, for our LMWG, there is a single charged group at high pH, which conceptually is removed on pH decrease and gelation. We have recently shown for our class of LMWG that even after the pH has been decreased and a gel has formed, there is some residual charge on the fibers,56 but significantly less than the other examples described above. In some cases, further removal of charge leads to fiber–fiber association and syneresis of the gel phase.57,58 We therefore suggest that fiber aggregation is easier for our relatively uncharged gelators as compared to other LMWGs. This may also be related to the association with water that presumably is more prevalent for charged LMWG than for uncharged LMWG.
The second question is why is there a discrepancy between the sizes determined by SANS and microscopy for the xerogels? As mentioned above, model fits to hypothetic fibers with diameters found by microscopy show that SANS is capable of measuring features in this size range if they were the primary fiber size over the whole sample area. Work by Zhang et al. has shown that when peptide amphiphile fibers laterally aggregate (as shown by microscopy),30 changes in the small-angle X-ray scattering are measured; this again implies that we might expect that SANS would be sensitive to the aggregation.
To further probe this, cryo-TEM of gels of 4 and 5 was carried out. From the data, it is clear that the gel consists of a network of fibers in the vitrified state (Figures 6d, S18, and S19, Supporting Information). To image the gels using this technique, it is necessary to dilute the gel. As such, the images cannot show the true network, but can be used to probe the fibrous structures that are present. Image analysis was used to determine the fiber widths, and the diameters were found to be 7.7 ± 2.0 nm for 4 and 7.1 ± 3.0 nm for 5. These are significantly smaller than the data from the SEM and AFM of the xerogel and close to the values determined from the SANS fitting. Hence, it appears that the SANS probes the primary fibers. These clearly aggregate to some degree even when in the vitrified gel phase, as shown by the cryo-TEM data (an example where two fibers are aggregating is highlighted in Figure 6d), but the aggregation is even more pronounced on drying. The aggregation is not observed in the SANS data either in the gel or xerogel state. What is clear is that the AFM and SEM data do not represent the fiber network in the gel state, as shown by the significant discrepancy between data from the cryo-TEM and SEM.
Conclusions
We have shown here the first in situ drying study for LMWGs. To maximize scattering intensity, deuteration of the LMWG is beneficial. Deuteration allows for sufficient scattering intensity to follow the drying in situ, and to probe the xerogels. Our data show that the method of drying is very important; comparison of the data before and after drying shows that in a number of cases there are significant differences in the scattering.
Our data show that SANS is capable of probing the primary fibers for these gels, but is insensitive to lateral association of the fibers, either in the wet gel phase or on drying. The cryo-TEM data shows the presence of fibers with radii which are consistent with the SANS data, as well as aggregates; essentially, the AFM and SEM only show large aggregates.
Hence, our data show that for such LMWGs, the SANS data is extremely useful and represents the primary fiber network. However, for those LWMGs where the fibers are hydrophobic and not heavily charged, microscopy on dried gels does not represent the network, but rather aggregation of the fibers. It is not possible to observe the primary fibers by AFM and SEM, although SANS can still determine that these larger structures are formed from thinner fibers. Microscopy on the dried gels is extremely common, but our data suggests that the images should therefore be treated with caution.
Acknowledgments
L.M., A.C., B.D., and E.D. thank the EPSRC for funding (EP/L021978/1). D.A. thanks the EPSRC for a Fellowship (EP/L021978/1). M.N. thanks the University of Liverpool for a DTA studentship. G.S. acknowledges funding support from an ERC Advanced Investigator Grant (PISA 320372). Experiments at the ISIS Pulsed Neutron and Muon Source were supported by a beamtime allocation (RB1520075, RB1520074) from the Science and Technology Facilities Council. This work benefitted from the SasView software, originally developed by the DANSE project under NSF Award DMR-0520547. We thank Dr. Michael Barrow (University of Liverpool) for his help with the TGA measurements.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biomac.7b00823.
Synthesis and characterization details for the LMWG, TGA data, SANS model fit parameters, SANS curves for additional LMWGs and indicating reproducibility, and additional SEM, AFM, and cryo-TEM images (PDF).
The authors declare no competing financial interest.
Supplementary Material
References
- Terech P.; Weiss R. G. Low Molecular Mass Gelators of Organic Liquids and the Properties of Their Gels. Chem. Rev. 1997, 97 (8), 3133–3159. 10.1021/cr9700282. [DOI] [PubMed] [Google Scholar]
- Du X.; Zhou J.; Shi J.; Xu B. Supramolecular Hydrogelators and Hydrogels: From Soft Matter to Molecular Biomaterials. Chem. Rev. 2015, 115 (24), 13165–307. 10.1021/acs.chemrev.5b00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estroff L. A.; Hamilton A. D. Water Gelation by Small Organic Molecules. Chem. Rev. 2004, 104 (3), 1201–1218. 10.1021/cr0302049. [DOI] [PubMed] [Google Scholar]
- Skilling K. J.; Citossi F.; Bradshaw T. D.; Ashford M.; Kellam B.; Marlow M. Insights into low molecular mass organic gelators: a focus on drug delivery and tissue engineering applications. Soft Matter 2014, 10 (2), 237–56. 10.1039/C3SM52244J. [DOI] [PubMed] [Google Scholar]
- Babu S. S.; Praveen V. K.; Ajayaghosh A. Functional pi-gelators and their applications. Chem. Rev. 2014, 114 (4), 1973–2129. 10.1021/cr400195e. [DOI] [PubMed] [Google Scholar]
- Fleming S.; Debnath S.; Frederix P. W.; Hunt N. T.; Ulijn R. V. Insights into the coassembly of hydrogelators and surfactants based on aromatic peptide amphiphiles. Biomacromolecules 2014, 15 (4), 1171–84. 10.1021/bm401720z. [DOI] [PubMed] [Google Scholar]
- Fichman G.; Gazit E. Self-assembly of short peptides to form hydrogels: design of building blocks, physical properties and technological applications. Acta Biomater. 2014, 10 (4), 1671–82. 10.1016/j.actbio.2013.08.013. [DOI] [PubMed] [Google Scholar]
- Liu X.; Fei J.; Wang A.; Cui W.; Zhu P.; Li J. Transformation of Dipeptide-Based Organogels into Chiral Crystals by Cryogenic Treatment. Angew. Chem., Int. Ed. 2017, 56 (10), 2660–2663. 10.1002/anie.201612024. [DOI] [PubMed] [Google Scholar]
- Zou Q.; Abbas M.; Zhao L.; Li S.; Shen G.; Yan X. Biological Photothermal Nanodots Based on Self-Assembly of Peptide-Porphyrin Conjugates for Antitumor Therapy. J. Am. Chem. Soc. 2017, 139 (5), 1921–1927. 10.1021/jacs.6b11382. [DOI] [PubMed] [Google Scholar]
- Wang J.; Liu K.; Xing R.; Yan X. Peptide self-assembly: thermodynamics and kinetics. Chem. Soc. Rev. 2016, 45 (20), 5589–5604. 10.1039/C6CS00176A. [DOI] [PubMed] [Google Scholar]
- Alakpa E. V.; Jayawarna V.; Lampel A.; Burgess; Karl V.; West; Christopher C.; Bakker; Sanne C. J.; Roy S.; Javid N.; Fleming S.; Lamprou; Dimitris A.; Yang J.; Miller A.; Urquhart; Andrew J.; Frederix; Pim W. J. M.; Hunt; Neil T.; Péault B.; Ulijn; Rein V.; Dalby; Matthew J. Tunable Supramolecular Hydrogels for Selection of Lineage-Guiding Metabolites in Stem Cell Cultures. Chem 2016, 1 (2), 298–319. 10.1016/j.chempr.2016.07.001. [DOI] [Google Scholar]
- Martin C.; Oyen E.; Mangelschots J.; Bibian M.; Ben Haddou T.; Andrade J.; Gardiner J.; Van Mele B.; Madder A.; Hoogenboom R.; Spetea M.; Ballet S. Injectable peptide hydrogels for controlled-release of opioids. MedChemComm 2016, 7 (3), 542–549. 10.1039/C5MD00440C. [DOI] [Google Scholar]
- Liu K.; Xing R.; Zou Q.; Ma G.; Mohwald H.; Yan X. Simple Peptide-Tuned Self-Assembly of Photosensitizers towards Anticancer Photodynamic Therapy. Angew. Chem., Int. Ed. 2016, 55 (9), 3036–9. 10.1002/anie.201509810. [DOI] [PubMed] [Google Scholar]
- Okesola B. O.; Vieira V. M.; Cornwell D. J.; Whitelaw N. K.; Smith D. K. 1,3:2,4-Dibenzylidene-D-sorbitol (DBS) and its derivatives--efficient, versatile and industrially-relevant low-molecular-weight gelators with over 100 years of history and a bright future. Soft Matter 2015, 11 (24), 4768–87. 10.1039/C5SM00845J. [DOI] [PubMed] [Google Scholar]
- Yan C.; Altunbas A.; Yucel T.; Nagarkar R. P.; Schneider J. P.; Pochan D. J. Injectable solid hydrogel: mechanism of shear-thinning and immediate recovery of injectable beta-hairpin peptide hydrogels. Soft Matter 2010, 6 (20), 5143–5156. 10.1039/c0sm00642d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muro-Small M. L.; Chen J.; McNeil A. J. Dissolution parameters reveal role of structure and solvent in molecular gelation. Langmuir 2011, 27 (21), 13248–53. 10.1021/la202702r. [DOI] [PubMed] [Google Scholar]
- Ikeda M.; Ochi R.; Kurita Y. S.; Pochan D. J.; Hamachi I. Heat-induced morphological transformation of supramolecular nanostructures by retro-Diels-Alder reaction. Chem. - Eur. J. 2012, 18 (41), 13091–6. 10.1002/chem.201201670. [DOI] [PubMed] [Google Scholar]
- Nebot V. J.; Smith D. K.. Functional Molecular Gels; The Royal Society of Chemistry, 2013. [Google Scholar]
- Terech P.; Weiss R. G.. Molecular Gels: Materials with Self-Assembled Fibrillar Networks; Springer: Dordrecht, The Netherlands, 2006. [Google Scholar]
- Onogi S.; Shigemitsu H.; Yoshii T.; Tanida T.; Ikeda M.; Kubota R.; Hamachi I. In situ real-time imaging of self-sorted supramolecular nanofibres. Nat. Chem. 2016, 8 (8), 743–52. 10.1038/nchem.2526. [DOI] [PubMed] [Google Scholar]
- da Silva R. M.; van der Zwaag D.; Albertazzi L.; Lee S. S.; Meijer E. W.; Stupp S. I. Super-resolution microscopy reveals structural diversity in molecular exchange among peptide amphiphile nanofibres. Nat. Commun. 2016, 7, 11561. 10.1038/ncomms11561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G.; Yan X.; Han C.; Huang F. Characterization of supramolecular gels. Chem. Soc. Rev. 2013, 42 (16), 6697–722. 10.1039/c3cs60080g. [DOI] [PubMed] [Google Scholar]
- Cui H.; Hodgdon T. K.; Kaler E. W.; Abezgauz L.; Danino D.; Lubovsky M.; Talmon Y.; Pochan D. J. Elucidating the assembled structure of amphiphiles in solution via cryogenic transmission electron microscopy. Soft Matter 2007, 3 (8), 945. 10.1039/b704194b. [DOI] [PubMed] [Google Scholar]
- Newcomb C. J.; Moyer T. J.; Lee S. S.; Stupp S. I. Advances in cryogenic transmission electron microscopy for the characterization of dynamic self-assembling nanostructures. Curr. Opin. Colloid Interface Sci. 2012, 17 (6), 350–359. 10.1016/j.cocis.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilbaud J. B.; Saiani A. Using small angle scattering (SAS) to structurally characterise peptide and protein self-assembled materials. Chem. Soc. Rev. 2011, 40 (3), 1200–10. 10.1039/C0CS00105H. [DOI] [PubMed] [Google Scholar]
- Hule R. A.; Nagarkar R. P.; Hammouda B.; Schneider J. P.; Pochan D. J. Dependence of Self-Assembled Peptide Hydrogel Network Structure on Local Fibril Nanostructure. Macromolecules 2009, 42 (18), 7137–7145. 10.1021/ma9003242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terech P.; Furman I.; Weiss R. G.; Bouas-Laurent H.; Desvergne J.-P.; Ramasseul R. Gels from Small Molecules in Organic Solvents: Structural Features of a Family of Steroid and Anthryl-based Organogelators. Faraday Discuss. 1995, 101, 345–358. 10.1039/fd9950100345. [DOI] [Google Scholar]
- Hamley I. W.; Dehsorkhi A.; Castelletto V.; Furzeland S.; Atkins D.; Seitsonen J.; Ruokolainen J. Reversible helical unwinding transition of a self-assembling peptide amphiphile. Soft Matter 2013, 9 (39), 9290. 10.1039/c3sm51725j. [DOI] [Google Scholar]
- Hule R. A.; Nagarkar R. P.; Altunbas A.; Ramay H. R.; Branco M. C.; Schneider J. P.; Pochan D. J. Correlations between structure, material properties and bioproperties in self-assembled β-hairpin peptide hydrogels. Faraday Discuss. 2008, 139, 251. 10.1039/b717616c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.; Greenfield M. A.; Mata A.; Palmer L. C.; Bitton R.; Mantei J. R.; Aparicio C.; de la Cruz M. O.; Stupp S. I. A self-assembly pathway to aligned monodomain gels. Nat. Mater. 2010, 9 (7), 594–601. 10.1038/nmat2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rughani R. V.; Salick D. A.; Lamm M. S.; Yucel T.; Pochan D. J.; Schneider J. P. Folding, Self-Assembly, and Bulk Material Properties of a De Novo Designed Three-Stranded β-Sheet Hydrogel. Biomacromolecules 2009, 10, 1295–1304. 10.1021/bm900113z. [DOI] [PubMed] [Google Scholar]
- Boothroyd S.; Miller A. F.; Saiani A. From fibres to networks using self-assembling peptides. Faraday Discuss. 2013, 166, 195. 10.1039/c3fd00097d. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Nieuwendaal R.; Dimitriadis E.; Hammouda B.; Douglas J.; Xu B.; Horkay F. Supramolecular Self-Assembly of a Model Hydrogelator: Characterization of Fiber Formation and Morphology. Gels 2016, 2 (4), 27. 10.3390/gels2040027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z.; Tantakitti F.; Palmer L. C.; Stupp S. I. Asymmetric Peptide Nanoribbons. Nano Lett. 2016, 16 (11), 6967–6974. 10.1021/acs.nanolett.6b03062. [DOI] [PubMed] [Google Scholar]
- Saiani A.; Mohammed A.; Frielinghaus H.; Collins R.; Hodson N.; Kielty C. M.; Sherratt M. J.; Miller A. F. Self-assembly and gelation properties of α-helix versus β-sheet forming peptides. Soft Matter 2009, 5 (1), 193–202. 10.1039/B811288F. [DOI] [Google Scholar]
- Shimada T.; Sakamoto N.; Motokawa R.; Koizumi S.; Tirrell M. Self-assembly process of peptide amphiphile worm-like micelles. J. Phys. Chem. B 2012, 116 (1), 240–3. 10.1021/jp209105z. [DOI] [PubMed] [Google Scholar]
- Terech P.; Allegraud J. J.; Garner C. M. Thermoreversible Gelation of Organic Liquids by Arylcyclohexanol Derivatives: a Structural Study. Langmuir 1998, 14, 3991–3998. 10.1021/la980160c. [DOI] [Google Scholar]
- Draper E. R.; Mears L. L. E.; Castilla A. M.; King S. M.; McDonald T. O.; Akhtar R.; Adams D. J. Using the hydrolysis of anhydrides to control gel properties and homogeneity in pH-triggered gelation. RSC Adv. 2015, 5 (115), 95369–95378. 10.1039/C5RA22253B. [DOI] [Google Scholar]
- Jamieson S. A.; Tong K. W.; Hamilton W. A.; He L.; James M.; Thordarson P. Small angle neutron scattering (SANS) studies on the structural evolution of pyromellitamide self-assembled gels. Langmuir 2014, 30 (46), 13987–93. 10.1021/la502546n. [DOI] [PubMed] [Google Scholar]
- Houton K. A.; Morris K. L.; Chen L.; Schmidtmann M.; Jones J. T.; Serpell L. C.; Lloyd G. O.; Adams D. J. On crystal versus fiber formation in dipeptide hydrogelator systems. Langmuir 2012, 28 (25), 9797–806. 10.1021/la301371q. [DOI] [PubMed] [Google Scholar]
- Chen L.; Morris K.; Laybourn A.; Elias D.; Hicks M. R.; Rodger A.; Serpell L.; Adams D. J. Self-assembly mechanism for a naphthalene-dipeptide leading to hydrogelation. Langmuir 2010, 26 (7), 5232–42. 10.1021/la903694a. [DOI] [PubMed] [Google Scholar]
- Krezel A.; Bal W. A formula for correlating pKa values determined in D2O and H2O. J. Inorg. Biochem. 2004, 98 (1), 161–166. 10.1016/j.jinorgbio.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Arnold O.; Bilheux J. C.; Borreguero J. M.; Buts A.; Campbell S. I.; Chapon L.; Doucet M.; Draper N.; Ferraz Leal R.; Gigg M. A.; Lynch V. E.; Markvardsen A.; Mikkelson D. J.; Mikkelson R. L.; Miller R.; Palmen K.; Parker P.; Passos G.; Perring T. G.; Peterson P. F.; Ren S.; Reuter M. A.; Savici A. T.; Taylor J. W.; Taylor R. J.; Tolchenov R.; Zhou W.; Zikovsky J. Mantid—Data analysis and visualization package for neutron scattering and SR experiments. Nucl. Instrum. Methods Phys. Res., Sect. A 2014, 764, 156–166. 10.1016/j.nima.2014.07.029. [DOI] [Google Scholar]
- Wignall G. D.; Bates F. S. Absolute Calibration of Small-Angle Neutron Scattering Data. J. Appl. Crystallogr. 1987, 20, 28. 10.1107/S0021889887087181. [DOI] [Google Scholar]
- www.sasview.org.
- Cardoso A. Z.; Mears L. L.; Cattoz B. N.; Griffiths P. C.; Schweins R.; Adams D. J. Linking micellar structures to hydrogelation for salt-triggered dipeptide gelators. Soft Matter 2016, 12 (15), 3612–21. 10.1039/C5SM03072B. [DOI] [PubMed] [Google Scholar]
- Pocker Y.; Green E. Hydrolysis of D-Glucono-δ-lactone. I. General Acid-Base Catalysis, Solvent Deuterium Isotope Effects, and Transition State Characterization. J. Am. Chem. Soc. 1973, 95 (1), 113–119. 10.1021/ja00782a019. [DOI] [PubMed] [Google Scholar]
- Adams D. J.; Butler M. F.; Frith W. J.; Kirkland M.; Mullen L.; Sanderson P. A new method for maintaining homogeneity during liquid–hydrogel transitions using low molecular weight hydrogelators. Soft Matter 2009, 5 (9), 1856. 10.1039/b901556f. [DOI] [Google Scholar]
- Morris K. L.; Chen L.; Raeburn J.; Sellick O. R.; Cotanda P.; Paul A.; Griffiths P. C.; King S. M.; O’Reilly R. K.; Serpell L. C.; Adams D. J. Chemically programmed self-sorting of gelator networks. Nat. Commun. 2013, 4, 1480. 10.1038/ncomms2499. [DOI] [PubMed] [Google Scholar]
- Micklitsch C. M.; Medina S. H.; Yucel T.; Nagy-Smith K. J.; Pochan D. J.; Schneider J. P. Influence of Hydrophobic Face Amino Acids on the Hydrogelation of β-Hairpin Peptide Amphiphiles. Macromolecules 2015, 48 (5), 1281–1288. 10.1021/ma5024796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C.; Li H.; Li Y.; Kewalramani S.; Palmer L. C.; Dravid V. P.; Stupp S. I.; Olvera de la Cruz M.; Bedzyk M. J. Electrostatic Control of Polymorphism in Charged Amphiphile Assemblies. J. Phys. Chem. B 2017, 121 (7), 1623–1628. 10.1021/acs.jpcb.6b11602. [DOI] [PubMed] [Google Scholar]
- Cui H.; Cheetham A. G.; Pashuck E. T.; Stupp S. I. Amino acid sequence in constitutionally isomeric tetrapeptide amphiphiles dictates architecture of one-dimensional nanostructures. J. Am. Chem. Soc. 2014, 136 (35), 12461–8. 10.1021/ja507051w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boothroyd S.; Saiani A.; Miller A. F. Controlling network topology and mechanical properties of co-assembling peptide hydrogels. Biopolymers 2014, 101 (6), 669–680. 10.1002/bip.22435. [DOI] [PubMed] [Google Scholar]
- Elsawy M. A.; Smith A. M.; Hodson N.; Squires A.; Miller A. F.; Saiani A. Modification of beta-Sheet Forming Peptide Hydrophobic Face: Effect on Self-Assembly and Gelation. Langmuir 2016, 32 (19), 4917–23. 10.1021/acs.langmuir.5b03841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A. D.; Wojciechowski J. P.; Robinson A. B.; Heu C.; Garvey C. J.; Ratcliffe J.; Waddington L. J.; Gardiner J.; Thordarson P. Controlling self-assembly of diphenylalanine peptides at high pH using heterocyclic capping groups. Sci. Rep. 2017, 7, 43947. 10.1038/srep43947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace M.; Iggo J. A.; Adams D. J. Using solution state NMR spectroscopy to probe NMR invisible gelators. Soft Matter 2015, 11 (39), 7739–47. 10.1039/C5SM01760B. [DOI] [PubMed] [Google Scholar]
- Wallace M.; Adams D. J.; Iggo J. A. Analysis of the mesh size in a supramolecular hydrogel by PFG-NMR spectroscopy. Soft Matter 2013, 9 (22), 5483. 10.1039/c3sm27793c. [DOI] [Google Scholar]
- Castilla A. M.; Wallace M.; Mears L. L.; Draper E. R.; Doutch J.; Rogers S.; Adams D. J. On the syneresis of an OPV functionalised dipeptide hydrogel. Soft Matter 2016, 12 (37), 7848–7854. 10.1039/C6SM01194B. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.