

ABSTRACT
Multiple mechanisms of epigenetic control that include DNA methylation, histone modification, noncoding RNAs, and mitotic gene bookmarking play pivotal roles in stringent gene regulation during lineage commitment and maintenance. Experimental evidence indicates that bivalent chromatin domains, i.e., genome regions that are marked by both H3K4me3 (activating) and H3K27me3 (repressive) histone modifications, are a key property of pluripotent stem cells. Bivalency of developmental genes during the G1 phase of the pluripotent stem cell cycle contributes to cell fate decisions. Recently, some cancer types have been shown to exhibit partial recapitulation of bivalent chromatin modifications that are lost along with pluripotency, suggesting a mechanism by which cancer cells reacquire properties that are characteristic of undifferentiated, multipotent cells. This bivalent epigenetic control of oncofetal gene expression in cancer cells may offer novel insights into the onset and progression of cancer and may provide specific and selective options for diagnosis as well as for therapeutic intervention.
KEYWORDS: bivalency, cancer, epigenetic control, nuclear structure, oncofetal gene expression
INTRODUCTION
Epigenetic control of gene expression plays a pivotal role in physiological responsiveness and is often compromised during onset and progression of cancer. Epigenetic changes are heritable but do not involve changes in DNA sequences. Within a given cell, there are many distinct carriers of epigenetic information that are relayed to progeny upon cell division. Epigenetic mechanisms include methylation of CpG residues, modifications of nucleosomal histone proteins, regulation of gene transcription and protein translation by noncoding RNA molecules, and mitotic retention of transcription factors (1–8). From an architectural perspective, epigenetic control is engaged at multiple levels of nuclear organization from sequence-specific regulatory elements to chromatin remodeling at the nucleosomal level to large-scale inter- and intrachromosomal interactions (9–14). These epigenetic mechanisms function in a complex but coordinated manner to orchestrate cellular responses to extracellular signals.
The cellular epigenetic landscape is dynamically modified by a number of posttranslational modifications of nucleosomal histones (1, 3, 15, 16). These modifications function in concert—a phenomenon described as the histone code—to establish context-dependent chromatin landscapes that control access of transcription factors to gene regulatory regions (1, 3, 15, 16). This review focuses on the bivalent chromatin landscape defined by addition of three methyl moieties to lysine 4 and lysine 27 residues of histone H3 (referred to as histone 3 lysine 4 me3 [H3K4me3] and H3K27me3 throughout this article). Chromatin bivalency, i.e., the presence of both activating H3K4me3 and repressive H3K27me3 modifications at gene promoters, was first observed in pluripotent stem cells (17–19). Emerging evidence indicates that bivalency poises genes for expression and is likely a key regulatory mechanism in trans-differentiation during lineage commitment as well as in dedifferentiation during onset and progression of cancer (Fig. 1) (20–24). We propose that partial recapitulation of the bivalent chromatin landscape of pluripotent cells in some cancer types—termed “oncofetal epigenetic control” in this review—can offer novel avenues for the development of specific and selective diagnostic and therapeutic approaches.
FIG 1.
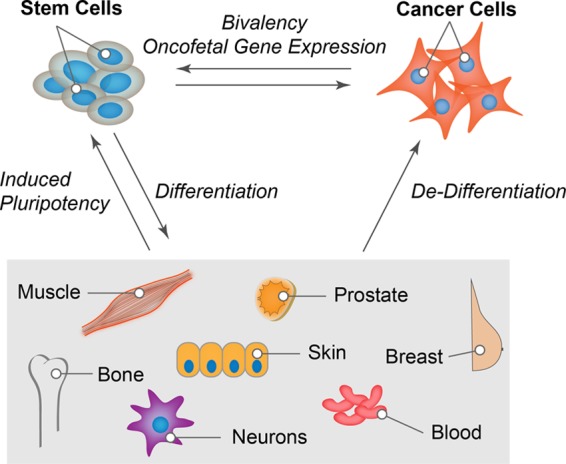
Emerging role of bivalent chromatin in establishing pluripotent or cancer state of cells. The schematic shows the capacity of pluripotent stem cells to give rise to various tissue types during development. Bivalency appears to contribute to cell fate decisions during the pluripotent stem cell cycle. Studies are needed to establish how much of the bivalent chromatin landscape is recapitulated in induced pluripotent cells. It is increasingly evident that a subpopulation of cancer cells exhibits partial recapitulation of bivalent chromatin domains. This oncofetal epigenetic control of gene expression may be a key determinant for cancer onset and progression.
BIVALENT EPIGENETIC LANDSCAPE
Histone posttranslational modifications as indicators and regulators of gene transcription.
A fundamental feature of any cell that undergoes significant phenotypic change is a permissive epigenetic environment that supports rapid changes in transcriptional output, which in turn contribute to diverse processes, including asymmetric cell division, cell fate/lineage commitment decisions, and developmentally regulated tissue formation (2, 25–27).
All four histone core proteins, histone 2A (H2A), H2B, H3, and H4, are subject to widespread covalent posttranslational modifications that include methylation, acetylation, phosphorylation, ribosylation, deimination, crotonylation, butyrylation, N-formylation, and proline isomerization (28–30). These modifications generally occur on lysine, arginine, serine, and threonine residues located at N-terminal “tails” of histone proteins, although several sites of modification in globular histone domains within the nucleosome core have also been identified (31). In general, the N-terminal regions of histone proteins contribute minimally to the overall structural integrity of the nucleosome (31, 32). Instead, these regions exert their regulatory activity through (i) regulation of higher-order chromatin structure by controlling interactions between nucleosomes, as well as between nucleosomes and DNA, and (ii) recruitment of regulatory proteins and complexes to specific genomic positions and combinations of posttranslational histone modifications.
Methylation of histone 3 lysine 4 (H3K4) is a highly evolutionarily conserved modification that has been linked to a number of biological processes, including DNA repair, meiotic recombination (33), heritability of transcriptional states through cell divisions (34), and, most notably, marking of transcriptionally active genes near the transcription start site (TSS) (35–38). In addition to the specific role of trimethylated H3K4 (H3K4me3) within promoter regions, other H3K4 modifications are more widespread; monomethylation (H3K4me1), dimethylation (H3K4me2), and H3K4 acetylation signals outside promoter regions have been identified as markers of functional regulatory elements, such as enhancers (37–39). The enzyme complexes that methylate H3K4 are highly conserved among eukaryotes, and mammals have six nonredundant complexes that regulate H3K4 methylation [Su(var)3 to Su(var)9, enhancer of zeste, Trithorax 1A (SET1A) and SET1B, and mixed-lineage leukemia 1 (MLL1) to MLL4] (40). Deletion(s) or disruption(s) of SET family genes through translocations (and subsequent loss of H3K4 methylation) leads to cancer in vertebrates (40) and life span changes in invertebrate models (41) (see “Formation and Regulation of Bivalent Chromatin Domains” below for a detailed discussion of these enzymes).
Although there is strong evidence that the presence of H3K4me3 is indicative of active gene expression, it is clear that the H3K4me3 modification on its own is not sufficient to regulate gene activation (42). Alteration of endogenous Drosophila histone H3.2 genes to carry a nonmethylatable residue in place of Lys4 (H3.2K4A) resulted in only minor effects on global transcriptional activity (43). Similarly, deletion of histone-lysine N-methyltransferase 2F (KMT2F) and KMT2G cofactor CFP1 (CXXC finger protein 1) resulted in minimal changes in transcript levels (44, 45), and deletion of SET1 in Saccharomyces cerevisiae had little impact on global transcription patterns (46) and, in some cases, resulted in a net upregulation of genes (47, 48). In addition, induced deposition of CFP1-dependent H3K4me3 at engineered, nonmethylated CpG islands was not sufficient to drive transcription in mouse fibroblasts (49). Taken together, these points suggest that the majority of H3K4me3 modified regulatory sites are not required for gene activation and that additional contextual cues are needed to regulate the initiation and maintenance of transcription (50).
Two marks are better than one: epigenetic bivalent domains provide transcriptional plasticity.
Trimethylated H3K27 (H3K27me3) is normally associated with regions of the genome that are transcriptionally repressed by polycomb repressor complex 2 (PRC2). In embryonic stem cells (ESCs), H3K27me3 colocalizes with H3K4me3 modifications primarily near the TSS of developmentally regulated genes (17, 51) to form distinct bivalent domains. These bivalent domains act to restrict the expression of developmental genes during self-renewal and poise developmentally important, lineage-specific transcriptional regulators for expression in response to differentiation cues (25, 52). One of the hallmarks of bivalent domains is that they are frequently associated with CpG islands (52). Several studies have shown that CpG domains, whether methylated or unmethylated, may be sufficient to recruit PRC2 to methylate H3K27 (24, 53, 54). The association of H3K27me3 with CpG islands would indicate a role for H3K27me3 as a marker for DNA methylation. Although embryonic stem cell DNA is largely devoid of methylation, DNA in promoters marked with H3K27me3 was more likely to become methylated during differentiation than that in promoters lacking H3K27me3 (55). However, some PRC2-mediated, tissue-specific bivalency was observed independently of CpG islands (26). Disruption of H3K27 methylation by replacement of wild-type (WT) nucleosomes with methylation-deficient H3K27R mutant nucleosomes resulted in developmental aberrations due to a loss of Hox gene repression, similarly to the effects observed with PRC2 loss of function or deletion of the functional catalytic subunit of PRC2: enhancer of zeste homolog 2 (EZH2) (56–58). It has been suggested that silencing of lineage-specific genes is mediated by PRC2 complexes and H3K27me3 in early development and is maintained by the recruitment of DNMT1 and DNA methylation at bivalent sites to compensate for the reduced expression of PRC2 components that is common in differentiated tissues (59, 60) (see “Formation and Regulation of Bivalent Chromatin Domains” below for a detailed discussion of how the PRC2 complex deposits H3K27me3).
Given the important role for bivalently marked promoters in stem cells, it has been suggested that these bivalent domains are linked to oncogenesis. Cancer cell lines have fewer bivalent domains than normal cells, with higher variation and lower stability of the cell lines (20). Although there is a reduction in the number of bivalent sites in cancer through loss of H3K27me3, DNA at bivalent promoters was observed to be hypermethylated (20, 61, 62), whereas in normal cells, those promoters tend to have lower levels of methylation, which is accompanied by increased gene expression (63, 64). In the context of cancer, bivalency seems to be crucial for keeping cells in a differentiated state as loss of bivalent control at the developmentally regulated HOX genes plays an important role in both oncogenesis and tumor suppression (22, 65).
The role of acetylation in promoting bivalent chromatin has been largely understudied. Dysregulation or aberrant deposition of histone acetylation is a common trait of several malignant tumors and has been a successful target for therapeutic intervention (66, 67). Many of the histone residues that are methylated (e.g., H3K4, H3K9, and H3K27) can alternatively be modified by acetylation, generally promoting gene activation and antagonizing gene repression (68–74). Specifically, H3K4 acetylation may play a role in establishing or maintaining bivalent domains in certain cell types. It has been demonstrated that deacetylation of H3K4ac by histone deacetylase 3 (HDAC3) is an important regulator of marker gene expression in hypoxia-induced epithelium-to-mesenchyme transition (EMT) (75). In breast cancer subtypes, H3K4ac was associated with estrogen-responsive and cancer-related genes at promoters that were also marked with H3K4me3 and H3K27me3 (76), suggesting that H3K4ac may be able to functionally replace H3K4me3 at certain promoters.
In search of true bivalency.
Despite the emerging body of literature suggesting a fundamental role(s) of bivalency in gene regulation, key questions remain. For example, a significant amount of effort has gone into identifying chromatin regions that are truly bivalent; however, technological limitations present challenges for definitively establishing bivalency. Each nucleosome contains two copies of the core histone proteins H2A, H2B, H3, and H4; thus, there could be comodification (H3K4me3/H3K27me3) of one or both histone copies in the nucleosome. Several studies have shown, however, that the majority of nucleosomes demonstrate asymmetric modification, with a H3K4me3 mark on one H3 tail and a H3K27me3 on the other (“sister”) tail (29, 77, 78). Although symmetric modification of H3K4me3 and H3K27me3 on the same histone tail is rarely observed, other modifications have been shown to be symmetrically distributed (79, 80). Further complicating the issue of identifying bivalent chromatin domains, traditional chromatin immunoprecipitation sequencing (ChIP-seq)-based techniques are limited by the contribution of mosaic signals from a mixed population of cells (such as tumors) and of allelic differences within the same cell.
However, recent studies using sequential or combinatorial ChIP strategies have identified chromatin regions that are occupied by H3K4me3/H3K27me3 as well as other combinations of posttranslational modifications on the same nucleosome (81, 82). Currently, these studies are limited in scope because sequential combinatorial ChIP requires much more material (and is thus not always possible to carry out in human tumor samples), as well as technical understanding and the ability to analyze the resulting data sets (thus restricting these approaches to use by a few highly experienced researchers). Recently, there has been a significant technological advance in the ability to examine bivalency at the single-molecule/single-nucleosome level. Shema et al. showed definitively bivalent nucleosomes purified from pluripotent stem cells (77). Although outside the native context of the cell nucleus, a combination of this approach with fluorescence-activated cell sorter analysis to purify various subpopulations of tumors should establish the extent to which bivalency is truly recapitulated in cancer.
FORMATION AND REGULATION OF BIVALENT CHROMATIN DOMAINS
The H3K4me3 and H3K27me3 modifications are catalyzed by different histone lysine methyltransferases (KMTs) that are individually associated with the Trithorax group (TrxG) and polycomb group (PcG) complexes, respectively. While the precise mechanism(s) that targets these complexes to specific chromatin elements for deposition of bivalent marks is not clear, much progress has been made in understanding the regulation of the methytransferases that mediate bivalent epigenetic modifications. Here, we briefly review the proteins that catalyze the deposition of bivalent chromatin with a focus on the factors that regulate these modifications.
Forming the “active” component of bivalency: H3K4 methylation and KMT2 histone lysine methyltransferases.
In mammals, methylation of histone H3 at lysine 4 is catalyzed by members of the histone-lysine N-methyltransferase 2 family (KMT2; also known as mixed-lineage leukemia [MLL]). The KMT2 enzymes are highly specific and catalyze the methyl transfer from S-adenosyl-l-methionine (SAM) to the ε-amino group of lysine 4 of histone H3, creating mono-, di-, and trimethylated H3K4me1/2/3 modifications. There are seven different KMT2 family members, six of which have been demonstrated to catalyze H3K4 methylation (KMT2A-2D and 2F-2G). Each KMT2 member has a characteristic catalytic SET domain (83–86). KMT2 enzymes form large multisubunit complexes that associate with distinctive sets of interacting proteins (87–92). Catalytic KMT2 complexes share a SET domain-mediated interaction with a group of proteins called the WRAD complex, which is composed of the following: WD repeat protein 5 (WDR5); retinoblastoma binding protein 5 (RBBP5); absent, small, homeotic discs 2-Like (ASH2L); and DumPY: shorter than wild-type 30 (DPY30) (93, 94). Depletion of individual WRAD subunits reduces KMT2 methyltransferase activity, indicating that the SET domain requires WRAD for methyltransferase activity (95–97). Interaction of KMT2 enzymes with WRAD is mediated via a WIN (WDR5 interacting) motif adjacent to the SET domain (98–102).
While different KMT2 members are important in regulating ESC pluripotency and early ESC differentiation, recent studies have clarified nonredundant functional roles of individual KMT2 proteins in mediating bivalent chromatin. Studies show that KMT2F and KMT2G (SETD1A and SETD1B, respectively) are likely responsible for H3K4me3 deposition globally at active promoters in ESCs (45). In contrast, KMT2B (MLL2) has been shown to serve as the principal H3K4 methyltransferase that marks bivalent promoters with H3K4me3 in mouse ESCs (103, 104). However, identification of the molecular mechanism(s) that guides methylation selectivity by the KMT2 enzymes remains elusive.
As discussed in the previous section, bivalent domains are frequently associated with CpG islands (105, 106). This observation may, in part, be explained by the finding that KMT2 proteins contain a DNA binding domain comprised of CXXC or zinc finger CXXC (ZF-CXXC) motifs that recognize hypomethylated CpGs and may play a role in targeting the DNA methylation complex(es) to bivalent domains (107). Furthermore, KMT2F and KMT2G are present in a complex with a CFP1 subunit which also includes a DNA binding domain that binds selectively to unmethylated CpGs (91). As bivalent domains are predominantly associated with CpG islands in ESCs, the CXXC portion of KMT2 proteins may come into play. Interestingly, CFP1 deficiency results in a drastic loss of H3K4 methylation levels at the promoters of highly expressed genes in ESCs but not at bivalent gene promoters (45). These findings together indicate that additional mechanisms are operative in directly linking DNA methylation with the enzymology of H3K4 trimethylation.
Forming the “repressive” component of bivalency: H3K27 trimethylation and polycomb repressive complex.
Trimethylation at H3K27 is catalyzed by PRC2 (19, 51, 105, 108–110), which is comprised of four core subunits: suppressor of zeste 12 protein homolog (SUZ12), embryonic ectoderm development (EED) protein, retinoblastoma binding protein 4 (RBBP4), and EZH1/EZH2 (EZH1/2). EZH1/2 is the catalytic subunit that serves as the histone lysine methyltransferase for H3K27 (reviewed in reference 111). In ESCs, genes with bivalent domains are targeted by pluripotency factors, including Oct4 and Nanog. Recruitment of PRC2 to these targets is directed by PRC2-associated protein Jarid2 or Mtf2 (110, 112–115). In addition to their role in establishing a bivalent chromatin landscape in pluripotent cells (19, 51, 108, 109), PRC2 and H3K27me3 are implicated in numerous physiological processes, including differentiation, tumorigenesis, senescence, and X-chromosome inactivation (53). Despite documentation of PRC2-mediated chromatin modification in ESCs, the determinants responsible for directing PRC2 occupancy at specific genomic elements remain a focus of active investigation.
There are also studies suggesting an interplay between PRC1 and PRC2 (116, 117). For example, a subset of genes repressed by PRC2 is also bound by the PRC1 polycomb complex (105). The recruitment and involvement of PRC1 at these loci may support gene silencing, at least in some biological contexts. The PRC1 complex catalyzes ubiquitination of histone H2A on lysine 119 (H2AK119ub), a modification that appears to contribute to transcriptional repression through RNA polymerase II pausing as well as by facilitating recruitment of PRC2. While PRC1 and PRC2 reinforce each other's recruitment to genes for repression, it is noteworthy that PRC1-mediated repression during early mouse development does not require activity of RING1B, a ubiquitin ligase and a core component of PRC1 (118). Unlike H2A ubiquitination, H2B ubiquitination (H2Bub1) has been shown to occur cotranscriptionally, is positively linked with gene expression, and accurately reflects the advance of RNA polymerase II in live cells (119, 120). Whether there is interplay between PRC1-mediated ubiquitination of H2A at lysine 119 and H2Bub at bivalent chromatin domains for tighter epigenetic control of gene expression remains to be established. Similarly, neither PRC1 nor PRC2 is required for recruitment of histone variant H2A.Z, which colocalizes with PRC2, to developmental genes in mouse embryonic stem cells (121). Together, these findings indicate that additional studies are required to identify the precise mechanistic underpinnings of transcriptional regulation by the two complexes.
REGULATORY CONSEQUENCES OF BIVALENT CHROMATIN
Research into the biological roles of bivalent chromatin is in its infancy. Establishing a direct link between bivalency and cellular functions has been difficult due to limitations of traditional genetic approaches that can modify only one histone modification at a time. Advances in clustered regularly interspaced short palindromic repeat (CRISPR)-based genome editing may lead to definitively understanding the functional consequences of bivalency. Nonetheless, some progress has been made in examining the effects of bivalency in biological control and in cancer. Here, we review examples that suggest a link between bivalency and cellular processes, including both transient (e.g., cell cycle control, responsiveness to cell signaling) and long-term (e.g., lineage commitment) processes, in normal and cancer cells (17, 20–26, 76, 122–125).
Transient gene regulation for lineage commitment: pluripotent cell cycle progression.
Pluripotent stem cells provide a unique model system to investigate the role(s) of bivalency in cell cycle and lineage commitment for several reasons. Pluripotent cells have (i) a largely bivalent chromatin landscape, (ii) unlimited proliferative potential, (iii) a shortened G1 phase (126, 127), and (iv) the ability to differentiate into any cell type. Recent findings from our group and others indicate that bivalent chromatin marks undergo dynamic reassignment during the pluripotent stem cell cycle. For example, bivalently marked genes that are maximally upregulated upon differentiation of pluripotent cells are enriched in H3K4me3 during mitosis, and after differentiation, H3K4me3 on these genes becomes cell cycle independent. Deposition of H3K4me3 is mediated by cell cycle-dependent recruitment to bivalent gene promoters of chromatin modifiers that are involved in H3K4 methylation and demethylation (125). Dalton and colleagues have shown that CDK2-dependent phosphorylation of the KMT2B (MLL2) histone methyltransferase results in its recruitment to developmental genes in G1 (128). Recruitment of KMT2B and deposition of H3K4me3 result in interactions between distal enhancers and proximal regulatory sites in promoter regions of developmental genes. These chromosomal interactions modify the higher-order architectural landscape and lead to brief activation of these genes that is restricted to G1 (128). Transient activation of developmental genes explains why stem cell differentiation is often initiated during the G1 cell cycle phase and provides a role for bivalent chromatin in cell fate determination following mitosis.
Role of bivalency in long-term regulation of cell phenotype: maintaining hematopoietic lineages.
Hematopoietic stem cells (HSCs) give rise to diverse cell types of both the myeloid and lymphoid lineages. The survival of this wide array of hematopoietic cells depends on a delicate balance between self-renewal and differentiation at each successive stage of lineage commitment. This balance is controlled in part by epigenetic regulation of gene expression.
As discussed above, PRC2 plays a key role in deposition of the H3K27me3 mark and thus establishes bivalent chromatin domains. Interestingly, HSCs exhibit monomethylation of, among other residues, H3K4 and H3K27 within enhancer regions of genes that are required for differentiation of HSCs to erythrocyte precursors. Furthermore, the majority of genes associated with trimethylation of these marks lose H3K4me3 after differentiation. These changes in the bivalent epigenetic landscape of HSCs are tightly associated with changes in gene expression (23), establishing a key role of bivalency in hematopoietic lineage commitment. It has also been shown that PRC1, the histone 2A (H2A) ubiquitin ligase, which functions as a transcriptional repressor, is required for maintenance of the self-renewing potential of HSCs.
Together, bivalent histone modifications play key roles in regulating the balance between HSC renewal and differentiation. They preserve the quiescent stem cell pool and support the development of the full repertoire of mature hematopoietic cells.
Mutations, loss of function, or mistargeting of key enzymes that mediate epigenetic modifications has been identified in both lymphoid and myeloid leukemias, further emphasizing the importance of epigenetic regulation in hematopoiesis. Knowledge of how these chromatin modifying enzymes and cofactors influence hematopoiesis presents opportunities for translational research (reviewed in “Bivalent Epigenetic Therapy” below).
Transient gene regulation in cancer: steroid signaling and bivalency.
The steroid hormone estradiol functions primarily by binding to the steroid nuclear receptors ESR1 (ERα) and ESR2 (ERβ), leading to mobilization of coregulators that form complexes altering chromatin structure in association with histone modifying enzymes (66, 67). While estradiol has biological roles in both normal proliferation and normal differentiation, providing the cellular plasticity needed for tissue remodeling throughout the reproductive years, unregulated estradiol exposure can result in constitutive activation of metabolic and cell cycle regulatory genes and increases the risk of breast cancer. There is strong clinical and epidemiological data linking estrogen exposure with an increased risk of developing breast cancer (68–74).
Epigenetic changes have been increasingly identified in the development of breast cancer. For example, the role of DNA methylation has been extensively studied and DNA hypermethylation is frequently associated with silencing of tumor suppressor genes, steroid receptor genes, and genes involved in cell cycle regulation (129–132). Additionally, alterations in patterns of histone modifications by several histone modifying enzymes, including KMT2B, KMT2F (H3K4), and EZH2 (H3K27me3), have been linked with cancer initiation and progression (133–136). Overexpression of the histone methyltransferase EZH2 is well documented in breast cancer, with increasing levels being associated with more-aggressive metastatic breast cancers (137–139). Overexpression of wild-type EZH2 has also been reported to serve as a transcriptional activator and/or repressor in ESR1-positive breast cancer by modulating estrogen signaling (140–144).
About 75% to 80% of breast cancers are ESR1 positive and these tumors generally exhibit increased levels of histone modifications compared to tumors in patients with a poorer prognosis (145). Epigenetic studies assessing the levels of H3K4me3 and H3K27me3 by our group, using three breast cancer cell lines, provide evidence of higher levels of H3K27me3 in two basal breast cancer cell lines (MCF10A and MDA-MB-231) with relatively unchanged H3K4me3 levels overall among the three cell lines (76). Importantly, analysis of these data reveals a bivalent epigenetic signature that is more prominent in the ESR1-positive MCF7 cell line and is similar to that identified in pluripotent stem cells (Fig. 2).
FIG 2.
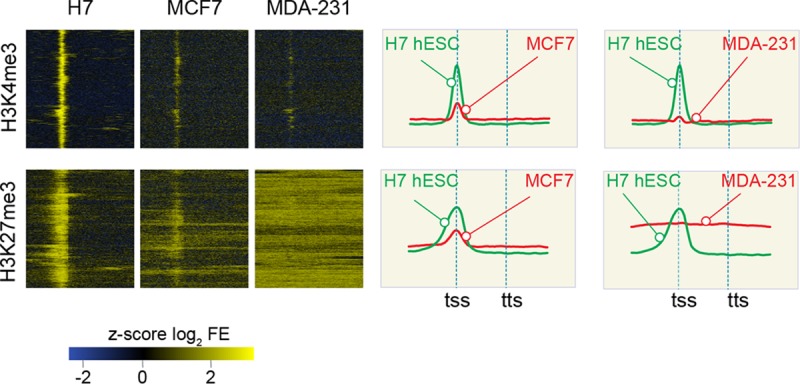
Partial recapitulation of the oncofetal epigenetic landscape, i.e., reemergence of genes bivalently marked in stem cells, in cancer cells. Heat maps (left) and summarization profiles (right) of H3K4me3 (top) and H3K27me3 (bottom) enrichment in the H7-hESC cell line and in the MCF7 and MDA-231 cell lines in 1,172 H7-hESC bivalent genes are shown. The gene transcription start site (tss) and transcription termination site (tts) are indicated. Note the similarity between MCF7 and H7-hESC with respect to H3K27me3 near promoters and the lack of the same in MDA-MB-231. FE, fold enrichment. (The data in the figure are adapted and modified from reference 76 with permission.)
The concept of partial recapitulation of a stem cell-like epigenetic signature, i.e., oncofetal epigenetic control, in cancer cells is further reinforced by single-cell studies that show the presence of a quiescent subpopulation that exhibits high levels of pluripotency transcription factors Nanog and Oct4 in MCF7 cells (146–149). Additional studies are required to establish whether this oncofetal epigenetic signature is contributed by the cancer stem cell subpopulation and reflects the dedifferentiated nature of cancer cells and whether it could be used to identify specific and selective diagnostic and therapeutic targets (76).
Bivalency for regulation of tumor phenotype: epithelium-to-mesenchyme transition in breast cancer.
The epithelium-to-mesenchyme transition (EMT) is a cellular transformation by which epithelial cells lose their cellular polarity and cell-cell adhesion properties and become mesenchyme-like, with enhanced motility and invasive capacity (150, 151). Importantly, this change in the cell state is not permanent. Cancer cells that have gone through the EMT process in the primary tumor may later revert to the epithelial state through a mesenchyme-epithelium transition (152, 153). Therefore, the EMT program is coordinately regulated; multiple signaling pathways, as well as several epigenetic mechanisms, are involved in activation/repression of EMT (154, 155).
The E-cadherin (CDH1) gene provides a model system to examine the coordinated nature of epigenetic regulation during EMT. The CDH1 gene is downregulated during EMT by the binding of the Zeb, Snail, Slug, or Twist transcription factor to its gene promoter region (156). The repression of the CDH1 gene by CpG hypermethylation is commonly observed in several human cancers, including breast, bladder, liver, gastric, and prostate cancer (157–160). In breast cancer, hypermethylation on the CDH1 promoter is positively correlated with EMT (161). Expression of CDH1 is also regulated by histone modification. For example, hepatocyte nuclear factor 3 (HNF3) promotes CDH1 expression and inhibits metastasis by cooperating with the p300 acetyltransferase in cancer cell lines (156). In contrast, the CDH1 gene is also controlled through deacetylation. Snail homologue 1 (SNAI 1) binds to its promoter and recruits HDACs to deacetylate the promoter, thus suppressing the expression of the gene (162, 163). SNAI 1 also recruits LSD1 (lysine-specific histone demethylase 1) or SUV39H1 (suppressor of variegation 3-9 homolog 1, histone methyltransferase) to the CDH1 promoter, where the enzyme either removes methyl groups from H3K4 (resulting in H3K4me2 enrichment) or adds methyl groups to H3K9 (resulting in H3K9me3 enrichment), respectively (164, 165). EZH2, a component of the PRC2 complex, was also found to be associated with silencing of the E-cadherin gene by H3K27me3 during tumor progression (166). These findings establish a complex but precise regulatory network regulating CDH1 expression during EMT and suggest that this gene may frequently be in a bivalent configuration.
BIVALENT EPIGENETIC THERAPY
Mutations in proteins responsible for mediating epigenetic changes are widespread in cancer and present attractive opportunities for therapeutic targets (see Table 1 for examples). Therapies have been developed to target gain-of-function mutations in DNA methylases, as well as histone modifiers and readers, and to impact DNA methylation, histone modification, and reading and to epigenetically impact the tumor phenotype. These include LSD1 inhibitors, EZH2 inhibitors, PRMT5 (protein arginine methyltransferase 5) inhibitors, DOT1L (disruptor of telomeric silencing 1-like) inhibitors, and BET (bromodomain and extraterminal) protein inhibitors (167). Loss-of-function mutations are more challenging to impact therapeutically; however, with a deep understanding of the pathways involved, drug development can focus on targeting synthetic lethal interactions. Synthetic lethality exploits the relationship between two genes wherein loss of one gene does not lead to cell death but loss of the two genes simultaneously does result in cell death. This strategy has been successfully applied to target loss-of-function mutations in chromatin remodeling complexes (for example, the synthetic lethal effect of an EZH2 inhibitor in SNF5-mutated rhabdoid tumors) and histone methyltransferases (168, 169).
TABLE 1.
Examples of the steadily growing repertoire of epigenetic therapies for a variety of malignancies that are currently being used or tested in clinical trialsa
| Drug | Target | Epigenetic class | Disease(s) treated |
|---|---|---|---|
| Azacitidine | DNA methyltransferase (DNMT1) | DNA methylation | AML |
| Decitadine guadecitadine | DNA methyltransferase (DNMT1) | DNA methylation | Recurrent fallopian tube carcinoma; recurrent ovarian carcinoma; recurrent primary peritoneal carcinoma; papillary thyroid carcinoma; follicular thyroid carcinoma; non-small-cell lung cancer; colorectal cancer; metastatic pancreatic adenocarcinoma |
| Vorinostat | Histone deacetylase | Histone modification | Cutaneous T-cell lymphoma; AML; glioma; prostate cancer |
| EPZ-5676 (pinometostat) | DOT1L (a histone methyltransferase) | Histone modification | MLL-rearranged leukemia |
| Tranylcypromine | LSD1 (a histone demethylase) | Histone modification | AML; refractory solid tumors |
| CPI-0610 | BET family proteins | Histone reader | Lymphoma; multiple myeloma; acute leukemias |
| GSK525762 | BET family proteins | Histone reader | NUT midline carcinoma |
| TEN-010 | BET family proteins | Histone reader | AML; refractory solid tumors |
| AG-120 | Mutant IDH1 | Metabolic pathway | AML; glioma; cholangiocarcinoma; refractory solid tumors |
| Tazemetostat | EZH2 (a histone methyltransferase) | Histone modification (direct); chromatin remodeling complexes (indirect) | B-cell lymphoma; mesothelioma; synovial sarcoma; rhabdoid tumors; INI1-negative tumors |
AML, acute myeloid leukemia.
Knowing that chromatin bivalency contributes in a significant way to oncogenesis, specific efforts are under way to target the enzymes that mediate construction of the bivalent signature in cancer. As described above, the members of the MLL (KMT) family of complexes are key mediators of H3K4 methylation. Oncogenic MLL translocations are frequently seen in acute leukemias and are particularly prevalent in a devastating form of infantile acute lymphoblastic leukemia (ALL). The majority of MLL translocations result in loss of the catalytic SET domain and occur with fusion partners capable of recruiting the H4K79 lysine methyltransferase DOT1L. Efforts to target the oncogenic MLL fusions have been largely focused on the development of DOT1L inhibitors, such as EPZ-5676, which is currently in clinical trials (170). It has additionally been found that H2B E3 ligase RNF20 is required for MLL-rearranged leukemogenesis, wherein there are increased levels of H3Bub in MLL target genes correlating with sites of H3K79me2, making RNF20 an attractive epigenetic target in MLL-rearranged leukemias (171). Interestingly, cancers characterized by an oncogenic MLL fusion must retain one wild-type MLL allele, as the normal H3K4 methyltransferase activity is essential for oncogenesis. Therefore, other efforts to target this epigenetic disease are focused on the function of WT MLL. There has been some investigation into using LSD1 inhibitors to restore the balance between MLL and LSD1 in H3K4 methylation (172). Other groups are using compounds that specifically inhibit the SET-WDR5 interaction in order to inhibit the WT MLL complex (42, 173).
As described above, PRC2, and its catalytic subunit EZH2, are largely responsible for maintaining the “repressive” half of the bivalent signature. Overexpression of and gain-of-function mutations in EZH2 have been described in a variety of cancers, most notably B-cell-derived hematopoietic malignancies (174). Efforts undertaken to inhibit EZH2 have resulted in the identification of tazemetostat and CPI-1205, small-molecule inhibitors of EZH2 which are currently being tested in clinical trials for treatment of a variety of PRC2-dependent malignancies, including B-cell lymphomas. These examples highlight the therapeutic potential of targeting the enzymology for bivalent chromatin.
BIVALENT CHROMATIN: NOVEL REGULATORY DIMENSIONS
Emerging literature on bivalent chromatin and its pivotal role in regulation of key biological processes indicates additional regulatory functions for bivalency in regulating cancer phenotypes. Here, we highlight two key areas where additional research will yield a mechanistic understanding of bivalency in establishing cancer phenotypes and may provide options for therapeutic intervention.
Oncofetal gene expression.
Oncofetal genes have been defined as highly expressed and stringently regulated during embryonic development and inactivated in most adult tissues but aberrantly reexpressed in many cancer types (reviewed in references 175 and 176). Notable examples of oncofetal gene products include alpha fetoprotein (AFP), carcinoembryonic antigen (CEA), high mobility group AT-hook protein 2 (HMGA2), abnormal cell lineage protein 28 homologue (LIN-28), insulin-like growth factor 2 mRNA binding protein (IGF2BP), 5T4 oncofetal trophoblast glycoprotein, sal-like protein 4 (SALL4), receptor tyrosine kinase-like orphan receptor 1 (ROR1), forkhead box M1 (FOXM1), NODAL, and teratocarcinoma-derived growth factor 1 (TDGF1; also called “Cripto growth factor”) (reviewed in references 177 to 186). Collectively, oncofetal proteins contribute to tumor phenotypes through regulation of stemness, proliferation, invasion, and metastasis. Decades of research have established a number of oncofetal proteins as effective biomarkers for cancer aggressiveness (reviewed in references 177 to 180, 183, and 185). Unfortunately, there has been limited success in using oncofetal proteins as therapeutic targets. Furthermore, the mechanistic underpinnings of their reexpression or regulation in cancer are not well understood (187–191).
Recent studies have provided some insights into the regulation of oncofetal genes in cancer. A number of oncofetal genes have been identified as downstream targets of let-7, a tumor suppressor microRNA that plays key roles in a variety of cancer types (192). Targets of let-7 include the well-characterized LIN-28 and HMGA2 oncofetal genes and the IGF2BP1 to -3 family member genes (178, 184, 193, 194). Each of these genes is involved in growth at the organism level, as well as in regulation of metabolism during development (178, 184, 193, 194). In cancer, each of these genes is reactivated and contributes to the tumor phenotype. For example, the LIN28B gene is associated with neuroblastoma (195), the HMGA2 gene is expressed in aggressive ovarian cancer (196), and IGF2BP family member genes are reactivated in a number of cancers, including breast, cervical, and hepatic cancers (197). These findings indicate that a common mechanism is operative in posttranscriptional regulation of several oncofetal genes and suggest that a common mechanism may also regulate transcriptional reactivation of these genes across cancer types.
While upstream transcriptional regulators of some oncofetal genes are known (198–201), surprisingly little is known about epigenetic regulation of their expression. Studies examining cancer-associated reexpression of various oncofetal genes have mostly focused on DNA methylation. For example, the AFP, CEA, SALL4, and Nodal genes are all unmethylated during development, become methylated in adult tissues, and are unmethylated or hypomethylated in several cancer types (202–205). As we discuss above in detail, many aspects of epigenetic control, including chromatin bivalency, are reacquired at the onset and during progression of cancer. We propose the hypothesis that once oncofetal genes become unmethylated at the onset of cancer, they acquire bivalent chromatin marks, poising them for increased expression throughout the progression of cancer. This hypothesis has the potential to explain the correlation between increased levels of various oncofetal proteins and tumor aggressiveness. It is equally possible that genes encoding oncofetal proteins are either transiently marked by bivalent histone modifications or not bivalently marked at all. Instead, their upstream regulators are bivalently marked, thus allowing the expression of these genes in cancer cells. Lack of a true cancer progression model also limits the identification of the transient nature of bivalency. Additional studies are needed to establish whether oncofetal genes acquire a bivalent chromatin landscape during tumorigenesis. Such a bivalent landscape of oncofetal genes will provide options for regulatory plasticity and may offer selective and specific therapeutic targets.
Cancer stem cells.
The quiescent nature of cancer stem cells results in escape of this cell population from the conventional chemotherapeutic approaches that often target actively dividing cancer cells (206–208). It will be important to investigate whether cancer stem cells—the cell population that represents a dedifferentiated state—reacquire bivalency in regulatory regions of oncogenes. There are some examples that point to such a mechanism. For example, the gene encoding E-cadherin permits dynamic regulation of gene expression during the EMT. Within the CD24−/CD44+ stem cell-enriched populations of human breast cancer cells, E-cadherin expression has been silenced and its promoter contains bivalent H3K4me3 and H3K27me3 modification. However, in CD24+ cells, which express high levels of E-cadherin, only the H3K4me3 mark is present on the E-cadherin promoter (209). To allow this dynamic switching between cell states, the key EMT transcription factor ZEB1 is also marked with bivalent chromatin (210). Systematic genome-wide studies using a purified cancer stem cell population are needed to examine how much of the bivalent chromatin landscape of pluripotent stem cells is reestablished in cancer stem cells. Such studies may also reveal the key differences between the two cell types in control of proliferation. Epigenetic therapies can then be developed to target bivalent chromatin and may be more effective in eradicating cancer stem cells.
OPTIONS AND OPPORTUNITIES
The complexity of gene regulation is reflected by the establishment and maintenance of bivalent chromatin through the interplay of various histone modifications, ranging from methylation of H3K4 and H3K27 residues to sequential addition of posttranslational modifications to or removal of posttranslational modifications from adjacent residues and/or histones, as well the coordination between these modifications and DNA methylation. Both redundancy and selectivity of enzymes and cofactors are required for deposition or removal of these marks and further highlight the intricate nature of bivalency. An additional level of control is presented by activation or repression of other genomic targets (mRNAs, long noncoding RNAs, and microRNAs) in a context-dependent manner that is dictated by extracellular signaling. Taking the findings together, this complexity provides options for cellular plasticity in a variety of biological or disease-related contexts.
However, several key issues remain unaddressed and require additional studies. (i) Experimental evidence discussed in this review provides a preliminary insight into the role of bivalency in cell fate decisions in pluripotent cells. Additional evidence is needed to establish whether bivalency is a prerequisite for lineage commitment and whether distinct gene subsets are bivalently marked prior to acquisition of distinct cell phenotypes. This knowledge will illuminate the significance of partial recapitulation of bivalency in some cancer types. (ii) In addition to H3K4me3/H3K27me3-mediated bivalency, other histone modifications may poise genes for rapid transcription. For example, trophoblast and endodermal stem cells demonstrate a pattern of bivalent H3K4me3/H3K9me3 modifications on lineage-specific genes that function independently of H3K4me3/H3K27me3 to regulate differentiation (211). Similarly, in mesenchymal stem cells, H3K4/H3K9me3 bivalent chromatin at several key adipogenic genes was demonstrated to keep lineage-specific genes poised, but repressed, until commitment to adipogenesis took place (21). It remains unclear how prevalent such “noncanonical bivalent domains” are and whether these are associated with gene regulation in specific cellular pathways. (iii) Partial recapitulation of pluripotent bivalency in some cancer types (termed “oncofetal epigenetic control” in this review) is an exciting recent observation. Further research is required to establish whether the same set of genes reacquires bivalency across tumor types. (iv) Our group has noted that the numbers of bivalent genes differed within the same cancer type as well as across cancer types. This variability may reflect the heterogeneous nature of tumor cell population and/or variability in cancer stem cell populations. It will be important to examine bivalency at the single-cell level in purified cancer stem cell populations. (v) Is oncofetal epigenetic control a consequence of or a prerequisite for cancer onset and progression? Despite the open-ended questions concerning these issues, bivalency is emerging as a key determinant of cellular plasticity, and its partial recapitulation in a number of cancer types offers new avenues for identification of biomarkers and for development of therapeutic approaches that are selective and specific.
ACKNOWLEDGMENTS
The work in our laboratories that is discussed in this review was supported by a Pfizer Investigator-Initiated Research Award (WS2049100), by an NIH Program Project grant (P01 CA 082834), by an NCI Consortium grant (U01 CA 196383), and by the Charlotte Perelman Fund for Cancer Research.
Biographies

Sayyed K. Zaidi received his Ph.D. from University of the Punjab, Pakistan, and postdoctoral training from University of Massachusetts Medical School, Worcester, MA, where he was a research assistant professor from 2008 to 2012. He is currently an associate professor in the Department of Biochemistry at the University of Vermont and an associate director for regional collaborations in the University of Vermont Cancer Center. Dr. Zaidi's research interests include epigenetic control of gene expression, mechanisms of leukemogenesis, mitotic gene bookmarking, and higher-order nuclear organization.

Seth E. Frietze received his Ph.D. from Harvard University. He served both as a postdoctoral fellow at the University of Southern California and as a visiting faculty fellow at Los Alamos National Laboratories. Dr. Frietze is currently an assistant professor in the Department of Medical Laboratory Sciences at the University of Vermont. His research interests over the last 10 years have focused on understanding the role of altered epigenetic programs during tumorigenesis.

Jonathan A. Gordon carried out his doctoral research at the University of Western Ontario, London, Ontario, Canada. He was previously a research assistant professor, Department of Cell Biology, University of Massachusetts Medical School, Worcester, MA. Currently, Dr. Gordon is an assistant professor in the Department of Biochemistry at University of Vermont. Dr. Gordon's research has focused on understanding the mechanisms of bone marrow-derived mesenchymal stem cell (MSC) commitment to osteogenic lineages and their involvement in malignant disease processes, such as metastatic niches and tumor microenvironments. The differentiation of MSCs as a normal process (such as bone development) or as an aberrant process due to association with neoplastic tissues is regulated by transcriptional regulators, early chromatin remodeling events, and epigenetic modifiers. These epigenetic processes have obvious implications for controlling MSC commitment, potentially leading to novel therapies to increase bone mass under chronic disease conditions or to interfere with unwanted differentiation in cancer-associated environments.

Jessica L. Heath received her bachelor of science degree in biology from Cornell University and her doctor of medicine degree from the State University of New York at Stony Brook. She then completed her pediatric residency training at Albany Medical Center, followed by fellowships in pediatric hematology-oncology and intensive research training in pediatric oncology at Duke University. She was a medical instructor in the Division of Pediatric Hematology-Oncology at Duke University prior to accepting her current position as assistant professor of pediatrics and biochemistry at the University of Vermont in 2015. Dr. Heath has been studying the molecular underpinnings of aggressive pediatric leukemias, with a focus on epigenetic mechanisms of disease, for the past 7 years. Her focus is on translational cancer biology, with the goal of developing novel targeted therapeutics for high-risk pediatric leukemias.

Terri Messier is a research analyst in the Department of Biochemistry at the University of Vermont and in the University of Vermont Cancer Center. She received her undergraduate education at the University of Vermont in the Department of Laboratory Sciences. Her biomedical research experience consists of over 30 years of work in both the academic and biotechnology sectors, including projects in several laboratories at the University of Vermont in Burlington, and as a group leader at Acadia Pharmaceuticals in San Diego, CA. Her research interests include development of improved detection and treatment modalities in cancer by studying both genetic and epigenetic changes that result in transcriptional reprograming of cancer cells.

Deli Hong received a master of science degree from the Department of Chemistry and Biochemistry, Worcester Polytechnic Institute. Between 2009 and 2010, he worked as a Research Associate in Kendall Knight's laboratory in the Department of Biochemistry and Molecular Pharmacology, University of Massachusetts Medical School. Currently, Mr. Hong is a Ph.D. student in the Graduate Program of Cell Biology, University of Massachusetts Medical School. His work is focused on understanding the mechanisms of breast cancer progression. Epithelium-to-mesenchyme transition (EMT) has been implicated as an essential process in breast cancer metastasis and chemoresistance. Understanding the epigenetic silencing of E-cadherin, a key mechanism activating EMT, can help the development of promising targets for the prevention of metastasis.

Joseph R. Boyd received his master's degree in bioinformatics and computational biology from Worcester Polytechnic Institute. He is currently the bioinformatician for the Stein-Lian research group at the University of Vermont. He has been processing and analyzing transcriptome sequencing (RNA-seq), ChIP-seq, and comparative high-throughput chromosome conformation capture (Hi-C) datasets for three years. His primary interest is in constructing application-specific interactive visualizations to enable biologists to analyze their omics-scale data. Joseph enjoys the challenges posed by the task of distilling meaningful information from the deluge of data available with the advent of next-generation sequencing.

Mingu Kang received his bachelor's degree in biology at the University of Oregon. After graduation, he worked for a year as a laboratory technician at the Korea Research Institute of Bioscience and Biotechnology (KRIBB). He received a master's degree in biochemistry and molecular medicine at George Washington University. Currently, he is a Ph.D. student in the Cellular, Molecular, and Biomedical Sciences Program at the University of Vermont. His primary focus is in epigenetic cell reprogramming related to the functions of various histone modifications during cancer progression. Specifically, a unique epigenetic feature known as bivalency provides flexibility in gene expression. He has been investigating the resulting control of cellular plasticity as a key regulatory mechanism in the reprograming of cancer cells in the Stein-Lian laboratory for a year.

Anthony N. Imbalzano is professor of biochemistry and molecular pharmacology and associate dean, Office for Postdoctoral Scholars, at the University of Massachusetts Medical School. He and his fellow laboratory staff members have had a long-term interest in the roles of SWI/SNF chromatin remodeling enzymes in controlling gene expression, chromatin structure, and high-order genome organization in differentiation and in cancer. Tony received his Ph.D. in microbiology and molecular genetics with Neal DeLuca at Harvard University and did postdoctoral studies with Bob Kingston at Massachusetts General Hospital.

Jane B. Lian is a professor in the biochemistry Department at the University of Vermont and codirector of a Translational Research Program of the University of Vermont Cancer Center. Dr. Lian received her Ph.D. from Boston University Medical School and held faculty positions at the Children's Hospital, Harvard Medical School, and at University of Massachusetts Medical School. Her research programs for over 3 decades have investigated the genetic and epigenetic mechanisms regulating osteoblast growth and differentiation. Dr. Lian contributed to discovery of the bone-specific c protein osteocalcin and the Runx2 transcription factor, essential for bone formation. Her investigations established Runx2 as a mediator of signaling pathways activated during osteogenesis and in tumor cells causing metastatic bone disease. Dr. Lian's studies have received recognition in the form of several prestigious awards, including awards from the American Academy of Orthopedic Surgeons and from the American Society for Bone and Mineral Res She has published over 400 peer-reviewed articles and numerous reviews.

Janet L. Stein received her Ph.D. in chemistry from Princeton University and pursued her postdoctoral training in the Department of Biochemistry and Molecular Biology at the University of Florida. She has held faculty positions in the Department of Immunology and Medical Microbiology at the University of Florida and the Department of Cell Biology at University of Massachusetts Medical School. She is currently a Professor in the Department of Biochemistry at the University of Vermont and a program leader at the UVM Cancer Center. Her major research contributions to science include insights into alterations in nuclear organization and chromatin in cancer and histone gene regulation and its influence on cell cycle control, the abbreviated cell cycle that characterizes pluripotent stem cells, and mechanisms for transcription factor targeting and mitotic retention at gene loci.

Gary S. Stein trained in biology and pathology and is dedicated to basic, translational, and clinical cancer investigation. He pioneered discovery of mechanisms controlling proliferation and differentiation, emphasizing compromised genetic and epigenetic regulation in cancer. His focus is on mechanisms and biomarkers, including noncoding RNAs associated with prostate, leukemia, and breast cancer prevention, and early detection, treatment, and survivorship. A priority is characterizing genetic and epigenetic regulation mediating cell cycle control and the abbreviated pluripotent cell cycle in human embryonic stem cells, induced pluripotent cells, and cancer stem cells. He is investigating breast and prostate tumor metastasis to bone, including microRNA-mediated control, and is defining mechanisms that govern the combinatorial organization and assembly of regulatory machinery in nuclear microenvironments and epigenetic control of cell fate and lineage commitment. Dr. Stein is currently the chairperson of the Department of Biochemistry at the University of Vermont and the Director of the University of Vermont Cancer Center.
REFERENCES
- 1.Chi P, Allis CD, Wang GG. 2010. Covalent histone modifications—miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer 10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldberg AD, Allis CD, Bernstein E. 2007. Epigenetics: a landscape takes shape. Cell 128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 3.Strahl BD, Allis CD. 2000. The language of covalent histone modifications. Nature 403:41–45. [DOI] [PubMed] [Google Scholar]
- 4.John S, Workman JL. 1998. Bookmarking genes for activation in condensed mitotic chromosomes. Bioessays 20:275–279. [DOI] [PubMed] [Google Scholar]
- 5.Zaidi SK, Young DW, Montecino MA, Lian JB, van Wijnen AJ, Stein JL, Stein GS. 2010. Mitotic bookmarking of genes: a novel dimension to epigenetic control. Nat Rev Genet 11:583–589. doi: 10.1038/nrg2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kadauke S, Blobel GA. 2013. Mitotic bookmarking by transcription factors. Epigenetics Chromatin 6:6. doi: 10.1186/1756-8935-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lodhi N, Ji Y, Tulin A. 2016. Mitotic bookmarking: maintaining post-mitotic reprogramming of transcription reactivation. Curr Mol Biol Rep 2:10–15. doi: 10.1007/s40610-016-0029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He S, Dunn KL, Espino PS, Drobic B, Li L, Yu J, Sun J-M, Chen HY, Pritchard S, Davie JR. 2008. Chromatin organization and nuclear microenvironments in cancer cells. J Cell Biochem 104:2004–2015. doi: 10.1002/jcb.21485. [DOI] [PubMed] [Google Scholar]
- 9.Zaidi SK, Young DW, Javed A, Pratap J, Montecino M, van Wijnen A, Lian JB, Stein JL, Stein GS. 2007. Nuclear microenvironments in biological control and cancer. Nat Rev Cancer 7:454–463. doi: 10.1038/nrc2149. [DOI] [PubMed] [Google Scholar]
- 10.Fritz AJ, Barutcu AR, Martin-Buley L, van Wijnen AJ, Zaidi SK, Imbalzano AN, Lian JB, Stein JL, Stein GS. 2016. Chromosomes at work: organization of chromosome territories in the interphase nucleus. J Cell Biochem 117:9–19. doi: 10.1002/jcb.25280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dundr M. 2012. Nuclear bodies: multifunctional companions of the genome. Curr Opin Cell Biol 24:415–422. doi: 10.1016/j.ceb.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schneider R, Grosschedl R. 2007. Dynamics and interplay of nuclear architecture, genome organization, and gene expression. Genes Dev 21:3027–3043. doi: 10.1101/gad.1604607. [DOI] [PubMed] [Google Scholar]
- 13.Stein GS, Montecino M, van Wijnen AJ, Stein JL, Lian JB. 2000. Nuclear structure-gene expression interrelationships: implications for aberrant gene expression in cancer. Cancer Res 60:2067–2076. [PubMed] [Google Scholar]
- 14.Malyavantham KS, Bhattacharya S, Berezney R. 2010. The architecture of functional neighborhoods within the mammalian cell nucleus. Adv Enzyme Regul 50:126–134. doi: 10.1016/j.advenzreg.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang H, Sabari BR, Garcia BA, Allis CD, Zhao Y. 2014. SnapShot: histone modifications. Cell 159:458–458.e451. doi: 10.1016/j.cell.2014.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruthenburg AJ, Li H, Patel DJ, Allis CD. 2007. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol 8:983–994. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES. 2006. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 18.Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, Bell GW, Otte AP, Vidal M, Gifford DK, Young RA, Jaenisch R. 2006. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 19.Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K, Koseki H, Fuchikami T, Abe K, Murray HL, Zucker JP, Yuan B, Bell GW, Herbolsheimer E, Hannett NM, Sun K, Odom DT, Otte AP, Volkert TL, Bartel DP, Melton DA, Gifford DK, Jaenisch R, Young RA. 2006. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bernhart SH, Kretzmer H, Holdt LM, Jühling F, Ammerpohl O, Bergmann AK, Northoff BH, Doose G, Siebert R, Stadler PF, Hoffmann S. 23 November 2016. Changes of bivalent chromatin coincide with increased expression of developmental genes in cancer. Sci Rep doi: 10.1038/srep37393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsumura Y, Nakaki R, Inagaki T, Yoshida A, Kano Y, Kimura H, Tanaka T, Tsutsumi S, Nakao M, Doi T, Fukami K, Osborne TF, Kodama T, Aburatani H, Sakai J. 2015. H3K4/H3K9me3 bivalent chromatin domains targeted by lineage-specific DNA methylation pauses adipocyte differentiation. Mol Cell 60:584–596. doi: 10.1016/j.molcel.2015.10.025. [DOI] [PubMed] [Google Scholar]
- 22.Hahn MA, Li AX, Wu X, Yang R, Drew DA, Rosenberg DW, Pfeifer GP. 2014. Loss of the polycomb mark from bivalent promoters leads to activation of cancer-promoting genes in colorectal tumors. Cancer Res 74:3617–3629. doi: 10.1158/0008-5472.CAN-13-3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cui K, Zang C, Roh TY, Schones DE, Childs RW, Peng W, Zhao K. 2009. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell 4:80–93. doi: 10.1016/j.stem.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodriguez J, Munoz M, Vives L, Frangou CG, Groudine M, Peinado MA. 2008. Bivalent domains enforce transcriptional memory of DNA methylated genes in cancer cells. Proc Natl Acad Sci U S A 105:19809–19814. doi: 10.1073/pnas.0810133105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Voigt P, Tee WW, Reinberg D. 2013. A double take on bivalent promoters. Genes Dev 27:1318–1338. doi: 10.1101/gad.219626.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jadhav U, Nalapareddy K, Saxena M, O'Neill NK, Pinello L, Yuan GC, Orkin SH, Shivdasani RA. 2016. Acquired tissue-specific promoter bivalency is a basis for PRC2 necessity in adult cells. Cell 165:1389–1400. doi: 10.1016/j.cell.2016.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Margueron R, Reinberg D. 2010. Chromatin structure and the inheritance of epigenetic information. Nat Rev Genet 11:285–296. doi: 10.1038/nrg2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen I, Poreba E, Kamieniarz K, Schneider R. 2011. Histone modifiers in cancer: friends or foes? Genes Cancer 2:631–647. doi: 10.1177/1947601911417176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Voigt P, LeRoy G, Drury WJ III, Zee BM, Son J, Beck DB, Young NL, Garcia BA, Reinberg D. 2012. Asymmetrically modified nucleosomes. Cell 151:181–193. doi: 10.1016/j.cell.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, Lu Z, Ye Z, Zhu Q, Wysocka J, Ye Y, Khochbin S, Ren B, Zhao Y. 2011. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tropberger P, Schneider R. 2013. Scratching the (lateral) surface of chromatin regulation by histone modifications. Nat Struct Mol Biol 20:657–661. doi: 10.1038/nsmb.2581. [DOI] [PubMed] [Google Scholar]
- 32.Morales V, Richard-Foy H. 2000. Role of histone N-terminal tails and their acetylation in nucleosome dynamics. Mol Cell Biol 20:7230–7237. doi: 10.1128/MCB.20.19.7230-7237.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Acquaviva L, Szekvolgyi L, Dichtl B, Dichtl BS, de La Roche Saint Andre C, Nicolas A, Geli V. 2013. The COMPASS subunit Spp1 links histone methylation to initiation of meiotic recombination. Science 339:215–218. doi: 10.1126/science.1225739. [DOI] [PubMed] [Google Scholar]
- 34.Muramoto T, Müller I, Thomas G, Melvin A, Chubb JR. 2010. Methylation of H3K4 is required for inheritance of active transcriptional states. Curr Biol 20:397–406. doi: 10.1016/j.cub.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 35.Eissenberg JC, Shilatifard A. 2010. Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev Biol 339:240–249. doi: 10.1016/j.ydbio.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shilatifard A. 2008. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr Opin Cell Biol 20:341–348. doi: 10.1016/j.ceb.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. 2007. High-resolution profiling of histone methylations in the human genome. Cell 129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 38.Guillemette B, Drogaris P, Lin HH, Armstrong H, Hiragami-Hamada K, Imhof A, Bonneil E, Thibault P, Verreault A, Festenstein RJ. 2011. H3 lysine 4 is acetylated at active gene promoters and is regulated by H3 lysine 4 methylation. PLoS Genet 7:e1001354. doi: 10.1371/journal.pgen.1001354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 40.Shilatifard A. 2012. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem 81:65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greer EL, Maures TJ, Hauswirth AG, Green EM, Leeman DS, Maro GS, Han S, Banko MR, Gozani O, Brunet A. 2010. Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature 466:383–387. doi: 10.1038/nature09195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cao F, Townsend EC, Karatas H, Xu J, Li L, Lee S, Liu L, Chen Y, Ouillette P, Zhu J, Hess JL, Atadja P, Lei M, Qin ZS, Malek S, Wang S, Dou Y. 2014. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol Cell 53:247–261. doi: 10.1016/j.molcel.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hödl M, Basler K. 2012. Transcription in the absence of histone H3.2 and H3K4 methylation. Curr Biol 22:2253–2257. doi: 10.1016/j.cub.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 44.Clouaire T, Webb S, Bird A. 2014. Cfp1 is required for gene expression-dependent H3K4 trimethylation and H3K9 acetylation in embryonic stem cells. Genome Biol 15:451. doi: 10.1186/s13059-014-0451-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clouaire T, Webb S, Skene P, Illingworth R, Kerr A, Andrews R, Lee JH, Skalnik D, Bird A. 2012. Cfp1 integrates both CpG content and gene activity for accurate H3K4me3 deposition in embryonic stem cells. Genes Dev 26:1714–1728. doi: 10.1101/gad.194209.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T. 2002. Active genes are tri-methylated at K4 of histone H3. Nature 419:407–411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- 47.Venkatasubrahmanyam S, Hwang WW, Meneghini MD, Tong AH, Madhani HD. 2007. Genome-wide, as opposed to local, antisilencing is mediated redundantly by the euchromatic factors Set1 and H2A.Z. Proc Natl Acad Sci U S A 104:16609–16614. doi: 10.1073/pnas.0700914104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Margaritis T, Oreal V, Brabers N, Maestroni L, Vitaliano-Prunier A, Benschop JJ, van Hooff S, van Leenen D, Dargemont C, Geli V, Holstege FC. 2012. Two distinct repressive mechanisms for histone 3 lysine 4 methylation through promoting 3′-end antisense transcription. PLoS Genet 8:e1002952. doi: 10.1371/journal.pgen.1002952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, Edsall LE, Kuan S, Luu Y, Klugman S, Antosiewicz-Bourget J, Ye Z, Espinoza C, Agarwahl S, Shen L, Ruotti V, Wang W, Stewart R, Thomson JA, Ecker JR, Ren B. 2010. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell 6:479–491. doi: 10.1016/j.stem.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Howe FS, Fischl H, Murray SC, Mellor J. 2017. Is H3K4me3 instructive for transcription activation? Bioessays 39:1–12. doi: 10.1002/bies.201600095. [DOI] [PubMed] [Google Scholar]
- 51.Pan G, Tian S, Nie J, Yang C, Ruotti V, Wei H, Jonsdottir GA, Stewart R, Thomson JA. 2007. Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell 1:299–312. doi: 10.1016/j.stem.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 52.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, Lee W, Mendenhall E, O'Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE. 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Riising EM, Comet I, Leblanc B, Wu X, Johansen JV, Helin K. 2014. Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol Cell 55:347–360. doi: 10.1016/j.molcel.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 54.Jermann P, Hoerner L, Burger L, Schubeler D. 2014. Short sequences can efficiently recruit histone H3 lysine 27 trimethylation in the absence of enhancer activity and DNA methylation. Proc Natl Acad Sci U S A 111:E3415–E3421. doi: 10.1073/pnas.1400672111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mohn F, Weber M, Rebhan M, Roloff TC, Richter J, Stadler MB, Bibel M, Schubeler D. 2008. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell 30:755–766. doi: 10.1016/j.molcel.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 56.Pengelly AR, Copur O, Jackle H, Herzig A, Muller J. 2013. A histone mutant reproduces the phenotype caused by loss of histone-modifying factor Polycomb. Science 339:698–699. doi: 10.1126/science.1231382. [DOI] [PubMed] [Google Scholar]
- 57.O'Carroll D, Erhardt S, Pagani M, Barton SC, Surani MA, Jenuwein T. 2001. The polycomb-group gene Ezh2 is required for early mouse development. Mol Cell Biol 21:4330–4336. doi: 10.1128/MCB.21.13.4330-4336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Simon JA, Kingston RE. 2013. Occupying chromatin: Polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol Cell 49:808–824. doi: 10.1016/j.molcel.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rose NR, Klose RJ. 2014. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim Biophys Acta 1839:1362–1372. doi: 10.1016/j.bbagrm.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Neri F, Krepelova A, Incarnato D, Maldotti M, Parlato C, Galvagni F, Matarese F, Stunnenberg HG, Oliviero S. 2013. Dnmt3L antagonizes DNA methylation at bivalent promoters and favors DNA methylation at gene bodies in ESCs. Cell 155:121–134. doi: 10.1016/j.cell.2013.08.056. [DOI] [PubMed] [Google Scholar]
- 61.Lange CP, Campan M, Hinoue T, Schmitz RF, van der Meulen-de Jong AE, Slingerland H, Kok PJ, van Dijk CM, Weisenberger DJ, Shen H, Tollenaar RA, Laird PW. 2012. Genome-scale discovery of DNA-methylation biomarkers for blood-based detection of colorectal cancer. PLoS One 7:e50266. doi: 10.1371/journal.pone.0050266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gal-Yam EN, Egger G, Iniguez L, Holster H, Einarsson S, Zhang X, Lin JC, Liang G, Jones PA, Tanay A. 2008. Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc Natl Acad Sci U S A 105:12979–12984. doi: 10.1073/pnas.0806437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu YC, Pfenning AR, Wang X, Claussnitzer M, Liu Y, Coarfa C, Harris RA, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, Hawkins RD, Lister R, Hong C, Gascard P, Mungall AJ, Moore R, Chuah E, Tam A, Canfield TK, Hansen RS, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh KH, Feizi S, Karlic R, Kim AR, et al. 2015. Integrative analysis of 111 reference human epigenomes. Nature 518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, Gnirke A, Jaenisch R, Lander ES. 2008. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454:766–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dhar SS, Lee SH, Chen K, Zhu G, Oh W, Allton K, Gafni O, Kim YZ, Tomoiga AS, Barton MC, Hanna JH, Wang Z, Li W, Lee MG. 2016. An essential role for UTX in resolution and activation of bivalent promoters. Nucleic Acids Res 44:3659–3674. doi: 10.1093/nar/gkv1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Collingwood TN, Urnov FD, Wolffe AP. 1999. Nuclear receptors: coactivators, corepressors and chromatin remodeling in the control of transcription. J Mol Endocrinol 23:255–275. doi: 10.1677/jme.0.0230255. [DOI] [PubMed] [Google Scholar]
- 67.Dilworth FJ, Chambon P. 2001. Nuclear receptors coordinate the activities of chromatin remodeling complexes and coactivators to facilitate initiation of transcription. Oncogene 20:3047–3054. doi: 10.1038/sj.onc.1204329. [DOI] [PubMed] [Google Scholar]
- 68.Chuffa LG, Lupi-Junior LA, Costa AB, Amorim JP, Seiva FR. 2017. The role of sex hormones and steroid receptors on female reproductive cancers. Steroids 118:93–108. doi: 10.1016/j.steroids.2016.12.011. [DOI] [PubMed] [Google Scholar]
- 69.Hill J, Hodsdon W. 2014. In utero exposure and breast cancer development: an epigenetic perspective. J Environ Pathol Toxicol Oncol 33:239–245. doi: 10.1615/JEnvironPatholToxicolOncol.2014011005. [DOI] [PubMed] [Google Scholar]
- 70.Liu Q, Wuu J, Lambe M, Hsieh SF, Ekbom A, Hsieh CC. 2002. Transient increase in breast cancer risk after giving birth: postpartum period with the highest risk (Sweden). Cancer Causes Control 13:299–305. doi: 10.1023/A:1015287208222. [DOI] [PubMed] [Google Scholar]
- 71.Mann M, Cortez V, Vadlamudi RK. 2011. Epigenetics of estrogen receptor signaling: role in hormonal cancer progression and therapy. Cancers (Basel) 3:1691–1707. doi: 10.3390/cancers3021691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Russo J, Russo IH. 2006. The role of estrogen in the initiation of breast cancer. J Steroid Biochem Mol Biol 102:89–96. doi: 10.1016/j.jsbmb.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stossi F, Likhite VS, Katzenellenbogen JA, Katzenellenbogen BS. 2006. Estrogen-occupied estrogen receptor represses cyclin G2 gene expression and recruits a repressor complex at the cyclin G2 promoter. J Biol Chem 281:16272–16278. doi: 10.1074/jbc.M513405200. [DOI] [PubMed] [Google Scholar]
- 74.Osmanbeyoglu HU, Lu KN, Oesterreich S, Day RS, Benos PV, Coronnello C, Lu X. 2013. Estrogen represses gene expression through reconfiguring chromatin structures. Nucleic Acids Res 41:8061–8071. doi: 10.1093/nar/gkt586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu MZ, Tsai YP, Yang MH, Huang CH, Chang SY, Chang CC, Teng SC, Wu KJ. 2011. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol Cell 43:811–822. doi: 10.1016/j.molcel.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 76.Messier TL, Boyd JR, Gordon JAR, Stein JL, Lian JB, Stein GS. 2016. Oncofetal epigenetic bivalency in breast cancer cells: H3K4 and H3K27 tri-methylation as a biomarker for phenotypic plasticity. J Cell Physiol 231:2474–2481. doi: 10.1002/jcp.25359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shema E, Jones D, Shoresh N, Donohue L, Ram O, Bernstein BE. 2016. Single-molecule decoding of combinatorially modified nucleosomes. Science 352:717–721. doi: 10.1126/science.aad7701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shliaha PV, Baird MA, Nielsen MM, Gorshkov V, Bowman AP, Kaszycki JL, Jensen ON, Shvartsburg AA. 2017. Characterization of complete histone tail proteoforms using differential ion mobility spectrometry. Anal Chem 89:5461–5466. doi: 10.1021/acs.analchem.7b00379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Drogaris P, Villeneuve V, Pomies C, Lee EH, Bourdeau V, Bonneil E, Ferbeyre G, Verreault A, Thibault P. 2012. Histone deacetylase inhibitors globally enhance h3/h4 tail acetylation without affecting h3 lysine 56 acetylation. Sci Rep 2:220. doi: 10.1038/srep00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mao H, Han G, Xu L, Zhu D, Lin H, Cao X, Yu Y, Chen CD. 2015. Cis-existence of H3K27me3 and H3K36me2 in mouse embryonic stem cells revealed by specific ions of isobaric modification chromatogram. Stem Cell Res Ther 6:132. doi: 10.1186/s13287-015-0131-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kinkley S, Helmuth J, Polansky JK, Dunkel I, Gasparoni G, Frohler S, Chen W, Walter J, Hamann A, Chung HR. 2016. reChIP-seq reveals widespread bivalency of H3K4me3 and H3K27me3 in CD4(+) memory T cells. Nat Commun 7:12514. doi: 10.1038/ncomms12514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sadeh R, Launer-Wachs R, Wandel H, Rahat A, Friedman N. 2016. Elucidating combinatorial chromatin states at single-nucleosome resolution. Mol Cell 63:1080–1088. doi: 10.1016/j.molcel.2016.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Del Rizzo PA, Trievel RC. 2011. Substrate and product specificities of SET domain methyltransferases. Epigenetics 6:1059–1067. doi: 10.4161/epi.6.9.16069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang X, Novera W, Zhang Y, Deng LW. 2017. MLL5 (KMT2E): structure, function, and clinical relevance. Cell Mol Life Sci 74:2333–2344. doi: 10.1007/s00018-017-2470-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Madan V, Madan B, Brykczynska U, Zilbermann F, Hogeveen K, Dohner K, Dohner H, Weber O, Blum C, Rodewald HR, Sassone-Corsi P, Peters AH, Fehling HJ. 2009. Impaired function of primitive hematopoietic cells in mice lacking the Mixed-Lineage-Leukemia homolog MLL5. Blood 113:1444–1454. doi: 10.1182/blood-2008-02-142638. [DOI] [PubMed] [Google Scholar]
- 86.Mas-Y-Mas S, Barbon M, Teyssier C, Déméné H, Carvalho JE, Bird LE, Lebedev A, Fattori J, Schubert M, Dumas C, Bourguet W, le Maire A. 2016. The Human Mixed Lineage Leukemia 5 (MLL5), a Sequentially and Structurally Divergent SET Domain-Containing Protein with No Intrinsic Catalytic Activity. PLoS One 11:e0165139. doi: 10.1371/journal.pone.0165139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nakamura T, Mori T, Tada S, Krajewski W, Rozovskaia T, Wassell R, Dubois G, Mazo A, Croce CM, Canaani E. 2002. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell 10:1119–1128. doi: 10.1016/S1097-2765(02)00740-2. [DOI] [PubMed] [Google Scholar]
- 88.Cho YW, Hong T, Hong S, Guo H, Yu H, Kim D, Guszczynski T, Dressler GR, Copeland TD, Kalkum M, Ge K. 2007. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J Biol Chem 282:20395–20406. doi: 10.1074/jbc.M701574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Goo YH, Sohn YC, Kim DH, Kim SW, Kang MJ, Jung DJ, Kwak E, Barlev NA, Berger SL, Chow VT, Roeder RG, Azorsa DO, Meltzer PS, Suh PG, Song EJ, Lee KJ, Lee YC, Lee JW. 2003. Activating signal cointegrator 2 belongs to a novel steady-state complex that contains a subset of trithorax group proteins. Mol Cell Biol 23:140–149. doi: 10.1128/MCB.23.1.140-149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA, Kay GF, Hayward NK, Hess JL, Meyerson M. 2004. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell 13:587–597. doi: 10.1016/S1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- 91.Lee JH, Skalnik DG. 2005. CpG-binding protein (CXXC finger protein 1) is a component of the mammalian Set1 histone H3-Lys4 methyltransferase complex, the analogue of the yeast Set1/COMPASS complex. J Biol Chem 280:41725–41731. doi: 10.1074/jbc.M508312200. [DOI] [PubMed] [Google Scholar]
- 92.Lee JH, Tate CM, You JS, Skalnik DG. 2007. Identification and characterization of the human Set1B histone H3-Lys4 methyltransferase complex. J Biol Chem 282:13419–13428. doi: 10.1074/jbc.M609809200. [DOI] [PubMed] [Google Scholar]
- 93.van Nuland R, Smits AH, Pallaki P, Jansen PW, Vermeulen M, Timmers HT. 2013. Quantitative dissection and stoichiometry determination of the human SET1/MLL histone methyltransferase complexes. Mol Cell Biol 33:2067–2077. doi: 10.1128/MCB.01742-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Odho Z, Southall SM, Wilson JR. 2010. Characterization of a novel WDR5-binding site that recruits RbBP5 through a conserved motif to enhance methylation of histone H3 lysine 4 by mixed lineage leukemia protein-1. J Biol Chem 285:32967–32976. doi: 10.1074/jbc.M110.159921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Roguev A, Schaft D, Shevchenko A, Pijnappel WW, Wilm M, Aasland R, Stewart AF. 2001. The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J 20:7137–7148. doi: 10.1093/emboj/20.24.7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dehé PM, Dichtl B, Schaft D, Roguev A, Pamblanco M, Lebrun R, Rodríguez-Gil A, Mkandawire M, Landsberg K, Shevchenko A, Shevchenko A, Rosaleny LE, Tordera V, Chávez S, Stewart AF, Géli V. 2006. Protein interactions within the Set1 complex and their roles in the regulation of histone 3 lysine 4 methylation. J Biol Chem 281:35404–35412. doi: 10.1074/jbc.M603099200. [DOI] [PubMed] [Google Scholar]
- 97.Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, Allis CD, Roeder RG. 2006. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol 13:713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- 98.Zhang P, Lee H, Brunzelle JS, Couture JF. 2012. The plasticity of WDR5 peptide-binding cleft enables the binding of the SET1 family of histone methyltransferases. Nucleic Acids Res 40:4237–4246. doi: 10.1093/nar/gkr1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Alicea-Velázquez NL, Shinsky SA, Loh DM, Lee JH, Skalnik DG, Cosgrove MS. 2016. Targeted disruption of the interaction between WD-40 repeat protein 5 (WDR5) and mixed lineage leukemia (MLL)/SET1 family proteins specifically inhibits MLL1 and SETd1A methyltransferase complexes. J Biol Chem 291:22357–22372. doi: 10.1074/jbc.M116.752626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Patel A, Dharmarajan V, Cosgrove MS. 2008. Structure of WDR5 bound to mixed lineage leukemia protein-1 peptide. J Biol Chem 283:32158–32161. doi: 10.1074/jbc.C800164200. [DOI] [PubMed] [Google Scholar]
- 101.Li Y, Han J, Zhang Y, Cao F, Liu Z, Li S, Wu J, Hu C, Wang Y, Shuai J, Chen J, Cao L, Li D, Shi P, Tian C, Zhang J, Dou Y, Li G, Chen Y, Lei M. 2016. Structural basis for activity regulation of MLL family methyltransferases. Nature 530:447–452. doi: 10.1038/nature16952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhou P, Wang Z, Yuan X, Zhou C, Liu L, Wan X, Zhang F, Ding X, Wang C, Xiong S, Wang Z, Yuan J, Li Q, Zhang Y. 2013. Mixed lineage leukemia 5 (MLL5) protein regulates cell cycle progression and E2F1-responsive gene expression via association with host cell factor-1 (HCF-1). J Biol Chem 288:17532–17543. doi: 10.1074/jbc.M112.439729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hu D, Garruss AS, Gao X, Morgan MA, Cook M, Smith ER, Shilatifard A. 2013. The Mll2 branch of the COMPASS family regulates bivalent promoters in mouse embryonic stem cells. Nat Struct Mol Biol 20:1093–1097. doi: 10.1038/nsmb.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hu D, Gao X, Cao K, Morgan MA, Mas G, Smith ER, Volk AG, Bartom ET, Crispino JD, Di Croce L, Shilatifard A. 2017. Not all H3K4 methylations are created equal: Mll2/COMPASS dependency in primordial germ cell specification. Mol Cell 65:460–475 e466. doi: 10.1016/j.molcel.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS, Adli M, Kasif S, Ptaszek LM, Cowan CA, Lander ES, Koseki H, Bernstein BE. 2008. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet 4:e1000242. doi: 10.1371/journal.pgen.1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lee SM, Lee J, Noh KM, Choi WY, Jeon S, Oh GT, Kim-Ha J, Jin Y, Cho SW, Kim YJ. 2017. Intragenic CpG islands play important roles in bivalent chromatin assembly of developmental genes. Proc Natl Acad Sci U S A 114:E1885–E1894. doi: 10.1073/pnas.1613300114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Allen MD, Grummitt CG, Hilcenko C, Min SY, Tonkin LM, Johnson CM, Freund SM, Bycroft M, Warren AJ. 2006. Solution structure of the nonmethyl-CpG-binding CXXC domain of the leukaemia-associated MLL histone methyltransferase. EMBO J 25:4503–4512. doi: 10.1038/sj.emboj.7601340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhao XD, Han X, Chew JL, Liu J, Chiu KP, Choo A, Orlov YL, Sung WK, Shahab A, Kuznetsov VA, Bourque G, Oh S, Ruan Y, Ng HH, Wei CL. 2007. Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell 1:286–298. doi: 10.1016/j.stem.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 109.Mikkelsen TS, Hanna J, Zhang X, Ku M, Wernig M, Schorderet P, Bernstein BE, Jaenisch R, Lander ES, Meissner A. 2008. Dissecting direct reprogramming through integrative genomic analysis. Nature 454:49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Landeira D, Sauer S, Poot R, Dvorkina M, Mazzarella L, Jorgensen HF, Pereira CF, Leleu M, Piccolo FM, Spivakov M, Brookes E, Pombo A, Fisher C, Skarnes WC, Snoek T, Bezstarosti K, Demmers J, Klose RJ, Casanova M, Tavares L, Brockdorff N, Merkenschlager M, Fisher AG. 2010. Jarid2 is a PRC2 component in embryonic stem cells required for multi-lineage differentiation and recruitment of PRC1 and RNA polymerase II to developmental regulators. Nat Cell Biol 12:618–624. doi: 10.1038/ncb2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Holoch D, Margueron R. 5 May 2017. Mechanisms regulating PRC2 recruitment and enzymatic activity. Trends Biochem Sci doi: 10.1016/j.tibs.2017.04.003. [DOI] [PubMed] [Google Scholar]
- 112.Peng JC, Valouev A, Swigut T, Zhang J, Zhao Y, Sidow A, Wysocka J. 2009. Jarid2/Jumonji coordinates control of PRC2 enzymatic activity and target gene occupancy in pluripotent cells. Cell 139:1290–1302. doi: 10.1016/j.cell.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shen X, Kim W, Fujiwara Y, Simon MD, Liu Y, Mysliwiec MR, Yuan GC, Lee Y, Orkin SH. 2009. Jumonji modulates polycomb activity and self-renewal versus differentiation of stem cells. Cell 139:1303–1314. doi: 10.1016/j.cell.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pasini D, Cloos PA, Walfridsson J, Olsson L, Bukowski JP, Johansen JV, Bak M, Tommerup N, Rappsilber J, Helin K. 2010. JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature 464:306–310. doi: 10.1038/nature08788. [DOI] [PubMed] [Google Scholar]
- 115.Zhang Z, Jones A, Sun CW, Li C, Chang CW, Joo HY, Dai Q, Mysliwiec MR, Wu LC, Guo Y, Yang W, Liu K, Pawlik KM, Erdjument-Bromage H, Tempst P, Lee Y, Min J, Townes TM, Wang H. 2011. PRC2 complexes with JARID2, MTF2, and esPRC2p48 in ES cells to modulate ES cell pluripotency and somatic cell reprogramming. Stem Cells 29:229–240. doi: 10.1002/stem.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.de Napoles M, Mermoud JE, Wakao R, Tang YA, Endoh M, Appanah R, Nesterova TB, Silva J, Otte AP, Vidal M, Koseki H, Brockdorff N. 2004. Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev Cell 7:663–676. doi: 10.1016/j.devcel.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 117.Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, Zhang Y. 2004. Role of histone H2A ubiquitination in Polycomb silencing. Nature 431:873–878. doi: 10.1038/nature02985. [DOI] [PubMed] [Google Scholar]
- 118.Illingworth RS, Moffat M, Mann AR, Read D, Hunter CJ, Pradeepa MM, Adams IR, Bickmore WA. 2015. The E3 ubiquitin ligase activity of RING1B is not essential for early mouse development. Genes Dev 29:1897–1902. doi: 10.1101/gad.268151.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fuchs G, Shema E, Vesterman R, Kotler E, Wolchinsky Z, Wilder S, Golomb L, Pribluda A, Zhang F, Haj-Yahya M, Feldmesser E, Brik A, Yu X, Hanna J, Aberdam D, Domany E, Oren M. 2012. RNF20 and USP44 regulate stem cell differentiation by modulating H2B monoubiquitylation. Mol Cell 46:662–673. doi: 10.1016/j.molcel.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fuchs G, Hollander D, Voichek Y, Ast G, Oren M. 2014. Cotranscriptional histone H2B monoubiquitylation is tightly coupled with RNA polymerase II elongation rate. Genome Res 24:1572–1583. doi: 10.1101/gr.176487.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Illingworth RS, Botting CH, Grimes GR, Bickmore WA, Eskeland R. 2012. PRC1 and PRC2 are not required for targeting of H2A.Z to developmental genes in embryonic stem cells. PLoS One 7:e34848. doi: 10.1371/journal.pone.0034848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Harikumar A, Meshorer E. 2015. Chromatin remodeling and bivalent histone modifications in embryonic stem cells. EMBO Rep 16:1609–1619. doi: 10.15252/embr.201541011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Xie W, Ling T, Zhou Y, Feng W, Zhu Q, Stunnenberg HG, Grummt I, Tao W. 2012. The chromatin remodeling complex NuRD establishes the poised state of rRNA genes characterized by bivalent histone modifications and altered nucleosome positions. Proc Natl Acad Sci U S A 109:8161–8166. doi: 10.1073/pnas.1201262109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cheong CY, Lufkin T. 2010. Transcriptional repression in ES cells. J Cell Biochem 110:288–293. [DOI] [PubMed] [Google Scholar]
- 125.Grandy RA, Whitfield TW, Wu H, Fitzgerald MP, VanOudenhove JJ, Zaidi SK, Montecino MA, Lian JB, van Wijnen AJ, Stein JL, Stein GS. 2015. Genome-wide studies reveal that H3K4me3 modification in bivalent genes is dynamically regulated during the pluripotent cell cycle and stabilized upon differentiation. Mol Cell Biol 36:615–627. doi: 10.1128/MCB.00877-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kapinas K, Grandy R, Ghule P, Medina R, Becker K, Pardee A, Zaidi SK, Lian J, Janet van Wijnen A, Stein G. 2013. The abbreviated pluripotent cell cycle. J Cell Physiol 228:9–20. doi: 10.1002/jcp.24104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Becker KA, Stein JL, Lian JB, van Wijnen AJ, Stein GS. 2010. Human embryonic stem cells are pre-mitotically committed to self-renewal and acquire a lengthened G1 phase upon lineage programming. J Cell Physiol 222:103–110. doi: 10.1002/jcp.21925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Singh AM, Sun Y, Li L, Zhang W, Wu T, Zhao S, Qin Z, Dalton S. 2015. Cell-cycle control of bivalent epigenetic domains regulates the exit from pluripotency. Stem Cell Rep 5:323–336. doi: 10.1016/j.stemcr.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.van Hoesel AQ, Sato Y, Elashoff DA, Turner RR, Giuliano AE, Shamonki JM, Kuppen PJ, van de Velde CJ, Hoon DS. 2013. Assessment of DNA methylation status in early stages of breast cancer development. Br J Cancer 108:2033–2038. doi: 10.1038/bjc.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Widschwendter M, Jones PA. 2002. DNA methylation and breast carcinogenesis. Oncogene 21:5462–5482. doi: 10.1038/sj.onc.1205606. [DOI] [PubMed] [Google Scholar]
- 131.Bistulfi G, Pozzi S, Ren M, Rossetti S, Sacchi N. 2006. A repressive epigenetic domino effect confers susceptibility to breast epithelial cell transformation: implications for predicting breast cancer risk. Cancer Res 66:10308–10314. doi: 10.1158/0008-5472.CAN-06-1052. [DOI] [PubMed] [Google Scholar]
- 132.Cheng AS, Culhane AC, Chan MW, Venkataramu CR, Ehrich M, Nasir A, Rodriguez BA, Liu J, Yan PS, Quackenbush J, Nephew KP, Yeatman TJ, Huang TH. 2008. Epithelial progeny of estrogen-exposed breast progenitor cells display a cancer-like methylome. Cancer Res 68:1786–1796. doi: 10.1158/0008-5472.CAN-07-5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yoo KH, Hennighausen L. 2012. EZH2 methyltransferase and H3K27 methylation in breast cancer. Int J Biol Sci 8:59–65. doi: 10.7150/ijbs.8.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Salz T, Deng C, Pampo C, Siemann D, Qiu Y, Brown K, Huang S. 2015. Histone methyltransferase hSETD1A is a novel regulator of metastasis in breast cancer. Mol Cancer Res 13:461–469. doi: 10.1158/1541-7786.MCR-14-0389. [DOI] [PubMed] [Google Scholar]
- 135.Lim S, Janzer A, Becker A, Zimmer A, Schule R, Buettner R, Kirfel J. 2010. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 31:512–520. doi: 10.1093/carcin/bgp324. [DOI] [PubMed] [Google Scholar]
- 136.Kim JH, Sharma A, Dhar SS, Lee SH, Gu B, Chan CH, Lin HK, Lee MG. 2014. UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res 74:1705–1717. doi: 10.1158/0008-5472.CAN-13-1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, Sabel MS, Livant D, Weiss SJ, Rubin MA, Chinnaiyan AM. 2003. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A 100:11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Collett K, Eide GE, Arnes J, Stefansson IM, Eide J, Braaten A, Aas T, Otte AP, Akslen LA. 2006. Expression of enhancer of zeste homologue 2 is significantly associated with increased tumor cell proliferation and is a marker of aggressive breast cancer. Clin Cancer Res 12:1168–1174. doi: 10.1158/1078-0432.CCR-05-1533. [DOI] [PubMed] [Google Scholar]
- 139.Bachmann IM, Halvorsen OJ, Collett K, Stefansson IM, Straume O, Haukaas SA, Salvesen HB, Otte AP, Akslen LA. 2006. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol 24:268–273. doi: 10.1200/JCO.2005.01.5180. [DOI] [PubMed] [Google Scholar]
- 140.Hervouet E, Cartron PF, Jouvenot M, Delage-Mourroux R. 2013. Epigenetic regulation of estrogen signaling in breast cancer. Epigenetics 8:237–245. doi: 10.4161/epi.23790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hwang C, Giri VN, Wilkinson JC, Wright CW, Wilkinson AS, Cooney KA, Duckett CS. 2008. EZH2 regulates the transcription of estrogen-responsive genes through association with REA, an estrogen receptor corepressor. Breast Cancer Res Treat 107:235–242. doi: 10.1007/s10549-007-9542-7. [DOI] [PubMed] [Google Scholar]
- 142.Shi B, Liang J, Yang X, Wang Y, Zhao Y, Wu H, Sun L, Zhang Y, Chen Y, Li R, Zhang Y, Hong M, Shang Y. 2007. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol Cell Biol 27:5105–5119. doi: 10.1128/MCB.00162-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yang X, Karuturi RK, Sun F, Aau M, Yu K, Shao R, Miller LD, Tan PB, Yu Q. 2009. CDKN1C (p57) is a direct target of EZH2 and suppressed by multiple epigenetic mechanisms in breast cancer cells. PLoS One 4:e5011. doi: 10.1371/journal.pone.0005011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Du J, Li L, Ou Z, Kong C, Zhang Y, Dong Z, Zhu S, Jiang H, Shao Z, Huang B, Lu J. 2012. FOXC1, a target of polycomb, inhibits metastasis of breast cancer cells. Breast Cancer Res Treat 131:65–73. doi: 10.1007/s10549-011-1396-3. [DOI] [PubMed] [Google Scholar]
- 145.Elsheikh SE, Green AR, Rakha EA, Powe DG, Ahmed RA, Collins HM, Soria D, Garibaldi JM, Paish CE, Ammar AA, Grainge MJ, Ball GR, Abdelghany MK, Martinez-Pomares L, Heery DM, Ellis IO. 2009. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res 69:3802–3809. doi: 10.1158/0008-5472.CAN-08-3907. [DOI] [PubMed] [Google Scholar]
- 146.Anjanappa M, Cardoso A, Cheng L, Mohamad S, Gunawan A, Rice S, Dong Y, Li L, Sandusky GE, Srour EF, Nakshatri H. 2017. Individualized breast cancer characterization through single-cell analysis of tumor and adjacent normal cells. Cancer Res 77:2759–2769. doi: 10.1158/0008-5472.CAN-16-3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Heselmeyer-Haddad K, Berroa Garcia LY, Bradley A, Ortiz-Melendez C, Lee WJ, Christensen R, Prindiville SA, Calzone KA, Soballe PW, Hu Y, Chowdhury SA, Schwartz R, Schaffer AA, Ried T. 2012. Single-cell genetic analysis of ductal carcinoma in situ and invasive breast cancer reveals enormous tumor heterogeneity yet conserved genomic imbalances and gain of MYC during progression. Am J Pathol 181:1807–1822. doi: 10.1016/j.ajpath.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, Zhou A, Eyob H, Balakrishnan S, Wang C-Y, Yaswen P, Goga A, Werb Z. 2015. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 526:131–135. doi: 10.1038/nature15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Akrap N, Andersson D, Bom E, Gregersson P, Stahlberg A, Landberg G. 2016. Identification of distinct breast cancer stem cell populations based on single-cell analyses of functionally enriched stem and progenitor pools. Stem Cell Rep 6:121–136. doi: 10.1016/j.stemcr.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Thiery JP. 2002. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 151.Radisky DC. 2005. Epithelial-mesenchymal transition. J Cell Sci 118:4325–4326. doi: 10.1242/jcs.02552. [DOI] [PubMed] [Google Scholar]
- 152.Tsai AG, Chen DM, Lin M, Hsieh JCF, Okitsu CY, Taghva A, Shibata D, Hsieh C-L. 25 January 2012. Heterogeneity and randomness of DNA methylation patterns in human embryonic stem cells. DNA Cell Biol doi: 10.1089/dna.2011.1477. [DOI] [PubMed] [Google Scholar]
- 153.Ocaña OH, Córcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, Barrallo-Gimeno A, Cano A, Nieto MA. 2012. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 22:709–724. doi: 10.1016/j.ccr.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 154.Serrano-Gomez SJ, Maziveyi M, Alahari SK. 2016. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol Cancer 15:18. doi: 10.1186/s12943-016-0502-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ye X, Weinberg RA. 2015. Epithelial-mesenchymal plasticity: a central regulator of cancer progression. Trends Cell Biol 25:675–686. doi: 10.1016/j.tcb.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Liu YN, Lee WW, Wang CY, Chao TH, Chen Y, Chen JH. 2005. Regulatory mechanisms controlling human E-cadherin gene expression. Oncogene 24:8277–8290. doi: 10.1038/sj.onc.1208991. [DOI] [PubMed] [Google Scholar]
- 157.Yoshiura K, Kanai Y, Ochiai A, Shimoyama Y, Sugimura T, Hirohashi S. 1995. Silencing of the E-cadherin invasion-suppressor gene by CpG methylation in human carcinomas. Proc Natl Acad Sci U S A 92:7416–7419. doi: 10.1073/pnas.92.16.7416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Graff JR, Herman JG, Lapidus RG, Chopra H, Xu R, Jarrard DF, Isaacs WB, Pitha PM, Davidson NE, Baylin SB. 1995. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res 55:5195–5199. [PubMed] [Google Scholar]
- 159.Bornman DM, Mathew S, Alsruhe J, Herman JG, Gabrielson E. 2001. Methylation of the E-cadherin gene in bladder neoplasia and in normal urothelial epithelium from elderly individuals. Am J Pathol 159:831–835. doi: 10.1016/S0002-9440(10)61758-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tamura G, Yin J, Wang S, Fleisher AS, Zou T, Abraham JM, Kong D, Smolinski KN, Wilson KT, James SP, Silverberg SG, Nishizuka S, Terashima M, Motoyama T, Meltzer SJ. 2000. E-cadherin gene promoter hypermethylation in primary human gastric carcinomas. J Natl Cancer Inst 92:569–573. doi: 10.1093/jnci/92.7.569. [DOI] [PubMed] [Google Scholar]
- 161.Lombaerts M, van Wezel T, Philippo K, Dierssen JW, Zimmerman RM, Oosting J, van Eijk R, Eilers PH, van de Water B, Cornelisse CJ, Cleton-Jansen AM. 2006. E-cadherin transcriptional downregulation by promoter methylation but not mutation is related to epithelial-to-mesenchymal transition in breast cancer cell lines. Br J Cancer 94:661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia De Herreros A. 2000. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 163.Peinado H, Ballestar E, Esteller M, Cano A. 2004. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol Cell Biol 24:306–319. doi: 10.1128/MCB.24.1.306-319.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lin Y, Wu Y, Li J, Dong C, Ye X, Chi YI, Evers BM, Zhou BP. 2010. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J 29:1803–1816. doi: 10.1038/emboj.2010.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Dong C, Wu Y, Wang Y, Wang C, Kang T, Rychahou PG, Chi YI, Evers BM, Zhou BP. 2013. Interaction with Suv39H1 is critical for Snail-mediated E-cadherin repression in breast cancer. Oncogene 32:1351–1362. doi: 10.1038/onc.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Cao Q, Yu J, Dhanasekaran SM, Kim JH, Mani RS, Tomlins SA, Mehra R, Laxman B, Cao X, Yu J, Kleer CG, Varambally S, Chinnaiyan AM. 2008. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 27:7274–7284. doi: 10.1038/onc.2008.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Dawson MA. 2017. The cancer epigenome: concepts, challenges, and therapeutic opportunities. Science 355:1147–1152. doi: 10.1126/science.aam7304. [DOI] [PubMed] [Google Scholar]
- 168.Kaelin WG., Jr 2005. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer 5:689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 169.Grassian AR, Scales TM, Knutson SK, Kuntz KW, McCarthy NJ, Lowe CE, Moore JD, Copeland RA, Keilhack H, Smith JJ, Wickenden JA, Ribich S. 2015. A medium-throughput single cell CRISPR-Cas9 assay to assess gene essentiality. Biol Proced Online 17:15. doi: 10.1186/s12575-015-0028-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Rau RE, Rodriguez BA, Luo M, Jeong M, Rosen A, Rogers JH, Campbell CT, Daigle SR, Deng L, Song Y, Sweet S, Chevassut T, Andreeff M, Kornblau SM, Li W, Goodell MA. 2016. DOT1L as a therapeutic target for the treatment of DNMT3A-mutant acute myeloid leukemia. Blood 128:971–981. doi: 10.1182/blood-2015-11-684225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wang E, Kawaoka S, Yu M, Shi J, Ni T, Yang W, Zhu J, Roeder RG, Vakoc CR. 2013. Histone H2B ubiquitin ligase RNF20 is required for MLL-rearranged leukemia. Proc Natl Acad Sci U S A 110:3901–3906. doi: 10.1073/pnas.1301045110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Feng Z, Yao Y, Zhou C, Chen F, Wu F, Wei L, Liu W, Dong S, Redell M, Mo Q, Song Y. 2016. Pharmacological inhibition of LSD1 for the treatment of MLL-rearranged leukemia. J Hematol Oncol 9:24. doi: 10.1186/s13045-016-0252-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Karatas H, Townsend EC, Cao F, Chen Y, Bernard D, Liu L, Lei M, Dou Y, Wang S. 2013. High-affinity, small-molecule peptidomimetic inhibitors of MLL1/WDR5 protein-protein interaction. J Am Chem Soc 135:669–682. doi: 10.1021/ja306028q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wang GG, Konze KD, Tao J. 2015. Polycomb genes, miRNA, and their deregulation in B-cell malignancies. Blood 125:1217–1225. doi: 10.1182/blood-2014-10-606822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wepsic HT. 1983. Overview of oncofetal antigens in cancer. Ann Clin Lab Sci 13:261–266. [PubMed] [Google Scholar]
- 176.Ahrlund-Richter L, Hendrix MJ. 2014. Oncofetal signaling as a target for cancer therapy. Semin Cancer Biol 29:1–2. doi: 10.1016/j.semcancer.2014.08.001. [DOI] [PubMed] [Google Scholar]
- 177.Stern PL, Brazzatti J, Sawan S, McGinn OJ. 2014. Understanding and exploiting 5T4 oncofoetal glycoprotein expression. Semin Cancer Biol 29:13–20. doi: 10.1016/j.semcancer.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 178.Lederer M, Bley N, Schleifer C, Huttelmaier S. 2014. The role of the oncofetal IGF2 mRNA-binding protein 3 (IGF2BP3) in cancer. Semin Cancer Biol 29:3–12. doi: 10.1016/j.semcancer.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 179.Hojjat-Farsangi M, Moshfegh A, Daneshmanesh AH, Khan AS, Mikaelsson E, Osterborg A, Mellstedt H. 2014. The receptor tyrosine kinase ROR1–an oncofetal antigen for targeted cancer therapy. Semin Cancer Biol 29:21–31. doi: 10.1016/j.semcancer.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 180.Klauzinska M, Castro NP, Rangel MC, Spike BT, Gray PC, Bertolette D, Cuttitta F, Salomon D. 2014. The multifaceted role of the embryonic gene Cripto-1 in cancer, stem cells and epithelial-mesenchymal transition. Semin Cancer Biol 29:51–58. doi: 10.1016/j.semcancer.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Yong KJ, Chai L, Tenen DG. 2013. Oncofetal gene SALL4 in aggressive hepatocellular carcinoma. N Engl J Med 369:1171–1172. doi: 10.1056/NEJMc1308785. [DOI] [PubMed] [Google Scholar]
- 182.Nicolè L, Sanavia T, Veronese N, Cappellesso R, Luchini C, Dabrilli P, Fassina A. 2017. Oncofetal gene SALL4 and prognosis in cancer: a systematic review with meta-analysis. Oncotarget 8:22968–22979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kirsammer G, Strizzi L, Margaryan NV, Gilgur A, Hyser M, Atkinson J, Kirschmann DA, Seftor EA, Hendrix MJ. 2014. Nodal signaling promotes a tumorigenic phenotype in human breast cancer. Semin Cancer Biol 29:40–50. doi: 10.1016/j.semcancer.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Cheng S-W, Tsai H-W, Lin Y-J, Cheng P-N, Chang Y-C, Yen C-J, Huang H-P, Chuang Y-P, Chang T-T, Lee C-T, Chao A, Chou C-Y, Chan S-H, Chow N-H, Ho C-L. 2013. Lin28B is an oncofetal circulating cancer stem cell-like marker associated with recurrence of hepatocellular carcinoma. PLoS One 8:e80053. doi: 10.1371/journal.pone.0080053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bella L, Zona S, de Moraes GN, Lam EWF. 2014. FOXM1: a key oncofoetal transcription factor in health and disease. Semin Cancer Biol 29:32–39. doi: 10.1016/j.semcancer.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 186.Feigenberg T, Gofrit ON, Pizov G, Hochberg A, Benshushan A. 2013. Expression of the H19 oncofetal gene in premalignant lesions of cervical cancer: a potential targeting approach for development of nonsurgical treatment of high-risk lesions. ISRN Obstet Gynecol 2013:137509. doi: 10.1155/2013/137509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Rijlaarsdam MA, Looijenga LH. 2014. An oncofetal and developmental perspective on testicular germ cell cancer. Semin Cancer Biol 29:59–74. doi: 10.1016/j.semcancer.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 188.Sarandakou A, Protonotariou E, Rizos D. 2007. Tumor markers in biological fluids associated with pregnancy. Crit Rev Clin Lab Sci 44:151–178. doi: 10.1080/10408360601003143. [DOI] [PubMed] [Google Scholar]
- 189.Adinolfi M, Lessof MH. 1985. Cancer, oncogenes and oncofetal antigens. Q J Med 54:193–204. [PubMed] [Google Scholar]
- 190.Neville AM, Mackay AM, Westwood J, Turberville C, Laurence DJ. 1975. Human tumour-associated and tumour-specific antigens: some concepts in relation to clinical oncology. J Clin Pathol Suppl (Assoc Clin Pathol) 6:102–112. doi: 10.1136/jcp.s1-6.1.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Coggin JH., Jr 1986. The implications of embryonic gene expression in neoplasia. Crit Rev Oncol Hematol 5:37–55. doi: 10.1016/S1040-8428(86)80052-X. [DOI] [PubMed] [Google Scholar]
- 192.Boyerinas B, Park SM, Shomron N, Hedegaard MM, Vinther J, Andersen JS, Feig C, Xu J, Burge CB, Peter ME. 2008. Identification of let-7-regulated oncofetal genes. Cancer Res 68:2587–2591. doi: 10.1158/0008-5472.CAN-08-0264. [DOI] [PubMed] [Google Scholar]
- 193.Bell JL, Wachter K, Muhleck B, Pazaitis N, Kohn M, Lederer M, Huttelmaier S. 2013. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): post-transcriptional drivers of cancer progression? Cell Mol Life Sci 70:2657–2675. doi: 10.1007/s00018-012-1186-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Fusco A, Fedele M. 2007. Roles of HMGA proteins in cancer. Nat Rev Cancer 7:899–910. doi: 10.1038/nrc2271. [DOI] [PubMed] [Google Scholar]
- 195.Schnepp RW, Diskin SJ. 2016. LIN28B: an orchestrator of oncogenic signaling in neuroblastoma. Cell Cycle 15:772–774. doi: 10.1080/15384101.2015.1137712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Wu J, Wei JJ. 2013. HMGA2 and high-grade serous ovarian carcinoma. J Mol Med (Berl) 91:1155–1165. doi: 10.1007/s00109-013-1055-8. [DOI] [PubMed] [Google Scholar]
- 197.Degrauwe N, Suva ML, Janiszewska M, Riggi N, Stamenkovic I. 2016. IMPs: an RNA-binding protein family that provides a link between stem cell maintenance in normal development and cancer. Genes Dev 30:2459–2474. doi: 10.1101/gad.287540.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Hsu CC, Chiang CW, Cheng HC, Chang WT, Chou CY, Tsai HW, Lee CT, Wu ZH, Lee TY, Chao A, Chow NH, Ho CL. 2011. Identifying LRRC16B as an oncofetal gene with transforming enhancing capability using a combined bioinformatics and experimental approach. Oncogene 30:654–667. doi: 10.1038/onc.2010.451. [DOI] [PubMed] [Google Scholar]
- 199.Kajiyama Y, Tian J, Locker J. 2002. Regulation of alpha-fetoprotein expression by Nkx2.8. Mol Cell Biol 22:6122–6130. doi: 10.1128/MCB.22.17.6122-6130.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Morford LA, Davis C, Jin L, Dobierzewska A, Peterson ML, Spear BT. 2007. The oncofetal gene glypican 3 is regulated in the postnatal liver by zinc fingers and homeoboxes 2 and in the regenerating liver by alpha-fetoprotein regulator 2. Hepatology 46:1541–1547. doi: 10.1002/hep.21825. [DOI] [PubMed] [Google Scholar]
- 201.Chiovaro F, Chiquet-Ehrismann R, Chiquet M. 2015. Transcriptional regulation of tenascin genes. Cell Adh Migr 9:34–47. doi: 10.1080/19336918.2015.1008333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Fan H, Cui Z, Zhang H, Mani SK, Diab A, Lefrancois L, Fares N, Merle P, Andrisani O. 2017. DNA demethylation induces SALL4 gene re-expression in subgroups of hepatocellular carcinoma associated with hepatitis B or C virus infection. Oncogene 36:2435–2445. doi: 10.1038/onc.2016.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Chen H, Egan JO, Chiu JF. 1997. Regulation and activities of alpha-fetoprotein. Crit Rev Eukaryot Gene Expr 7:11–41. doi: 10.1615/CritRevEukarGeneExpr.v7.i1-2.20. [DOI] [PubMed] [Google Scholar]
- 204.Tran R, Kashmiri SV, Kantor J, Greiner JW, Pestka S, Shively JE, Schlom J. 1988. Correlation of DNA hypomethylation with expression of carcinoembryonic antigen in human colon carcinoma cells. Cancer Res 48:5674–5679. [PubMed] [Google Scholar]
- 205.Arai D, Hayakawa K, Ohgane J, Hirosawa M, Nakao Y, Tanaka S, Shiota K. 2015. An epigenetic regulatory element of the Nodal gene in the mouse and human genomes. Mech Dev 136:143–154. doi: 10.1016/j.mod.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 206.Nassar D, Blanpain C. 2016. Cancer stem cells: basic concepts and therapeutic implications. Annu Rev Pathol 11:47–76. doi: 10.1146/annurev-pathol-012615-044438. [DOI] [PubMed] [Google Scholar]
- 207.Dawood S, Austin L, Cristofanilli M. 2014. Cancer stem cells: implications for cancer therapy. Oncology (Williston Park) 28:1101–1107, 1110. [PubMed] [Google Scholar]
- 208.Clevers H. 2011. The cancer stem cell: premises, promises and challenges. Nat Med 17:313–319. [DOI] [PubMed] [Google Scholar]
- 209.Maruyama R, Choudhury S, Kowalczyk A, Bessarabova M, Beresford-Smith B, Conway T, Kaspi A, Wu Z, Nikolskaya T, Merino VF, Lo PK, Liu XS, Nikolsky Y, Sukumar S, Haviv I, Polyak K. 2011. Epigenetic regulation of cell type-specific expression patterns in the human mammary epithelium. PLoS Genet 7:e1001369. doi: 10.1371/journal.pgen.1001369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, D'Alessio AC, Young RA, Weinberg RA. 2013. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 154:61–74. doi: 10.1016/j.cell.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Rugg-Gunn PJ, Cox BJ, Ralston A, Rossant J. 2010. Distinct histone modifications in stem cell lines and tissue lineages from the early mouse embryo. Proc Natl Acad Sci U S A 107:10783–10790. doi: 10.1073/pnas.0914507107. [DOI] [PMC free article] [PubMed] [Google Scholar]