

ABSTRACT
CsrA of Escherichia coli is an RNA-binding protein that globally regulates a wide variety of cellular processes and behaviors, including carbon metabolism, motility, biofilm formation, and the stringent response. CsrB and CsrC are small RNAs (sRNAs) that sequester CsrA, thereby preventing CsrA-mRNA interaction. RpoE (σE) is the extracytoplasmic stress response sigma factor of E. coli. Previous RNA sequencing (RNA-seq) studies identified rpoE mRNA as a CsrA target. Here, we explored the regulation of rpoE by CsrA and found that CsrA represses rpoE translation. Gel mobility shift, footprint, and toeprint studies identified three CsrA binding sites in the rpoE leader transcript, one of which overlaps the rpoE Shine-Dalgarno (SD) sequence, while another overlaps the rpoE translation initiation codon. Coupled in vitro transcription-translation experiments showed that CsrA represses rpoE translation by binding to these sites. We further demonstrate that σE indirectly activates the transcription of csrB and csrC, leading to increased sequestration of CsrA, such that repression of rpoE by CsrA is reduced. We propose that the Csr system fine-tunes the σE-dependent cell envelope stress response. We also identified a 51-amino-acid coding sequence whose stop codon overlaps the rpoE start codon and demonstrate that rpoE is translationally coupled with this upstream open reading frame (ORF51). The loss of coupling reduces rpoE translation by more than 50%. Identification of a translationally coupled ORF upstream of rpoE suggests that this previously unannotated protein may participate in the cell envelope stress response. In keeping with existing nomenclature, we named ORF51 rseD, resulting in an operon arrangement of rseD-rpoE-rseA-rseB-rseC.
IMPORTANCE CsrA posttranscriptionally represses genes required for bacterial stress responses, including the stringent response, catabolite repression, and the RpoS (σS)-mediated general stress response. We show that CsrA represses the translation of rpoE, encoding the extracytoplasmic stress response sigma factor, and that σE indirectly activates the transcription of csrB and csrC, resulting in reciprocal regulation of these two global regulatory systems. These findings suggest that extracytoplasmic stress leads to derepression of rpoE translation by CsrA, and CsrA-mediated repression helps reset RpoE abundance to prestress levels once envelope damage is repaired. The discovery of an ORF, rseD, translationally coupled with rpoE adds further complexity to translational control of rpoE.
KEYWORDS: CsrA, RpoE, sigma factors, stress response, translational control, translational coupling
INTRODUCTION
Bacterial survival depends upon the capacity to rapidly and profoundly alter physiology and behavior in response to changing environmental conditions. These responses involve regulated adjustments in the expression of numerous genes via coordinated global regulatory networks that are subject to the action of a limited repertoire of trans-acting factors. The Csr (carbon storage regulator) system, which is also referred to as Rsm (repressor of secondary metabolites) in some organisms, is one such network that globally controls gene expression posttranscriptionally (reviewed in references 1–3). Depending on the particular bacterial species, this system regulates a wide variety of cellular processes, including carbon metabolism, the general stress response, motility, biofilm formation, quorum sensing, mRNA decay, and virulence (1–3). Recent transcriptomics studies revealed that the Csr system controls expression of several hundred genes, including numerous regulatory factors, establishing a general framework for understanding how Csr governs lifestyle decisions on a global scale (4–7).
CsrA is the central component of the Csr system in Escherichia coli. This homodimeric protein contains two identical RNA binding surfaces that can bridge two sites within an RNA target (8, 9). GGA is a highly conserved motif within CsrA binding sites and is often present in the loop of RNA hairpins (10, 11). CsrB and CsrC, two small RNA (sRNA) antagonists of CsrA, are capable of sequestering multiple CsrA dimers from its mRNA targets (10, 12). The transcription of csrB and csrC, in turn, is activated by the BarA-UvrY two-component signal transduction system in response to short-chain carboxylic acids (13, 14) and by the alarmone guanosine tetraphosphate (ppGpp) (4, 15). The decay of CsrB and CsrC is also tightly regulated. CsrD, a membrane-bound GGDEF-EAL domain protein that does not metabolize cyclic di-GMP (c-di-GMP), is required for the initiation of decay by RNase E cleavage (16, 17). Glucose activates CsrB and CsrC decay by binding of CsrD to the unphosphorylated form of EIIAGlc, which predominates during glucose uptake by the phosphotransferase system (PTS) (18). Thus, the availability of preferred carbon depresses CsrB and CsrC levels and increases CsrA availability, while the depletion of preferred carbon and accumulation of end products cause CsrB and CsrC accumulation, CsrA sequestration, and derepression of stationary-phase and stress response genes. CsrA indirectly activates csrB and csrC transcription (13, 19) and indirectly represses csrD expression (16, 20), creating negative feedback loops, which have been reported to improve signaling response dynamics of the Csr system (21). Furthermore, since CsrA represses its own translation and indirectly activates its transcription (22), it is apparent that the level of CsrA is tightly controlled in the cell.
CsrA can both repress and activate the expression of its target genes, depending on the location of the CsrA binding sites. The most common CsrA-mediated regulatory mechanism involves CsrA binding to multiple sites that overlap the cognate Shine-Dalgarno (SD) sequence and/or translation initiation region, such that bound CsrA represses translation initiation by blocking ribosome binding (1–3, 23, 24). The best-characterized activation mechanism involves CsrA binding at the extreme 5′ end of the flhDC transcript, such that bound CsrA prevents 5′-end-dependent RNase E cleavage of the flhDC transcript (25).
RpoE (σE) is the extracytoplasmic stress response sigma factor of E. coli and is required for viability, as well as the response to stress in the cell envelope. The activity of σE is controlled by the membrane bound anti-sigma factor RseA in conjunction with the periplasmic protein RseB (reviewed in reference 26). RseA binds to σE and prevents it from interacting with core RNA polymerase. RseA is a proteolytically unstable protein and in the absence of envelope stress is degraded at a basal level, providing cells with sufficient free σE to support viability. Upon envelope stress, the degradation rate of RseA increases dramatically, releasing σE to direct transcription. RseB binds to RseA and modulates its susceptibility to proteolysis. The genes encoding these proteins are cotranscribed in the rpoE-rseA-rseB-rseC operon. The transcription of this operon is driven by at least two promoters, one of which is recognized by σE itself (27). In addition to regulation by RseA and RseB, the σE RNA polymerase holoenzyme is activated by ppGpp during periods of nutrient limitation (28–30). We previously identified rpoE mRNA as a CsrA target by RNA sequencing (RNA-seq) (4). In this work, we determined that CsrA represses rpoE translation by binding to three sites in the rpoE leader RNA. We further found that the translation of rpoE is coupled to a previously unannotated 51-amino-acid protein of unknown function. Our findings extend the known interactions between the Csr system and stress response systems to include the σE cell envelope stress response.
RESULTS
CsrA represses rpoE expression.
rpoE encodes the extracytoplasmic stress response sigma factor σE. rpoE mRNA was identified as a direct CsrA target by RNA-seq (4). Visual inspection of the rpoE leader region led to the identification of four potential CsrA binding sites, each containing a critical GGA motif (Fig. 1). Transcription initiation occurs from at least two promoters, P1 and P2, with P2 being σE dependent (27). We first examined the expression of chromosomally integrated P1-P2-rpoE′-′lacZ and P2-rpoE′-′lacZ translational fusions (Fig. 2A) in wild-type (WT) and CsrA-deficient (csrA::kan mutant) strains. This mutant csrA::kan allele contains a transposon insertion following the 50th codon of the 61-amino-acid coding sequence, resulting in a 62-amino-acid fusion protein that retains about 12% of the RNA binding affinity of WT CsrA (25). A null mutation was not used because it causes very slow growth and rapidly accumulates suppressor mutations (A. Potts, C. Vakulskas, P. Babitzke, T. Romeo, unpublished data). Depending on the stage of growth (Fig. 2B), the expression levels of these fusions were 2- to 3-fold higher in the csrA::kan mutant strain, indicating that CsrA represses rpoE expression (Fig. 2C). We next examined the expression of a leader fusion in which the rpoE P1-P2 promoter region was replaced with the lacUV5 promoter (PlacUV5). Note that this fusion retained the rpoE leader region (Fig. 2A). Because CsrA does not affect transcription from PlacUV5 (4), an effect of the csrA::kan mutation is inferred to reflect CsrA-dependent posttranscriptional effects of the rpoE leader transcript. As was observed with the P1-P2-rpoE′-′lacZ and P2-rpoE′-′lacZ fusions, expression of the PlacUV5-rpoE′-′lacZ fusion was ∼2-fold higher in the csrA::kan mutant strain (Fig. 2D), consistent with CsrA-dependent repression occurring posttranscriptionally. When experiments were conducted with a P1-P2-rpoE-lacZ transcriptional fusion (Fig. 2A), in which the rpoE leader region containing the putative CsrA binding sites was absent, expression was somewhat lower in the csrA::kan mutant strain, especially during exponential-phase growth (Fig. 2E). These results suggest that CsrA indirectly activates rpoE transcription from P1 and/or P2 during exponential phase, which partially counteracts the direct effect that CsrA has on posttranscriptional repression.
FIG 1.

Nucleotide sequence and features of the rpoE promoter and leader region. The −35 and −10 elements of promoters P1 and P2, and the corresponding transcription start sites (+1), are in bold. P2 is a σE-dependent promoter (27, 37), although the sigma factor that drives the expression from P1 is not known. Additional upstream promoters (38) are not shown, and promoter numbering is as in reference 28. The ORF16 and ORF51 translation start and stop codons are boxed in yellow. ORF51 stop codon mutations are shown in cyan above the corresponding codon. CsrA binding sites BS1 to BS3 are marked with a line, with critical GGA motifs in red. Mutations in BS1 and BS3 are shown in cyan below the corresponding GGA motif. A fourth GGA motif that we determined is not part of an authentic CsrA binding site is also shown in red. The rpoE Shine-Dalgarno (SD) sequence and start codon are marked with a line below each sequence. The ORF51 stop and rpoE start codons overlap by two nucleotides. We named this ORF rseD. Numbering is with respect to the start of rpoE translation.
FIG 2.

CsrA represses rpoE expression posttranscriptionally. (A) Schematic representation of the fusions used in this analysis. Relative positions of the rpoE promoters (P1 and P2), PlacUV5, and the start codons (ATG) driving the translation of each fusion are shown. The rpoE promoter and leader regions are depicted with a thin black line, while the rpoE and lacZ coding sequences are depicted with thick black and red lines, respectively. The dashed line indicates that the corresponding sequence is absent. (B) Representative growth curve of a wild-type (WT) strain. (C to E) β-Galactosidase activity (units per milligram of protein) ± the standard deviation was determined throughout growth. Experiments were performed at least three times. (C) Expression of the P1-P2-rpoE′-′lacZ and P2-rpoE′-′lacZ translational fusions in WT and csrA::kan mutant strains. (D) Expression of the PlacUV5-rpoE′-′lacZ leader fusion in WT and csrA::kan mutant strains. (E) Expression of the P1-P2-rpoE-lacZ transcriptional fusion in WT and csrA::kan mutant strains.
CsrA binds specifically to rpoE leader RNA.
Quantitative gel mobility shift assays were performed to determine whether CsrA bound to an rpoE transcript containing all four of the potential CsrA binding sites. A distinct band with lower mobility was observed between 2 and 16 nM CsrA, indicating that CsrA formed a tight complex with this transcript. Nonlinear least-squares analysis of these data yielded an apparent Kd (dissociation constant) value of 5 ± 1 nM (Fig. 3A, WT). At higher CsrA concentrations, a distinct complex with even lower mobility was observed. These data suggest that the first shifted complex contained one CsrA dimer bound per transcript, while the second complex contained two CsrA dimers. The specificity of CsrA-rpoE RNA interaction was also investigated by performing competition experiments with specific (rpoE) and nonspecific (phoB) unlabeled RNA competitors. Whereas unlabeled rpoE RNA was an effective competitor, phoB RNA was not (Fig. 3B). We conclude that CsrA binds to the rpoE leader transcript with high affinity and specificity.
FIG 3.

Gel mobility shift analysis of CsrA-rpoE leader RNA interaction. (A) Labeled rpoE RNA (40 pM) was incubated with the concentration of CsrA shown at the top of each lane. Positions of bound (b) and free (f) RNA are marked with arrowheads. Experiments were performed at least twice, and representative gels are shown. CsrA formed a high-affinity complex with WT rpoE RNA and a second complex at higher protein concentrations. CsrA binding was greatly reduced when the GGA motif of binding site 1 (BS1 mut) was changed to CCA, and it was eliminated when combined with a GGA-to-CCA mutation in BS3 (BS1,3 mut). (B) RNA competition experiment demonstrating binding specificity. Labeled rpoE RNA (0.1 nM) was combined with the indicated concentration of unlabeled specific (rpoE) or nonspecific (phoB) competitor RNA and incubated with the concentration of CsrA shown at the top of each lane. Positions of bound (b) and free (f) RNA are marked. Experiments were performed twice, and a representative gel is shown.
RNase T1 cleaves RNA following single-stranded G residues that are not occluded by bound protein. Thus, we performed CsrA-rpoE leader RNA footprint experiments using RNase T1 as a G-specific probe to identify the authentic CsrA binding sites in the rpoE leader transcript. Bound CsrA resulted in strong protection of RNase T1-mediated cleavage of the G residues between −60 and −51 (BS1). Protection of the G residues between −18 and −13 (BS2) and between −2 and +3 (BS3) was also observed at the highest CsrA concentration used (Fig. 4A). Notably, bound CsrA did not protect the GGA sequence centered at position −30 (Fig. 1 and 4A), indicating that this GGA motif is not part of an authentic CsrA binding site. We also performed CsrA toeprint experiments as a complementary method to observe the positions of bound CsrA. In this assay, the position of bound CsrA will inhibit primer extension by reverse transcriptase, resulting in a toeprint band near the 3′ edge of the bound protein. A strong CsrA-dependent toeprint band was observed just downstream from BS3, confirming that CsrA binds to this site (Fig. 4B).
FIG 4.

CsrA-rpoE RNA footprint and toeprint analyses. (A) CsrA-rpoE RNA footprint. Labeled rpoE RNA was treated with RNase T1 in the presence of the CsrA concentration shown at the top of the lane. A partial alkaline hydrolysis ladder (OH), an RNase T1 digestion ladder (T1) generated under partial denaturing conditions so that every G residue can be observed, and a control lane without RNase T1 treatment (C) are marked. The positions of CsrA binding sites BS1, BS2, and BS3 are shown. The positions of the rpoE start codon (Met) and Shine-Dalgarno (SD) sequence are also marked. Numbering is with respect to the start of rpoE translation. (B) CsrA-rpoE RNA toeprints were performed in the absence or presence of 1 μM CsrA. Positions of BS1, BS2, BS3, and the CsrA toeprint (carat) are marked. Sequencing lanes to reveal A, C, G, and U residues are labeled. Numbering is with respect to the start of rpoE translation.
We next tested binding to a transcript in which the GGA motif of BS1 was changed to CCA using a gel mobility shift assay. This mutation resulted in a severe CsrA binding defect (Fig. 3A, BS1 mut). CsrA binding was eliminated when the BS1 mutation was combined with a GGA-to-CCA mutation in BS3 (Fig. 3A, BS1,3 mut). We conclude that CsrA binds to 3 sites in the rpoE leader transcript, including those that overlap the rpoE SD sequence (BS2) and translation initiation codon (BS3).
CsrA represses rpoE translation.
The positions of BS2 and BS3 suggested that bound CsrA would be capable of repressing the translation of rpoE. Thus, we used the in vitro coupled transcription-translation PURExpress system to determine whether CsrA represses rpoE translation. Plasmids carrying rpoE′-′lacZ translational fusions were used in this analysis, all of which were driven by identical T7 RNA polymerase (RNAP) promoters. Three plasmids gave rise to transcripts that initiated near the P2 transcription start site. One of the plasmids contained the WT sequence, a second contained a GGA-to-CCA mutation in BS1, and the third contained GGA-to-CCA mutations in both BS1 and BS3 (Fig. 5A). Note that we did not test a fusion with a mutation in BS2, because this GGA motif is part of the rpoE SD sequence. A fourth plasmid in which T7 RNAP drove the expression of a phoB′-′lacZ translational fusion was used as a negative control, because we previously established that CsrA does not repress expression of this fusion (31). The addition of increasing CsrA concentrations resulted in ∼90% repression of the WT fusion and 50% repression of the BS1 mutant fusion, whereas CsrA-mediated repression was lost with the fusion containing mutations in both BS1 and BS3 (Fig. 5A). We also tested the expression of WT and BS1 mutant fusions in which T7 RNAP gave rise to transcripts that initiated near the P1 transcription start site (Fig. 5B). In this case, CsrA repressed the expression of the WT fusion to a similar extent as we observed from the P2-derived WT fusion; however, CsrA-mediated repression was nearly absent with a BS1 mutant fusion in which the GGA motif was changed to GCA. From these data, we conclude that CsrA represses the translation of rpoE and that BS1 and BS3 contribute to repression.
FIG 5.

CsrA represses translation of rpoE. (A and B) Schematic representations of the fusions used in this analysis are shown at the top. T7 RNAP drives transcription from the P2 (A) or P1 (B) transcription start sites. The start codon (ATG) driving the translation of each fusion is shown. The rpoE promoter and leader regions are depicted with a thin black line, while the rpoE and lacZ coding sequences are depicted with thick black and red lines, respectively. GGA motif mutations in BS1 and/or BS3, as well as an ORF51 stop codon mutant, are indicated with a red X. Relative β-galactosidase activity ± standard deviation as a function of CsrA concentration from at least three experiments was determined in vitro with PURExpress. A phoB′-′lacZ translational fusion was used as a negative control. (A) Expression of a WT T7(P2)-rpoE′-′lacZ translational fusion, as well as mutant fusions containing GGA-to-CCA mutations in BS1, or both BS1 and BS3. (B) Expression of a WT T7(P1)-rpoE′-′lacZ translational fusion, as well as mutant fusions containing a GGA-to-GCA mutation in BS1, or a stop codon mutation in codon 12 of ORF51.
σE activates transcription of csrB and csrC.
Previous studies demonstrated that the circuitry of the Csr system is linked to the stringent response and the catabolite repression systems (4, 32). In the case of the stringent response, CsrA represses the expression of relA, which encodes a guanosine pentaphosphate [(p)ppGpp] synthetase, while ppGpp and DksA activate the transcription of csrB and csrC (4). In the case of catabolite repression, cyclic AMP (cAMP)-cAMP receptor protein (CRP) represses csrB and csrC transcription, while CsrA represses the translation of crp (32). Thus, we tested whether rpoE affected the expression of csrB and/or csrC by comparing the expression of these genes in a WT strain, in a strain with elevated σE activity due to a deletion of the anti-sigma factor RseA, and in a strain lacking σE activity due to a deletion of the rpoE gene. Although rpoE is essential for E. coli viability, it can be deleted from strains containing a hicB deletion (33). We performed primer extension analysis on total cellular RNA isolated from WT and ΔhicB, ΔhicB ΔrpoE, and ΔrseA mutant strains. We identified a single primer extension product for each transcript corresponding to the previously identified transcription start sites derived from σ70-dependent promoters (12, 34). Of particular interest, both csrB and csrC transcript levels were dramatically reduced in the absence of σE (Fig. 6). Since neither of the promoters driving the transcription of csrB and csrC resembles a σE-dependent promoter, the observed σE-dependent activation of csrB and csrC transcription is indirect. This conclusion is consistent with our inability to transcribe templates containing these promoters in vitro using E. coli core RNAP and σE (data not shown).
FIG 6.

Deletion of rpoE reduces transcription of csrB and csrC. (A) Nucleotide sequences of the csrB and csrC promoter regions. Promoter −35 and −10 elements, as well as the transcription start sites (+1), are shown. (B and C) Primer extension analysis of csrB (B) and csrC (C) of total cellular RNA extracted from stationary-phase cultures. Lane 1, WT; lane 2, ΔhicB mutant; lane 3, ΔhicB ΔrpoE mutant; lane 4, ΔrseA mutant. Transcription start sites (+1) are marked. (D) β-Galactosidase activity (units of milligram of protein) ± standard deviation of csrB-lacZ and csrC-lacZ transcriptional fusions was determined during stationary phase in WT (black) and ΔhicB (red), ΔhicB ΔrpoE (gray), and ΔrseA (blue) mutant strains. Experiments were performed three times.
Our primer extension results indicated that σE indirectly activates the transcription of csrB and csrC. Thus, we would have expected that the absence of RseA would lead to increased transcription of these two genes; however, this result was not apparent in our primer extension studies. Hence, we tested the expression of csrB-lacZ and csrC-lacZ transcriptional fusions in WT and ΔhicB, ΔhicB ΔrpoE, and ΔrseA mutant strains. Although not as pronounced as for the primer extension analysis, we observed decreased expression of both fusions in the ΔhicB ΔrpoE genetic background (Fig. 6D). Moreover, the expression of both fusions was increased in the ΔrseA genetic background, indicating that the absence of σE or RseA has opposite effects on csrB and csrC expression, as expected. Taken together, our results indicate that the Csr and σE-dependent stress response systems are reciprocally regulated.
An ORF within the rpoE leader region is translationally coupled with rpoE.
We identified ORF sequences capable of encoding products of 16 and 51 amino acids from transcripts derived from P1; these ORFs had not been previously annotated (Fig. 1). Thus, we generated chromosomally integrated P1-ORF16′-′lacZ and P1-ORF51′-′lacZ translational fusions to determine whether these ORFs were expressed. Although both of the ORFs were expressed, the expression of ORF51 was much higher, especially during exponential-phase growth (Fig. 2B and 7A). Since ORF16 is followed by three tandem stop codons and is far upstream of the CsrA binding sites and rpoE (Fig. 1), we did not explore this ORF further.
FIG 7.

Translational coupling between ORF51 and rpoE increases rpoE translation. (A to C) Schematic representations of the fusions used in this analysis are shown at the top. Relative positions of the rpoE promoters (P1 and P2) and the start codons (ATG) driving translation of each fusion are shown. The rpoE promoter and leader regions are depicted with a thin black line, while the rpoE and lacZ coding sequences are depicted with thick black and red lines, respectively. Stop codon mutations in ORF51 (B) or a potential ORF (C) are marked with an X. β-Galactosidase activity (units of milligram of protein) ± standard deviation was determined throughout growth. Experiments were performed at least three times. (A) Expression of ORF16′-′lacZ and ORF51′-′lacZ translational fusions. (B) Expression of WT or ORF51 stop codon mutant P1-P2-rpoE′-′lacZ translational fusions in WT and csrA::kan mutant strains. (C) Expression of WT or ORF stop codon mutant P2-rpoE′-′lacZ translational fusions in WT and csrA::kan mutant strains.
The ORF51 coding sequence extends throughout the CsrA binding region. Moreover, the ORF51 stop codon overlaps the rpoE start codon by two nucleotides, a sequence arrangement typical of translational coupling, a mechanism in which translation of the downstream coding sequence is at least partially dependent on translation of the upstream coding sequence (Fig. 1) (reviewed in references 35 and 36). Thus, we introduced a stop codon at the codon for amino acid 12 of the ORF51 coding sequence in the context of the P1-P2-rpoE′-′lacZ translational fusion to determine whether the translation of rpoE was affected by translation of ORF51. This stop codon was positioned 65 nucleotides (nt) upstream of the GGA motif within BS1. The stop codon mutation resulted in 2- to 3-fold reduced expression of the fusion (Fig. 7B). These results indicate that ORF51 and rpoE are translationally coupled from transcripts derived from P1 and that coupling is responsible for more than half of the rpoE translation initiation events.
Since all three CsrA binding sites are within the ORF51 coding sequence, we reasoned that translation of this ORF would displace bound CsrA, perhaps leading to reduced CsrA-mediated repression of rpoE translation. Hence, we compared expression of the WT and ORF51 stop codon mutant fusions in WT and csrA::kan mutant backgrounds. However, we found that CsrA repressed the expression of the two fusions to a comparable level (Fig. 7B). Thus, while it is likely that bound CsrA would be displaced by a ribosome translating ORF51, such displacement did not interfere with CsrA-mediated repression of rpoE translation. This result is consistent with PURExpress results showing that CsrA repressed rpoE translation of WT and ORF51 stop codon mutant fusions to similar extents (Fig. 5B).
In addition to the ORF51 start codon, there are two internal Met codons, one of which is downstream from the P2 transcription start site; this ATG codon overlaps the GGA motif in BS1 (Fig. 1). Thus, we tested whether transcripts derived from P2 were subject to translational coupling in WT and csrA::kan mutant backgrounds. In this case, we found that expression of the P2-rpoE′-′lacZ translational fusion was unaffected when we introduced a stop codon in what would be the fifth codon of this potential ORF, indicating that rpoE is not translationally coupled to an upstream ORF when transcription initiates from P2 (Fig. 7C). Since ORF51 is expressed, and its expression increases translation of rpoE via translational coupling, we have named this gene rseD, resulting in an operon arrangement of rseD-rpoE-rseA-rseB-rseC.
DISCUSSION
CsrA has been shown to repress the translation of several genes in E. coli and other organisms (1–4, 22–24, 31, 32). In most cases, CsrA binds to the SD sequence and/or initially translated region, thereby inhibiting binding of 30S ribosomal subunits. Since translating ribosomes can protect the mRNA from ribonucleases, translational repression often leads to decreased mRNA stability. In this study, we found that CsrA represses the translation of rpoE, which encodes the E. coli extracytoplasmic stress response sigma factor σE (Fig. 2 and 5). CsrA binds to three sites in rpoE leader RNA (Fig. 3 and 4). Since BS2 and BS3 overlap the rpoE SD sequence and translation initiation codon, respectively, bound CsrA would occlude the ribosome binding site, providing an explanation for translation repression. We also found that σE activates the transcription of csrB and csrC (Fig. 6), which encode the two sRNA antagonists of CsrA activity. Since the deletion of rpoE affects transcription from the previously identified σ70-dependent promoters and these sequences do not resemble σE-dependent promoters, our results indicate that σE activates csrB and csrC transcription indirectly. Our inability to obtain transcription from these promoters with RNAP containing σE is consistent with this interpretation.
Previous studies demonstrated that the circuitry of the Csr system is linked to the stringent response and the catabolite repression systems (4, 32). CsrA represses the translation of relA, which reduces production of (p)ppGpp during the stringent response (4). ppGpp and DksA, in turn, activate the transcription of csrB and csrC (4). Thus, the Csr and stringent response systems are reciprocally regulated. When cells are starved for amino acids, CsrB and CsrC levels increase via activation by ppGpp and DksA, leading to sequestration of free CsrA and derepression of relA translation. When amino acids become available, the concentration of ppGpp decreases, resulting in reduced transcription of csrB and csrC. In conjunction with CsrB and CsrC turnover via CsrD-EIIAGlc-mediated targeting of the sRNAs for cleavage by RNase E (16–18), the concentration of free CsrA increases, which restores translational repression of relA.
Our studies provide a second closely related example of this kind of reciprocal regulation in which CsrA is linked to a transcriptional global regulatory system (Fig. 8). In this case, CsrA directly represses rpoE translation, while σE indirectly activates the transcription of csrB and csrC. As such, the Csr system adds an extra layer of regulation of σE activity. During envelope stress, increased free σE will indirectly activate the transcription of csrB and csrC, reducing translational repression of rpoE by CsrA and increasing σE production. Once damage to the cell envelope is repaired, RseA will be stabilized and sequester σE, and the levels of CsrB and CsrC will decline via degradation and reduced transcription. As a consequence, free CsrA increases, rapidly restoring translational repression of rpoE.
FIG 8.

Regulatory circuitry of the Csr, stringent response, and σE stress response systems. CsrA represses translation of rpoE, while σE activates transcription of csrB and csrC. CsrB/C sRNAs bind to and antagonize CsrA. CsrA represses its own translation and activates its own transcription. CsrD targets CsrB/C for cleavage by RNase E, although the mechanism has not been established. CsrA represses csrD expression and activates the transcription of csrB and csrC. RseA binds to and inhibits σE activity. Cell envelope stress leads to degradation of RseA, leading to σE-dependent transcription of stress response genes. σE activates its own transcription, while ppGpp activates σE-directed transcription. ppGpp also activates transcription of csrB and csrC. Solid and dashed lines indicate direct and indirect effects, respectively.
Our findings reveal additional complexity in the regulatory network connecting σE, the Csr system, and the stringent response (Fig. 8). During nutrient limitation, σE is regulated independently of RseA and envelope stress by the stringent factors ppGpp and DksA (28–30). When ppGpp levels rise, ppGpp and DksA activate transcription directed by σE, which should indirectly increase the transcription of CsrB and CsrC. In addition, ppGpp and DksA increase the transcription of csrB and csrC, likely independently of σE (4, 15). CsrB and CsrC will then sequester CsrA, alleviating the repression of rpoE and relA translation. The regulatory network will reset when ppGpp levels drop, as nutrients are restored. The interconnected reciprocal regulatory loops among the Csr, σE, and stringent response systems have the effect of reinforcing the activation of these starvation-responsive pathways. The extent of interdependence of these pathways, such as whether envelope stress impacts regulation of other Csr targets by activating csrB and csrC expression, remains to be explored.
In addition to reciprocal regulation of the Csr and σE-dependent cell envelope stress response systems, we identified a previously unannotated open reading frame, ORF51 (rseD), upstream of rpoE that is cotranscribed from P1 and translationally coupled with rpoE (Fig. 5 and 7); the stop codon for rseD and the rpoE start codon overlap (Fig. 1). Translational coupling within a polycistronic transcript is a process in which translation of a downstream cistron at least partially depends on the translation of a cistron immediately upstream (35, 36). In this case, following termination of rseD, the same ribosome can reinitiate translation of rpoE. We found that >50% of rpoE translation occurs via coupling (Fig. 7B). rseD does not appear to have any known conserved protein domains, so the function remains unknown. The rseD-rpoE-rseA-rseB-rseC operon arrangement makes it tempting to speculate that rseD functions in the cell envelope stress response. The answer to this intriguing possibility will require additional experimentation.
Similar small proteins whose stop codon overlaps the start codon of rpoE are found upstream of rpoE in several Enterobacteriaceae species, including Salmonella enterica, Shigella flexneri, Citrobacter freundii, Citrobacter rodentium, and E. coli (Fig. 9). Interestingly, Klebsiella pneumoniae and Enterobacter cloacae have related sequences, but the ORF is interrupted by stop codons. In more distantly related species, the presence of an overlapping ORF varies. For example, in Yersinia enterocolitica, a translationally coupled ORF with 26% identity and 40% similarity to E. coli rseD is found upstream of rpoE. Pseudomonas aeruginosa has an ORF preceding the rpoE homolog algU, in which the stop codon of the ORF overlaps the second codon of algU and should be subject to translational coupling. However, the Pseudomonas ORF bears no similarity to rseD.
FIG 9.
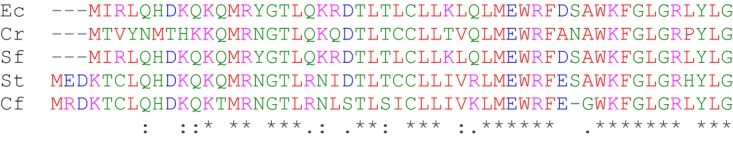
rseD is conserved in some Enterobacteriaceae. An alignment of rseD sequences from Escherichia coli MG1655 (Ec), Citrobacter rodentium ICC168 (Cr), Shigella flexneri 2a strain 301 (Sf), Salmonella enterica enterica serovar Typhimurium strain LT2 (St), and Citrobacter freundii ATCC 8090 (Cf) created using Clustal Omega is shown. Asterisks, sequence identities; colons, strong similarities; periods, weak similarities. Amino acids with the same color have similar chemical properties.
The potential role of translational coupling in the σE stress response is of interest. Transcription of the rpoE operon appears to be complex. Two promoters were originally mapped, with P2 being dependent on σE and showing increased activity under stress conditions (27, 37). Recent work suggests that additional promoters upstream of P1, all σE independent, may also contribute to expression of the operon (38). Under conditions in which σE activity is low, i.e., there is no envelope stress or nutrient limitation, rseD will boost the translation of rpoE from transcripts originating from P1 or other upstream promoters, thereby increasing the amount of σE produced. rseD might also be particularly important to maintain cellular levels of σE during shutoff of the envelope stress response when RseA is stabilized (39). During the stress response, transcription from P2 by the σE holoenzyme will increase, while translation from these mRNAs will rely solely on initiation signals for the rpoE gene. The additional levels of regulation by both rseD and CsrA could serve to better fine-tune the σE response, ensuring that the appropriate amounts of σE are available under different growth and stress conditions.
The Csr system mediates global changes in gene expression in response to the metabolic state of the cell. Like ppGpp, Csr acts to shift the program of gene expression from one optimized for rapid growth to one needed for stress survival (2, 3, 40). Our findings here suggest a model in which Csr and ppGpp integrate σE into this switch by reducing basal σE expression during rapid growth via repression by CsrA, by enhancing the production of σE during metabolic stress via sequestration of CsrA by CsrB and CsrC, and by activation of σE-dependent transcription by ppGpp and DksA (28–30). The primary role of the σE response in E. coli is to maintain the biosynthetic systems that deliver outer membrane porins and lipopolysaccharides (LPS) to the outer membrane (41). During sudden envelope stress when a rapid robust response is required, regulation by Csr and ppGpp is overridden by the degradation of RseA, resulting in a large increase in free σE available to direct transcription. During metabolic stress when such a robust response is not necessary, the Csr and ppGpp systems may serve to enhance the σE response to maintain envelope homeostasis, preloading the cell with proteins needed to ensure the integrity of the outer membrane and facilitating envelope remodeling in response to changing nutrient availability. In addition, the finding that σE increases the transcription of CsrB and CsrC, presumably leading to sequestration of CsrA, provides a mechanism for the cell to activate a cytoplasmic program of stress survival in the face of envelope stress, thereby promoting homeostasis throughout the bacterium.
The importance of integrating responses to the state of the bacterial cell envelope with response to the metabolic state of the cell is supported by similarities between the E. coli systems and regulatory interactions found in Pseudomonas species. The σE homologue AlgU is regulated by the CsrA homologue RsmA. RsmA binds to the algU operon mRNA and represses the expression of AlgU (42). However, in addition to indirectly regulating the sRNA inhibitors of RsmA (RsmX and RsmZ), as seen for σE in E. coli, AlgU also directs the transcription of RsmA, enhancing its effect on target genes (43, 44). Both the Csr and σE systems are found in many other bacterial species, including important pathogens, and are often linked to virulence (3, 45). It will be of interest to determine whether interconnections between Csr/Rsm and RpoE/AlgU systems have been widely selected for bacterial strategies to integrate cytoplasmic and envelope functions.
MATERIALS AND METHODS
Bacterial strains and plasmids.
All bacterial strains used in this study are listed in Table 1. E. coli strain S17-1 λpir+ (46) was used for conditional-replication, integration, and modular (CRIM)-based plasmid construction (47). Plasmids pLFT, pLFX, and pUV5 (4) were used to generate translational, transcriptional, and leader fusions, respectively. Plasmid pYH195 contains a P1-P2-rpoE′-′lacZ translation fusion (nucleotides [nt] −292 to +26 cloned into the PstI and BamHI sites of pLFT). Plasmid pYH300 contains a P2-rpoE′-′lacZ translation fusion (nt −147 to +26 cloned into the PstI and BamHI sites of pLFT). Plasmid pYH282 contains a P1-P2-rpoE-lacZ transcriptional fusion (nt −292 to −64 cloned into the PstI and EcoRI sites of pLFX). Plasmid pYH224 contains a PlacUV5-rpoE′-′lacZ leader fusion (nt −75 to +26 cloned into the EcoRI and BamHI sites of pUV5), such that the P1 and P2 promoters were replaced with the lacUV5 promoter. Plasmid pYH298 contains a P1-ORF16′-′lacZ translational fusion (nt −292 to −179 cloned into the PstI and BamHI sites of pLFT). Plasmid pYH297 contains a P1-ORF51′-′lacZ translational fusion (nt −292 to −140 cloned into the PstI and BamHI sites of pLFT). Mutations in the CsrA binding sites BS1 and/or BS3, or stop codon mutations in ORF51′ in the context of P1-P2-rpoE′-′lacZ or P2-rpoE′-′lacZ translational fusions were introduced using the QuikChange protocol (Agilent Technologies). WT and mutant fusions were integrated into the chromosomal λ att site of E. coli strain CF7789, as described previously (47). P1-mediated transduction was used to introduce the csrA::kan allele from TRMG1655 (13) into strains containing integrated WT and mutant fusions.
TABLE 1.
E. coli strains used in this study
| Strain | Descriptiona | Source or reference |
|---|---|---|
| CAG45170 | MC1061 rpoE::cam sup+ λRpoHP3::lac Cmr | 51 |
| CF7789 | ΔlacI-lacZ (MluI) | M. Cashel |
| PLB2026 | CF7789 yfiC::kan ΔrseA Kmr | This study |
| PLB2027 | CF7789 ΔhicB::kan Kmr | This study |
| PLB2039 | CF7789 P1-P2-rpo′-′lacZ Apr | This study |
| PLB2045 | CF7789 csrA::kan P1-P2-rpoE′-′lacZ Apr Kmr | This study |
| PLB2121 | CF7789 ΔhicB::kan rpoE::cam Cmr Kmr | This study |
| PLB2124 | CF7789 csrB-lacZ Apr | This study |
| PLB2126 | CF7789 csrC-lacZ Apr | This study |
| PLB2128 | CF7789 ΔhicB::kan csrB-lacZ Apr Kmr | This study |
| PLB2130 | CF7789 ΔhicB::kan csrC-lacZ Apr Kmr | This study |
| PLB2136 | CF7789 nadB::Tn10 ΔrseA csrB-lacZ Apr Tcr | This study |
| PLB2138 | CF7789 nadB::Tn10 ΔrseA csrC-lacZ Apr Tcr | This study |
| PLB2140 | CF7789 ΔhicB::kan rpoE::cam csrB-lacZ Apr Kmr Cmr | This study |
| PLB2142 | CF7789 ΔhicB::kan rpoE::cam csrC-lacZ Apr Kmr Cmr | This study |
| PLB2144 | CF7789 PlacUV5-rpoE′-′lacZ Apr | This study |
| PLB2145 | CF7789 csrA::kan PlacUV5-rpoE′-′lacZ Apr Kmr | This study |
| PLB2701 | CF7789 P1-P2-rpoE-lacZ Apr | This study |
| PLB2702 | CF7789 csrA::kan P1-P2-rpoE-lacZ Apr Kmr | This study |
| PLB2755 | CF7789 P1-ORF51′-′lacZ Apr | This study |
| PLB2756 | CF7789 P1-ORF16′-′lacZ Apr | This study |
| PLB2759 | CF7789 csrA::kan P1-ORF51′-′lacZ Apr | This study |
| PLB2760 | CF7789 csrA::kan P1-ORF16′-′lacZ Apr | This study |
| PLB2762 | CF7789 P1-P2-rpoE′-′lacZ (TAA stop codon) Apr | This study |
| PLB2763 | CF7789 csrA::kan P1-P2-rpoE′-′lacZ (TAA stop codon) Apr Kmr | This study |
| PLB2778 | CF7789 P1-P2-rpoE′-′lacZ (TGA stop codon) Apr | This study |
| PLB2779 | CF7789 P2-rpoE′-′lacZ Apr | This study |
| PLB2780 | CF7789 P2-rpoE′-′lacZ (TAA stop codon) Apr | This study |
| PLB2788 | CF7789 csrA::kan P2-rpoE′-′lacZ Apr Kmr | This study |
| PLB2789 | CF7789 csrA::kan P2-rpoE′-′lacZ (TAA stop codon) Apr Kmr | This study |
| PLB2799 | CF7789 csrA::kan P1-P2-rpoE′-′lacZ (TGA stop codon) Apr Kmr | This study |
| S17-1 λpir | recA thi pro hsdR-M+ RP4: 2-Tc::Mu Km::Tn7 λpir+ | 46 |
| SEA2000 | MG1655 rpoHP3::lacZ ΔlacX74 ΔrseA nadB::Tn10 Tcr | 28 |
| SEA6462 | MG1655 yfiC::kan ΔrseA Kmr | 52 |
| SEA6491 | MG1655 λrpoHP3-lacZ ΔlacX74 ΔhicB::kan Kmr | This study |
| TRCF7789 | CF7789 csrA::kan Kmr | 13 |
All fusions were integrated into the λ att site via the CRIM system (47). The rpoE sequences in each fusion are relative to the rpoE translation initiation codon. The csrB and csrC sequences in each fusion are relative to the start of transcription. Cmr, chloramphenicol resistance; Kmr, kanamycin resistance; Apr, ampicillin resistance; Tcr, tetracycline resistance.
E. coli bacterial strains MC1061 (48) and MG1655 (49) have been described. Plasmid pYH227 contains the csrC sequence from −289 to +188 relative to the start of transcription, and PYH229 contains the csrB sequence from −343 to +135 relative to the start of transcription. P1-mediated transduction was used to produce PLB2026 (CF7789 ΔrseA), PLB2027 (CF7789 ΔhicB), and PLB2121 (CF7789 ΔhicB ΔrpoE) using strains SEA6462 (ΔrseA), SEA6491 (ΔhicB), and CAG45170 (ΔrpoE) as the donors.
Plasmids pBW1 and pBW3 contain a csrB-lacZ transcriptional fusion (nt −282 to +5) and a csrC-lacZ transcriptional fusion (nt −289 to +4) cloned into the PstI and EcoRI sites of pLFX, respectively. These fusions were integrated into the chromosomal λ att site of strain CF7789, as described previously (47). P1-mediated transduction was then used to introduce the ΔhicB, ΔrpoE, and ΔrseA mutant alleles from strains SEA6491, CAG45170, and SEA2000, respectively.
β-Galactosidase assay.
Bacterial cultures containing lacZ fusions were grown at 30°C in Luria-Bertani (LB) broth supplemented with 100 μg/ml ampicillin and 50 μg/ml kanamycin for csrA::kan mutant strains. Cells were harvested at various times throughout growth. β-Galactosidase activity was measured as described previously (22). At least three independent experiments were performed for each strain.
Gel mobility shift assay.
Quantitative gel mobility shift assays followed a published procedure (22). His-tagged CsrA (CsrA-H6) was purified as described previously (50). WT and mutant RNAs (nt −72 to +26) were synthesized with the RNAMaxx kit (Agilent Technologies) using PCR-generated DNA templates. Gel-purified RNA was dephosphorylated and then 5′-end-labeled using T4 polynucleotide kinase (New England BioLabs) and [γ-32P]ATP (7,000 Ci/mmol). Labeled RNAs were renatured by heating for 1 min at 90°C followed by slow cooling to room temperature. Binding reaction mixtures (10 μl) contained 40 pM labeled RNA, 10 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 100 mM KCl, 40 ng of yeast RNA, 7.5% glycerol, 0.1 mg/ml xylene cyanol, and various concentrations of purified CsrA-H6. Reaction mixtures were incubated for 30 min at 37°C to allow CsrA-RNA complex formation and then fractionated through 15% polyacrylamide gels. Free and bound RNA species were visualized with a Typhoon 9410 phosphorimager (GE Healthcare). CsrA-RNA interactions were quantified as described previously (22).
Footprint assay.
CsrA-rpoE RNA footprint assays followed a published procedure (22). WT labeled rpoE RNA (nt −133 to +26) was labeled as described for the gel mobility shift assay. The reaction mixtures were identical to those in the gel shift assay except that the concentration of labeled RNA was increased to 2 nM, and 1 μg of acetylated bovine serum albumin (BSA) was added to each reaction mixture. After a 30-min incubation at 37°C to allow for CsrA-RNA complex formation, RNase T1 (0.016 U) was added, and incubation was continued for 15 min at 37°C. The reactions were stopped by adding 10 μl of stop solution (95% formamide, 0.025% SDS, 20 mM EDTA, 0.025% bromophenol blue, 0.025% xylene cyanol). Samples were heated for 5 min at 90°C and fractionated through standard 6% (vol/vol) polyacrylamide-8 M urea sequencing gels. Cleaved patterns were examined using a phosphorimager.
Toeprint assay.
CsrA-RNA toeprint assays followed a published procedure (22). Gel-purified rpoE RNA (150 nM) (nt −133 to +26 of rpoE plus a 3′ extension derived from lacZ) in Tris-EDTA (TE) buffer was hybridized to a 5′-end-labeled DNA oligonucleotide (150 nM) complementary to the lacZ 3′ extension by heating for 3 min at 85°C, followed by slow cooling to room temperature. Toeprint reaction mixtures (10 μl) contained 2 μl of the hybridization mixture (30 nM final concentration), 0 or 1 μM CsrA-H6, 375 μM each dinucleoside triphosphate (dNTP), and 10 mM dithiothreitol (DTT) in avian myeloblastosis virus (AMV) reverse transcriptase buffer. Mixtures were incubated for 30 min at 37°C to allow CsrA-RNA complex formation. AMV reverse transcriptase (0.5 U) was then added, and incubation was continued for 15 min at 37°C. The reactions were terminated by the addition of 6 μl of gel loading buffer. Samples were heated to 90°C for 5 min and fractionated through standard 6% (vol/vol) polyacrylamide-8 M urea sequencing gels. Toeprint patterns were visualized with a phosphorimager.
Coupled transcription-translation assay.
In vitro coupled transcription-translation assays using PURExpress (New England BioLabs) followed a published procedure (31). Plasmid pYH258 contains a T7 promoter driving transcription of the rpoE translational fusion from near the P2 transcription start site (nt −75 to +26 relative to the rpoE start codon). Plasmids pYH259 and pYH260 are identical to pYH258, except that they contain a GGA-to-CCA mutation in BS1 or in both BS1 and BS3, respectively (Fig. 1). pYH310 is identical to pYH258, except that it contains a TAA stop codon in ORF51 (Fig. 1). Plasmid pYH309 contains a T7 promoter that drives transcription from near the P1 transcription start site (nt −215 to +26 relative to the rpoE start codon). Plasmids pYH314 and pYH315 are identical to pYH309, except that they contain a TGA stop codon in ORF51 and a GGA-to-GCA mutation in BS1, respectively (Fig. 1). These plasmids were used as the templates for coupled transcription-translation reactions using the PURExpress in vitro protein synthesis kit, according to the manufacturer's instructions. Each 6.7-μl reaction mixture contained 250 ng of plasmid DNA template, various concentrations of purified CsrA-His6, 1 U of RNase inhibitor (Promega), 2.5 mM DTT, 2.7 μl of solution A, and 2 μl of solution B. The mixtures were incubated for 2.5 h at 37°C, and β-galactosidase activity was determined according to the manufacturer's instructions.
Primer extension assay.
Total cellular RNA was isolated from CF7789, PLB2026, PLB2027, and PLB2121 cultures during stationary phase (LB medium, 30°C) using the RNeasy kit (Qiagen). Ten micrograms of total RNA was hybridized to 150 nM 32P-end-labeled DNA oligonucleotide complementary to either nt +60 to +78 relative to csrB transcription, or to nt +52 to +71 relative to csrC transcription, for 3 min at 80°C. Reaction mixtures (10 μl) containing 2 μl of hybridization mixture, 375 μM each dNTP, 10 mM DTT, 200 μg/ml BSA (Promega), 1× SuperScript III buffer, and 5 units of SuperScript III reverse transcriptase (Life Technologies) were incubated for 15 min at 42°C. Reactions were terminated by the addition of 10 μl of stop solution (95% formamide, 20 mM EDTA, 0.025% SDS, 0.025% xylene cyanol, and 0.025% bromophenol blue). Samples were fractionated through standard 6% polyacrylamide sequencing gels and visualized with a phosphorimager. Sequencing reactions were performed using pYH227 for csrC and pYH229 for csrB as DNA templates and the same end-labeled DNA oligonucleotides as primers.
ACKNOWLEDGMENTS
This work was funded by NIH grant R01 GM059969 to T.R. and P.B. and NIH grant R01 GM097365 to S.E.A.
We thank Elena Sineva and Hongmarn Park for helpful discussions and Brendan Wilcox for technical assistance.
REFERENCES
- 1.Babitzke P, Romeo T. 2007. CsrB sRNA family: sequestration of RNA-binding regulatory proteins. Curr Opin Microbiol 10:156–163. doi: 10.1016/j.mib.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 2.Romeo T, Vakulskas CA, Babitzke P. 2013. Posttranscriptional regulation on a global scale: Form and function of Csr/Rsm systems. Env Microbiol 15:313–324. doi: 10.1111/j.1462-2920.2012.02794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vakulskas CA, Potts AH, Babitzke P, Ahmer BMM, Romeo T. 2015. Regulation of bacterial virulence by Csr (Rsm) systems. Microbiol Mol Biol Rev 79:193–224. doi: 10.1128/MMBR.00052-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Edwards AN, Patterson-Fortin LM, Vakulskas CA, Mercante JW, Potrykus K, Camacho MI, Fields JA, Thompson SA, Georgellis D, Cashel M, Babitzke P, Romeo T. 2011. Circuitry linking the Csr and stringent response global regulatory systems. Mol Microbiol 80:1561–1580. doi: 10.1111/j.1365-2958.2011.07663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holmqvist E, Wright PR, Li L, Bischler T, Barquist L, Reinhardt R, Backofen R, Vogel J. 2016. Global RNA recognition patterns of post-transcriptional regulators Hfq and CsrA revealed by UV crosslinking in vivo. EMBO J 35:991–1011. doi: 10.15252/embj.201593360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sowa SW, Gelderman G, Leistra AN, Buvanendiran A, Lipp S, Pitaktong A, Vakulskas CA, Romeo T, Baldea M, Contreras LM. 2017. Integrative FourD omics approach profiles the target network of the carbon storage regulatory system. Nucleic Acids Res 45:1673–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Potts AH, Vakulskas CA, Pannuri A, Yakhnin H, Babitzke P, Romeo T. Global role of the bacterial post-transcriptional regulator CsrA revealed by integrated transcriptomics. Nat Commun, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schubert M, Lapouge K, Duss O, Oberstrass FC, Jelesarov I, Haas D, Allain FH-T. 2007. Molecular basis of messenger RNA recognition by the specific bacterial repressing clamp RsmA/CsrA. Nat Struct Mol Biol 14:807–813. doi: 10.1038/nsmb1285. [DOI] [PubMed] [Google Scholar]
- 9.Mercante J, Edwards AN, Dubey AK, Babitzke P, Romeo T. 2009. Molecular geometry of CsrA (RsmA) binding to RNA and its implications for regulated expression. J Mol Biol 392:511–528. doi: 10.1016/j.jmb.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu MY, Gui G, Wei B, Preston JF III, Oakford L, Yüksel Ü Giedroc DP, Romeo T. 1997. The RNA molecule CsrB binds to the global regulatory protein CsrA and antagonizes its activity in Escherichia coli. J Biol Chem 272:17502–17510. doi: 10.1074/jbc.272.28.17502. [DOI] [PubMed] [Google Scholar]
- 11.Dubey AK, Baker CS, Romeo T, Babitzke P. 2005. RNA sequence and secondary structure participate in high-affinity CsrA-RNA interaction. RNA 11:1579–1587. doi: 10.1261/rna.2990205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weilbacher T, Suzuki K, Dubey AK, Wang X, Gudapaty S, Morozov I, Baker CS, Georgellis D, Babitzke P, Romeo T. 2003. A novel sRNA component of the carbon storage regulatory system of Escherichia coli. Mol Microbiol 48:657–670. doi: 10.1046/j.1365-2958.2003.03459.x. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki K, Wang X, Weilbacher T, Pernestig AK, Melefors Ö Georgellis D, Babitzke P, Romeo T. 2002. Regulatory circuitry of the CsrA/CsrB and BarA/UvrY systems of Escherichia coli. J Bacteriol 184:5130–5140. doi: 10.1128/JB.184.18.5130-5140.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chavez RG, Alvarez AF, Romeo T, Georgellis D. 2010. The physiological stimulus for the BarA sensor kinase. J Bacteriol 192:2009–2012. doi: 10.1128/JB.01685-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zere TR, Vakulskas CA, Leng Y, Pannuri A, Potts AH, Dias R, Tang D, Kolaczkowski B, Georgellis D, Ahmer BM, Romeo T. 2015. Genomic targets and features of BarA-UvrY (-SirA) signal transduction systems. PLoS One 10:e0145035. doi: 10.1371/journal.pone.0145035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suzuki K, Babitzke P, Kushner SR, Romeo T. 2006. Identification of a novel regulatory protein (CsrD) that targets the global regulatory RNAs CsrB and CsrC for degradation by RNase E. Genes Dev 20:2605–2617. doi: 10.1101/gad.1461606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vakulskas CA, Leng Y, Abe H, Amaki T, Okayama A, Babitzke P, Suzuki K, Romeo T. 2016. Antagonistic control of the turnover pathway for the global regulatory sRNA CsrB by the CsrA and CsrD proteins. Nucleic Acids Res 44:7896–7910. doi: 10.1093/nar/gkw484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leng Y, Vakulskas CA, Zere TR, Pickering BS, Watnick PI, Babitzke P, Romeo T. 2016. Regulation of CsrB/C sRNA decay by EIIAGlc of the phosphoenolpyruvate: carbohydrate phosphotransferase system. Mol Microbiol 99:627–639. doi: 10.1111/mmi.13259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Camacho MI, Alvarez AF, Chavez RG, Romeo T, Merino E, Georgellis D. 2015. Effects of the global regulator CsrA on the BarA/UvrY two-component signaling system. J Bacteriol 197:983–991. doi: 10.1128/JB.02325-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jonas K, Edwards AN, Simm R, Romeo T, Römling U, Melefors O. 2008. The RNA binding protein CsrA controls cyclic di-GMP metabolism by directly regulating the expression of GGDEF proteins. Mol Microbiol 70:236–257. doi: 10.1111/j.1365-2958.2008.06411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adamson DN, Lim HN. 2013. Rapid and robust signaling in the CsrA cascade via RNA-protein interactions and feedback regulation. Proc Natl Acad Sci U S A 110:13120–13125. doi: 10.1073/pnas.1308476110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yakhnin H, Yakhnin AV, Baker CS, Sineva E, Berezin I, Romeo T, Babitzke P. 2011. Complex regulation of the global regulatory gene csrA: CsrA-mediated translational repression, transcription from five promoters by Eσ70 and EσS, and indirect transcriptional activation by CsrA. Mol Microbiol 81:689–704. doi: 10.1111/j.1365-2958.2011.07723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baker CS, Morozov I, Suzuki K, Romeo T, Babitzke P. 2002. CsrA regulates glycogen biosynthesis by preventing translation of glgC in Escherichia coli. Mol Microbiol 44:1599–1610. doi: 10.1046/j.1365-2958.2002.02982.x. [DOI] [PubMed] [Google Scholar]
- 24.Park H, McGibbon LC, Potts AH, Yakhnin H, Romeo T, Babitzke P. 2017. Translational repression of the RpoS antiadapter IraD by CsrA is mediated via translational coupling to leader peptide synthesis. mBio 8:e01355-17. doi: 10.1128/mBio.01355-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yakhnin AV, Baker CS, Vakulskas CA, Yakhnin H, Berezin I, Romeo T, Babitzke P. 2013. CsrA activates flhDC expression by protecting flhDC mRNA from RNase E-mediated cleavage. Mol Microbiol 87:851–866. doi: 10.1111/mmi.12136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ades SE. 2008. Regulation by destruction: design of the σE envelope stress response. Curr Opin Microbiol 11:535–540. doi: 10.1016/j.mib.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 27.Rouvière PE, De Las Peñas A, Mecsas J, Lu CZ, Rudd KE, Gross CA. 1995. rpoE, the gene encoding the second heat-shock sigma factor, σE, in Escherichia coli. EMBO J 14:1032–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Costanzo A, Ades SE. 2006. Growth phase-dependent regulation of the extracytoplasmic stress factor, σE, by guanosine 3′,5′-bispyrophosphate (ppGpp). J Bacteriol 188:4627–4634. doi: 10.1128/JB.01981-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Costanzo A, Nicoloff H, Barchinger SE, Banta AB, Gourse RL, Ades SE. 2008. ppGpp and DksA likely regulate the activity of the extracytoplasmic stress factor σE in Escherichia coli by both direct and indirect mechanisms. Mol Microbiol 67:619–632. doi: 10.1111/j.1365-2958.2007.06072.x. [DOI] [PubMed] [Google Scholar]
- 30.Gopalkrishnan S, Nicoloff H, Ades SE. 2014. Co-ordinated regulation of the extracytoplasmic stress factor, sigmaE, with other Escherichia coli sigma factors by (p)ppGpp and DksA may be achieved by specific regulation of individual holoenzymes. Mol Microbiol 93:479–493. doi: 10.1111/mmi.12674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park H, Yakhnin H, Connolly M, Romeo T, Babitzke P. 2015. CsrA participates in a PNPase autoregulatory mechanism by selectively repressing translation of pnp transcripts that have been previously processed by RNase III and PNPase. J Bacteriol 197:3751–3759. doi: 10.1128/JB.00721-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pannuri A, Vakulskas CA, Zere T, McGibbon LC, Edwards AN, Georgellis D, Babitzke P, Romeo T. 2016. Circuitry linking the catabolite repression and Csr global regulatory systems of Escherichia coli. J Bacteriol 198:3000–3015. doi: 10.1128/JB.00454-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Button JE, Silhavy TJ, Ruiz N. 2007. A suppressor of cell death caused by the loss of σE downregulates extracytoplasmic stress responses and outer membrane vesicle production in Escherichia coli. J Bacteriol 189:1523–1530. doi: 10.1128/JB.01534-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gudapaty S, Suzuki K, Wang X, Babitzke P, Romeo T. 2001. Regulatory interactions of Csr components: the RNA binding protein CsrA activates csrB transcription in Escherichia coli. J Bacteriol 183:6017–6027. doi: 10.1128/JB.183.20.6017-6027.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laursen BS, Sorensen HP, Mortensen KK, Sperling-Petersen HU. 2005. Initiation of protein synthesis in bacteria. Microbiol Mol Biol Rev 69:101–123. doi: 10.1128/MMBR.69.1.101-123.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Babitzke P, Baker CS, Romeo T. 2009. Regulation of translation initiation by RNA binding proteins. Annu Rev Microbiol 63:27–44. doi: 10.1146/annurev.micro.091208.073514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raina S, Missiakas D, Georgopoulos C. 1995. The rpoE gene encoding the σE (σ24) heat shock sigma factor of Escherichia coli. EMBO J 14:1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klein G, Stupak A, Biernacka D, Wojtkiewicz P, Lindner B, Raina S. 2016. Multiple transcriptional factors regulate transcription of the rpoE gene in Escherichia coli under different growth conditions and when the lipopolysaccharide biosynthesis is defective. J Biol Chem 291:22999–23019. doi: 10.1074/jbc.M116.748954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ades SE, Grigorova IL, Gross CA. 2003. Regulation of the alternative sigma factor σE during initiation, adaptation, and shutoff of the extracytoplasmic heat shock response in Escherichia coli. J Bacteriol 185:2512–2519. doi: 10.1128/JB.185.8.2512-2519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Magnusson LU, Farewell A, Nyström T. 2005. ppGpp: a global regulator in Escherichia coli. Trends Microbiol 13:236–242. doi: 10.1016/j.tim.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 41.Rhodius VA, Suh WC, Nonaka G, West J, Gross CA. 2006. Conserved and variable functions of the σE stress response in related genomes. PLoS Biol 4:e2. doi: 10.1371/journal.pbio.0040002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martínez-Granero F, Navazo A, Barahona E, Redondo-Nieto M, Rivilla R, Martín M. 2012. The Gac-Rsm and SadB signal transduction pathways converge on AlgU to downregulate motility in Pseudomonas fluorescens. PLoS One 7:e31765. doi: 10.1371/journal.pone.0031765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stacey SD, Pritchett CL. 2016. Pseudomonas aeruginosa AlgU contributes to posttranscriptional activity by increasing rsmA expression in a mucA22 strain. J Bacteriol 198:1812–1826. doi: 10.1128/JB.00133-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stacey SD, Williams DA, Pritchett CL. 2017. The Pseudomonas aeruginosa two-component regulator AlgR directly activates rsmA expression in a phosphorylation independent manner. J Bacteriol 199:e00048-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rowley G, Spector M, Kormanec J, Roberts M. 2006. Pushing the envelope: extracytoplasmic stress responses in bacterial pathogens. Nat Rev Microbiol 4:383–394. doi: 10.1038/nrmicro1394. [DOI] [PubMed] [Google Scholar]
- 46.de Lorenzo V, Herrero M, Jakubzik U, Timmis KN. 1990. Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in Gram-negative eubacteria. J Bacteriol 172:6568–6572. doi: 10.1128/jb.172.11.6568-6572.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haldimann A, Wanner BL. 2001. Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J Bacteriol 183:6384–6393. doi: 10.1128/JB.183.21.6384-6393.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Casadaban MJ, Cohen SN. 1980. Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J Mol Biol 138:179–207. doi: 10.1016/0022-2836(80)90283-1. [DOI] [PubMed] [Google Scholar]
- 49.Jensen KF. 1993. The Escherichia coli K-12 “wild types” W3110 and MG1655 have an rph frameshift mutation that leads to pyrimidine starvation due to low pyrE expression levels. J Bacteriol 175:3401–3407. doi: 10.1128/jb.175.11.3401-3407.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mercante J, Suzuki K, Cheng X, Babitzke P, Romeo T. 2006. Comprehensive alanine-scanning mutagenesis of Escherichia coli CsrA defines two subdomains of critical functional importance. J Biol Chem 281:31832–31842. doi: 10.1074/jbc.M606057200. [DOI] [PubMed] [Google Scholar]
- 51.De Las Peñas A, Connolly L, Gross CA. 1997. The σE-mediated response to extracytoplasmic stress in Escherichia coli is transduced by RseA and RseB, two negative regulators of σE. Mol Microbiol 24:373–385. doi: 10.1046/j.1365-2958.1997.3611718.x. [DOI] [PubMed] [Google Scholar]
- 52.Nicoloff H, Gopalkrishnan S, Ades SE. 2017. Appropriate regulation of the σE-dependent envelope stress response is necessary to maintain cell envelope integrity and stationary-phase survival in Escherichia coli. J Bacteriol 199:e00089-17. doi: 10.1128/JB.00089-17. [DOI] [PMC free article] [PubMed] [Google Scholar]