

ABSTRACT
The CRISPR-Cas (clustered regularly interspaced short palindromic repeat–CRISPR-associated protein) system is unique to prokaryotes and provides the majority of bacteria and archaea with immunity against nucleic acids of foreign origin. CRISPR RNAs (crRNAs) are the key element of this system, since they are responsible for its selectivity and effectiveness. Typical crRNAs consist of a spacer sequence flanked with 5′ and 3′ handles originating from repeat sequences that are important for recognition of these small RNAs by the Cas machinery. In this investigation, we studied the type I-C CRISPR-Cas system in Porphyromonas gingivalis, a human pathogen associated with periodontitis, rheumatoid arthritis, cardiovascular disease, and aspiration pneumonia. We demonstrated the importance of the 5′ handle for crRNA recognition by the effector complex and consequently activity, as well as secondary trimming of the 3′ handle, which was not affected by modifications of the repeat sequence.
IMPORTANCE Porphyromonas gingivalis, a clinically relevant Gram-negative, anaerobic bacterium, is one of the major etiologic agents of periodontitis and has been linked with the development of other clinical conditions, including rheumatoid arthritis, cardiovascular disease, and aspiration pneumonia. The presented results on the biogenesis and functions of crRNAs expand our understanding of CRISPR-Cas cellular defenses in P. gingivalis and of horizontal gene transfer in bacteria.
KEYWORDS: CRISPR, CRISPR-Cas, Porphyromonas gingivalis, biogenesis, crRNA
INTRODUCTION
The CRISPR-Cas (clustered regularly interspaced short palindromic repeat–CRISPR-associated protein) system defends prokaryotes against potentially deleterious nucleic acids. In contrast to other prokaryotic defense mechanisms, e.g., restriction-modification or phage receptor downregulation, it provides adaptive and hereditary immunity (1), and it may be considered an example of Lamarckian inheritance. The CRISPR-Cas system is widely distributed among bacteria and archaea (2) and is composed of genomic and proteomic elements. Genomic components include a number of short fragments homologous to the targeted sequences (spacers) with short repeated sequences interspaced between them (repeats). A leader sequence is located upstream of the array of repeat-spacer units. This plays an important role during acquisition of new spacers as well as in transcription of the whole CRISPR array (3). The CRISPR array is often accompanied by an operon encoding Cas proteins, which functionalize the nucleic acid stretches. The diversity of CRISPR-Cas systems is considerable, as there are two main classes and six types distinguished (4–6).
Although each CRISPR-Cas type has its own characteristics, there is a common mode of action (adaptation-processing-nucleic acid degradation). During the first stage, nucleic acids are cleaved into fragments and incorporated into the CRISPR array as new spacers proximal to the leader element (7), being inserted between repeat sequences duplicated from a single unit (8, 9). In the second stage, the CRISPR array is transcribed and the resulting RNA matures into functional CRISPR RNAs (crRNAs). Most commonly, the whole array is transcribed, with the leader sequence acting as a promoter, and the long transcript (pre-crRNA) is cleaved and truncated by Cas6 protein homologs (10–12). In some CRISPR-Cas types, the crRNA is further truncated at its 3′ end inside an effector complex (CRISPR ribonucleoprotein [crRNP]) (13). Alternatively, in some prokaryotes, crRNAs are generated by direct transcription from a promoter embedded within the repeat sequence (14). In the final stage, crRNA is incorporated into the system's crRNP. The crRNP composition, specificity, and mechanism of action depend on the CRISPR type, yet in every case crRNPs scan nucleic acids for a sequence complementary to the spacer element contained within the scanning crRNA (protospacer). Once the region is found, the crRNA anneals to the nucleic acid (15). The resulting structure, called an R-loop, triggers conformational change in the crRNP and leads to cleavage of the nucleic acid (16).
To protect the prokaryote's own nucleic acids from degradation, type I, II, V, and VI CRISPR-Cas systems use mechanisms based on protospacer adjacent motifs (PAMs). A PAM is a short nucleotide stretch flanking the protospacer required for successful recognition of the site (17, 18). Elements interspaced between the repeat regions within the CRISPR array lack a PAM and thus do not constitute a target for the system. Type III systems use a PAM-independent safety mechanism. Instead, interaction between the 5′ handle of the crRNA and the repeat sequence in the CRISPR array protects self-DNA from cleavage (19). For type IV, no experimental data on such a safety mechanism have been provided so far.
The crRNAs are the key elements of each CRISPR-Cas system, as they carry spacer-based information required for specific DNA recognition and consequent cleavage. Aside from the spacer sequence, the crRNAs also contain fragments of repeats from both sides, which are critical for incorporation of the crRNA into the crRNP. The 5′ handles in most of the systems are 8 nucleotides (nt) long, but in types I-C and I-D they are 11 and 13 nt long, respectively (20, 21). The 3′ handle often contains a stem-loop structure, and its length may vary depending on the system. Primary pre-crRNA processing in the majority of types I and III is performed by Cas6, with the exception of type I-C, in which Cas5d carries out the processing (10).
Porphyromonas gingivalis is a Gram-negative, rod-shaped, anaerobic bacterium that belongs to the phylum Bacteroidetes. It is one of the major etiologic agents of periodontitis (22), which affects up to 30% of adults (23). Besides periodontitis, P. gingivalis has also been associated with other clinical conditions, including rheumatoid arthritis, cardiovascular disease, and aspiration pneumonia (24–27).
The P. gingivalis genome contains CRISPR arrays, which for strain W83 have been assigned the numbers 30, 36.1, 36.2, and 37 (28, 29). CRISPR30 contains 23 spacers, whereas the others contain 7 spacers each. All four CRISPR arrays are transcriptionally active (29, 30). Two Cas operons are present, one of type I-C and one of type III-B, neighboring CRISPR30 and CRISPR37, respectively. The type I-C system is active in vivo and uses a canonical NGG PAM at the 3′ end of the protospacer (29). We were not able to show the activity of the type III-B operon, consistent with the fact that it lacks the cmr1 gene, which is essential for the activity (29, 31).
In this investigation, we studied crRNA biogenesis, the role of repeat regions in formation of crRNA, and structural requirements for crRNA activity in vivo for the P. gingivalis type I-C CRISPR30. We showed that the 5′ handle of the crRNA is required for its activity (and presumably interaction with the effector Cas protein complex), while the 3′ handle is less important. We also showed that in the case of partial disruption of crRNA processing, the system is still able, to some extent, to compensate for the lack of mature crRNAs and to maintain activity.
RESULTS
In silico analysis.
Four CRISPR arrays were identified in the P. gingivalis genome (2, 28, 32), of which CRISPR30 and CRISPR37 are located in close proximity to the cas operons of types I-C and III-B, respectively. It was shown that CRISPR-Cas type I-C is functional, whereas type III-B activity was not observed (29). The activity of the CRISPR-Cas system is inseparably linked with crRNA biogenesis. Maturation of crRNAs is dependent on the sequence and often the secondary structure of the repeat (33). The role of the type I-C repeat structure has been determined in Bacillus halodurans, Thermus thermophilus, Streptococcus pyogenes, and Xanthomonas oryzae (20, 34, 35). The common element in these systems is a single G/C-rich hairpin with a tetra- or pentaloop structure on top and a single-stranded 5′-AUUGAAAC/U-3′ sequence at the 3′ end. To determine the CRISPR30 repeat structure in P. gingivalis, we employed the Mfold online RNA folding tool (36). The predicted structure formed by the repeat includes two hairpins in the form of a short G/C-rich hairpin located on top of a second, longer A/U-rich hairpin.
CRISPR30 pre-crRNA cleavage site.
To determine the cleavage site within the CRISPR30 repeat sequence, we explored a data set containing small RNA sequences from P. gingivalis W83 (30) and extracted all reads referring to CRISPR30 (positions 2102526 to 2104069). These reads were aligned with the CRISPR30 reference sequence using ClustalX, and the alignment was verified by eye. This enabled us to sort sequences into subgroups containing repeat sequences or specific spacers. Sequences within each subgroup were realigned and analyzed to identify possible gaps in the sequence coverage that could indicate crRNA processing sites within the repeat sequence (see the supplemental material). The last nucleotide covered by reads closest to the 3′ end of the repeat sequence (2102531, 2103980, 2103850, and 2103124a; repeat alignment) was A23. The first nucleotide covered by reads closest to the 5′ end of spacer sequences was U24. This included reads marked 2103893 (spacer 3 alignment), 2103827 (spacer 4 alignment), 2103233 (spacer 10 alignment), 2103101 (spacer 15 alignment), 2102971 (spacer 17 alignment), and 2102904 (spacer 18 alignment). No reads overlapping A23 and U24 were detected.
Role of the repeat in CRISPR-Cas activity.
As mentioned above, repeat elements are important for CRISPR-Cas activity, because they are responsible for crRNA biogenesis and incorporation into crRNPs. To identify sequences and/or structural motifs required for processing and interference, we prepared a series of P. gingivalis mutants with altered repeat elements by replacing a complete CRISPR30 array as shown in Fig. 1A. Briefly, each mutant carried a different modified single repeat sequence, and their type I-C CRISPR-Cas activity was verified using previously developed functional assay (29). Biogenesis of crRNAs from the artificial CRISPR30 array was analyzed using Northern blotting.
FIG 1.

CRISPR-Cas cassette in wild-type P. gingivalis and its mutants. (A) Modification of the CRISPR array. The native CRISPR30 array (top) with 23 spacers (sp1N to sp23N) was replaced with an artificial construct (bottom) containing 5 spacers: 1 native (sp4N) and 4 artificial (sp3A, sp5A, sp6A, and sp7A). Arrows indicate introduced restriction sites, which allowed for further modification of the repeat region (R). (B) Modification of the cas operon. The knockout mutant with a deletion of the cas operon (ΔCas) was prepared. The region encoding CRISPR30 Cas proteins in P. ginigvalis was replaced with an erythromycin resistance gene (ermF).
Preparation of P. gingivalis mutants.
Studying the role of the repeat element in type I-C CRISPR-Cas interference in vivo required preparation of a series of P. gingivalis mutants, each of which contained a mutation in a single repeat sequence. To facilitate this process, we designed suicide plasmid-based genetic constructs containing an easily modifiable artificial CRISPR30 array by the introduction of additional unique restriction sites (Fig. 1A). This also allowed replacement of the native CRISPR30 with the modified form by means of homologous recombination. Spacers of artificial origin were included in the novel CRISPR array to avoid interference with natural spacers. Native spacer 4 remained unmodified to serve as a reference and enable comparison between native and artificial CRISPR30 arrays. The single repeat sequence located between artificial spacers 5 and 6 was chosen for modification. Based on the predicted secondary structure of the CRISPR30 repeat sequence (Fig. 2A), we designed several alternative repeats (detailed in Fig. 2B). The general idea behind the selection of alternative repeat variants was either to disturb secondary RNA structures (mutants 1, 3, 4, and 6) or to retain them but alter the sequence (mutants 2 and 5). Three elements were chosen for examination: the long stem (mutants 1, 2, and 3), the short stem (mutants 4 and 5), and the top loop (mutant 6). Furthermore, a construct for preparation of a mutant with the type I-C operon deleted was also prepared (Fig. 1B). The main purpose of this mutant was to serve as an additional control in the functional assay. P. gingivalis mutants resulting from transformation with the suicide vectors were selected against erythromycin and verified by CRISPR30 array sequencing.
FIG 2.

Secondary RNA structures of CRISPR30 P. gingivalis repeat predicted using Mfold software. (A) Wild-type structure with major elements marked. The arrow indicates the putative primary crRNA processing site. (B) Modified structures (1 to 6). Modified nucleotides are indicated with uppercase letters and additional lines.
Functional analysis.
To investigate the effect of modifying the repeat on crRNA activity, wild-type and mutant bacteria were analyzed using the in vivo CRISPR-Cas activity assay. The assay is based on the conjugal transfer of plasmids containing the tetracycline resistance gene (tetQ) and different protospacer variants. Following conjugation, bacteria were selected using tetracycline. If the plasmid carrying tetQ is degraded due to CRISPR-Cas interference, cells are unable to survive in the presence of the antibiotic. Conversely, if the CRISPR-Cas system is not able to recognize a particular protospacer, the plasmid remains intact and bacteria can survive in the presence of the antibiotic. Interference was investigated with plasmids containing protospacers recognized by both natural and artificial spacers. Additionally, a plasmid lacking a protospacer sequence was used as a control. The results (Fig. 3) showed that for wild-type bacteria, interference was observed for native protospacers 4, 5, and 6. Meanwhile, the mutant with an artificial CRISPR cassette with an unmodified repeat sequence (artificial CRISPR backbone [ACbb]) recognized native protospacer 4 as well as artificial protospacers 5 and 6. ACbb was constructed to study the role of CRISPR arrays by introduction of restriction sites allowing for simple mutagenesis. All mutants with a modified repeat sequence recognized native protospacers 4 and artificial protospacer 5 but not artificial protospacer 6. No interference was observed for the mutant lacking the cas operon with any of the protospacer variants.
FIG 3.

In vivo assessment of CRISPR-Cas system activity. Plasmids containing different protospacer sequences were conjugated into P. gingivalis W83 wild-type bacteria or mutants with modified CRISPR-Cas cassettes. Subsequent tetracycline selection revealed which bacteria lost their resistance to antibiotics due to the CRISPR-Cas interference. Consequently, the number of bacteria able to form colonies (CFU) in the presence of tetracycline was reversely correlated to the activity of CRISPR-Cas interference. Values on the y axis represent the numbers of bacterial colonies. Each experiment was repeated at least three times. Error bars represent standard deviations. PS, protospacer; N, native; A, artificial.
Biogenesis of crRNAs.
To acquire insight into the biogenesis of crRNAs with altered repeat sequences in bacteria, we performed Northern blot analysis using the biotinylated oligonucleotide probes complementary to the repeat sequence, and spacer sequences neighboring modified repeats (sp5A and sp6A) (Fig. 4). Probe detection of the repeat sequence revealed a characteristic pattern. Visible bands corresponded in size to single (∼65 nt), double (∼130 nt), and triple (∼195 nt) repeat-spacer units. For the majority of the mutants (2, 4, 5, and 6,) a quadruple (∼260 nt) repeat-spacer unit was also observed. In addition, variant 1 also displayed a single discrete band just below the double repeat-spacer unit due to deletion within the repeat region. The probe detecting native spacer 4, which is common to wild-type bacteria and all artificial CRISPR mutants, revealed the same band pattern for all variants except the ΔCas mutant. This pattern included three distinct bands representing single and double repeat-spacer units, as well as a single band of <50 nt. Northern blots carried out with a probe complementary to artificial spacer 5 detected two distinct RNA fragments of <50 nt, with less dense bands corresponding in size to the double repeat-spacer. In contrast, the probe complementary to artificial spacer 6 did not reveal the presence of RNA species smaller than 50 nt for variants in which the repeat sequence was altered. With the ΔCas mutant, no bands were detected by any probe, confirming that the presence of crRNAs is entirely dependent on the I-C CRISPR-Cas system.
FIG 4.

Northern blot analysis of crRNAs in wild-type P. gingivalis and its modified variants. Certain crRNAs were detected with biotinylated probes complementary to the repeat and spacers sp4N, sp5A, and sp6A. WT, wild type; ACbb, artificial CRISPR backbone; ΔCas, mutant with deleted cas operon; 1 to 6, mutants with modified repeat. On the right side of each blot, schematic structures of possible crRNA maturation intermediates are included.
DISCUSSION
Biogenesis of crRNAs is essential for CRISPR-Cas activity and differs considerably between CRISPR-Cas types. Cleavage of the long pre-crRNA precursor into single repeat-spacer units is guided by interaction of the repeat sequence with a nuclease, often dependent on the secondary RNA structure (37). In contrast to most type I CRISPR-Cas systems, type I-C was shown to utilize Cas5d as a nuclease performing primary processing (20), recognizing the structural motif formed within the repeat element. The secondary RNA structures of type I-C repeats described so far contain a G/C-rich hairpin with a stem of 7 to 9 bp, a top loop, and a single-stranded sequence at the 3′ end containing the conserved 5′-AUUGAAAC/U-3′ motif (20, 34, 35). A consistent model for CRISPR30 of P. gingivalis was obtained using in silico modeling, yet some differences are evident (Fig. 2). The repeat region seems to form a long A/T-rich stem, with a shorter G/C-rich hairpin on the top loop. The predicted hairpin structure encompasses the complete repeat region, which lacks the single-stranded sequence at the 3′ end. Nevertheless, the conserved motif at the top loop is retained. In previously described type I-C repeat structures, Cas5d cleaves pre-crRNA at the 3′ base of the hairpin (20, 34, 35), but our analysis of deep-sequencing data for small RNA-enriched P. gingivalis W83 transcripts suggests a probable location of primary processing cleavage between A23 and U24 in the stem region (Fig. 5). One may, however, assume that due to high A/U content, the hairpin structure is metastable and the single-stranded region is at least partially exposed.
FIG 5.

Predicted structure of P. gingivalis CRISPR30 crRNA. The spacer sequence is flanked with 5′ and 3′ handles derived from the repeat regions. While the 5′ handle seems to be important for incorporation of the crRNA into the effector complex, the 3′ handle is essential for pre-crRNA maturation. Further, the region potentially trimmed during effector complex formation is marked.
The previous studies on crRNA biogenesis in type I-C systems focused on processing of different repeat variants in vitro. These showed that the 3′ end of the repeat is essential for pre-crRNA digestion (20, 38). The predicted secondary structure of the repeat region of P. gingivalis CRISPR30 shows differences from previously described repeat structures. Thus, to understand the role of each region of the hairpin, we developed an in vivo model that utilized a set of P. gingivalis mutants with altered repeat sequence between artificial spacers 5 and 6 (Fig. 2). The activity of CRISPR30 was evaluated in P. gingivalis mutants using functional analysis and compared to that of the wild-type strain. Native spacer 4 remained intact and was used as an internal control. No difference in nucleic acid degradation between the wild-type bacteria and mutated variants was observed for this spacer, confirming the validity of the developed model.
Activity of the artificial spacer 5, located upstream of the repeat, was not affected, regardless of the change implemented in the repeat region (including complete disruption). On the other hand, every modification of the repeat region disabled activity of artificial spacer 6, located downstream of the repeat. To fully study this phenomenon, we analyzed the crRNA biogenesis in different mutants using Northern blotting. Consistently with the results of the functional assay, we did not observe any differences in the pattern of pre-crRNA and crRNA fragments for the native spacer 4 region, whereas for the ΔCas no crRNA at any stage of maturation was visible, further confirming the importance of Cas proteins in biogenesis of crRNA. The lack of any crRNA transcripts can be caused by their low stability in the absence of Cas proteins, but it cannot be excluded that Cas proteins may act as transcription factors (35). Interestingly, despite the artificial spacer 5 activity remaining unchanged for modified repeat variants, Northern blot analysis revealed a lack of mature crRNA. This suggests that the cleavage between artificial spacers 5 and 6 might have been hampered due to alteration of the repeat sequence or structure. In turn, smaller fragments corresponding roughly in size to the spacer without the repeat are present, suggesting downstream processing of the crRNA. For biologically inactive artificial spacer 6, the noncleaved, duplicated repeat-spacer unit was observable, while shorter fragments were not detected. Figure 6 presents the proposed mechanism of crRNA biogenesis in artificial CRISPR mutants with and without modified repeat sequence. It must also be noted that in addition to Cas5d the P. gingivalis type I-C operon also encodes Cas6, which is a pre-crRNA nuclease in most of type I systems, as previously reported (39, 40). One may assume that both of these proteins could be involved in crRNA processing in P. gingivalis.
FIG 6.
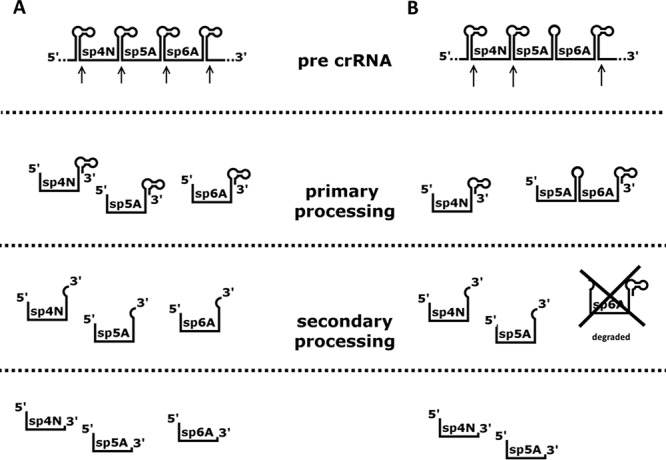
Proposed mechanism of crRNA biogenesis in artificial CRISPR mutants. Arrows indicate primary processing cleavage sites. (A) In the case of unmodified repeat sequence between artificial spacers 5 and 6, primary processing of pre-crRNA results in series of single repeat-spacer units, which are further trimmed at their 3′ ends during secondary processing. (B) Any modification into repeat sequence between artificial spacers 5 and 6 abolished cleavage within modified repeat sequence. This is evident by the lack of observable single repeat-spacer units for probes complementary to those spacers. Secondary processing, which is probably independent from the primary cleavage, separates spacer 5 crRNA from the spacer 6 fragment, which is degraded during the process. As a result, functional spacer 5 crRNA molecule is generated.
Together, the results from the functional assay and Northern blot analysis confirm that the 5′ terminus, but not the 3′ terminus, of crRNA is essential for its activity. This is supported by structural data available for type I interference complexes (CASCADE), where the 5′ end of the crRNA is embedded in close proximity to a Cas8 protein, which is referred to as the large subunit of CASCADE, involved in PAM recognition (15, 41).
Furthermore, the presence of short (<50-nt) fragments for sp5A crRNA (active), which were lacking for the artificial spacer 6 crRNA (inactive), suggests two options. Presumably, crRNAs may be further trimmed in a mechanism independent from the repeat hairpin. Trimming of the 3′ end of crRNA is common in type III as well as in many type I systems (including types I-A, I-B, and I-D) (11, 21, 42, 43). It can therefore be assumed that spacer 5-repeat-spacer 6-repeat molecules are incorporated into the CASCADE complex via the 5′ terminus of the sp5A region, whereas the sp6A crRNA region lacks the 5′ handle and is biologically inactive. Further, the artificial spacer 5 crRNAs are trimmed to yield the spacer region deprived of the repeat sequence. Whether the spacer region by itself can serve as the active crRNA remains to be shown, yet one may assume such a scenario, as even the complete removal of the repeat structures located at the 3′ end did not affect sp5A crRNA activity.
To summarize, we showed the role of the repeat element in the CRISPR cassette of the P. gingivalis CRISPR30 in biogenesis and biological activity of corresponding crRNAs. Our results indicate that a 5′ handle of crRNA is essential for the activity. On the other hand, the repeat element localizing to the 3′ handle seems less important. It also appears that each tested modification of the structure or the sequence of the repeat element hampers its activity, most likely by impeding the ability to interact with the primary processing nuclease. These results are important for understanding the biology of P. gingivalis and the CRISPR-Cas system.
MATERIALS AND METHODS
In silico analysis.
Secondary structures of the CRISPR30 repeat sequence and its derivatives were predicted using the Mfold web server (version 2.3) (36). Alignment of sequences from transcriptome sequencing was performed using ClustalX (version 2.1) (44).
Purification of nucleic acids.
Plasmids from E. coli were purified using a GeneJET plasmid miniprep kit (Thermo Fisher Scientific) in accordance with the manufacturer's instructions. Genomic DNA from P. gingivalis was isolated using a genomic minikit (A&A Biotechnology, Poland) in accordance with the manufacturer's instructions. Total RNA from P. gingivalis was isolated using Fenozol (A&A Biotechnology) according to the manufacturer's protocol, with slight modifications. Briefly, 10 ml of an overnight P. gingivalis culture was centrifuged at 5,000 × g and 4°C for 10 min. The pellet was resuspended in 1 ml of Fenozol and centrifuged (12,000 × g and 4°C for 10 min). The aqueous phase was transferred to a new tube and 200 μl of chloroform was added. The sample was vortexed and centrifuged (12,000 × g and 4°C for 15 min), and the aqueous phase was transferred to a new tube, mixed with 500 μl of propan-2-ol, and incubated for 16 h at −20°C. After centrifugation (12,000 × g and 4°C for 10 min) and a washing with 1 ml of 75% ethanol, RNA was dissolved in 50 μl of ultrapure diethyl pyrocarbonate (DEPC)-treated water (Life Technologies, USA) and treated with Turbo DNase (Life Technologies, USA) according to the manufacturer's instructions. The RNA concentration was determined with a NanoDrop spectrophotometer (Thermo Fisher Scientific). Samples were stored at −80°C.
Preparation of plasmids containing protospacers.
Different protospacer sequences were inserted into the shuttle vector pT-COW as described previously (29). Briefly, synthetic oligonucleotides (listed in Table 1) encoding protospacer sequences were annealed to form double-stranded DNA with single-stranded overhangs. These overhangs enabled sticky-end ligation with pT-COW plasmid previously digested with HindIII and SalI restriction enzymes (Thermo Fisher Scientific). The resulting plasmids were used to transform chemically competent E. coli TOP10 bacteria (Thermo Fisher Scientific). Transformants were selected against ampicillin and the constructs were verified by DNA sequencing.
TABLE 1.
Oligonucleotides used for construction of inserts containing protospacer sequences
| Spacer name | Sequences (5′→3′) |
|---|---|
| Native spacer 4 | AGC TAA ATT TCA GCA TTG TAT TGA ACT GAA CAT ATA GAG AAT CAG G; TCG ACC TGA TTC TCT ATA TGT TCA GTT CAA TAC AAT GCT GAA ATT T; TCG ATT TGA TTC TCT ATA TGT TCA GTT CAA TAC AAT GCT GAA ATT T |
| Native spacer 5 | AGC TAA AAA AGT TTT AAG ATT AGC AAA CAT TTT ACC ATC TTG TAG G; TCG ACC TAC AAG ATG GTA AAA TGT TTG CTA ATC TTA AAA CTT TTT T; TCG ATT TAC AAG ATG GTA AAA TGT TTG CTA ATC TTA AAA CTT TTT T |
| Native spacer 6 | AGC TCC TTT TAC TAC ATT GAA AAA ATC GTC TTC GTC TGC TAA AAA; TCG ATT TTT AGC AGA CGA AGA CGA TTT TTT CAA TGT AGT AAA AGG; TCG ATT TTT AGC AGA CGA AGA CGA TTT TTT CAA TGT AGT AAA TTT |
| Artificial spacer 5 | AGC TCC TTT TAT CGT AAA AAT TCG GAT CCA TTC TAT TTA AGA AAA A; TCG ATT TTT CTT AAA TAG AAT GGA TCC GAA TTT TTA CGA TAA AAG G; TCG ATT TTT CTT AAA TAG AAT GGA TCC GAA TTT TTA CGA TAA ATT T |
| Artificial spacer 6 | AGC TCC TTA ATC ATT CAA AAT GCG GCC GCC TTA TTA TTT AAT AAA; TCG ATT TAT TAA ATA ATA AGG CGG CCG CAT TTT GAA TGA TTA AGG; TCG ATT TAT TAA ATA ATA AGG CGG CCG CAT TTT GAA TGA TTA TTT |
Bacterial culture.
E. coli was grown under aerobic conditions (shaking at 200 rpm) in LB medium (Bioshop, Canada). LB plates were supplemented with 2% agar (Bioshop, Canada). Bacterial stocks were stored at −80°C in medium supplemented with glycerol. When needed, ampicillin (100 μg/ml) was added.
P. gingivalis was cultured under anaerobic conditions (80% N2, 10% CO2, and 10% H2) using an A85 anaerobic workstation (Whitley Scientific, UK). For liquid cultures, tryptic soy broth (TSB; Fluka, Switzerland) supplemented with 0.5% yeast extract (Bioshop, Canada), l-cysteine (0.5 mg/ml; Bioshop, Canada), menadione (0.5 μg/ml; ICN Biomedicals, USA), and hemin (5 μg/ml; ICN Biomedicals) was used. TSB plates were additionally supplemented with 5% sheep blood and 2% agar. To prepare bacterial stocks, cells from culture plates were transferred to sterile 10% skimmed milk (Bioshop) in water and stored at −80°C. When needed, tetracycline (1.0 μg/ml), erythromycin (5.0 μg/ml), and/or gentamicin (150 μg/ml) was added. Electrocompetent P. gingivalis cells were prepared according to the protocol described by Bélanger et al. (45).
Preparation of P. gingivalis mutants.
Inserts for the preparation of P. gingivalis mutants with a modified CRISPR array or with the cas operon removed (ΔCas) were synthesized by GeneArt (Thermo Fisher Scientific) and incorporated into the pMK-RQ plasmid (Fig. 1). The repeat sequence within the plasmids was modified by cloning. Briefly, the purchased backbone plasmid containing the unmodified repeat sequence was digested with BamHI and NotI restriction enzymes (New England BioLabs) and gel purified. Inserts were prepared by hybridization of synthetic nucleotides (Table 2) (Genomed, Poland) as described previously (29) and ligated using T4 DNA ligase (Thermo Fisher Scientific), and the resulting plasmids were used to transform E. coli TOP10 bacteria (Thermo Fisher Scientific). Positive transformants were selected using ampicillin, plasmids were isolated, and their identity was confirmed by DNA sequencing.
TABLE 2.
Oligonucleotides used for construction of P. gingivalis mutants with altered repeat sequences
| Mutant | Sequences (5′→3′)a |
|---|---|
| 1 | ggc cgc att ttg aat gat tag ttt cct gta tgg tgc aaa ttt ctt aaa tag aat g; gat cca ttc tat tta aga aat ttg cac cat aca gga aac taa tca ttc aaa atg c |
| 2 | ggc cgc att ttg aat gat tag AAA tTT AAc ctg tat ggt gcT TAA gTT TAt tct taa ata gaa tg; gat cca ttc tat tta aga aTA AAc TTA Agc acc ata cag gTT AAa TTT cta atc att caa aat gc |
| 3 | ggc cgc att ttg aat gat tag AAA GTT AAc ctg tat ggt gca att gaa att tct taa ata gaa tg; gat cca ttc tat tta aga aat ttc aat tgc acc ata cag gTT AAC TTT cta atc att caa aat gc |
| 4 | ggc cgc att ttg aat gat tag ttt taa ttc ctg tat CCt gca att gaa att tct taa ata gaa tg; gat cca ttc tat tta aga aat ttc aat tgc aGG ata cag gaa tta aaa cta atc att caa aat gc |
| 5 | ggc cgc att ttg aat gat tag ttt taa ttG Gtg tat CCt gca att gaa att tct taa ata gaa tg; gat cca ttc tat tta aga aat ttc aat tgc aGG ata caC Caa tta aaa cta atc att caa aat gc |
| 6 | ggc cgc att ttg aat gat tag ttt taa ttc gtg caa ttg aaa ttt ctt aaa tag aat g; gat cca ttc tat tta aga aat ttc aat tgc acg aat taa aac taa tca ttc aaa atg c |
The repeat region is marked with bold letters; uppercase letters represent regions altered compared to the original sequence.
Mutants were prepared by homologous recombination. Briefly, 1 μg of vector containing the artificial CRISPR cassette with the desired modifications was linearized by digestion with KpnI restriction enzyme (New England BioLabs) according to the manufacturer's instructions in a 10-μl reaction mixture. The digested vector was cooled on ice, mixed with 100 μl of electrocompetent P. gingivalis W83 cells, and transferred to a cuvette, and transformation was carried out with a Micropulser device (Bio-Rad) using a 2.5-kV pulse for 4 ms. Immediately after pulsing, 1 ml of prewarmed TSB medium was added and bacteria were cultured at 37°C under anaerobic conditions for 24 h. Subsequently, bacteria were plated on TSB plates with erythromycin and cultured at 37°C in anaerobic conditions for 7 to 14 days. Single colonies were collected and verified by DNA sequencing.
Assessment of CRISPR-Cas activity.
The CRISPR-Cas activity of each mutant was evaluated based on the outcome of the conjugal transfer of plasmids carrying particular protospacers, which determined antibiotic resistance. Briefly, CRISPR-Cas-mediated degradation of plasmids resulted in the loss of antibiotic resistance and consequently a lack of growth. For conjugation, the E. coli S 17-1 donor strain was transformed with pT-COW plasmids containing different protospacer sequences and selection was performed with ampicillin. Multiple colonies for each protospacer variant were mixed to prepare conjugation stocks that were stored at −80°C. Each of the E. coli S 17-1 samples was separately cultured on an LB plate containing ampicillin. P. gingivalis mutants were cultured on TSB plates with erythromycin. Wild-type P. gingivalis cells were cultured on plates without antibiotics. Bacteria were harvested from plates, mixed together in equal quantities, seeded on a fresh TSB plate without antibiotics, and incubated anaerobically overnight at 37°C. Subsequently, cultures were reseeded on TSB plates containing gentamicin and tetracycline. After 7 to 11 days of incubation under anaerobic conditions at 37°C, plates were imaged using a DSC-HX400V camera (Sony) with a white backlight. Colonies were counted using OpenCFU software (46), and the results were verified by eye.
Northern blotting.
Bacterial RNA from P. gingivalis (4 μg) was separated on a 15% polyacrylamide gel (19:1 acrylamide-bisacrylamide) in the presence of 8 M urea. The gel was prerun at 200 V for 1 h and samples were denatured at 65°C and separated at 180 V. After separation, RNA was transferred to an Immobilon-Ny+ membrane (Merck Millipore) using the wet transfer method with a Mini Trans-Blot cell (Bio-Rad). Membranes were then fixed by cross-linking using a Hoefer UVC 500 UV cross-linker (Hoefer, USA) system at 70,000 μJ/cm2. Hybridization was performed in a Shake ‘n’ Stack hybridization oven (Thermo Fisher Scientific) with 5 ml of hybridization buffer (7% SDS, 200 mM phosphate buffer [pH 7.2]). Incubation was carried out at 46°C for 30 min. Hybridization buffer was then supplemented with a 500 μM concentration of the specific biotinylated probe (Table 3) and incubated overnight. Subsequently, membranes were washed twice with 15 ml of wash buffer (2× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate], pH 7.0, and 1% SDS) for 15 min each time. Membranes were then incubated for 2 h with 5 ml of wash buffer (2× SSC, 0.1% SDS) supplemented with streptavidin-conjugated horseradish peroxidase (HRP; diluted 1:25,000; GE Healthcare). The signal was visualized with 1 ml of Immobilon Western chemiluminescent HRP (Millipore) and recorded using a ChemiDoc imager (Bio-Rad).
TABLE 3.
Probes used for Northern blot analysis
| Recognized sequence | Sequence (5′→3′) |
|---|---|
| Native spacer 4 | GAT TCT CTA TAT GTT CAG TTC AAT ACA ATG CTG AAA-biotin |
| Native spacer 5 | ACA AGA TGG TAA AAT GTT TGC TAA TCT TAA AAC TTT-biotin |
| Native spacer 6 | TTA GCA GAC GAA GAC GAT TTT TTC AAT GTA GTA AA-biotin |
| Artificial spacer 5 | TTC TTA AAT AGA ATG GAT CCG AAT TTT TAC GAT AAA-biotin |
| Artificial spacer 6 | ATT AAA TAA TAA GGC GGC CGC ATT TTG AAT GAT TA-biotin |
| Repeat | GTT TTA ATT CCT GTA TGG TGC AAT TGA AAT-biotin |
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from the National Science Center, Poland (2011/01/D/NZ6/00269), to K.P. The Faculty of Biochemistry, Biophysics and Biotechnology of Jagiellonian University is a beneficiary of structural funds from the European Union (grant no. POIG.02.01.00-12-064/08, Molecular Biotechnology for Health). The Faculty of Biochemistry, Biophysics and Biotechnology is a partner of the Leading National Research Center (KNOW), supported by the Ministry of Science and Higher Education.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We have no competing interests.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00275-17.
REFERENCES
- 1.Westra ER, Swarts DC, Staals RH, Jore MM, Brouns SJ, van der Oost J. 2012. The CRISPRs, they are a-changin': how prokaryotes generate adaptive immunity. Annu Rev Genet 46:311–339. doi: 10.1146/annurev-genet-110711-155447. [DOI] [PubMed] [Google Scholar]
- 2.Grissa I, Vergnaud G, Pourcel C. 2007. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics 8:172. doi: 10.1186/1471-2105-8-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jansen R, Embden JD, Gaastra W, Schouls LM. 2002. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43:1565–1575. doi: 10.1046/j.1365-2958.2002.02839.x. [DOI] [PubMed] [Google Scholar]
- 4.Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, van der Oost J, Koonin EV. 2011. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol 9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJ, Charpentier E, Haft DH, Horvath P, Moineau S, Mojica FJ, Terns RM, Terns MP, White MF, Yakunin AF, Garrett RA, van der Oost J, Backofen R, Koonin EV. 2015. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol 13:722–736. doi: 10.1038/nrmicro3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shmakov S, Smargon A, Scott D, Cox D, Pyzocha N, Yan W, Abudayyeh OO, Gootenberg JS, Makarova KS, Wolf YI, Severinov K, Zhang F, Koonin EV. 2017. Diversity and evolution of class 2 CRISPR-Cas systems. Nat Rev Microbiol 15:169–182. doi: 10.1038/nrmicro.2016.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pourcel C, Salvignol G, Vergnaud G. 2005. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151:653–663. doi: 10.1099/mic.0.27437-0. [DOI] [PubMed] [Google Scholar]
- 8.Yosef I, Goren MG, Qimron U. 2012. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res 40:5569–5576. doi: 10.1093/nar/gks216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Díez-Villaseñor C, Guzmán NM, Almendros C, García-Martínez J, Mojica FJ. 2013. CRISPR-spacer integration reporter plasmids reveal distinct genuine acquisition specificities among CRISPR-Cas I-E variants of Escherichia coli. RNA Biol 10:792–802. doi: 10.4161/rna.24023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reeks J, Naismith JH, White MF. 2013. CRISPR interference: a structural perspective. Biochem J 453:155–166. doi: 10.1042/BJ20130316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hatoum-Aslan A, Maniv I, Marraffini LA. 2011. Mature clustered, regularly interspaced, short palindromic repeats RNA (crRNA) length is measured by a ruler mechanism anchored at the precursor processing site. Proc Natl Acad Sci U S A 108:21218–21222. doi: 10.1073/pnas.1112832108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hale CR, Majumdar S, Elmore J, Pfister N, Compton M, Olson S, Resch AM, Glover CV, Graveley BR, Terns RM, Terns MP. 2012. Essential features and rational design of CRISPR RNAs that function with the Cas RAMP module complex to cleave RNAs. Mol Cell 45:292–302. doi: 10.1016/j.molcel.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hatoum-Aslan A, Samai P, Maniv I, Jiang W, Marraffini LA. 2013. A ruler protein in a complex for antiviral defense determines the length of small interfering CRISPR RNAs. J Biol Chem 288:27888–27897. doi: 10.1074/jbc.M113.499244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, Heidrich N, Ampattu BJ, Gunderson CW, Seifert HS, Schoen C, Vogel J, Sontheimer EJ. 2013. Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis. Mol Cell 50:488–503. doi: 10.1016/j.molcel.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sashital DG, Wiedenheft B, Doudna JA. 2012. Mechanism of foreign DNA selection in a bacterial adaptive immune system. Mol Cell 46:606–615. doi: 10.1016/j.molcel.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jore MM, Lundgren M, van Duijn E, Bultema JB, Westra ER, Waghmare SP, Wiedenheft B, Pul U, Wurm R, Wagner R, Beijer MR, Barendregt A, Zhou K, Snijders AP, Dickman MJ, Doudna JA, Boekema EJ, Heck AJ, van der Oost J, Brouns SJ. 2011. Structural basis for CRISPR RNA-guided DNA recognition by Cascade. Nat Struct Mol Biol 18:529–536. doi: 10.1038/nsmb.2019. [DOI] [PubMed] [Google Scholar]
- 17.Semenova E, Jore MM, Datsenko KA, Semenova A, Westra ER, Wanner B, van der Oost J, Brouns SJ, Severinov K. 2011. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc Natl Acad Sci U S A 108:10098–10103. doi: 10.1073/pnas.1104144108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marraffini LA, Sontheimer EJ. 2010. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 463:568–571. doi: 10.1038/nature08703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nam KH, Haitjema C, Liu X, Ding F, Wang H, DeLisa MP, Ke A. 2012. Cas5d protein processes pre-crRNA and assembles into a cascade-like interference complex in subtype I-C/Dvulg CRISPR-Cas system. Structure 20:1574–1584. doi: 10.1016/j.str.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scholz I, Lange SJ, Hein S, Hess WR, Backofen R. 2013. CRISPR-Cas systems in the cyanobacterium Synechocystis sp. PCC6803 exhibit distinct processing pathways involving at least two Cas6 and a Cmr2 protein. PLoS One 8:e56470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL. 1998. Microbial complexes in subgingival plaque. J Clin Periodontol 25:134–144. doi: 10.1111/j.1600-051X.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- 23.Cobb CM, Williams KB, Gerkovitch MM. 2009. Is the prevalence of periodontitis in the U S A in decline? Periodontol 2000 50:13–24. doi: 10.1111/j.1600-0757.2008.00284.x. [DOI] [PubMed] [Google Scholar]
- 24.de Pablo P, Chapple IL, Buckley CD, Dietrich T. 2009. Periodontitis in systemic rheumatic diseases. Nat Rev Rheumatol 5:218–224. doi: 10.1038/nrrheum.2009.28. [DOI] [PubMed] [Google Scholar]
- 25.Friedewald VE, Kornman KS, Beck JD, Genco R, Goldfine A, Libby P, Offenbacher S, Ridker PM, Van Dyke TE, Roberts WC. 2009. The American Journal of Cardiology and Journal of Periodontology editors' consensus: periodontitis and atherosclerotic cardiovascular disease. Am J Cardiol 104:59–68. doi: 10.1016/j.amjcard.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 26.Terpenning MS, Taylor GW, Lopatin DE, Kerr CK, Dominguez BL, Loesche WJ. 2001. Aspiration pneumonia: dental and oral risk factors in an older veteran population. J Am Geriatr Soc 49:557–563. doi: 10.1046/j.1532-5415.2001.49113.x. [DOI] [PubMed] [Google Scholar]
- 27.Benedyk M, Mydel PM, Delaleu N, Płaza K, Gawron K, Milewska A, Maresz K, Koziel J, Pyrc K, Potempa J. 2016. Gingipains: critical factors in the development of aspiration pneumonia caused by Porphyromonas gingivalis. J Innate Immun 8:185–198. doi: 10.1159/000441724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watanabe T, Nozawa T, Aikawa C, Amano A, Maruyama F, Nakagawa I. 2013. CRISPR regulation of intraspecies diversification by limiting IS transposition and intercellular recombination. Genome Biol Evol 5:1099–1114. doi: 10.1093/gbe/evt075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burmistrz M, Dudek B, Staniec D, Rodriguez Martinez JI, Bochtler M, Potempa J, Pyrc K. 2015. Functional analysis of Porphyromonas gingivalis W83 CRISPR-Cas systems. J Bacteriol 197:2631–2641. doi: 10.1128/JB.00261-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phillips P, Progulske-Fox A, Grieshaber S, Grieshaber N. 2014. Expression of Porphyromonas gingivalis small RNA in response to hemin availability identified using microarray and RNA-seq analysis. FEMS Microbiol Lett 351:202–208. doi: 10.1111/1574-6968.12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L, Terns RM, Terns MP. 2009. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 139:945–956. doi: 10.1016/j.cell.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nelson KE, Fleischmann RD, DeBoy RT, Paulsen IT, Fouts DE, Eisen JA, Daugherty SC, Dodson RJ, Durkin AS, Gwinn M, Haft DH, Kolonay JF, Nelson WC, Mason T, Tallon L, Gray J, Granger D, Tettelin H, Dong H, Galvin JL, Duncan MJ, Dewhirst FE, Fraser CM. 2003. Complete genome sequence of the oral pathogenic bacterium Porphyromonas gingivalis strain W83. J Bacteriol 185:5591–5601. doi: 10.1128/JB.185.18.5591-5601.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lange SJ, Alkhnbashi OS, Rose D, Will S, Backofen R. 2013. CRISPRmap: an automated classification of repeat conservation in prokaryotic adaptive immune systems. Nucleic Acids Res 41:8034–8044. doi: 10.1093/nar/gkt606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garside EL, Schellenberg MJ, Gesner EM, Bonanno JB, Sauder JM, Burley SK, Almo SC, Mehta G, MacMillan AM. 2012. Cas5d processes pre-crRNA and is a member of a larger family of CRISPR RNA endonucleases. RNA 18:2020–2028. doi: 10.1261/rna.033100.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koo Y, Ka D, Kim EJ, Suh N, Bae E. 2013. Conservation and variability in the structure and function of the Cas5d endoribonuclease in the CRISPR-mediated microbial immune system. J Mol Biol 425:3799–3810. doi: 10.1016/j.jmb.2013.02.032. [DOI] [PubMed] [Google Scholar]
- 36.Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kunin V, Sorek R, Hugenholtz P. 2007. Evolutionary conservation of sequence and secondary structures in CRISPR repeats. Genome Biol 8:R61. doi: 10.1186/gb-2007-8-4-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hochstrasser ML, Taylor DW, Kornfeld JE, Nogales E, Doudna JA. 2016. DNA targeting by a minimal CRISPR RNA-guided cascade. Mol Cell 63:840–851. doi: 10.1016/j.molcel.2016.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. 2008. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carte J, Wang R, Li H, Terns RM, Terns MP. 2008. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev 22:3489–3496. doi: 10.1101/gad.1742908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Westra ER, van Erp PB, Künne T, Wong SP, Staals RH, Seegers CL, Bollen S, Jore MM, Semenova E, Severinov K, de Vos WM, Dame RT, de Vries R, Brouns SJ, van der Oost J. 2012. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Mol Cell 46:595–605. doi: 10.1016/j.molcel.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lintner NG, Kerou M, Brumfield SK, Graham S, Liu H, Naismith JH, Sdano M, Peng N, She Q, Copié V, Young MJ, White MF, Lawrence CM. 2011. Structural and functional characterization of an archaeal clustered regularly interspaced short palindromic repeat (CRISPR)-associated complex for antiviral defense (CASCADE). J Biol Chem 286:21643–21656. doi: 10.1074/jbc.M111.238485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Richter H, Zoephel J, Schermuly J, Maticzka D, Backofen R, Randau L. 2012. Characterization of CRISPR RNA processing in Clostridium thermocellum and Methanococcus maripaludis. Nucleic Acids Res 40:9887–9896. doi: 10.1093/nar/gks737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 45.Bélanger M, Rodrigues P, Progulske-Fox A. 2007. Genetic manipulation of Porphyromonas gingivalis. Curr Protoc Microbiol Chapter 13:Unit13C.2. [DOI] [PubMed] [Google Scholar]
- 46.Geissmann Q. 2013. OpenCFU, a new free and open-source software to count cell colonies and other circular objects. PLoS One 8:e54072. doi: 10.1371/journal.pone.0054072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.