

ABSTRACT
The Escherichia coli dnaXE145A mutation was discovered in connection with a screen for multicopy suppressors of the temperature-sensitive topoisomerase IV mutation parE10. The gene for the clamp loader subunits τ and γ, dnaX, but not the mutant dnaXE145A, was found to suppress parE10(Ts) when overexpressed. Purified mutant protein was found to be functional in vitro, and few phenotypes were found in vivo apart from problems with partitioning of DNA in rich medium. We show here that a large number of the replication forks that initiate at oriC never reach the terminus in dnaXE145A mutant cells. The SOS response was found to be induced, and a combination of the dnaXE145A mutation with recBC and recA mutations led to reduced viability. The mutant cells exhibited extensive chromosome fragmentation and degradation upon inactivation of recBC and recA, respectively. The results indicate that the dnaXE145A mutant cells suffer from broken replication forks and that these need to be repaired by homologous recombination. We suggest that the dnaX-encoded τ and γ subunits of the clamp loader, or the clamp loader complex itself, has a role in the restart of stalled replication forks without extensive homologous recombination.
IMPORTANCE The E. coli clamp loader complex has a role in coordinating the activity of the replisome at the replication fork and loading β-clamps for lagging-strand synthesis. Replication forks frequently encounter obstacles, such as template lesions, secondary structures, and tightly bound protein complexes, which will lead to fork stalling. Some pathways of fork restart have been characterized, but much is still unknown about the actors and mechanisms involved. We have in this work characterized the dnaXE145A clamp loader mutant. We find that the naturally occurring obstacles encountered by a replication fork are not tackled in a proper way by the mutant clamp loader and suggest a role for the clamp loader in the restart of stalled replication forks.
KEYWORDS: DnaX, DNA replication, DNA repair, replication fork restart
INTRODUCTION
The τ and γ subunits of the Escherichia coli DNA polymerase holoenzyme are part of the clamp loader complex, a multisubunit complex responsible for loading of the β-clamp of the DNA polymerase (see references 1 and 2 for reviews). Both the clamps and clamp loaders are well conserved across the three domains of life with regard to both structure and function (2).
The clamp loader is a circular heteropentameric complex, which in E. coli consists of three τ/γ subunits and one δ and δ′ subunit (3, 4). Two other subunits, χ and ψ, are associated with the clamp loader complex but are not necessary for assembly or clamp-loading activity (5, 6). Both τ and γ are encoded by the dnaX gene; τ is the full-length protein, while the γ protein is a shorter frameshifted version (7–9). The full-length τ contains domains for interaction with the α subunit of the DNA polymerase and the DnaB helicase; hence, it couples the DNA synthesis on the two strands and DNA synthesis to the unwinding of the DNA (10, 11). The γ protein lacks the ability to perform these interactions but can function in the loading of a β-clamp (5).
Once the β-clamp is loaded, replication can proceed with high processivity. However, the progression of replication forks is often impeded by DNA-bound proteins, DNA damage, or DNA secondary structures during the elongation process, which can lead to arrest and potential disintegration of the replication fork (reviewed in references 12 and 13). Four different models for replication fork disintegration have been explained to date; collapse, regress-split, rear-ending, and breakage (see Fig. 6A in reference 14). One mechanistically explicit model for replication fork collapse is when the replication fork encounters a nick in the template strand and thereby disconnects the nascent DNA duplex from the rest of the chromosome, resulting in a double-strand end (DSE) which is lethal to the cell and must be repaired by recombination enzymes (15, 16). RecBCD recognizes the DSE and degrades the DNA until it encounters a Chi site, a specific sequence which occurs about every 5 kb on the E. coli chromosome. At this site, the RecA protein is loaded and a nucleoprotein filament that invades a homologous DNA molecule is formed (see references 17 and 18 for reviews). The RecA invasion leads to the formation of a D-loop, which is a substrate for the PriA protein, which then can act with several different partners to reload the DnaB replicative helicase and promote replication restart (13, 19).
FIG 6.
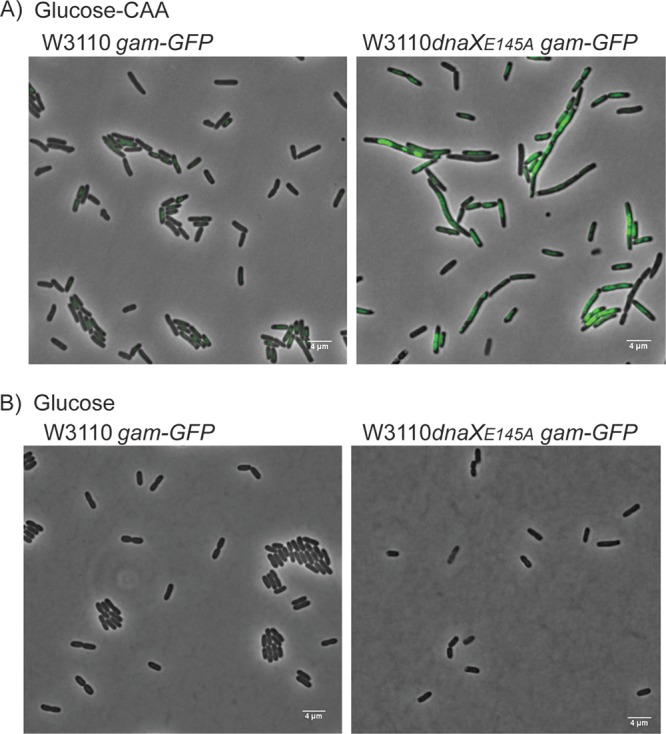
Formation of Gam-GFP foci during growth with multiforked chromosomes indicates that rear-ending causes DSEs in the dnaXE145A mutant. Representative microscopy images of Gam-GFP (pseudocolored green) in W3110 (EH52) and dnaXE145A (EH53) cells grown in glucose-CAA medium (A) and glucose medium (B). The cells were grown to an OD of ∼0.15 before Gam-GFP was induced by adding 100 ng/ml tetracycline. Growth was continued for 45 to 60 min, at which time the cells were spread onto agarose pads (1% in PBS with 100 ng/ml tetracycline) and investigated under the microscope.
The regress-split model involves reversal (or regression) of the replication fork, where the leading and lagging strands anneal and extrude as a duplex as the fork reverses (20). The replication fork reversal model was first recognized in mammalian cells (21) and later in E. coli replication mutants, such as dnaB(Ts) and rep mutants (22, 23). According to this model, regression of the fork creates a Holliday junction stabilized by RuvABC. The regression can continue to a Chi site where RecA is loaded by RecBCD after DSE digestion, which leads to recombination with subsequent restart of the fork by PriA. Alternatively, a more “simple” pathway can be utilized, not involving RecA, in which RecBCD degrades the DSE, displaces RuvABC, and allows what here is denoted as direct restart by PriA (see reference 24 for a review). However, if the regressed fork is not processed properly, RuvABC will simply cleave it (regress-split), leaving a lethal DSE (20). Biochemical investigations have shown that a replication fork stalled at a leading-strand lesion can undergo regression catalyzed either by RecA or RecG (see reference 13 for a review).
Disintegration of a replication fork by rear-ending is a scenario in which a newly fired replication fork catches up with a replication fork in front on chromosomes with multifork replication (25, 26). This can occur, for instance, if the leading fork pauses/stalls for a significant time period. The risk of rear-ending increases with increasing replication complexity, meaning that the more highly branched a chromosome is, the higher the danger of rear-ending (25, 26). This type of disintegration leaves DSEs that must be repaired through homologous recombination.
The fourth and final model for replication fork disintegration is breakage. The term implies that a rupture occurs at one of the two single-stranded regions of the unwound parental DNA at the replication fork. This means that the result is the same as for the replication fork collapse model, although fork disintegration is caused by a preexisting nick in the parental DNA (prior to unwinding) (14, 27).
Previously, it has been shown that the introduction of a high-copy-number plasmid containing the dnaX gene can suppress the temperature sensitivity of parE10(Ts) cells, which lack topoisomerase IV (TopoIV) activity at the nonpermissive temperature (28). Although several proposals were made, the actual mechanism for rescue of parE10(Ts) by τ and γ remained unclear. The dnaXE145A mutation was picked up in a screen for mutants that could not suppress the temperature sensitivity of the parE10(Ts) cells. This mutation led to cells with abnormal morphology when grown in rich medium, a so-called par phenotype (29). The par phenotype is characterized by long filamentous cells with large nucleoids in the middle, and it can be a result of a defect in the partitioning of daughter chromosomes. However, the phenotype might also be a result of defects in DNA replication with subsequent induction of the SOS response, which inhibits cell division (30). In a previous work by Espeli et al., the dnaXE145A mutant was found to have normal growth and DNA synthesis rates, and the purified mutant proteins were found to be able to load the β-clamp in vitro (29). Hence, it was concluded that the observed par phenotype might be a result of a lack of TopoIV activity (29). We have in this work characterized this mutant further.
We find that the cells containing the dnaXE145A mutation exhibit extensive fork disintegration with subsequent DSE formation and induction of the SOS response, in addition to the previously reported par phenotype (29). The phenotype was alleviated in cells with no more than two ongoing forks per chromosome and enhanced in a Δrep background where the replication forks stall more frequently and each chromosome harbors abnormally high numbers of forks. We also found that the dnaXE145A mutant cells exhibited increased levels of recombinational repair.
Our results suggest that the dnaX-encoded τ and γ subunits of the clamp loader or the clamp loader complex itself has a role in maintaining the integrity of the replication fork, possibly by aiding the restart of stalled or disintegrated replication forks.
RESULTS
Flow cytometry and marker frequency analysis indicate that many of the replication forks initiated at oriC do not reach the terminus in the dnaXE145A mutant.
In order to assess the in vivo effects of the dnaXE145A mutation on the replication process, we analyzed the DNA content and mass distributions of the mutant cells and the corresponding wild-type cells with flow cytometry. The cells were grown in minimal medium supplemented with glucose and Casamino Acids (glucose-CAA medium). At an optical density (OD) of 0.15, cells were harvested and fixed with ethanol directly or treated with rifampin and cephalexin for three to four generations before fixation. Rifampin and cephalexin inhibit the initiation of replication and cell division, respectively, so that all ongoing replication is allowed to finish, but new initiations and cell division are inhibited. The number of fully replicated chromosomes after drug treatment then normally represents the number of origins present in the cells at the time of drug addition. Since all origins are initiated in synchrony in E. coli, the numbers after treatment with rifampin and cephalexin should correspond to 2n and 2n + 1, where n is a number between zero and three and depends on the generation where replication is initiated (31, 32). If the cells have trouble with initiation or elongation of replication, this can be seen as abnormalities in the DNA histograms of the drug-treated cells (the so-called run-out histograms).
We calculated the cell cycle of the wild-type cells from analysis of the flow histograms, as described in reference 33. These cells had a cell cycle in which the initiation of replication occurred at the two origins of a partially replicated chromosome shortly after cell division (Fig. 1A). The cells thus had a short period with multiforked replication, i.e., six replication forks on a single chromosome, and for the rest of the cell cycle contained two chromosomes with two replication forks each. Correspondingly, wild-type cells contained mainly four chromosomes after treatment with cephalexin and rifampin (i.e., under run-out conditions) (Fig. 1B, right). This type of “regular” cell cycle pattern could not be shown for the dnaXE145A mutant cells, as the run-out histogram contained abnormal numbers of peaks not abiding the rule of 2n or 2n + 1 (Fig. 1C, right). Such a phenotype, together with findings that the cells had a reduced DNA concentration (DNA/mass) (Table 1), could point to a deficiency in initiation of replication, i.e., that origins are fired too seldom and not in synchrony. However, the peaks were not well separated, which is indicative of replication elongation trouble (i.e., that some of the replication forks were not able to reach the terminus under run-out conditions) (34, 35) and/or that DNA degradation and homologous recombination occurred (36).
FIG 1.

Flow cytometry and microarray frequency analysis show that replication forks do not reach the terminus in the dnaXE145A mutant cells. (A) Cartoon showing the cell cycle of wild-type cells (W3110) growing in glucose-CAA medium at 37°C. The blue arrow shows the duration of the replication period relative to the duration of the cell cycle. (B and C) Flow cytometry histograms of wild-type (W3110) and dnaXE145A mutant (LJ60) cells, respectively. Exponentially grown cells are shown in the left histograms, whereas cells treated with rifampin and cephalexin for 3 to 4 generations (replication run-out samples) are shown in the right histograms. The number of cells is shown on the ordinate; 10,000 cells were measured, and one tick on the ordinate represents 100 cells. (D and E) Comparative genomic hybridization of chromosomal DNA from exponentially growing wild-type (W3110) and dnaXE145A mutant (LJ60) cells grown in glucose-CAA (D) and LB (E) relative to DNA from run-out samples. Data points (gray dots) show the ratio of dnaXE145A versus wild-type rifampin run-out DNA. Local regression (Lowess) of the log2 relative abundance is shown with f = 0.0075 (gray line) or f = 0.5 (solid black line for dnaXE145A cells and dashed black line for wild-type cells).
TABLE 1.
DNA content, mass, DNA/mass, and oriC-to-ter ratio of dnaX and rep mutant cellsa
| Strain | Growth mediumb | Doubling time (min) | DNA content relative to wild type | Mass relative to wild type | DNA/mass | oriC-to-ter ratio |
|---|---|---|---|---|---|---|
| Wild type (W3110) | Glu-CAA | 37 ± 2 | 1 | 1 | 1 | 2.27 ± 0.197 |
| dnaXE145A mutant (LJ60) | Glu-CAA | 41 ± 2 | 1.22 ± 0.03 | 1.33 ± 0.03 | 0.92 ± 0.03 | 4.07 ± 0.422 |
| dnaXE145A dnaAA345S mutant (KS1002/1) | Glu-CAA | 40 ± 3 | 0.98 ± 0.02 | 1.69 ± 0.10 | 0.58 ± 0.02 | 1.47 ± 0.254 |
| Δrep mutant (IF136) | Glu-CAA | 50 ± 1 | 1.23 ± 0.01 | 1.49 ± 0.02 | 0.83 ± 0.01 | 4.42 ± 0.485 |
| dnaXE145AΔrep mutant (IBP91) | Glu-CAA | 59 ± 2 | 1.53 ± 0.05 | 1.57 ± 0.15 | 0.98 ± 0.12 | 7.30 ± 0.537 |
| Wild type (W3110) | Glucose | 63 ± 2 | 1 | 1 | 1 | ND |
| dnaXE145A mutant (LJ60) | Glucose | 66 ± 1 | 1.03 ± 0.02 | 1.00 ± 0.06 | 1.04 ± 0.06 | ND |
The experiment was repeated three times, and ± represents the standard deviation. ND, not determined.
Glu-CAA, minimal medium supplemented with glucose and Casamino Acids.
In order to investigate in more detail whether the dnaXE145A mutant cells had problems with initiation or elongation of replication, we performed a marker frequency analysis by microarray on genomic DNA purified from exponentially growing cells grown in glucose-CAA medium or LB medium (see Materials and Methods). Sample DNA was hybridized against DNA from nonreplicating cells. The data were normalized to give the average number of copies of chromosomal loci per chromosomal terminus region. During exponential growth, the relative abundance of chromosomal loci diminishes exponentially with increasing distance from the origin (37). A semilog plot of gene dosage versus chromosomal position should thus reveal a pair of straight lines descending from an apex at oriC (38). Here, we also observed a large difference in the mutant cells from the wild-type cells under both glucose-CAA and LB medium conditions (Fig. 1D and E, respectively). Whereas the plot of the wild-type cells shows straight lines from oriC to ter (Fig. 1D and E, dashed lines), this was not the case for the mutant cells (Fig. 1D and E, data points and solid lines). Instead, they exhibit an increased gene dosage of oriC proximal genes compared to the wild-type cells under both growth conditions. To confirm the result, we performed quantitative PCR of the oriC and ter regions (see Materials and Methods) and found that the oriC-to-ter ratio was higher in the mutant cells than in the wild-type cells (Table 1).
As mentioned, the dnaXE145A mutant cells were found to have a lower than normal DNA concentration (Table 1) and can be said to produce too little DNA relative to mass and thus to “underreplicate.” Since the microarray and quantitative PCR (qPCR) data show that the oriC-to-ter ratio is abnormally high, this underreplication must be because of problems with elongation and not because of too-few initiations. Thus, the results indicate that many of the replication forks initiated at oriC do not reach the terminus.
dnaXE145A cells show chromosomal fragmentation in the absence of RecBCD.
Since many replication forks were unable to reach the terminus in the dnaXE145A mutant cells, we wished to investigate whether this scenario could be caused by replication fork disintegration. We therefore analyzed genomic DNA from exponentially growing dnaXE145A cells with pulsed-field gel electrophoresis (PFGE). This method allows the separation of large DNA fragments that can arise in the cells after double-strand breaks, and such breaks are known to arise at disintegrated forks (14, 22, 39, 40). Before the analysis, the dnaXE145A mutation was transferred to a recBC(Ts) background to enable the inactivation of RecBCD and thereby avoid repair and degradation of DNA fragments (41). Fragmented chromosomes are mainly seen in the compression zone around 2 Mb (Fig. 2A), while whole chromosomes will stay in the wells. Our analysis showed that the dnaXE145A cells without RecBC function exhibit an increased fragmentation of the chromosome compared to the controls (Fig. 2). This result suggests that the cells either suffer from double-strand breaks due to a direct disintegration of replication forks or that they are dependent on RecBCD to restart replication forks in a way similar to that of certain mutants performing replication fork reversal (24).
FIG 2.
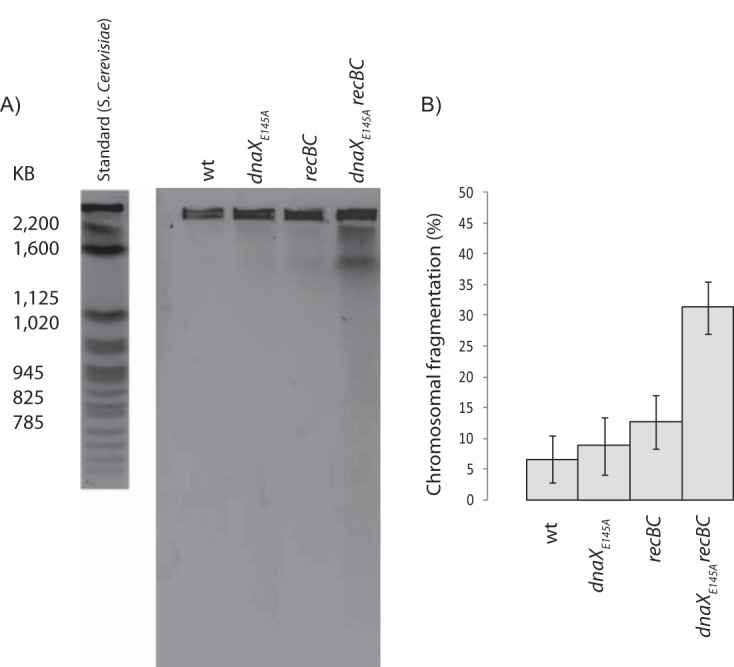
The dnaXE145A mutant cells exhibit fragmentation of the chromosome. (A) PFGE of the wild-type cells (AB1157) (lane 1), dnaXE145A cells (lane 2), wild-type cells with inactivated RecBC (grown at 42°C) (SK129) (lane 3), and dnaXE145A cells with inactivated RecBC (grown at 42°C) (IBP26) (lane 4). A standard of DNA from Saccharomyces cerevisiae is shown to the left of the gel. (B) Quantification of the observed fragmentation after pulsed-field gel electrophoresis. The calculated values are averages from the results from three experiments, and the error bars represent the standard deviations.
SOS response is induced in the dnaXE145A mutant cells.
If some types of stalled replication forks require τ and γ subunits to restart forks via a relatively simple pathway, and the mutant cells instead rely on homologous recombination and repair of double-strand breaks to salvage an otherwise straightforward situation, it might be expected that the SOS response becomes induced in the mutant cells. The SOS response is induced by the presence of single-stranded DNA (ssDNA)-RecA filaments, which cause autocleavage of the LexA repressor that normally binds to LexA boxes in the promoter regions of SOS genes, including the sfiA (sulA) gene (see reference 42 for a review on the SOS response). To check this, we transferred the dnaXE145A mutation into a strain where the lacZ gene was inserted as a reporter gene behind the sfiA promoter (ALO1186) and measured the transcription from this promoter with a β-galactosidase assay (43). The dnaXE145A cells exhibited about 50% increased transcription of this gene compared to the control cells when grown in glucose-CAA medium (Table 2), meaning that the SOS response was induced. For comparison, we measured SOS induction in control cells that had been exposed to a low concentration of ciprofloxacin (5 ng/ml) for two generations. Ciprofloxacin is a potent antibiotic that causes double-strand breaks (DSBs) by inhibiting the religation step performed by the two topoisomerases gyrase and TopoIV. The transcription of sfiA in these cells amounted to an approximate 3-fold increase compared to the untreated control (Table 2).
TABLE 2.
SOS response is induced in dnaXE145A mutant cellsa
| Strain and description | Miller units ± SD | Relative value |
|---|---|---|
| MG1655 lacZ::Tn5 sfiA-lacZ (ALO1186) | 29.0 ± 1.36 | 1 |
| ALO1186 dnaXE145A (IF137) | 44.1 ± 5.10 | 1.52 |
| MG1655 lacZ::Tn5 sfiA-lacZ (ALO1186) treated with 5 ng/ml ciprofloxacin | 84.6 ± 10.90 | 2.92 |
The experiments were repeated three times.
In order to test the idea that the dnaXE145A mutant cells rely on recombinational repair to a much higher extent than wild-type cells, we investigated their viability when lacking RecBCD and RecA activity. Indeed, the viability was significantly lowered in the dnaXE145A recBC and dnaXE145A recA double mutants (Fig. 3A). This might indicate that the presence of double-strand breaks is the reason for induction of the SOS response in the dnaXE145A mutant cells, and that the cells need the RecBCD and RecA proteins for repair by homologous recombination.
FIG 3.
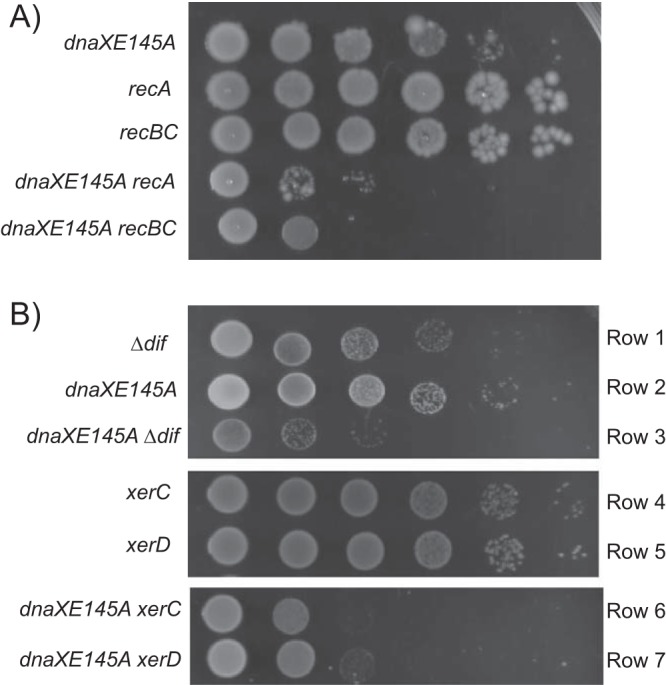
The dnaXE145A cells exhibit decreased viability when combined with a deletion of recA, recBC, or dif, xerC, or xerD. (A) Viability tests of the dnaXE145A recA938 (IBP108) and dnaXE145A recBC(Ts) (IBP26) mutant cells. The different cultures were serially diluted and 5 μl of each dilution spotted onto LB plates and incubated at 37°C for 24 h. The spots are, from left to right, undiluted culture and 10−2, 10−3, 10−4, 10−5, and 10−6 dilutions. (B) Viability tests of dnaXE145A mutant cells lacking the dif site (IBP72), xerC (EH100), or xerD (IBP69). The test was performed on minimal medium plates supplemented with glucose-CAA and incubated at 37°C for 24 h.
Repair by homologous recombination sometimes results in the formation of chromosomal dimers (44) that need to be resolved before segregation and cell division can occur. In E. coli, this is accomplished by the activities of XerC and XerD proteins, which perform site-specific recombination at a specific recombination site, dif, located in the terminal region on the chromosome (45, 46). We combined the dnaXE145A mutation with the deletion of dif and investigated the viability of the double mutant. The wild-type cells lacking dif exhibited a slight growth defect, confirming that such dimers also arise at a certain (low) frequency in otherwise normal cells (47) (Fig. 3B, row 1) (reviewed in reference 48). We found that the double mutant was extremely difficult to construct and scarcely viable (Fig. 3B, row 3). The combination of the dnaXE145A mutation with deletion of xerC or xerD instead of dif improved the viability slightly (Fig. 3B, rows 6 and 7), and this probably reflects that some dimer resolution activity can be performed by XerC without XerD and vice versa.
The results indicate that recombination and dimer formation occur in most dnaXE145A cells and suggest that the DSE repair that goes on in the mutant cells is similar to that in all cells and ends with a chromosomal dimer.
dnaXE145A mutant cells exhibit extensive chromosome degradation when lacking RecA function under run-out conditions.
It has previously been shown that whole chromosomes are missing in recA(Ts) cells at nonpermissive temperatures compared to the permissive temperature under replication run-out conditions (rifampin and cephalexin treatment) (36). The exact underlying mechanism is not clear, but it is proposed that RecBCD degrades one of the newly replicated chromosome arms of a partially replicated chromosome if a DSE was generated at the replication fork (see Fig. 6C in reference 49) (50, 51). The cell is unable to repair the break to synthesize the full chromosome without RecA, and RecBCD may continue digestion of the DSE beyond Chi sites (where recombination would normally occur) in the absence of RecA (52). Thus, cells that experience frequent formation of DSEs would exhibit more severe chromosome degradation than cells that do not.
We therefore combined the dnaXE145A mutation with the recA(Ts) mutation and compared the chromosome content during replication run-out in permissive versus nonpermissive temperature (i.e., with and without RecA function) (Fig. 4). The dnaXE145A recA(Ts) double mutant exhibited extensive chromosome loss during run-out in the absence of RecA (compare right and middle images in row 3, Fig. 4). At permissive temperature, the mutant cells typically contained from about three to nine chromosomes, whereas at the nonpermissive temperature, the chromosome content was reduced to between zero and four. The recA(Ts) single mutant also exhibited chromosome loss, as reported previously (36, 49), but to a much lesser extent than the double mutant (seen as more cells in the 3-chromosome peak and fewer cells in the 4-chromosome peak than at permissive temperature) (Fig. 4, row 2).
FIG 4.

The dnaXE145A mutant cells exhibit a significant degree of chromosomal degradation under run-out conditions when lacking RecA. Flow cytometry histograms of wild-type (N1331) (WT), recA(Ts) (N1332), and dnaXE145A recA(Ts) (IBP107) cells grown in glucose-CAA. The portions of the cultures with added rifampin and cephalexin (for run-out of replication) were split into two batches, in which one was kept at permissive temperature (30°C) (middle panels) and the other was elevated to 42°C to determine loss of RecA function (right panels). Chromosome equivalents per cell are shown on the abscissa. Fifty thousand cells were measured.
We also constructed a dnaXE145A ruvB double mutant to investigate what happens when Holliday junctions cannot be stabilized and resolved in cells that are abnormally dependent on recombinational repair. The flow cytometry run-out histogram showed that there is a significant degree of chromosomal degradation also in these cells (see Fig. S1B in the supplemental material). The mechanism behind this is unknown, but we speculate that since DSBs cannot be fully repaired, we have a scenario which resembles that in the dnaXE145A recA cells (i.e., inability to perform DSB repair leads to degradation by RecBCD).
The above-described results support our previous inferences, namely, that double-strand breaks arise in the dnaXE145A mutant cells.
Replication elongation issues diminish in the dnaXE145A mutant when the cells contain maximally two replication forks per chromosome.
According to the model of Bidnenko et al. (53), chromosomal dimers can be produced after recombinational repair of forks that rear-end. Rear-ending of forks can occur, for instance, on chromosomes which harbor multifork replication (i.e., more than two replication forks) if one or both of the oldest forks stall long enough for the newly formed forks at oriC genes to catch up (38, 53–55). It is thus possible that replication fork rear-ending contributes to their loss and leads to recombinational repair and chromosome dimer formation in the dnaXE145A mutant. If so, this might indicate that frequent stalling and/or insufficient replication fork restart are an issue in the mutant cells.
To investigate this scenario, we analyzed the dnaXE145A mutant cells during conditions under which they maximally harbor two replications forks per chromosome during run-out in rifampin and cephalexin (i.e., no new forks can approach from behind). This can be achieved by using mutants which underinitiate (too few forks are launched) or by growing the cells in poor medium. The dnaAA345S mutant has been shown to have replication initiation deficiencies (origins fire too seldomly and not in synchrony) and to contain only two forks per chromosome (see Fig. 6B in reference 56 and Fig. S2) (57). We therefore combined the dnaXE145A mutation with the dnaAA345S mutation and analyzed the cells by flow cytometry. These cells had a cell cycle in which the two oriC genes were not initiated simultaneously and they exhibited maximally two replication forks at each of the two chromosomes at any given time, as expected from previous studies (Fig. 5B) (56, 57). More importantly, very little of the dnaXE145A run-out phenotype could be detected in the run-out histograms (clearly separated peaks were formed) (Fig. 5B, right).
FIG 5.

The flow cytometry run-out phenotype of the dnaXE145A mutant cells is significantly improved during growth with reduced number of replication forks per chromosome but is worsened when the mutant cells lack Rep helicase. Schematic cell cycle cartoons (left) obtained from analysis of exponential (middle) and run-out (right) flow cytometry histograms of the indicated strains. The blue arrow shows the duration of the replication period relative to the duration of the cell cycle. For the dnaAA345S mutant, which initiates one origin before the other, the early replication is indicated in blue and the late replication is indicated in red. (A and B) Wild-type (W3110) (A) and dnaXE145A dnaAA345S (KS1002/1) (B) cells grown in glucose-CAA at 37°C. See Fig. S2 for the single dnaAA345S mutant. (C and D) Wild-type (W3110) (C) and dnaXE145A (LJ60) (D) cells grown in glucose medium at 37°C. (E and F) W3110 Δrep mutant (IF136) and dnaXE145A Δrep mutant (IBP91) cells grown in glucose-CAA medium at 37°C. The number of cells is indicated on the ordinate; 10,000 cells were measured, and one tick on the ordinate represents 100 cells.
To verify that much of the elongation issues in the dnaXE145A mutant cells disappear when the number of forks per chromosome is reduced, we also grew the cells in minimal medium supplemented with only glucose. Indeed, the effect of the dnaXE145A mutation more or less disappeared, and the mutant cells were capable of completing replication run-out (Fig. 5C and D).
These results show that the dnaXE145A mutant cells can perform proper replication elongation (i.e., forks can reach the terminus) when they do not grow with multiforked replication.
Replication elongation trouble is further elevated in dnaXE145A cells lacking Rep helicase.
In certain replication mutants, such as DnaB(Ts), replisome(Ts), and Δrep, stalled replication forks are proposed to be restarted by a mechanism involving replication fork reversal (22, 23). The Rep helicase is an accessory helicase which interacts with DnaB and is thought to have a role in clearing protein complexes and unwinding the DNA in front of the replication fork (58, 59). It was first shown that cells lacking the Rep protein have an extended C-period (replication period) (60) and exhibit an increased oriC-to-ter ratio (61), which indicates a slowing down of replication fork progression. Apparently, the Rep helicase activity is necessary in order to quickly clear protein complexes from the DNA and prevent replication fork stalling (62, 63). It was suggested that cells lacking the Rep protein undergo frequent replication fork reversal (22). In vitro studies indicate that Rep also has an important role in unwinding the lagging strand upon uncoupling of leading- and lagging-strand replication after fork stalling at an obstruction on the leading strand (12, 13). The well-characterized and frequent replication fork stalling and restart of rep mutants were here exploited in order to gain more information about the possible roles of the τ or γ protein in replication fork restart. We combined the dnaXE145A mutation with a deletion of the rep gene (60) and investigated the single- and double-mutant cells with flow cytometry and marker frequency analysis of the oriC and ter regions (Fig. 5E and F and Table 1).
When grown in glucose-CAA medium, the Δrep single mutant cells had, as previously reported, an increased doubling time and an elevated oriC-to-ter ratio compared to the wild-type cells (Table 1). The replication pattern was also different from that of the wild-type cells. While the wild-type cells had two and four chromosomes after replication run-out (Fig. 5A), the Δrep mutant cells had mainly four and eight chromosomes (Fig. 5E). This corresponds to a complicated cell cycle with substantial overlap of replication between generations (due to a significantly increased C-period), as the cells contain up to 14 replication forks per chromosome (Fig. 5E, left), in contrast to wild-type cells, which contain maximally six replication forks (Fig. 5A, left). Although an increased number of forks per chromosome elevates the danger of rear-ending, the Δrep mutant cells had no problem with completion of replication run-out, in accordance with previous findings (64).
In the dnaXE145A rep double mutant, a further increase in the oriC-to-ter ratio was observed beyond the already high ratio caused by the lack of Rep protein (Table 1). Moreover, the double mutant had a much wider DNA distribution in the DNA histograms of the exponential cells than the single mutants (Fig. 5F, middle) and was not able to complete the replication of the chromosomes under run-out conditions (Fig. 5E, right). The flow cytometry phenotype is similar to that of the dnaX single mutant (Fig. 1B), only worse (i.e., even more replication forks were unable to complete rounds of replication). The result supports the idea that the dnaXE145A mutant cells deal poorly with elevated numbers of replication forks, perhaps because replication fork restart mechanisms do not function properly, and as a consequence, replication forks may collide (rear-end).
Imaging of the double-strand end binding Gam-GFP protein indicates that rear-ending occurs in the dnaXE145A mutant cells during growth with multiforked replication.
The results so far are consistent with replication fork pausing in the dnaXE145A mutant, which may lead to rear-ending and double-strand-break formation. In order to visualize DSEs in living cells directly, we used a Gam-green fluorescent protein (Gam-GFP) construct kindly provided by S. Rosenberg (65). The Gam protein originates from the Mu phage and binds and protects DSEs of linear DNA, thus inhibiting exonuclease activity (66) and, subsequently, recombinational repair (65). Gam-GFP can therefore be used to visualize DSEs in living cells because it forms fluorescent foci upon DNA DSE binding.
We compared microscopy images of exponentially growing wild-type and dnaXE145A cells in which Gam-GFP was induced about one C-period before imaging (45 to 60 min). The experiment was conducted both during growth in glucose-CAA and during growth in glucose medium. In the first, the cells grow with a replication pattern including six-forked chromosomes (Fig. 5A), whereas in the glucose medium, the cells harbor maximally two replication forks (Fig. 5C and D). Under the hypothesis that DSEs are generated by rear-ending in the dnaXE145A mutant, we would expect to see persisting Gam-GFP foci during growth in glucose-CAA but not during growth in glucose (since Gam-GFP was induced for less than a C-period). Indeed, this was found to be the case. In glucose-CAA medium, the dnaXE145A cells exhibited a strong GFP signal (and extensive filamentation) compared to the wild-type cells upon induction of Gam-GFP (Fig. 6A). The same effect was not seen for dnaXE145A cells growing slowly in glucose medium (Fig. 6B). Microscopy images were in this case similar to those for the wild-type cells, as no strong Gam-GFP signal was observed. To confirm that DSEs can be detected in cells growing in poor medium, we additionally performed an experiment in which the control cells were treated with 1 μg/ml ciprofloxacin for 30 min. Indeed, persistent foci were formed (Fig. S3). Thus, it appears likely that DSEs in the dnaXE145A cells are formed due to rear-ending.
DISCUSSION
Frequent replication fork rear-ending may be the reason for the chromosomal fragmentation observed in dnaXE145A mutant cells.
The dnaXE145A mutant proteins were previously reported to have no defects in an in vitro replication assay (29), suggesting that the clamp loading activity of the mutant τ or γ protein necessary for lagging-strand synthesis is not compromised. In the present work, we obtained evidence suggesting that double-strand breaks occur in the dnaXE145A mutant. The mutant cells exhibited extensive chromosome fragmentation and degradation upon inactivation of RecBC and RecA, respectively (Fig. 2 and 4). Moreover, flow cytometry analysis of replication run-out samples and microarray marker frequency data indicates that replication forks do not reach the terminus (Fig. 1). It is therefore reasonable to assume that the replication forks disintegrate somewhere along the replichores. When the number of forks per chromosome was reduced to two, however, the deficiencies of the dnaXE145A mutant appeared to be alleviated, as the cells could perform proper run-out of replication (Fig. 5). Results from experiments with induction of Gam-GFP indicated that DSEs were formed only when dnaXE145A cells grew with multiforked replication, but not during slow growth with only two replication forks per chromosome (Fig. 6). Together, these results support the idea that chromosome fragmentation in the dnaXE145A mutant is caused by rear-ending of replication forks.
The τ or γ proteins may be involved in a direct restart of stalled replication forks.
We show here that the SOS response is induced in the dnaXE145A mutant cells, and that they suffer a prominent reduction in viability upon deletion of recBC and recA genes, as well as the chromosome dimer resolution site, dif. This indicates that the mutant cells are especially dependent upon recombinational repair, and that the repair leads to the generation of chromosome dimers.
Replication forks may stall for natural reasons, such as DNA-bound proteins, DNA secondary structures, and transcription, most of which may normally be taken care of by various restart mechanisms. Restart could occur, for instance, through a relatively simple pathway involving replication fork reversal, in which RecBCD digests the DSE (resulting from the annealing of the leading and lagging strand) and the fork is restarted by primosomal proteins directly, as described previously. However, if the τ or γ proteins are part of the required mechanism, the pathway will not work. Futile attempts at utilizing the dysfunctional pathway will lead to prolonged replication fork stalling and an increased likelihood of disintegration of the stalled fork. Disintegrated stalled forks, which are repaired by recombination, will contribute to the formation of chromosome dimers (67). Thus, the dependence of dnaXE145A mutant cells on chromosome dimer resolution supports the idea that a replication fork reversal-type restart mechanism (which does not generate dimers) cannot be performed in the dnaXE145A mutant cells.
In this work, we used the rep deletion as a tool to shed more light on whether the τ or γ proteins could be involved in a type of direct restart of replication forks. Rep helicase has been reported to clear protein-DNA barriers in front of the fork (63). A lack of Rep helicase thus results in increased fork stalling/pausing (64), and the stalled replication forks restart by a direct restart pathway which includes replication fork reversal (24). The replication elongation issues observed in the dnaXE145A mutant cells were further enhanced in the dnaXE145A Δrep double mutant, i.e., a large portion of the cells were not able to finish replication of the chromosomes under run-out conditions (Fig. 5F). In contrast, the Δrep single mutant had an increased C-period and increased replication complexity, probably due to increased fork stalling and restart, but the run-out histogram of these cells indicates that the chromosomes were successfully replicated all the way to the terminus (Fig. 5E). This result shows that increased replication complexity (and thus increased danger of rear-ending) due to frequent fork stalling is normally not a problem in cells with wild-type τ and γ proteins. The dnaXE145A mutant cells, on the other hand, may be unable to perform the restart mechanisms that are normally at play in Δrep mutants to support a successful elongation of replication.
It cannot be ruled out, however, that the mutant complex simply is less stable and generates a replisome that is more prone to disintegration. We find this less likely, because mutant complexes with reduced stability tend to function poorly in vitro. Moreover, if the replisome itself was weak, we would have expected to observe deficiencies in the ability to finish chromosomal replication also during slow growth, as replisomes frequently encounter obstacles during a normal replication cycle. We suspect that rear-ending (caused by frequent stalling and ineffective restart) represents a much more serious type of threat to the replication fork than that produced by naturally occurring obstacles (such as tightly bound proteins, transcription, secondary DNA structures, etc.), since it requires extensive DNA repair by homologous recombination and presumably a full reassembly of the replisome components before restart can be accomplished.
We therefore propose that the observed par phenotype in these cells, which was also in a previous work shown to be less extensive in poor media (29), is a result of inadequate restart of stalled replication forks leading to extensive rear-ending, recombination activity, and induction of the SOS response.
How does the E145A mutation affect DnaX?
The dnaXE145A mutation is a mutation of a surface residue (3) from glutamic acid to alanine at position 145, which resides in domain I of τ/γ (68). The τ and γ proteins have several interaction partners within the replisome, and one possibility is that the defect in the mutant τ and γ proteins is a result of the interruption of one of these interactions. However, the residues that have been shown to interact with other parts of the replisome are located in domains III, IV, and V. The interaction with the α subunit of the DNA polymerase occurs through the C-terminal domain (domain V) (68), while the domain IV is responsible for interaction with the DnaB helicase (69). These two domains are unique to the τ subunit (68). The δ and ψ subunits of the clamp loader also bind to τ or γ, but these interactions occur through domain III (70), which is also the region that enables DnaX to oligomerize (71). Thus, the defect of the dnaXE145A mutation is not likely to be a result of a disruption of any of the known interactions in the replisome. It is therefore reasonable to assume that the mutation affects so-far-unknown roles of the τ and γ proteins. This role could, for instance, also be clamp loading but perhaps with the extra complication of loading at a fork undergoing restart.
The multicopy dnaX suppression of parE10(Ts) might also be a result of an increased need for replication fork restart.
As mentioned, the dnaX gene has previously been shown to be a high-copy-number suppressor of the parE10(Ts) mutant at the nonpermissive temperature (28), and the dnaXE145A mutant was found in a screen for dnaX mutants that could no longer suppress a parE10(Ts) mutant at the nonpermissive temperature (29). In a previous work (29), it was suggested that the τ and γ proteins were needed to ensure proper localization of the two topoisomerase IV (TopoIV) subunits so that TopoIV became active only at the end of replication, i.e., when the two daughter chromosomes need to be decatenated, and that this was disrupted in the dnaXE145A mutant cells. However, suppression of the parE10(Ts) mutant could also be a result of the role of τ or γ in the restart of stalled replication forks. Several lines of evidence suggest that TopoIV also has roles during elongation, prior to the decatenation of the two daughter chromosomes at the end of replication. More specifically, TopoIV likely binds and resolves intertwined newly replicated DNA molecules (precatenanes) progressively behind the replication fork, thereby ensuring that the daughter DNA molecules can be properly segregated (72–74). The formation of precatenanes has been debated, but the results obtained in the recent work by Cebrián et al. (75) indicate that precatenanes do form in E. coli as replication progresses. The precatenanes will be in equilibrium with the positive supercoiling that arises in front of the fork, and if TopoIV fails to resolve them, this could lead to an accumulation of positive supercoiling in front of the fork (as well as partitioning defects). Such topological constraints will, naturally, impede fork progression, and increased positive supercoiling has been reported to lead to replication fork reversal in vitro (76). The fact that a parE10(Ts) mutant is dependent on PriA (77), one of the main actors in replication fork restart, also supports this assumption. We speculate that overexpression of DnaX in such cells might contribute to the restart of such stalled replication forks and hence alleviate the observed phenotypes.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
All strains used are Escherichia coli K-12 and are listed in Table 3. Cells were grown in AB minimal medium (78) supplemented with 10 μg/ml thiamine, 100 μg/ml uridine, and 0.4% glucose and 0.5% Casamino Acids (glucose-CAA medium), in AB minimal medium (78) supplemented with 10 μg/ml thiamine, 100 μg/ml uridine, and 0.4% glucose (glucose medium), or in Luria-Bertani (LB) medium at the indicated temperatures. The growth rates were determined by measuring the optical density of the cultures at 450 nm (glucose-CAA medium and glucose medium) or 600 nm (LB medium). All strains constructed in this work were made by P1 transduction, and the donor and acceptor strains are listed in Table 3. The KS1002/1 strain was made by P1 transduction of the dnaAA345S::mini-Tn10 Cmr allele from SMG379 (57) in to W3110dnaXE145A (LJ60). The strain IBP11 was made by amplifying the chloramphenicol gene with the primers 5′-TATATGTGGCGAATTCCGGTGATTGATGAAAAGCAAGAAAAGCACTGAAGGGAACACTTAACGGCTGACATGG and 5′-ACGCCAGAATGTACTGGCTGTACGCAATAATTGCAACTGCGATTGTTGTGGTCTTGAGCGATTGTGTAGGCTG containing regions homologous to a region in close proximity of the dnaX gene and insertion of this fragment into the chromosome as described in reference 79.
TABLE 3.
Strains
| Strain | Relevant genotype | Reference(s) or source |
|---|---|---|
| W3110 | Wild type | 29 |
| LJ60 | W3110 dnaXE145A | 29 |
| IBP11 | W3110 dnaXE145A-cam | This work |
| AB1157 | Wild type | 87 |
| SK129 | AB1157 recB270 recC271(Ts) | 41 |
| N1331 | recA+ | 88, 89 |
| N1332 | recA(Ts) | 88, 89 |
| ALS972 | recA938::cam | 90 |
| IF01 | CM735 recA938::cam | CM735 × P1 ALS972 (this work) |
| IBP109 | W3110 recA938::cam | W3110 × P1 IF01 (this work) |
| IBP108 | W3110 dnaXE145A recA938::cam | LJ60 × P1 IF01 (this work) |
| SMG379 | MG1655 dnaAA345S | 57 |
| IBP107 | dnaXE145A-cam recA(Ts) | N1332 × P1 IBP11 (this work) |
| IBP26 | AB1157 dnaXE145A-cam recB270 recC271(Ts) | SK129 × P1 IBP11 (this work) |
| EH182 | W3110 dnaAA345S | W3110 × P1 SMG379 |
| KS1002/1 | W3110 dnaXE145A dnaAA345S | LJ60 × P1 SMG379 (this work) |
| ALO1186 | MG1655 lacZ::Tn5 sfiA-lacZ | 91 |
| IF137 | MG1655 lacZ::Tn5 sfiA-lacZ dnaXE145A-cam | ALO1186 × P1 IBP11 (this work) |
| SS1211 | MG1655 Δrep::cam | 60 |
| NL40 | DS941 difΔ6::Kmr | 92 |
| DS984 | AB1157 xerC::mini-Mu cam | D. Sherratt (93) |
| DS9008 | AB1157 xerD::mini-Tn10 kan | D. Sherratt (94) |
| EH99 | W3110 xerC::mini-Mu cam | W3110 × P1 DS984 (this work) |
| EH101 | W3110 xerD::mini-Tn10 kan | W3110 × P1 DS9008 (this work) |
| IF136 | W3110 Δrep::cam | W3110 × P1 SS1211 (this work) |
| IBP91 | W3110 dnaXE145A Δrep::cam | LJ60 × P1 SS1211 (this work) |
| IBP72 | W3110 dnaXE145A difΔ6::Kmr | LJ60 × P1 NL40 (this work) |
| EH100 | W3110 dnaXE145A xerC::mini-Mu cam | LJ60 × P1 DS984 (this work) |
| IBP69 | W3110 dnaXE145A xerD::mini-Tn10 kan | LJ60 × P1 DS9008 (this work) |
| SMR14323 | MG1655 ΔaraBAD567 Δattλ::PBAD zfd2509.2::PN25-tetR FRT-Kan-FRT | S. Rosenberg (65) |
| SMR13957 | MG1655 Δattλ::PN25-tetR FRT ΔattTn7::FRT-cat-FRT PN25-tetO gam-gfp | S. Rosenberg (65) |
| EH52 | W3110 ΔattTn7::FRT-cat-FRT PN25-tetO gam-gfp zfd2509.2::PN25-tetR FRT-Kan-FRT | W3110 × P1 SMR14323 × P1 SMR13957 (this work) |
| EH53 | W3110 dnaXE145A ΔattTn7::FRT-cat-FRT PN25-tetO gam-gfp zfd2509.2::PN25-tetR FRT-Kan-FRT | LJ60 × P1 SMR14323 × P1 SMR13957 (this work) |
| JW1849 | Δ(araD-araB)567 ΔlacZ4787(::rrnB-3) λ− ΔruvB785::kan rph-1 Δ(rhaD-rhaB)568 hsdR514 | Keio collection (95) |
| EH178 | W3110 ΔruvB785::Kmr | W3110 × P1 JW1849 |
| EH179 | W3110 dnaXE145A ΔruvB785::Kmr | LJ60 × P1 JW1849 |
Flow cytometry analysis and cell cycle estimation.
Exponentially growing cells (OD, ∼0.15) were either harvested directly (by fixation in 70% ethanol) or treated with 300 μg/ml rifampin and 10 μg/ml cephalexin for three to four generations before fixation to inhibit new rounds of initiation or cell division, respectively (32). Flow cytometry was performed with a LSR II flow cytometer (BD Biosciences). Total protein content in the cells was stained with fluorescein isothiocyanate (FITC; Sigma-Aldrich) and used to calculate the average mass (80). The DNA was stained with Hoechst 33258 (Sigma-Aldrich) (81). Whereas one example histogram is shown in each panel of the figures, the values of DNA, mass, and DNA concentration (Table 1) are the averages calculated from the results from three independent experiments. Cell cycle parameters and the numbers of origins and replication forks per cell were obtained by analysis of the DNA distributions obtained by flow cytometry, as described in reference 33.
Quantitative PCR.
To obtain the oriC-to-ter ratio using quantitative PCR, chromosomal DNA was purified from 10 to 15 ml of exponential cultures with the DNeasy blood and tissue kit (Qiagen). Quantitative PCR on 0.5 to 2 ng of DNA was performed as described in reference 82. The data from the samples were normalized to the data obtained from E. coli MG1655 wild-type cells treated with rifampin and cephalexin, where the oriC-to-ter ratio is 1:1.
Marker frequency analysis by microarray.
Experiments were essentially carried out as described in reference 83. The cultures were grown to early exponential phase, and 10 ml of culture was collected by centrifugation. As a hybridization control, DNA from cells treated with rifampin and cephalexin was used. This treatment will lead to fully replicated chromosomes. Cells were sonicated using a UP 400s ultrasonic processor (Hielscher Ultrasonics GmbH) to give DNA fragments of around 400 bp. DNA was isolated with phenol-chloroform after RNase A treatment. Four hundred nanograms of DNA was labeled with Cy3-dCTP or Cy5-dCTP using the Klenow fragment and random primers of the BioPrime kit from Invitrogen. The labeled DNAs were mixed and hybridized for about 36 h at 55°C to E. coli whole-genome microarrays from Oxford Gene Technology. Arrays were scanned on an Agilent SureScan high-resolution scanner. Spot intensities were extracted using the Feature Extraction software 10.5.1.1 from Applied Biosystems, with a linear dye normalization correction method. Ratios of dye intensities were calculated and normalized to the array wide average. Data of a genomic deletion between positions 1395000 and 1433300 were excluded. The mean value of signal ratios (sample versus hybridization control) for probes between positions 1608000 and 1633000 were considered the terminal copy number and set to 1. Because probes on the microarray used are annotated according to E. coli MG1655, the sequences were remapped to the corresponding region on the E. coli W3110 genome and plotted accordingly.
Preparation of DNA in agarose plugs for PFGE.
Overnight cultures of recBC(Ts) strains at 22°C (permissive temperature) were diluted to an OD600 of 0.005 and grown at 42°C (nonpermissive temperature) in LB medium to an OD600 of 0.15 (exponential phase). A volume of culture corresponding to about 2.5 × 108 cells (counted by a Coulter Counter Multisizer from Beckman) was then centrifuged and resuspended twice in Tris-NaCl (10 mM Tris-HCl [pH 7.6], 1 M NaCl; initially 1 ml). Equal amounts of cell suspension (cells resuspended in Tris-NaCl) and molten clean cut agarose (Bio-Rad) were brought to 42°C and combined. A total of 90 μl of the agarose-cell suspension was dispensed to wells of disposable molds (Bio-Rad), solidified, expelled into a 50-ml tube containing 2.5 ml EC lysis buffer (6 mM Tris-HCl [pH 7.6], 1 M NaCl, 100 mM EDTA [adjusted to pH 7.6 with NaOH], 1% N-lauryl sarcosine, 1 mg/ml lysozyme, and 20 μg/ml RNase), and incubated overnight at 37°C. After removal of the EC lysis buffer, the plugs were rinsed with TE buffer (10 mM Tris-HCl [pH 8.0], 1 mM EDTA [pH 8.0]), and 2.5 ml of ESP buffer was added (500 mM EDTA [adjusted to pH 9 to 9.5 with NaOH], 1% N-lauryl sarcosine, proteinase K [50 μg/ml]), followed by incubation at 37°C overnight (or 48 to 72 h). The plugs were then washed with TE buffer at 25°C, with three washes of 2 h each using 30 ml of TE buffer. The steps mentioned above were taken from a standard protocol for preparation of plugs for pulsed-field gel electrophoresis (PFGE) of bacteria DNA (84).
CHEF-DR III PFGE and quantification of fragmented DNA.
A CHEF-DR III pulsed-field gel electrophoresis system (Bio-Rad) was used to resolve the DNA. The run time was set to 21 h; the temperature was 14°C; the initial and final switch times were 60 and 120 s, respectively; volts per centimeter was set to 6; the included angle was 120; and 0.5× Tris-borate-EDTA (TBE) was used as the running buffer. The gel was stained with SYBR Gold nucleic acid gel stain (Life Technologies) and quantified using the GeneTools (Syngene) software using the rolling disc method for background subtraction. SYBR Gold nucleic acid gel stain (Life Technologies) gives a linear relationship between fluorescence intensity and DNA content over at least two orders of magnitude (85), also applied previously (86). The percentage of chromosomal fragmentation was found by first measuring the DNA present in the well and directly beneath the well (nonfragmented DNA and most likely chromosomes with a single nick, respectively) and then measuring the DNA in the rest of the lane. The fragmented DNA value was then divided by the total DNA value. Quantification of the chromosomal fragmentation of a rep recBC mutant with this method was in agreement with already published results (∼50%) (22).
Replication run-out in the absence of RecA function.
Cultures of cells growing exponentially (to an OD of ∼0.15) at 30°C were split, and rifampin (450 g/ml) and cephalexin (10 g/ml) both were added to each culture. One portion was kept at the permissive temperature (30°C), whereas the other was shifted to the nonpermissive temperature (42°C) to determine the loss RecA function (36, 49). Cells were harvested before drug treatment and after 3 to 4 generation times in the presence of the drugs and fixed for analysis with flow cytometry to compare the replication fork run-out at each temperature. A total of 50,000 cells were recorded.
β-Galactosidase assay to measure induction of the SOS response.
Cellular levels of β-galactosidase were determined as described in reference 43 in cells permeabilized by toluene.
Viability tests.
Overnight cultures were diluted to approximately the same OD in medium, and 5 μl was spotted onto LB or minimal medium plates and incubated for 12 to 24 h at the indicated temperature.
Gam-GFP induction and fluorescence microscopy imaging.
The cells were grown to an OD of ∼0.15 before Gam-GFP was induced by adding 100 ng/ml tetracycline. Growth was continued for 45 to 60 min, at which time the cells were immobilized on a 17 by 28-mm agarose pad (1% containing phosphate-buffered saline [PBS] with 100 ng/ml tetracycline) and covered with a no. 1.5 coverslip. Images were acquired with a Leica DM6000 microscope equipped with a Leica EL6000 metal halide lamp and a Leica DFC350 FX monochrome charge-coupled-device (CCD) camera. Phase-contrast imaging was performed with an HCX PL APO 100×/1.46 numerical aperture (NA) objective. Fluorescence imaging was done using narrow-bandpass (BP) filter sets (excitation at BP 470/40 and emission at BP 525/50 for GFP).
In the experiments, care was taken to utilize the exact same settings for GFP imaging of wild-type and dnaXE145A cells (i.e., the same intensity, exposure time, etc.) to avoid misinterpretation of the images. Using the publicly available ImageJ software, only brightness/contrast was adjusted in the postprocessing of images, and it was done so with the exact same cutoff values for images of dnaXE145A and wild-type cells.
Supplementary Material
ACKNOWLEDGMENTS
We thank Anne Wahl for excellent technical assistance. We are grateful to the Department of Radiation Biology, The Norwegian Radium Hospital, OUS, for sharing their flow cytometry facility and to Trond Stokke, Kirsti Solberg Landsverk, and Idun Dale Rein for technical assistance with the LSR II. We also thank the Microarray Core Facility at Helse Sør-Øst/University of Oslo for their support. We thank K. Marians, A. Kuzminov, S. Rosenberg, and T. Katayama for sending strains.
We declare no conflicts of interest.
This work was supported by grants from The Norwegian Research Council (grants 197102 and 159310), Helse Sør-Øst (grants 2011028 and 2012062), and the German Research Council.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00412-17.
REFERENCES
- 1.Hedglin M, Kumar R, Benkovic SJ. 2013. Replication clamps and clamp loaders. Cold Spring Harb Perspect Biol 5:a010165. doi: 10.1101/cshperspect.a010165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Indiani C, O'Donnell M. 2006. The replication clamp-loading machine at work in the three domains of life. Nat Rev Mol Cell Biol 7:751–761. doi: 10.1038/nrm2022. [DOI] [PubMed] [Google Scholar]
- 3.Jeruzalmi D, O'Donnell M, Kuriyan J. 2001. Crystal structure of the processivity clamp loader gamma (gamma) complex of E. coli DNA polymerase III. Cell 106:429–441. doi: 10.1016/S0092-8674(01)00463-9. [DOI] [PubMed] [Google Scholar]
- 4.Onrust R, Finkelstein J, Naktinis V, Turner J, Fang L, O'Donnell M. 1995. Assembly of a chromosomal replication machine: two DNA polymerases, a clamp loader, and sliding clamps in one holoenzyme particle. I. Organization of the clamp loader. J Biol Chem 270:13348–13357. [DOI] [PubMed] [Google Scholar]
- 5.Onrust R, O'Donnell M. 1993. DNA polymerase III accessory proteins. II. Characterization of delta and delta′. J Biol Chem 268:11766–11772. [PubMed] [Google Scholar]
- 6.Xiao H, Dong Z, O'Donnell M. 1993. DNA polymerase III accessory proteins. IV. Characterization of chi and psi. J Biol Chem 268:11779–11784. [PubMed] [Google Scholar]
- 7.Blinkowa AL, Walker JR. 1990. Programmed ribosomal frameshifting generates the Escherichia coli DNA polymerase III gamma subunit from within the tau subunit reading frame. Nucleic Acids Res 18:1725–1729. doi: 10.1093/nar/18.7.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flower AM, McHenry CS. 1990. The gamma subunit of DNA polymerase III holoenzyme of Escherichia coli is produced by ribosomal frameshifting. Proc Natl Acad Sci U S A 87:3713–3717. doi: 10.1073/pnas.87.10.3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsuchihashi Z, Kornberg A. 1990. Translational frameshifting generates the gamma subunit of DNA polymerase III holoenzyme. Proc Natl Acad Sci U S A 87:2516–2520. doi: 10.1073/pnas.87.7.2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dallmann HG, Kim S, Pritchard AE, Marians KJ, McHenry CS. 2000. Characterization of the unique C terminus of the Escherichia coli tau DnaX protein. Monomeric C-tau binds alpha AND DnaB and can partially replace tau in reconstituted replication forks. J Biol Chem 275:15512–15519. [DOI] [PubMed] [Google Scholar]
- 11.Kim DR, McHenry CS. 1996. In vivo assembly of overproduced DNA polymerase III. Overproduction, purification, and characterization of the alpha, alpha-epsilon, and alpha-epsilon-theta subunits. J Biol Chem 271:20681–20689. [DOI] [PubMed] [Google Scholar]
- 12.Heller RC, Marians KJ. 2006. Replisome assembly and the direct restart of stalled replication forks. Nat Rev Mol Cell Biol 7:932–943. doi: 10.1038/nrm2058. [DOI] [PubMed] [Google Scholar]
- 13.Yeeles JT, Poli J, Marians KJ, Pasero P. 2013. Rescuing stalled or damaged replication forks. Cold Spring Harb Perspect Biol 5:a012815. doi: 10.1101/cshperspect.a012815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rotman E, Khan SR, Kouzminova E, Kuzminov A. 2014. Replication fork inhibition in seqA mutants of Escherichia coli triggers replication fork breakage. Mol Microbiol 93:50–64. doi: 10.1111/mmi.12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuzminov A. 1995. Collapse and repair of replication forks in Escherichia coli. Mol Microbiol 16:373–384. doi: 10.1111/j.1365-2958.1995.tb02403.x. [DOI] [PubMed] [Google Scholar]
- 16.Kuzminov A. 2001. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc Natl Acad Sci U S A 98:8241–8246. doi: 10.1073/pnas.131009198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lusetti SL, Cox MM. 2002. The bacterial RecA protein and the recombinational DNA repair of stalled replication forks. Annu Rev Biochem 71:71–100. doi: 10.1146/annurev.biochem.71.083101.133940. [DOI] [PubMed] [Google Scholar]
- 18.Michel B, Leach D. 2012. Homologous recombination–enzymes and pathways. EcoSal Plus 5. doi: 10.1128/ecosalplus.7.2.7. [DOI] [PubMed] [Google Scholar]
- 19.Sandler SJ, Marians KJ. 2000. Role of PriA in replication fork reactivation in Escherichia coli. J Bacteriol 182:9–13. doi: 10.1128/JB.182.1.9-13.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morgan AR, Severini A. 1990. Interconversion of replication and recombination structures: implications for terminal repeats and concatemers. J Theor Biol 144:195–202. doi: 10.1016/S0022-5193(05)80318-2. [DOI] [PubMed] [Google Scholar]
- 21.Higgins NP, Kato K, Strauss B. 1976. A model for replication repair in mammalian cells. J Mol Biol 101:417–425. doi: 10.1016/0022-2836(76)90156-X. [DOI] [PubMed] [Google Scholar]
- 22.Seigneur M, Bidnenko V, Ehrlich SD, Michel B. 1998. RuvAB acts at arrested replication forks. Cell 95:419–430. doi: 10.1016/S0092-8674(00)81772-9. [DOI] [PubMed] [Google Scholar]
- 23.Seigneur M, Ehrlich SD, Michel B. 2000. RuvABC-dependent double-strand breaks in dnaBts mutants require recA. Mol Microbiol 38:565–574. doi: 10.1046/j.1365-2958.2000.02152.x. [DOI] [PubMed] [Google Scholar]
- 24.Michel B, Boubakri H, Baharoglu Z, LeMasson M, Lestini R. 2007. Recombination proteins and rescue of arrested replication forks. DNA Repair (Amst) 6:967–980. doi: 10.1016/j.dnarep.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 25.Khan SR, Mahaseth T, Kouzminova EA, Cronan GE, Kuzminov A. 2016. Static and dynamic factors limit chromosomal replication complexity in Escherichia coli, avoiding dangers of runaway overreplication. Genetics 202:945–960. doi: 10.1534/genetics.115.184697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuzminov A. 2016. Chromosomal replication complexity: a novel DNA metrics and genome instability factor. PLoS Genet 12:e1006229. doi: 10.1371/journal.pgen.1006229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuzminov A. 1995. Instability of inhibited replication forks in E coli. Bioessays 17:733–741. doi: 10.1002/bies.950170810. [DOI] [PubMed] [Google Scholar]
- 28.Levine C, Marians KJ. 1998. Identification of dnaX as a high-copy suppressor of the conditional lethal and partition phenotypes of the parE10 allele. J Bacteriol 180:1232–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Espeli O, Levine C, Hassing H, Marians KJ. 2003. Temporal regulation of topoisomerase IV activity in E coli. Mol Cell 11:189–201. doi: 10.1016/S1097-2765(03)00013-3. [DOI] [PubMed] [Google Scholar]
- 30.Burton P, Holland IB. 1983. Two pathways of division inhibition in UV-irradiated E. coli. Mol Gen Genet 190:309–314. doi: 10.1007/BF00330656. [DOI] [PubMed] [Google Scholar]
- 31.Boye E, Lobner-Olesen A. 1991. Bacterial growth control studied by flow cytometry. Res Microbiol 142:131–135. doi: 10.1016/0923-2508(91)90020-B. [DOI] [PubMed] [Google Scholar]
- 32.Skarstad K, Boye E, Steen HB. 1986. Timing of initiation of chromosome replication in individual Escherichia coli cells. EMBO J 5:1711–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stokke C, Flåtten I, Skarstad K. 2012. An easy-to-use simulation program demonstrates variations in bacterial cell cycle parameters depending on medium and temperature. PLoS One 7:e30981. doi: 10.1371/journal.pone.0030981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kouzminova EA, Kuzminov A. 2006. Fragmentation of replicating chromosomes triggered by uracil in DNA. J Mol Biol 355:20–33. doi: 10.1016/j.jmb.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 35.Kouzminova EA, Kuzminov A. 2008. Patterns of chromosomal fragmentation due to uracil-DNA incorporation reveal a novel mechanism of replication-dependent double-stranded breaks. Mol Microbiol 68:202–215. doi: 10.1111/j.1365-2958.2008.06149.x. [DOI] [PubMed] [Google Scholar]
- 36.Skarstad K, Boye E. 1993. Degradation of individual chromosomes in recA mutants of Escherichia coli. J Bacteriol 175:5505–5509. doi: 10.1128/jb.175.17.5505-5509.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sueoka N, Yoshikawa H. 1965. The chromosome of Bacillus subtilis. I. Theory of marker frequency analysis. Genetics 52:747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simmons LA, Breier AM, Cozzarelli NR, Kaguni JM. 2004. Hyperinitiation of DNA replication in Escherichia coli leads to replication fork collapse and inviability. Mol Microbiol 51:349–358. doi: 10.1046/j.1365-2958.2003.03842.x. [DOI] [PubMed] [Google Scholar]
- 39.Kuzminov A. 1999. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol Mol Biol Rev 63:751–813, table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Michel B, Ehrlich SD, Uzest M. 1997. DNA double-strand breaks caused by replication arrest. EMBO J 16:430–438. doi: 10.1093/emboj/16.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kushner SR. 1974. Differential thermolability of exonuclease and endonuclease activities of the recBC nuclease isolated from thermosensitive recB and recC mutants. J Bacteriol 120:1219–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kreuzer KN. 2013. DNA damage responses in prokaryotes: regulating gene expression, modulating growth patterns, and manipulating replication forks. Cold Spring Harb Perspect Biol 5:a012674. doi: 10.1101/cshperspect.a012674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller JH. 1992. A short course in bacterial genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 44.Austin S, Ziese M, Sternberg N. 1981. A novel role for site-specific recombination in maintenance of bacterial replicons. Cell 25:729–736. doi: 10.1016/0092-8674(81)90180-X. [DOI] [PubMed] [Google Scholar]
- 45.Blakely G, Colloms S, May G, Burke M, Sherratt D. 1991. Escherichia coli XerC recombinase is required for chromosomal segregation at cell division. New Biol 3:789–798. [PubMed] [Google Scholar]
- 46.Kuempel PL, Henson JM, Dircks L, Tecklenburg M, Lim DF. 1991. dif, a recA-independent recombination site in the terminus region of the chromosome of Escherichia coli. New Biol 3:799–811. [PubMed] [Google Scholar]
- 47.Ting H, Kouzminova EA, Kuzminov A. 2008. Synthetic lethality with the dut defect in Escherichia coli reveals layers of DNA damage of increasing complexity due to uracil incorporation. J Bacteriol 190:5841–5854. doi: 10.1128/JB.00711-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lesterlin C, Barre FX, Cornet F. 2004. Genetic recombination and the cell cycle: what we have learned from chromosome dimers. Mol Microbiol 54:1151–1160. doi: 10.1111/j.1365-2958.2004.04356.x. [DOI] [PubMed] [Google Scholar]
- 49.Pedersen IB, Helgesen E, Flatten I, Fossum-Raunehaug S, Skarstad K. 2017. SeqA structures behind Escherichia coli replication forks affect replication elongation and restart mechanisms. Nucleic Acids Res 45:6471–6485. doi: 10.1093/nar/gkx263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Horiuchi T, Fujimura Y. 1995. Recombinational rescue of the stalled DNA replication fork: a model based on analysis of an Escherichia coli strain with a chromosome region difficult to replicate. J Bacteriol 177:783–791. doi: 10.1128/jb.177.3.783-791.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuzminov A, Stahl FW. 1997. Stability of linear DNA in recA mutant Escherichia coli cells reflects ongoing chromosomal DNA degradation. J Bacteriol 179:880–888. doi: 10.1128/jb.179.3.880-888.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuzminov A, Schabtach E, Stahl FW. 1994. Chi sites in combination with RecA protein increase the survival of linear DNA in Escherichia coli by inactivating exoV activity of RecBCD nuclease. EMBO J 13:2764–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bidnenko V, Ehrlich SD, Michel B. 2002. Replication fork collapse at replication terminator sequences. EMBO J 21:3898–3907. doi: 10.1093/emboj/cdf369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grigorian AV, Lustig RB, Guzman EC, Mahaffy JM, Zyskind JW. 2003. Escherichia coli cells with increased levels of DnaA and deficient in recombinational repair have decreased viability. J Bacteriol 185:630–644. doi: 10.1128/JB.185.2.630-644.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nordman J, Skovgaard O, Wright A. 2007. A novel class of mutations that affect DNA replication in E. coli. Mol Microbiol 64:125–138. doi: 10.1111/j.1365-2958.2007.05651.x. [DOI] [PubMed] [Google Scholar]
- 56.Morigen, Odsbu I, Skarstad K. 2009. Growth rate dependent numbers of SeqA structures organize the multiple replication forks in rapidly growing Escherichia coli. Genes Cells 14:643–657. doi: 10.1111/j.1365-2443.2009.01298.x. [DOI] [PubMed] [Google Scholar]
- 57.Gon S, Camara JE, Klungsoyr HK, Crooke E, Skarstad K, Beckwith J. 2006. A novel regulatory mechanism couples deoxyribonucleotide synthesis and DNA replication in Escherichia coli. EMBO J 25:1137–1147. doi: 10.1038/sj.emboj.7600990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brüning JG, Howard JL, McGlynn P. 2014. Accessory replicative helicases and the replication of protein-bound DNA. J Mol Biol 426:3917–3928. doi: 10.1016/j.jmb.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 59.Yancey-Wrona JE, Wood ER, George JW, Smith KR, Matson SW. 1992. Escherichia coli Rep protein and helicase IV. Distributive single-stranded DNA-dependent ATPases that catalyze a limited unwinding reaction in vitro. Eur J Biochem 207:479–485. [DOI] [PubMed] [Google Scholar]
- 60.Lane HE, Denhardt DT. 1974. The rep mutation. III. Altered structure of the replicating Escherichia coli chromosome. J Bacteriol 120:805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Colasanti J, Denhardt DT. 1987. The Escherichia coli rep mutation. X. Consequences of increased and decreased Rep protein levels. Mol Gen Genet 209:382–390. [DOI] [PubMed] [Google Scholar]
- 62.Atkinson J, Gupta MK, Rudolph CJ, Bell H, Lloyd RG, McGlynn P. 2011. Localization of an accessory helicase at the replisome is critical in sustaining efficient genome duplication. Nucleic Acids Res 39:949–957. doi: 10.1093/nar/gkq889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guy CP, Atkinson J, Gupta MK, Mahdi AA, Gwynn EJ, Rudolph CJ, Moon PB, van Knippenberg IC, Cadman CJ, Dillingham MS, Lloyd RG, McGlynn P. 2009. Rep provides a second motor at the replisome to promote duplication of protein-bound DNA. Mol Cell 36:654–666. doi: 10.1016/j.molcel.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gupta MK, Guy CP, Yeeles JT, Atkinson J, Bell H, Lloyd RG, Marians KJ, McGlynn P. 2013. Protein-DNA complexes are the primary sources of replication fork pausing in Escherichia coli. Proc Natl Acad Sci U S A 110:7252–7257. doi: 10.1073/pnas.1303890110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shee C, Cox BD, Gu F, Luengas EM, Joshi MC, Chiu LY, Magnan D, Halliday JA, Frisch RL, Gibson JL, Nehring RB, Do HG, Hernandez M, Li L, Herman C, Hastings PJ, Bates D, Harris RS, Miller KM, Rosenberg SM. 2013. Engineered proteins detect spontaneous DNA breakage in human and bacterial cells. eLife 2:e01222. doi: 10.7554/eLife.01222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Abraham ZH, Symonds N. 1990. Purification of overexpressed gam gene protein from bacteriophage Mu by denaturation-renaturation techniques and a study of its DNA-binding properties. Biochem J 269:679–684. doi: 10.1042/bj2690679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Michel B, Recchia GD, Penel-Colin M, Ehrlich SD, Sherratt DJ. 2000. Resolution of Holliday junctions by RuvABC prevents dimer formation in rep mutants and UV-irradiated cells. Mol Microbiol 37:180–191. doi: 10.1046/j.1365-2958.2000.01989.x. [DOI] [PubMed] [Google Scholar]
- 68.Gao D, McHenry CS. 2001. tau binds and organizes Escherichia coli replication through distinct domains. Partial proteolysis of terminally tagged tau to determine candidate domains and to assign domain V as the alpha binding domain. J Biol Chem 276:4433–4440. [DOI] [PubMed] [Google Scholar]
- 69.Gao D, McHenry CS. 2001. tau binds and organizes Escherichia coli replication proteins through distinct domains. Domain IV, located within the unique C terminus of tau, binds the replication fork, helicase, DnaB. J Biol Chem 276:4441–4446. [DOI] [PubMed] [Google Scholar]
- 70.Gao D, McHenry CS. 2001. Tau binds and organizes Escherichia coli replication proteins through distinct domains. Domain III, shared by gamma and tau, binds delta delta′ and chi psi. J Biol Chem 276:4447–4453. [DOI] [PubMed] [Google Scholar]
- 71.Glover BP, Pritchard AE, McHenry CS. 2001. tau binds and organizes Escherichia coli replication proteins through distinct domains: domain III, shared by gamma and tau, oligomerizes DnaX. J Biol Chem 276:35842–35846. doi: 10.1074/jbc.M103719200. [DOI] [PubMed] [Google Scholar]
- 72.Khodursky AB, Zechiedrich EL, Cozzarelli NR. 1995. Topoisomerase IV is a target of quinolones in Escherichia coli. Proc Natl Acad Sci U S A 92:11801–11805. doi: 10.1073/pnas.92.25.11801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lesterlin C, Gigant E, Boccard F, Espeli O. 2012. Sister chromatid interactions in bacteria revealed by a site-specific recombination assay. EMBO J 31:3468–3479. doi: 10.1038/emboj.2012.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang X, Reyes-Lamothe R, Sherratt DJ. 2008. Modulation of Escherichia coli sister chromosome cohesion by topoisomerase IV. Genes Dev 22:2426–2433. doi: 10.1101/gad.487508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cebrián J, Castan A, Martinez V, Kadomatsu-Hermosa MJ, Parra C, Fernandez-Nestosa MJ, Schaerer C, Hernandez P, Krimer DB, Schvartzman JB. 2015. Direct evidence for the formation of precatenanes during DNA replication. J Biol Chem 290:13725–13735. doi: 10.1074/jbc.M115.642272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Postow L, Ullsperger C, Keller RW, Bustamante C, Vologodskii AV, Cozzarelli NR. 2001. Positive torsional strain causes the formation of a four-way junction at replication forks. J Biol Chem 276:2790–2796. doi: 10.1074/jbc.M006736200. [DOI] [PubMed] [Google Scholar]
- 77.Grompone G, Bidnenko V, Ehrlich SD, Michel B. 2004. PriA is essential for viability of the Escherichia coli topoisomerase IV parE10(Ts) mutant. J Bacteriol 186:1197–1199. doi: 10.1128/JB.186.4.1197-1199.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Clark DJ, Maaløe O. 1967. DNA replication and the division cycle in Escherichia coli. J Mol 23:99–112. doi: 10.1016/S0022-2836(67)80070-6. [DOI] [Google Scholar]
- 79.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wold S, Skarstad K, Steen HB, Stokke T, Boye E. 1994. The initiation mass for DNA replication in Escherichia coli K-12 is dependent on growth rate. EMBO J 13:2097–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Torheim NK, Boye E, Lobner-Olesen A, Stokke T, Skarstad K. 2000. The Escherichia coli SeqA protein destabilizes mutant DnaA204 protein. Mol Microbiol 37:629–638. doi: 10.1046/j.1365-2958.2000.02031.x. [DOI] [PubMed] [Google Scholar]
- 82.Waldminghaus T, Skarstad K. 2010. ChIP on Chip: surprising results are often artifacts. BMC Genomics 11:414. doi: 10.1186/1471-2164-11-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Johnsen L, Flåtten I, Morigen Dalhus B, Bjoras M, Waldminghaus T, Skarstad K. 2011. The G157C mutation in the Escherichia coli sliding clamp specifically affects initiation of replication. Mol Microbiol 79:433–446. doi: 10.1111/j.1365-2958.2010.07453.x. [DOI] [PubMed] [Google Scholar]
- 84.Hillier AJ, Davidson BE. 1995. Pulsed field gel electrophoresis. Methods Mol Biol 46:149–164. [DOI] [PubMed] [Google Scholar]
- 85.Gradzka I, Iwanenko T. 2005. A non-radioactive, PFGE-based assay for low levels of DNA double-strand breaks in mammalian cells. DNA Repair (Amst) 4:1129–1139. doi: 10.1016/j.dnarep.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 86.Repar J, Briski N, Buljubasic M, Zahradka K, Zahradka D. 2013. Exonuclease VII is involved in “reckless” DNA degradation in UV-irradiated Escherichia coli. Mutat Res 750:96–104. doi: 10.1016/j.mrgentox.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 87.Howard-Flanders P, Simson E, Theriot L. 1964. A locus that controls filament formation and sensitivity to radiation in Escherichia coli K-12. Genetics 49:237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lloyd RG, Johnson S. 1979. Kinetics of recA function in conjugational recombinant formation. Mol Gen Genet 169:219–228. doi: 10.1007/BF00271674. [DOI] [PubMed] [Google Scholar]
- 89.Lloyd RG, Low B, Godson GN, Birge EA. 1974. Isolation and characterization of an Escherichia coli K-12 mutant with a temperature-sensitive recA− phenotype. J Bacteriol 120:407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Winans SC, Elledge SJ, Krueger JH, Walker GC. 1985. Site-directed insertion and deletion mutagenesis with cloned fragments in Escherichia coli. J Bacteriol 161:1219–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Morigen, Løbner-Olesen A, Skarstad K. 2003. Titration of the Escherichia coli DnaA protein to excess datA sites causes destabilization of replication forks, delayed replication initiation and delayed cell division. Mol Microbiol 50:349–362. doi: 10.1046/j.1365-2958.2003.03695.x. [DOI] [PubMed] [Google Scholar]
- 92.Leslie NR, Sherratt DJ. 1995. Site-specific recombination in the replication terminus region of Escherichia coli: functional replacement of dif. EMBO J. 14:1561–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Colloms SD, Sykora P, Szatmari G, Sherratt DJ. 1990. Recombination at ColE1 cer requires the Escherichia coli xerC gene product, a member of the lambda integrase family of site-specific recombinases. J Bacteriol 172:6973–6980. doi: 10.1128/jb.172.12.6973-6980.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Blakely G, May G, McCulloch R, Arciszewska LK, Burke M, Lovett ST, Sherratt DJ. 1993. Two related recombinases are required for site-specific recombination at dif and cer in E. coli K12. Cell 75:351–361. doi: 10.1016/0092-8674(93)80076-Q. [DOI] [PubMed] [Google Scholar]
- 95.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.