

ABSTRACT
Ebolavirus and Marburgvirus comprise two genera of negative-sense single-stranded RNA viruses that cause severe hemorrhagic fevers in humans. Despite considerable research efforts, the molecular events following Ebola virus (EBOV) infection are poorly understood. With the view of identifying host factors that underpin EBOV pathogenesis, we compared the transcriptomes of EBOV-infected human, pig, and bat kidney cells using a transcriptome sequencing (RNA-seq) approach. Despite a significant difference in viral transcription/replication between the cell lines, all cells responded to EBOV infection through a robust induction of extracellular growth factors. Furthermore, a significant upregulation of activator protein 1 (AP1) transcription factor complex members FOS and JUN was observed in permissive cell lines. Functional studies focusing on human cells showed that EBOV infection induces protein expression, phosphorylation, and nuclear accumulation of JUN and, to a lesser degree, FOS. Using a luciferase-based reporter, we show that EBOV infection induces AP1 transactivation activity within human cells at 48 and 72 h postinfection. Finally, we show that JUN knockdown decreases the expression of EBOV-induced host gene expression. Taken together, our study highlights the role of AP1 in promoting the host gene expression profile that defines EBOV pathogenesis.
IMPORTANCE Many questions remain about the molecular events that underpin filovirus pathophysiology. The rational design of new intervention strategies, such as postexposure therapeutics, will be significantly enhanced through an in-depth understanding of these molecular events. We believe that new insights into the molecular pathogenesis of EBOV may be possible by examining the transcriptomic response of taxonomically diverse cell lines (derived from human, pig, and bat). We first identified the responsive pathways using an RNA-seq-based transcriptomics approach. Further functional and computational analysis focusing on human cells highlighted an important role for the AP1 transcription factor in mediating the transcriptional response to EBOV infection. Our study sheds new light on how host transcription factors respond to and promote the transcriptional landscape that follows viral infection.
KEYWORDS: Ebola virus, FOS, JUN, RNA-seq, growth factor, host-pathogen interaction, AP1, Ebolavirus
INTRODUCTION
The filoviruses of the genera Ebolavirus and Marburgvirus are among the most deadly viruses known to humankind. Five species of the genus Ebolavirus are formally recognized, including Zaire ebolavirus, Reston ebolavirus, Taï Forest ebolavirus, Sudan ebolavirus, and Bundibugyo ebolavirus. With the exception of Reston ebolavirus, all Ebolavirus species cause serve hemorrhagic fevers in humans, often with high case fatality rates (1). While outbreaks of Ebola virus (EBOV) were sporadic prior to 2013, West Africa experienced its largest Ebola virus disease (EVD) epidemic in history between late 2013 and 2015 (2). This epidemic resulted in over 11,000 confirmed human fatalities, predominantly in Guinea, Liberia, and Sierra Leone (3).
While most filoviruses cause severe hemorrhagic fevers in humans and nonhuman primates (NHP), other species, including the reservoir and intermediate spillover hosts, appear to be less susceptible. Bats, order Chiroptera, are considered the most likely reservoir species for EBOV (4). In comparison to human and NHP models, experimental infection of fruit and insectivorous bats with EBOV causes only subclinical infections (5). The zoonotic transmission of Reston Ebolavirus from infected pigs to humans, reported in 2008 (6), raised the possibility that pigs could act as an intermediate host for more pathogenic Ebola virus species. Subsequent experimental infection of pigs with EBOV produced a nonfatal respiratory syndrome, characterized by severe lung pathology with oronasal shedding (7). Importantly, the transmission of EBOV from infected pigs to NHP was also reported (8), thus demonstrating that pigs can act as a spillover host for pathogenic Ebolavirus species. Despite their importance as potential reservoir and spillover hosts, relatively little research has focused on the molecular pathogenesis of EBOV within either pigs or bats.
Much of our knowledge concerning the pathogenesis of EVD comes from humans and NHP (namely cynomolgus and rhesus macaques). In these species, EBOV initially targets monocytes, dendritic cells, and macrophages. Within macrophages, the release of proinflammatory cytokines, including interleukin-1β (IL-1β), IL-6, IL-8, tumor necrosis factor alpha (TNF-α), and macrophage inflammatory protein-1α (MIP-1α), is observed (9–12). This proinflammatory response increases the permeability of the vascular endothelium, which destroys the endothelial barrier and facilitates secondary infection of endothelial cells (13–15). Viral replication subsequently occurs in almost all cell types, with the exception of lymphocytes (16). Infected macrophages also induce disseminated intravascular coagulation through the expression of cell surface tissue factor (TF) (17). EBOV infection disrupts cell-cell adhesion through differential regulation of the integrin cell adhesion molecules. Indeed, overexpression of the membrane-bound EBOV glycoprotein (GP) causes the downregulation of cell adhesion molecules, namely, the β1-integrins (18). Further studies using pseudovirions bearing the EBOV GP revealed that the α5β1-integrins are regulators of the endosomal cathepsins, which are required for viral entry (19).
The importance of growth factor signaling in EBOV pathogenesis has recently been demonstrated in human hepatocytes (Huh7 cells) following infection with EBOV (20). Kindrachuk and coworkers (20) demonstrated the upregulation of both transforming growth factor β (TGF-β) and vascular endothelial growth factor (VEGF) signaling at 24 h postinfection (hpi). The authors proposed a model where the infection of epithelial cells results in a loss of cell-cell adhesion, increased growth factor signaling, and a subsequent epithelial-to-mesenchymal-like transition. As a consequence, the infected epithelial cells lose their cell polarity and cell-cell adhesion and gain migratory/invasive properties, which have downstream effects on the surrounding vasculature (20). Growth factors mediate these cellular processes by enhancing the activity of transcription factors, which transactivate specific sets of genes responsible for biological processes. Although the importance of growth factor signaling in EBOV pathogenesis is now recognized, little is known concerning the involvement of the downstream transcription factors.
One transcription factor that is highly responsive to growth factor and cytokine signaling is activator protein 1 (AP1). The AP1 transcription factor complex is formed through a noncovalent dimerization between FOS and JUN family members. The gene encoding FOS (formally known as c-FOS) is a member of the FOS family of nuclear oncogenes. Other members include the genes for FosB and Fos-related antigens 1 and 2 (FRA1 and FRA2). To form the AP1 complex, FOS proteins must dimerize with JUN family proteins, including JUN (formally known as c-JUN), JunB, and JunD (21). While different combinations of FOS-JUN heterodimers have different abilities to transactivate AP1-dependent genes, the FOS-JUN heterodimer is considered a more stable complex with stronger DNA binding than JUN-JUN homodimers (22). FOS-JUN heterodimers are capable of binding to the palindromic DNA motif 5′-TGA(G/C)TCA-3′ to regulate gene expression. The expression of FOS and JUN mRNA is largely controlled through growth factor or cytokine signaling (23), and subsequent regulation of AP1 activity occurs through dimer composition and posttranslational events, such as phosphorylation. AP1 activity is increased in response to various extracellular signals, including the growth factors platelet-derived growth factor (PDGF), TGF-β, and TGF-α (21, 24), as well as the proinflammatory cytokines TNF-α and IL-1 (25–27).
AP1 transactivates target gene expression to regulate a plethora of physiological functions, including cellular proliferation, differentiation, transformation, and death (28, 29). In the context of EVD, many AP1 target genes may directly influence the pathophysiology commonly associated with EBOV infection. For instance, TNF-α- and IL-1β-mediated expression of tissue factor (F3) is controlled, in part, by AP1 transactivation activity (30). This suggests that AP1 contributes to the disseminated intravascular coagulation seen in EVD. Other AP1 targets of relevance include genes related to cell adhesion and extracellular matrix stability, namely, genes for matrix metalloproteinases (MMP; i.e., MMP9) (31) and integrins (32, 33). The latter is particularly relevant to EVD in the context of the loss of vascular integrity, which is associated with the distribution of cell-cell adhesion. AP1 can also directly transactivate its own FOS and JUN family members, thereby positively autoregulating its own expression (34). Despite its central role in mediating growth factor- and cytokine-induced gene expression, no studies have investigated the role of AP1 in EBOV pathogenesis.
Despite significant research efforts, many of the molecular events underpinning EBOV infection remain poorly understood. We propose that examining cells from taxonomically diverse species may provide new insights into molecular pathogenesis and the cell/tissue tropism of this virus. To this end, we examined the transcriptional response of three cell lines derived from human (HEK293T), pig (PK15A), and the African straw-colored fruit bat, Eidolon helvum (EhKiT), 6 and 24 h following infection by EBOV. When the host response across different cell lines is compared, it is important to compare like with like, that is, similar cell lineages. For this reason, we chose to examine kidney-derived immortalized cells. Kidney cells were chosen over liver-derived hepatocytes or monocytes/macrophages because these cell types were not available for all three species. The present study sheds new light on the transcriptomic response that underpins EBOV pathology and highlights that the AP1 transcription factor is an important mediator of this response.
RESULTS
Growth kinetics of EBOV across taxonomically diverse mammalian cell lines.
The transcriptome sequencing (RNA-seq) data were utilized to examine both viral and host gene expression profiles in parallel. The transcription of EBOV within kidney cell lines from human (HEK293T), bat (EhKiT), and pig (PK15A) was examined at 6 and 24 hpi. This was achieved by reference mapping the RNA-seq reads against the EBOV genome (Mayinga strain) and subsequently calculating the read depth at each position. As expected and previously reported (35), there was a decrease in transcript abundance from 3′ to 5′ across the genome in all cell lines, indicative of polar sequential transcription (Fig. 1A). At 6 hpi, only a small number of reads were found to map to the EBOV genome, regardless of cell line (Fig. 1A). However, at 24 hpi, a significant increase in transcription was observed, most notably, in PK15A cells. Considerable differences in the transcription of EBOV were observed across the three cell lines (Fig. 1A). The transcription of EBOV was the highest in PK15A cells, followed by HEK293T cells. The number of EBOV reads observed in the EhKiT cell line was remarkably low at 24 hpi compared to that in the other two cell lines. However, even in EhKiT cells we observed an obvious increase in the amount of viral mRNA from 6 to 24 hpi (Fig. 1A), thus confirming that EhKiT cells can be infected with EBOV and viral transcription can occur.
FIG 1.

EBOV growth kinetics within taxonomically diverse cell lines. (A) Transcription of EBOV in EhKiT, HEK293T and PK15A cells was quantified at 6 hpi and 24 hpi by mapping RNA-seq reads against the EBOV genome sequence (represented as blue arrows at the top). The coverage was normalized per million reads mapped to the host genomes. The normalized coverage was averaged across the three biological replicates at each time point. Note the different scales on the y axes. (B) Log10 number of copies of EBOV NP mRNA determined by TaqMan PCR across the three cell lines at 6 and 24 hpi. The mean ± standard error from three independent biological replicates is presented, with each replicate being performed as a duplicate PCR. (C) Western blot of EBOV NP protein in the three cell lines at 0, 6, and 24 hpi. Western blotting was performed from the three independent biological replicates, with a representative image being shown here. (D) The replication of EBOV was quantified as the number of TCID50 per milliliter at 24, 48, and 72 hpi for each cell line. The mean ± standard error from three independent biological replicates is presented.
To further verify the differences in viral transcription between the cell lines, a TaqMan PCR specific to the EBOV nucleocapsid protein (NP) gene was performed on all RNA samples from the RNA-seq experiments. At 6 hpi the three cell lines had similar abundances of EBOV NP mRNA (Fig. 1B). However, at 24 hpi a significant increase in the level of EBOV NP transcription in PK15A cells and, to a lesser extent, HEK293T cells was observed (Fig. 1B). Only a modest increase in EBOV NP transcription was observed in EhKiT cells using TaqMan PCR (Fig. 1B). No amplification was observed in the uninfected controls for any of the cell lines (data not shown). The translation of NP was quantified across the three cell lines by Western blotting. No detection of NP was observed in EhKiT cells, while larger quantities were observed in PK15A cells than HEK293T cells at 24 hpi (Fig. 1C).
The replication of EBOV was compared between the cell lines by titrating virus from the cell culture supernatant at 24, 48, and 72 hpi. All cell lines were initially infected at a multiplicity of infection (MOI) of 5. The highest virus titer was observed in HEK293T cells at all time points (Fig. 1D). A steady increase in viral replication was observed in these cells over the time course. Surprisingly, despite demonstrating the highest levels of transcription and translation of viral proteins, PK15A cells had lower titers than HEK293T cells at all time points. Low titers of EBOV were observed in EhKiT cells across the time course, with a slight increase toward 72 hpi (Fig. 1D).
Diverse transcriptional responses between cell lines.
Next we used the RNA-seq transcriptomics data to compare the host gene expression profiles between the EBOV-infected cell lines. RNA-seq experiments were performed in triplicate at each time point for all cell lines. Summary statistics showing the number of sequence reads trimmed, reads mapped, assembled genes, and BLAST hits are given in Table S1 in the supplemental material. EBOV infection resulted in the differential expression of hundreds of genes. Broadly, HEK293T and PK15A cells responded to EBOV in a similar manner, with more genes being upregulated and downregulated at 24 hpi than at 6 hpi (Fig. 2). In contrast, EhKiT cells had more genes differentially expressed at 6 hpi than at 24 hpi (Fig. 2). Indeed, EhKiT cells responded to EBOV at 6 hpi by downregulating more than 600 genes (Fig. 2). A full list of the gene expression profiles is provided in Table S2.
FIG 2.
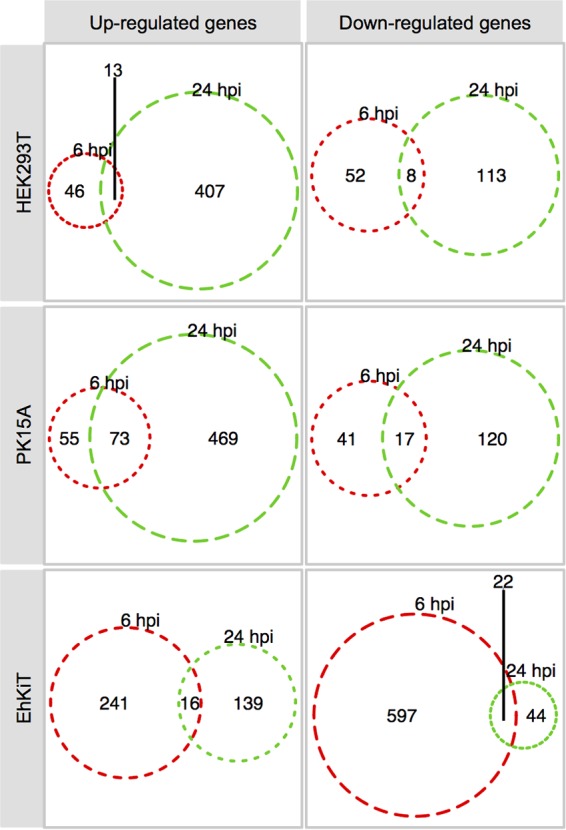
Differential host gene expression. Venn diagram summary of genes significantly differentially expressed at 6 hpi and/or 24 hpi within HEK293T (human), PK15A (pig), and EhKiT (bat) cells. Differential expression analysis was determined from three independent biological replicates. Genes were deemed significantly differentially expressed if the (adjusted) P value was <0.05 and the mean fold change in regulation, either up or down, was ≥2.
A simple three-way comparison of differentially expressed gene sets from HEK293T, PK15A, and EhKiT cells by use of a Venn diagram demonstrated little overlap. Only six genes were upregulated (PI15, NAV2, ADAMTS1, MYLK, FOS, HSPG2) and one gene was downregulated (DDIT4) in all cell lines at 6 and/or 24 hpi (Fig. 3A and B).
FIG 3.

Biological processes induced by EBOV across human, bat, and pigs cells. (A and B) The intersection of genes upregulated (A) and downregulated (B) across three cell lines following EBOV infection. The genes included are a combined set of genes differentially expressed at 6 and/or 24 hpi. (C) GO enrichment analysis was performed on gene sets up- and downregulated at 6 and/or 24 hpi. A subset of biological processes significantly enriched in the upregulated gene sets is presented here. Enrichment scores for each GO term are provided in the heatmap. The relationship between parent and child GO terms is represented on the right, with child term GO identifiers being indented on the right. Biological processes enriched in the downregulated gene sets are presented in Table S3 in the supplemental material. MAPK, mitogen-activated protein kinase.
Gene set enrichment analysis.
Gene Ontology (GO) enrichment analysis (36) was used to categorize the up- and downregulated gene sets (combined for 6 and 24 hpi) into biological processes for each cell line (full results are available in Table S3). A large number of GO terms were found to be enriched for up- and downregulated gene sets (Fig. 3C). For the upregulated genes, many high-level parent GO terms, such as cell differentiation, regulation of cell proliferation, and regulation of cell migration, were enriched in all cell lines. However, more specific child GO terms were often unique to each cell line. A closer look at the cell differentiation ontology demonstrates enrichment in all cell lines for genes broadly associated with vasculature development, blood vessel development, and angiogenesis. The genes associated with these vascular processes included genes for growth factors (PDGFA/B, TGFB, VEGF, CTGF), matricellular proteins (cysteine-rich angiogenic inducer 61 [CYR61], thrombospondin 1 [THBS1], nephroblastoma overexpressed [NOV]), cell adhesion proteins (neural cell adhesion molecule [NRCAM]), and transcription factors (FOS, JUN) (Fig. 4A). The expression of these genes varied both across the cell lines and between the time points (6 hpi versus 24 hpi).
FIG 4.

Expression dynamics of significantly differentially expressed genes involved in angiogenesis (GO:0001525) (A), extracellular matrix organization (GO:0030198) (B), and regulation of coagulation (GO:0050818) (C). Each tile represents the mean log2 fold change in regulation from three independent biological replicates. Gray tiles represent genes that were not identified within the PK15A or EhKiT cell line or had an infinity fold change (i.e., no reads at 0 hpi). Blue tiles represent genes which had one or more AP1 binding sites within their promoters.
All three cell lines also showed the upregulation of genes associated with extracellular matrix organization (Fig. 3C). This response was perhaps the most apparent in HEK293T cells at 24 hpi. Many of the genes associated with this process were also involved in the angiogenesis pathway (Fig. 4B). This is not surprising, given that remodeling of the extracellular matrix is essential for development of vascular structures. Integrins (ITGB3, ITGA8), matrix metalloproteinase-2 (MMP2), and cell adhesion proteins (cadherin [CDH1], fibronectin [FN1], versican [VCAN]) were all upregulated in one or more cell lines (Fig. 4B) and play important roles in extracellular matrix organization.
Significant enrichment for genes involved in the regulation of coagulation was observed in the upregulated gene sets from PK15A and EhKiT cells but not HEK293T cells (Fig. 3C). The genes contributing to this biological process differed between the PK15A and EhKiT cell lines (Fig. 4C). Indeed, PK15A cells upregulated coagulation factor F7 and its receptor, tissue factor (F3). In contrast, EhKiT cells upregulated coagulation factor F2 (thrombin), whereas this gene was downregulated in HEK293T cells (Fig. 4C).
The cell lines, however, also showed unique responses. For example, the type I interferon (IFN) signaling pathway was enriched only in the upregulated gene set from PK15A cells. This included the well-characterized interferon-stimulated genes (ISG) consisting of the interferon-induced protein with tetratricopeptide repeat 1 and 3 genes (IFIT1 and IFIT3, respectively), the myxovirus resistance 2 gene (MX2), the 2′-5′-oligoadenylate synthetase 1 and 3 genes (OAS1 and OAS3, respectively), and interferon-stimulated exonuclease gene 20 kDa (ISG20), along with chemokine genes: the chemokine (C-C motif) ligand 20, 22, and 25 genes (CCL20, CCL22, and CCL25, respectively) and the chemokine (C-X-C motif) ligand gene (CXCL2). The upregulation of interferon genes in PK15A cells was somewhat surprising, given our understanding that EBOV generally inhibits interferon responses through its VP35 and VP24 proteins.
Comparison of interferon responses across cell lines.
As described above, PK15A cells demonstrated a robust upregulation of genes involved in the type I interferon signaling pathway. This response was not observed in either HEK293T or EhKiT cells (Fig. 5A). Given that some cell lines may lose their ability to activate IFN pathways, we next compared the IFN production and signaling capacity of PK15A, HEK293T, and EhKiT cells following poly(I·C) and universal type I IFN stimulation. The expression of IFNB1 and IFIT1 mRNA was compared between the cell lines at 2, 4, and 6 h posttreatment. While we found that all three cell lines had the ability to produce IFNB1 mRNA following poly(I·C) transfection, the magnitude of IFNB1 production was vastly different between the cell lines (Fig. 5B). PK15A cells produced significantly more IFNB1 than EhKiT cells and HEK293T cells. In line with this finding, IFN signaling (measured as IFIT1 mRNA expression) following poly(I·C) transfection was also significantly greater in PK15A cells than in the cells of the other two cell lines (Fig. 5C). As expected, the stimulation of cells with universal type I interferon did not induce IFNB1 mRNA in any cell type (Fig. 5D). However, universal type I interferon stimulation did induce IFIT1 mRNA expression across all three cell lines at a similar level (Fig. 5E). Taken together, these findings suggest that while the capacity and magnitude of type I IFN signaling are similar across the cell lines, IFN production is considerably greater in PK15A cells than in HEK293T and EhKiT cells.
FIG 5.

Comparison of interferon response across cell lines. (A) Heatmap representation of individual genes contributing to the enriched GO term cytokine-mediated signaling pathway (GO:0019221). Each tile represents the mean log2 fold change in regulation from three independent biological replicates. Gray tiles represent genes that had no human ortholog or had an infinity fold change (i.e., no reads at 0 hpi). (B and C) Relative IFNB1 (B) and IFIT1 (C) mRNA expression (as the log2 fold change) for HEK293T, PK15A, and EhKiT cells following transfection with poly(I·C) for 2, 4, and 6 h. The mean ± standard error from three biological replicates is presented, with each replicate being performed as a duplicate PCR. (D and E) Relative IFNB1 (D) and IFIT1 (E) mRNA expression (as the log2 fold change) for HEK293T, PK15A, and EhKiT cells following treatment with type I universal interferon (IFN). The mean ± standard error from three biological replicates is presented, with each replicate being performed as a duplicate PCR.
Overrepresentation of TFBS.
The transcriptional response of a cell is largely mediated through the activity of specific transcription factors, thereby relaying extracellular signals to changes in gene expression. To identify which transcription factors are the most active following EBOV infection, we examined whether there was an overrepresentation of transcription factor binding sites (TFBS) in the genes upregulated in EBOV-infected cells compared to nondifferentially expressed background genes. Using oPOSSUM (version 3.0) single-site analysis (37, 38), we found that the most highly overrepresented TFBS in the upregulated gene sets from HEK293T cells were targets for specificity protein 1 (SP1) and activator protein 1 (AP1), the levels of regulation of both of which were greater than 1 standard deviation of the mean for all genes with overrepresentation of TFBS on the basis of the Z-score and Fisher score (Fig. 6A). Parallel analysis of the PK15A cell data set also demonstrated an overrepresentation of TFBS in the gene for AP1 within the upregulated genes (Fig. 6B). In contrast, the most overrepresented TFBS among the genes upregulated in EhKiT cells were those targeted by REL (an NF-κB family member) and SPIB (Fig. 6C).
FIG 6.

Transcription factor binding site (TFBS) analysis of upregulated gene sets. The overrepresentation of TFBS in the genes upregulated in HEK293T (A), PK15A (B), and EhKiT (C) at 24 hpi is indicated. The significance of enriched TFBS was assessed by combining the Z-score and the Fisher score. Dashed lines represent the mean + 1 standard deviation of the Z-score and the Fisher score for all TFBS. The AP1 transcription factor is circled in red.
Upregulation of AP1 transcription factor complex members.
Above we demonstrated an overrepresentation of TFBS in AP1 among the genes upregulated in HEK293T and PK15A cells. The AP1 transcription factor complex, formed through a noncovalent dimerization between FOS and JUN family members, plays a critical role in promoting the expression of genes involved in important biological processes, such as cell proliferation, differentiation, and apoptosis (28). Indeed, previous studies have demonstrated that AP1 promotes the expression of genes involved in extracellular matrix organization, angiogenesis, and the coagulation cascade (31–33). Our GO enrichment analysis revealed that many genes associated with these biological processes were upregulated across the cell lines (Fig. 3C). Furthermore, TFBS analysis (described above) showed that many of the genes contributing to angiogenesis (Fig. 4A), extracellular matrix organization (Fig. 4B), and the regulation of coagulation (Fig. 4C) had TFBS in AP1. This finding suggests a link between the activity of AP1 and the transcriptional landscape associated with EBOV infection. With this in mind, we focused our attention on the role of AP1 in EBOV and, in particular, on the transcription, translation, and regulation of AP1 activity. Due to the paucity of reagents for pig and bat cells, we concentrated these efforts on the human cells.
RNA-seq analysis demonstrated that the expression of both FOS and JUN mRNAs was highly upregulated in HEK293T and PK15A cells and, to a lesser degree, in EhKiT cells (Fig. 4A). In fact, FOS was one of only six genes that were upregulated in all cell lines (Fig. 3A). It is important to note that growth factors which are known to promote the expression of FOS mRNA, such as PDGFA/B (24), were also upregulated in cells of the HEK293T cell line (Fig. 4B).
Next we examined whether FOS and JUN mRNAs were also upregulated in HEK293T cells and cells of other human cell lines, including A549 and HeLa cells, following EBOV infection using quantitative PCR (qPCR). Previous studies have shown that both JUN and FOS mRNAs are upregulated in human Huh7 cells following EBOV infection (39). A549, HeLa, and HEK293T cells were infected with EBOV (MOI = 5) as described above for cells of the other cell lines. Using real-time quantitative PCR, we found that FOS was significantly upregulated in HEK293T, A549, and HeLa cells at 48 hpi (Fig. 7). FOS was also significantly upregulated at 24 hpi in HEK293T and HeLa cells but not A549 cells. Interestingly, we observed the upregulation of JUN in HEK293T cells only at 48 hpi. No significant upregulation of JUN was seen in either A549 or HeLa cells (Fig. 7). It is interesting to note that in HEK293T cells the differential expression of FOS and JUN determined by RNA-seq appeared to be higher than that determined by quantitative PCR at the 24-h time point. Nevertheless, the PCR analysis of the three human cells lines examined here confirms that FOS is upregulated across diverse human cell lines in response to EBOV.
FIG 7.

Relative expression of FOS and JUN in A549, HeLa, and HEK293T cells infected with EBOV. The mean fold change in expression ± standard error from three biological independent replicates is presented, with each replicate being performed as a duplicate PCR. *, P < 0.05; **, P < 0.01.
EBOV induces the phosphorylation of FOS and JUN.
In addition to the increased expression of mRNA for AP1 components FOS and JUN, the ability of FOS-JUN heterodimers to transactivate AP1-dependent gene expression is further regulated by the phosphorylation of individual FOS and JUN proteins. Indeed, the phosphorylation of FOS on Ser32 and Thr232 by extracellular signal-regulated kinases (ERK) is required for protein stability and nuclear localization (40). Similarly, the transcriptional activity of JUN is regulated by phosphorylation on Ser63 and Ser73 by c-Jun N-terminal kinases (JNK) (41). Using antibodies specific for both total and phosphorylated FOS (phos-FOS) and phosphorylated JUN (phos-JUN), we examined the expression of these proteins at 24 and 48 h following EBOV infection of HEK293T cells using Western blotting. As a positive control, HEK293T cells were first stimulated with phorbol 12-myristate 13-acetate (PMA) for 6 h, and the abundance of total and phosphorylated FOS and JUN was detected by Western blotting (Fig. 8A). PMA induced the expression of both FOS and phos-FOS, but FOS and phos-FOS were not detected in dimethyl sulfoxide (DMSO)-treated control cells. Unlike FOS, JUN and phos-JUN could be detected in unstimulated (DMSO-treated) cells. The total FOS amount appeared to be below the level of detection at 0, 24, and 48 hpi; however, an increase in the level of expression of phos-FOS was observed at 48 hpi (Fig. 8A). In contrast to FOS expression, the expression of total JUN was detected in both uninfected and EBOV-infected cells (Fig. 8A). The expression of both total JUN and phos-JUN increased with time at 24 and 48 hpi in EBOV-infected HEK293T cells (Fig. 8A).
FIG 8.
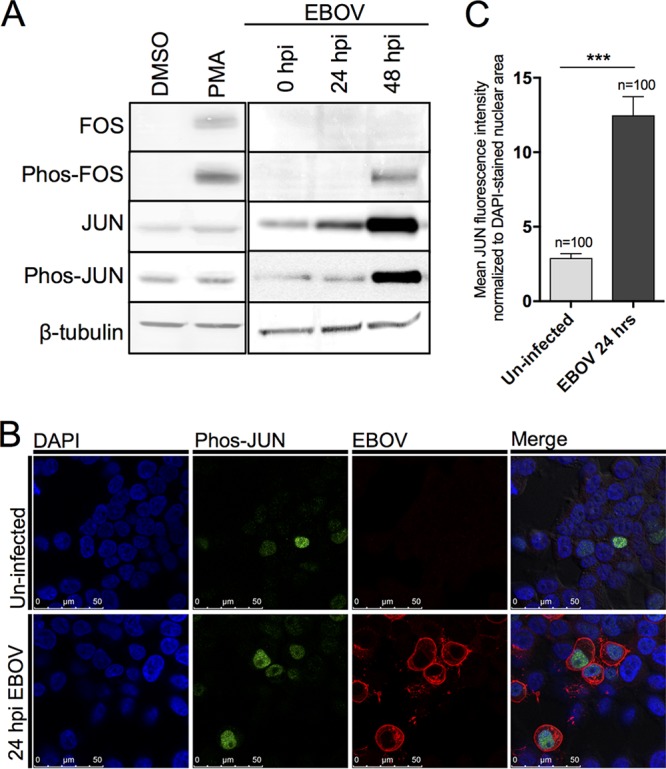
Upregulation and nuclear localization of FOS and JUN in HEK293T cells. (A) Expression of the total and phosphorylated forms of the FOS and JUN proteins was assessed by Western blotting. β-Tubulin was used as a loading control. PMA stimulation was used as a positive control. Western blots were performed from independent biological triplicates, with a representative image being shown here. (B) The nuclear abundance of phosphorylated JUN (phos-JUN) following EBOV infection for 24 hpi was assessed by immunofluorescence microscopy using specific antibodies. EBOV was detected using antiserum against the EBOV GP protein, and cell nuclei were stained with DAPI. (C) The nuclear abundance of phosphorylated JUN was determined by quantifying the fluorescence of phos-JUN (green) within the nuclei (blue) of EBOV-infected (red) or randomly chosen uninfected control cells. This quantification was performed on 100 infected cells and 100 control cells across at least five fields of view in two independent biological replicates. ***, P < 0.001.
EBOV-infected cells translocate phosphorylated JUN to the nucleus.
In order to activate AP1-dependent gene expression, phosphorylated FOS and JUN must translocate to the nucleus, where nuclear abundance correlates with AP1 target gene expression. With this in mind, we examined the nuclear accumulation of phos-FOS and phos-JUN following EBOV infection. Immunofluorescence microscopy revealed a significant increase in the nuclear abundance of phos-JUN in EBOV-infected cells compared to that in uninfected cells (Fig. 8B). Next we accurately quantified the abundance of phos-JUN within infected (n = 100) and uninfected (n = 100) cells by normalizing the phos-JUN fluorescence to that of the DAPI (4′,6-diamidino-2-phenylindole dihydrochloride)-stained area in the nucleus. Infected cells had an approximately 5-fold increase in mean fluorescence intensity compared to uninfected cells (Fig. 8C). In contrast to phos-JUN, immunofluorescence microscopy could not detect phos-FOS in either uninfected or infected cells (data not shown). This finding agrees with the Western blot results (Fig. 8A), where phos-FOS could be detected only at 48 hpi or by PMA stimulation.
AP1 transactivation in HEK293T cells.
It is generally accepted that increased levels of the AP1 transcription factor complex increase the level of transactivation of AP1 target gene expression. Given the significant upregulation of both FOS and JUN mRNA and JUN protein and the nuclear accumulation of phosphorylated JUN, we next examined whether EBOV infection increases AP1 transactivation activity via a dual-luciferase reporter assay. Compared to the AP1 activity in uninfected control cells, a significant increase in AP1 activity was observed in EBOV-infected cells at 24, 48, and 72 hpi (Fig. 9). The AP1 activity increased over the time course, which aligns with the increased expression of phosphorylated JUN and FOS described above.
FIG 9.
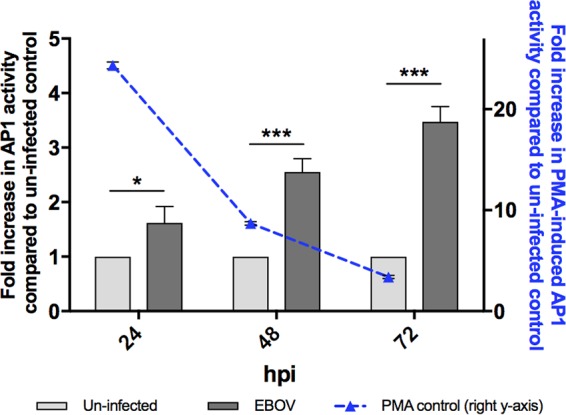
AP1 activity is induced by EBOV in HEK293T cells. The transactivation activity of AP1 was assessed following EBOV infection for 24, 48, and 72 hpi using a dual-luciferase reporter assay in HEK293T cells. Assays with PMA-induced controls (50 ng/ml) were performed at each time point, and the results are represented on the right y axis. The mean fold increase (relative to that for the uninfected control) ± standard error from three independent biological replicates is presented, with each replicate being performed as a duplicate luciferase assay. *, P < 0.05; ***, P < 0.001.
Knockdown of JUN mRNA reduces EBOV-induced expression of CYR61.
Next we examined the role of AP1 components FOS and JUN in promotion of the expression of AP1 target genes in response to EBOV infection in HEK293T cells. Small interfering RNAs (siRNAs) targeting human FOS and JUN and a nontargeting siRNA control were individually transfected into HEK293T cells, and the levels of expression of FOS and JUN mRNAs relative to the level of expression of the nontargeting control siRNA were determined by qPCR. The siRNAs reduced the expression of JUN and FOS to approximately 35 and 50% of their original expression, respectively (Fig. 10A). Despite the use of optimization experiments, we could not increase the knockdown efficiency of either siRNA. Next we examined the EBOV-induced expression of AP1 target genes CYR61 and ATF3 following siRNA knockdown of JUN and FOS. The knockdown of FOS did not have any significant effect on the EBOV-induced expression of either CYR61 or ATF3 (Fig. 10B and C). However, JUN knockdown significantly reduced the expression of CYR61 at 24 h following EBOV infection (Fig. 10B). JUN knockdown also reduced the expression of ATF3 following EBOV infection; however, the difference was not statistically significant (Fig. 10C). Taken together, these results suggest that JUN may play a role in promoting the expression of the AP1 target genes which are upregulated following EBOV infection in HEK293T cells.
FIG 10.

siRNA knockdown of FOS and JUN. (A) Percent relative expression of FOS and JUN mRNA following transfection with siRNAs targeting their respective genes. The level of expression (relative to that of nontargeting control siRNA) was normalized to the level of GAPDH expression. (B and C) The fold change in CYR61 (B) and ATF3 (C) expression at 24 h following EBOV infection for cells transfected with nontargeting control siRNA or siRNAs targeting FOS and JUN. For all panels, the mean ± standard error from three independent biological replicates is presented, with each replicate being performed as a duplicate PCR. *, P < 0.05.
Purified EBOV also induces AP1.
Given that growth factors and cytokines which are known to promote AP1 expression may be present within the viral inoculum (derived from the cell culture supernatant), we next examined if purified EBOV also induces the expression of AP1 components and target genes within HEK293T cells. All our previous experiments described above used cell culture supernatant as the inoculum. For these experiments, the cells were infected with purified EBOV (MOI = 5) and the relative expression of FOS, JUN, ATF3, and CYR61 mRNA was examined at 24 and 48 hpi. The expression of total and phosphorylated JUN was also examined at these time points by Western blotting. Compared to their expression in uninfected control cells (and normalized to the expression of GAPDH [glyceraldehyde-3-phosphate dehydrogenase] mRNA), the expression of FOS, ATF3, and CYR61 mRNAs was most highly upregulated at 48 hpi and, to a lesser degree, at 24 hpi (Fig. 11A). The upregulation of these genes appeared to be delayed compared to the earlier results obtained using a nonpurified EBOV inoculum. While only a modest upregulation of JUN mRNA was observed at 48 hpi (Fig. 11A), we observed increased expression of total and phosphorylated JUN protein at 48 hpi (Fig. 11B). This finding was similar to the expression observed for the JUN protein using nonpurified virus.
FIG 11.

Response of HEK293T cells to purified EBOV. (A) Relative expression of FOS, JUN, ATF3, and CYR61 mRNA following infection by purified EBOV for 24 and 48 h. The mean ± standard error from three independent biological replicates is presented, with each replicate being performed as a duplicate PCR. (B) The levels of expression of total and phosphorylated forms of the JUN protein were assessed by Western blotting. β-Tubulin was used as a loading control. Western blot analysis was performed in duplicate from two independent experiments, with a representative image being shown here. *, P < 0.05; ***, P < 0.001.
DISCUSSION
While this study was largely comparative in nature, we focused much of our downstream attention on examining the role of AP1 in EBOV infection of human cells. We chose to focus on AP1 because the gene for its major component, FOS, was upregulated in all cell lines, albeit to a lesser degree in bat cells, and it therefore likely represents a broadly relevant response that is not species specific. Furthermore, as a transcription factor, AP1 acts as a central mediator promoting the expression of downstream pathways, many of which may have biological relevance to EVD. The comparative aspects of this study, however, should not be ignored and will form the basis of other hypothesis-driven studies in the future. Indeed, while all three cell lines were permissive to EBOV infection, they also displayed significant differences in their ability to support EBOV replication. Moreover, the transcriptional response of the cell lines was vastly different. Indeed, the pig (PK15A) and bat (EhKiT) cells responded to EBOV through the upregulation of immune, inflammatory, and coagulation pathways. Such responses were not seen in the human (HEK293T) cells.
During the course of this study, Ng and coworkers published the results of a study, which, like our data, showed that cells from E. helvum were largely refractory to infection by EBOV (42). Their study went on to show that the reduced permissiveness of these cells was caused by a mutation in the EBOV entry receptor, Niemann-Pick C1 (NPC1). It is interesting to note that despite the fact that EhKiT cells are largely refractory to infection due to a mutation in NPC1, they still up- and downregulated more transcripts at 6 hpi than the more susceptible cell lines. In our study, a small increase in the amount of viral mRNA was observed between 6 and 24 hpi. Furthermore, we observed a minor increase in virus replication between 24 and 72 hpi. While it is possible that the virus titer at 24 hpi may, in part, represent the inoculum, the increased replication and transcription of EBOV after 24 h suggest that E. helvum may not be completely refractory to infection.
The transcriptional response of a cell is largely dictated by the activity of specific transcription factors. As described above, the significant upregulation of FOS and JUN mRNAs, notably, in human and pig cells, initially implied that AP1 might be activated in EBOV-infected cells. Through a combination of bioinformatic approaches, we confirmed that the transcriptional response of EBOV-infected cells largely reflects the increased activity of the AP1 transcription factor, particularly within the human and pig cells. A recent study examining the host response of human Huh7 cells to EBOV and Marburg virus (MARV) also found enrichment for TFBS in AP1 among the upregulated genes (39), thus demonstrating that the increased activity of AP1 may be broadly relevant to filoviruses other than EBOV. Using functional studies, we showed that the AP1 components FOS and JUN were translated, phosphorylated, and translocated into the nucleus (JUN only) within EBOV-infected cells. Despite the rapid upregulation of FOS and JUN mRNAs, we found a lag in the phosphorylation of the FOS and JUN proteins. While the sensitivity of the antibodies to detect the phosphorylated proteins may have affected this delay (as seen in the phos-FOS Western blot), it was also possible that the lower expression of mitogen-activated protein kinase pathways (responsible for the phosphorylation of AP1 components) may have contributed to this delay. In line with the upregulation of phosphorylated FOS and JUN at 48 hpi, a significant increase in the transactivation activity of AP1 was demonstrated at 48 and 72 h following EBOV infection in human cells. While it is possible that the expression/activation of AP1 components increased in response to increasing viral replication and cell stress, a positive-feedback loop of AP1 may have also contributed to the high level of AP1 activity observed at 72 hpi. Indeed, both FOS and JUN are targets themselves for the AP1 transcription factor and therefore would be expected to further promote the AP1 response as the infection progressed. It is important to acknowledge that other studies have also reported the upregulation of both JUN (39, 43) and FOS (20, 39) in human Huh7 cells infected with EBOV. The increased expression and activity of AP1 have also been reported in human cells infected with other viruses, including influenza virus (44), herpesvirus (45), and hepatitis C virus (46).
In human cells, AP1 is induced by a variety of extracellular stimuli, including cytokines, stress, UV, and growth factors (29). In the context of EBOV, growth factors may be of particular relevance. Kindrachuk and coworkers (20) demonstrated the upregulation of both TGF-β and VEGF signaling at 24 hpi in EBOV-infected Huh7 cells. The broad gene expression data generated in our study also highlight the importance of growth factor signaling. Indeed, the upregulation of numerous growth factors, including that of members of the TGF, PDGF, VEGF, connective tissue growth factor (CTGF), and fibroblast growth factor (FGF) families, was observed. The infection experiments for our RNA-seq analyses were performed using (Vero) cell culture supernatant as the inoculum. Given that this inoculum could contain various growth factors, it was important for us to demonstrate that purified EBOV also induces the expression of FOS and JUN. These experiments showed that the upregulation of FOS and JUN was delayed when cells were infected with purified EBOV. This finding suggests that growth factors within the inoculum may increase the AP1 response following EBOV infection. However, in this context it is also important to note that JUN phosphorylation was more abundant within the nucleus of EBOV-infected cells (compared to the nucleus of noninfected cells; Fig. 8B), thus demonstrating that EBOV infection is required for AP1 activation.
Turning our attention to the role that AP1 target genes may have on the pathophysiology of EBOV, it is interesting to note that many of the AP1 target genes found to be upregulated have important biological roles in vascular development (including angiogenesis), extracellular matrix organization, and coagulation. CYR61 is an AP1 target gene of particular note, as secreted extracellular matrix signaling protein CYR61 can directly impact the mesenchymal transition by downregulating epithelial markers (i.e., E-cadherin), upregulating mesenchymal markers (i.e., FN1), and stimulating cell migration (47). In parallel with the changes in the extracellular matrix, EBOV-infected cells upregulate genes associated with vascular remodeling and angiogenesis, including matricellular proteins (CYR61, thrombospondin 1 [THBS1], nephroblastoma overexpressed [NOV]) and cell adhesion proteins (neural cell adhesion molecule [NRCAM]). Given that increased vascular permeability through destruction of the endothelial barrier is a common feature of EBOV pathology, the upregulation of these genes may contribute.
To determine if EBOV-induced host gene expression such as that described above is influenced by AP1 activity, we examined the effect of knocking down the genes for AP1 components FOS and JUN on the expression of two EBOV-induced AP1 target genes, CYR61 and ATF3. While the knockdown of FOS had no effect on either gene, the knockdown of JUN significant reduced the expression of CYR61 and, to a lesser degree, ATF3. It should be noted that the siRNAs were more effective at knocking down JUN than at knocking down FOS, and this could have impacted the results described above. Nevertheless, these experiments do show that AP1 plays a functional role in promoting EBOV-induced host gene expression.
While the upregulation of AP1 components was the major focus of our study, other responses observed across the cell lines are also of interest. The robust interferon response of the PK15A cells was unexpected. In many cell types, EBOV is particularly effective at evading immune system processes (recently reviewed in reference 48), including blocking interferon production and signaling (49). Further experiments showed that PK15A cells upregulated IFNB1 mRNA significantly more than HEK293T and EhKiT cells in response to the viral mimic poly(I·C). This superior interferon production response of PK15A cells may have allowed these cells to partially overcome the antagonistic effects of EBOV VP35 on the interferon production pathway. However, it should be acknowledged that despite the robust interferon response observed in EBOV-infected PK15A cells, a high level of EBOV replication was still observed in these cells.
Viral transcription was higher in PK15A cells than HEK293T cells. Using Western blotting, we also showed that the translation of EBOV NP was higher in PK15A cells. However, in contrast to the viral transcription and translation results, the replication of infectious EBOV was the highest in HEK293T cells. Given the strong interferon-stimulated gene (ISG) response observed for the PK15A cells, we propose that the viral replication processes (i.e., assembly and budding) within PK15A cells may have been reduced by the host ISG response. Indeed, the expression of IFIT1, an ISG that can act to inhibit viral replication by sequestering specific viral nucleic acids (50), was up in PK15A cells alone. Other ISGs that can affect viral replication include OAS1/2/3 and GBP1, all of which were upregulated in PK15A cells but not HEK293T cells. Further research is required to fully elucidate the interferon response of PK15A cells to EBOV in the future and the relationship between the response of PK15A cells and the host response of pigs to EBOV infection.
In conclusion, we report the results of a comparative transcriptomic analysis of the response of three taxonomically diverse cell lines to EBOV. It should be acknowledged that as an in vitro-based study, the responses observed across the three cell lines need to be further examined in whole tissues and other cell types. Indeed, even within our study we observed variation in the upregulation of the AP1 components in different human cell lines (such as the HEK293T, HeLa, and A549 cell lines). Nevertheless, examination of the responses of a single cell line can provide valuable information concerning how cell type-specific responses (such as the interferon response reported in PK15A cells) can contribute to differences in viral cell and tissue tropism. When the results of this study are taken together, our study highlights the importance of the AP1 transcription factor in the host response to EBOV across different cell types. Further studies should now be directed to understand how this transcription factor promotes the host response within an in vivo system.
MATERIALS AND METHODS
Cell culture.
The transcriptome response to EBOV was investigated in three cell lines derived from human, pig, and the African straw-colored fruit bat (E. helvum). Serum antibodies against EBOV and MARV have previously been detected in a number of African fruit bats, including E. helvum (51, 52). We chose to examine kidney-derived immortalized cells from human (HEK293T; source, Peter MacCallum Cancer Centre, Melbourne, Australia), pig (PK15A; source, National Animal Disease Center, Ames, IA), and bat (EhKiT; source, CSIRO Culture Collection, Geelong, Australia). All kidney-derived cell lines had been immortalized with the simian virus 40 antigen (53). Cells were cultured in either Dulbecco modified Eagle medium (DMEM)–Ham's F-12 medium (EhKiT cells), DMEM (HEK293T cells), or minimal essential medium (MEM) (PK15A cells) (Pierce) containing 10% fetal calf serum (FCS) and 10 mM HEPES. In addition, human-derived HeLa cells (cervical cancer cells; source, ATCC, Manassas, VA) and A549 cells (lung epithelial cells; source, European Collection of Authenticated Cell Cultures, Salisbury, United Kingdom) were also used in this study. HeLa and A549 cells were cultured in MEM (Gibco) containing 10 mM HEPES and 10% FCS. All cells were cultured at 37°C.
Virus infection for RNA-seq analysis.
All virus infections were performed within a biosafety level 4 (BSL-4) laboratory at the Australian Animal Health Laboratory. For RNA-seq experiments, approximately 6 × 106 EhKiT, HEK293T, and PK15A cells were either mock infected (0 hpi) or infected with EBOV (Mayinga/1976 strain; GenBank accession number NC_002549.1) for 6 or 24 h at a multiplicity of infection (MOI) of 5. All infection experiments for RNA-seq analysis were conducted as independent triplicates. To infect the cells, culture medium was removed, the virus inoculum was added to the monolayer, and the monolayer was incubated at 37°C for 30 min. After this time, the virus inoculum was removed and replaced with fresh culture medium. The experiment was conducted in triplicate in 25-cm2 flasks. Cells were harvested by trypsinization, and the pellets were resuspended in RLT buffer (Qiagen) containing 1% β-mercaptoethanol. During preliminary analysis of the transcriptome data, we observed a small number of reads with sequence homology to the Mycoplasma hyorhinis genome within the samples obtained at 6 and 24 h. The virus stocks were subsequently treated with ciprofloxacin, and new virus stocks were generated in Vero E6 cells. A specific PCR was used to confirm that the new virus stocks were free of Mycoplasma contamination (data not shown). All infection experiments following the initial RNA-seq analysis were performed using the Mycoplasma-free virus stock.
Virus titrations.
To quantify virus replication across the three cell lines, additional infection experiments were performed in 96-well plates. Approximately 60,000 cells/well were seeded and infected with EBOV Mayinga/1976 for 24, 48, and 72 h at an MOI of 5. Each infection was performed in duplicate as described above. Virus titration was performed as an 8-fold 1:10 dilution, of which 25 μl was added to 2 × 104 Vero E6 cells/well in a 96-well plate in quadruplicate. The plates were incubated for 5 days and then fixed in 10% neutral buffered formalin for 48 h and stained using an in-house-prepared rabbit anti-Reston EBOV NP antibody and an Alexa Fluor 488-labeled goat anti-rabbit immunoglobulin secondary antibody (Life Technologies). Infected cells were visualized using an Evos microscope, and titers were determined using the Reed-Muench calculation (54).
Purification of EBOV and infection.
In order to generate purified virus stocks, two T-150 flasks of confluent Vero E6 cells were infected with Ebola Zaire Kikwit-95 at an MOI of 0.005 and maintained for 7 days in a 37°C incubator with 5% CO2. After the incubation, the supernatant was transferred to 50-ml tubes and the tubes were centrifuged at 4,000 rpm for 10 min at 4°C to remove cell debris. The clarified supernatant was transferred to new 50-ml tubes and the tubes were kept on ice while ultracentrifuge tubes were prepared. One milliliter of a filter-sterilized (pore size, 0.22-μm) 60% sucrose solution was added to the bottom of six 17-ml Beckman Coulter Ultra-Clear ultracentrifuge tube (Beckman Coulter, Australia), followed by addition of 2 ml of filter-sterilized (pore size, 0.22-μm) 20% sucrose solution and, finally, 14 ml of clarified supernatant. The samples were ultracentrifuged at 76,700 × g for 2 h at 4°C using a Beckman Coulter Optima L-100 XP ultracentrifuge. After the 2-h spin, the top 15 ml of medium–20% sucrose solution was removed, being careful not to disturb the 60%-20% sucrose solution interface. Once the unwanted supernatant had been removed, 1 ml of the 60%-20% sucrose interface from each centrifuge tube was removed and the 60%-20% sucrose interfaces from each of the centrifuge tubes were pooled, diluted in 40 ml phosphate-buffered saline (PBS), and temporarily stored on ice. Three fresh ultracentrifuge tubes were prepared by adding 2 ml of filter-sterilized (pore size, 0.22-μm) 20% sucrose solution to the bottom of the tube, followed by addition of 15 ml of the PBS-diluted virus preparation. The tubes were ultracentrifuged again at 76,700 × g for 2 h at 4°C. After the centrifugation, all the liquid was carefully removed from each tube, while ensuring that the virus pellet was not disturbed. The pellets from each tube were resuspended in 5 ml PBS, pooled, and aliquoted into 500-μl aliquots. These aliquots were titrated by the 50% tissue culture infective dose (TCID50) assay (as described above) and stored at −80°C until needed.
HEK293T cells were infected with purified EBOV as described above using an MOI of 5 for 24 and 48 h. Monolayers were harvested in RLT buffer (for gene expression analysis) or 5% SDS (for Western blot analysis). Noninfected HEK293T cells served as the control. The infection of cells was performed as independent triplicate experiments.
RNA isolation and sequencing.
Total RNA was isolated from cells in RLT buffer using an RNeasy kit (Qiagen) with DNase I treatment per the manufacturer's instructions. RNA isolation was performed once in the BSL-4 facility, and then the resulting RNA was resuspended in RLT buffer and reisolated in a BSL-2 facility. The quality and quantity of RNA were assessed for all samples using a Bioanalyzer system (Agilent). mRNA was sequenced as 100-bp paired-end reads across three lanes on a HiSeq 2000 sequencer (Illumina) by the Australian Genome Research Facility, Parkville, Victoria, Australia. The resulting reads were trimmed for adapters, and quality was assessed using the FastQC tool.
Read mapping and differential expression testing.
Sequence reads were first trimmed for adapter sequences, and poor-quality reads were removed or trimmed (see Table S1 in the supplemental material). For each sample, forward and reverse reads were mapped against the E. helvum (GenBank assembly accession number GCA_000465285.1), human (hg19), or pig (susScr3) genome with the Bowtie2 (version 2.2.2)/TopHat (version 2.0.13) program (55). A maximum of 2 nucleotide mismatches per read was tolerated. Using the alignment outputs (BAM files), transcripts were assembled for each sample and merged into a single transcriptome for each species using the Cufflinks/Cuffmerge (version 2.1.1) program (56). Differential expression testing was performed for each cell line by comparing the level of expression at 0 hpi with that at 6 and 24 hpi using the Cuffdiff (version 2.1.1) program. Only genes with adjusted P values of <0.05 and a fold change in regulation of ≥2 were considered significantly differentially expressed.
Forward and reverse reads for each sample obtained at 6 and 24 hpi were also mapped against the EBOV genome (GenBank accession number NC_002549.1) using the Bowtie2 (version 2.2.2) program. The resulting BAM files were converted to SAM files, sorted, and indexed using the SAMtools (version 0.1.18) program. To quantify EBOV genome coverage, BAM files were converted to BED files, and coverage was determined using the genomeCoverageBed command in BEDTools (version 2.19.1) software. To account for differences in the total number of reads between samples, we normalized the EBOV per base coverage to the total number of reads mapped to the host genome. The average coverage per million mapped reads was then calculated across the three replicates for each cell line.
Comparison of human, pig, and bat data sets.
To compare the transcriptome responses between the three species, we first identified orthologous genes between human and bat and between human and pig. To identify orthologs, the entire EhKiT and PK15A cell Cufflinks-assembled transcriptomes were compared to the HEK293T cell Cufflinks-assembled transcriptome using BLASTn analysis. Only EhKiT and PK15A cell transcripts with hits of <e−2 to HEK293T cell transcripts were considered orthologs. Next, EhKiT and PK15A cell transcripts with human orthologs were annotated with gene names according to their human ortholog.
Gene Ontology and TF enrichment analysis.
Official gene identifiers were retrieved for all significantly differentially expressed transcripts. Gene identifiers for transcripts that were significantly upregulated at one or more time points were combined into a single list. A list of significantly downregulated gene identifiers was also compiled. Biological process GO enrichment was performed separately on the up- and downregulated transcript lists using the unranked target and background analysis in the GOrilla tool (36). Background gene lists for each cell line were compiled by retrieving all gene identifiers from the reference assembled transcriptomes for each species. The strength of the enrichment was calculated as an enrichment score with the GOrilla tool (36).
We identified transcription factor binding sites (TFBS) in upregulated human, pig, and bat genes using oPOSSUM (version 3.0) human single site analysis (37, 38). We examined whether there was an overrepresentation of TFBS 5,000 bp upstream and/or downstream of the start codon of genes upregulated in the cells (at 6 and/or 24 h) compared to the representation for nondifferentially expressed background genes. The strength of enrichment of TFBS was determined using the Fisher score and Z-score, both of which were obtained in oPOSSUM (37, 38).
Poly(I·C) transfection and universal interferon treatments.
The interferon responses of PK15A, HEK293T, and EhKiT cells were compared following poly(I·C) transfection and universal type I interferon treatment. Cells were seeded at a density of 5 × 104 cells in 96-well microplates and left overnight at 37°C to adhere. For interferon treatments, the cells were stimulated (in independent triplicate experiments) with 1,000 units of universal type I IFN (UIFN; PBL Assay Science) in 100 μl of serum-free medium Control cells were mock stimulated with serum-free medium only. Poly(I·C) (InvivoGen) transfections were performed as previously described (in independent triplicate experiments) (57). Control cells were mock transfected with the Lipofectamine 2000 reagent only. For both poly(I·C) and interferon stimulation, cells were harvested into RLT lysis buffer (Qiagen) at 2, 4, and 6 h posttreatment.
Real-time quantitative PCR.
RNA was isolated from cells lysed in RLT lysis buffer as previously described (58). Primers for SYBR green reactions were designed using Primer3 (Table S4). Reaction parameters were identical for all genes. A two-step SYBR green reaction was performed. Total RNA was reverse transcribed using SuperScript III reverse transcriptase (Life Technologies) that had been primed with oligo(dT) per the manufacturer's instructions. For each independent replicate, duplicate SYBR green real-time PCRs were performed in a 25-μl reaction mixture containing 1× Express SYBR green master mix (Life Technologies), 200 nM forward and reverse primers, and 20 ng of the template. Cycling parameters were 95°C for 10 min and then 40 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 1 min, followed by melt curve analysis. Differential expression was normalized to GAPDH expression and calculated using the relative quantification method as previously described (59). Quantification of the EBOV NP gene was performed using a specific TaqMan PCR as previously described (60).
Western blotting.
Expression of the FOS and JUN proteins by HEK293T cells was assessed using Western blotting. Cells (150,000/well) were seeded into 24-well plates. Cells were either mock infected or infected for 24 h with EBOV at an MOI of 5 as described above. Cells were lysed and harvested in 5% (wt/vol) SDS, boiled for 10 min, subjected to SDS-PAGE, and transferred to a polyvinylidene difluoride membrane. The membranes were blocked with 5% (wt/vol) skim milk for 1 h. The membranes were washed three times (10 min each) with Tris-buffered saline–Tween 20 (TBST) and then incubated for 1 h with primary antibody diluted in TBST. The following antibodies (all of which were obtained from Cell Signaling Technology) were used: c-Fos (antibody number 9F6) rabbit monoclonal antibody (MAb; 1:1,000), phospho-c-Fos (Ser32) (antibody number D82C12) XP rabbit MAb (1:1,000), c-Jun (antibody number 60A8) rabbit MAb (1:1,000), phospho-c-Jun (Ser73) (antibody number D47G9) XP rabbit MAb (1:1,000), and β-tubulin (9F3) rabbit MAb (1:1,000). For the immunodetection of viral nucleoprotein, we used rabbit polyclonal anti-Reston EBOV NP antiserum (1:5,000; CSIRO). Membranes were washed three times with TBST and then incubated for 1 h with horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin secondary antibody (Bio-Rad) diluted in TBST. The membranes were finally washed twice with TBST and once with Tris-buffered saline and developed with ECL Plus chemiluminescence substrate (Thermo) per the manufacturer's instructions. The membranes were scanned at 473 nm on a Typhoon FLA9000 gel imaging scanner (GE Healthcare). All Western blots were performed in duplicate, with representative images being presented.
Immunofluorescence microscopy.
HEK293T cells (150,000 cells/well) were seeded onto 13-mm-diameter coverslips in 24-well plates. Cells were either mock infected or infected with EBOV for 24 h at an MOI of 5 as described above. Following infection, medium was removed and cells were fixed in 4% (wt/vol) paraformaldehyde–PBS for 48 h. The coverslips were then washed three times in PBS, permeabilized with 0.1% (wt/vol) Triton X-100 for 10 min, blocked with 0.5% (wt/vol) bovine serum albumin (BSA) for 30 min, and incubated with either phospho-c-Fos (Ser32) (D82C12) XP rabbit MAb (1:800) or phospho-c-Jun (Ser73) (D47G9) XP rabbit MAb (1:800) in combination with anti-EBOV GP serum (1:500) (CSIRO) in 0.5% (wt/vol) BSA for 1 h. The cells were washed three times with PBS and treated with Alexa Fluor 488-labeled (1:200) and Alexa Fluor 586-labeled secondary antibodies (Life Technologies) in 0.5% (wt/vol) BSA for 1 h. Finally, the cells were washed twice with PBS and once with H2O and stained with DAPI (4′,6-diamidino-2-phenylindole dihydrochloride; Life Technologies) for 10 min. The coverslips were washed in PBS, mounted in Vectashield antifade mounting medium (Vector Laboratories), and imaged with a Leica SP5 confocal microscope using sequential scanning between channels. Acquisition parameters were not changed between samples, in order to perform a quantitative comparison of the fluorescence intensity. Image data were collected using Leica LAS AF software (Leica Microsystems, Germany).
Image analysis.
Image analysis and quantification of the nuclear fluorescence on confocal microscopy images were performed using NIH ImageJ (version 1.49) software. For each image, the individual fluorescent channels were loaded separately and merged into a stack. The nuclear area (identified by the DAPI fluorescence) was then traced manually for all cells within the image. This was repeated for at least five independent images to obtain quantitative data on over 100 cells. The nuclear area and the mean fluorescence for each channel were then calculated. The Alexa Fluor 488 channel was normalized over the nuclear area for each cell, and the average Alexa Fluor 488 fluorescence was calculated. To compare the nuclear abundance of phospho-JUN and phospho-FOS in EBOV-infected cells, we compared the average Alexa Fluor 488 fluorescence for the infected cells (identified through Alexa Fluor 586 staining) to that for uninfected control cells. A two-tailed unpaired t test was used to test statistical significance.
AP1 luciferase assay.
To quantify the transactivation activity of AP1 following EBOV infection, a dual-luciferase reporter assay (Promega) was utilized. HEK293T cells (60,000 cells/well) were transfected with the pGL4.44(luc2P/AP1 RE/Hygro) vector, which contains six copies of an AP1 response element (AP1 RE) which drives transcription of the luciferase reporter gene luc2P. Cells were also transfected with control plasmid pGL4.75(hRluc/CMV), which encodes the luciferase reporter gene hRluc. At approximately 24 h posttransfection (hpt), transfected cells were infected with EBOV as described above. At 24, 48, and 72 hpi, the dual-luciferase reporter assay was performed per the manufacturer's instructions. Phorbol 12-myristate 13-acetate (PMA; Promega) is known to induce the AP1 response in human cells and was used as a positive control at 50 ng/ml. All luciferase assays were performed in six replicates, and a two-tailed unpaired t test was used to test statistical significance.
siRNA knockdown.
We examined the effect of JUN and FOS on EBOV-induced host gene expression using siRNAs targeting human FOS and JUN and a nontargeting siRNA control. HEK293T cells were seeded into 96-well plates as described above, and after 24 h, cells were individually transfected with 100 nm On-TargetPlus SMARTpool human JUN (catalog number 3725) and On-TargetPlus SMARTpool human FOS (catalog number 2353) siRNAs (Dharmacon) per the manufacturer's instructions using the DharmaFECT 1 reagent (Dharmacon). Cells were also transfected with an On-TargetPlus nontargeting pool control siRNA exactly as described above. After 24 h of transfection, cells were infected with EBOV at an MOI of 5 for 24 h. Cells were harvested in RLT buffer, and RNA was extracted for PCR as described above. Each siRNA was transfected as three independent biological replicates, and PCR for each replicate was performed as a technical duplicate reaction. A two-tailed unpaired t test was used to test statistical significance.
Accession number(s).
The reads obtained in this study have been deposited in the NCBI Sequence Read Archive and assigned BioProject accession number PRJNA305831.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Wojtek Michalski and Stacey Leech for critical reviews of the manuscript. We also acknowledge assistance from Lawrence Mok, Diane Green, and Catherine Williams with cell culture and confocal microscopy. We also acknowledge use of the facilities of the Australian Microscopy & Microanalysis Research Facility-linked laboratory at the CSIRO Australian Animal Health Laboratory.
J.W.W., G.A.M., and M.L.B. designed the research; J.W.W., S.T., M.T., V.B., R.K., B.S., A.J.M., A.P.W., and A.W.P. performed the research; J.W.W., M.D., M.T., R.K., and V.B. analyzed the data; and J.W.W. and M.L.B. wrote the paper.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01174-17.
REFERENCES
- 1.Feldmann H, Geisbert TW. 2011. Ebola haemorrhagic fever. Lancet 377:849–862. doi: 10.1016/S0140-6736(10)60667-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gatherer D. 2014. The 2014 Ebola virus disease outbreak in West Africa. J Gen Virol 95:1619–1624. doi: 10.1099/vir.0.067199-0. [DOI] [PubMed] [Google Scholar]
- 3.World Health Organization. 2016. Ebola situation report. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 4.Jones KE, Patel NG, Levy MA, Storeygard A, Balk D, Gittleman JL, Daszak P. 2008. Global trends in emerging infectious diseases. Nature 451:990–994. doi: 10.1038/nature06536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swanepoel R, Leman PA, Burt FJ, Zachariades NA, Braack LEO, Ksiazek TG, Rollin PE, Zaki SR, Peters CJ. 1996. Experimental inoculation of plants and animals with Ebola virus. Emerg Infect Dis 2:321–325. doi: 10.3201/eid0204.960407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barrette RW, Metwally SA, Rowland JM, Xu LZ, Zaki SR, Nichol ST, Rollin PE, Towner JS, Shieh WJ, Batten B, Sealy TK, Carrillo C, Moran KE, Bracht AJ, Mayr GA, Sirios-Cruz M, Catbagan DP, Lautner EA, Ksiazek TG, White WR, McIntosh MT. 2009. Discovery of swine as a host for the Reston ebolavirus. Science 325:204–206. doi: 10.1126/science.1172705. [DOI] [PubMed] [Google Scholar]
- 7.Kobinger GP, Leung A, Neufeld J, Richardson JS, Falzarano D, Smith G, Tierney K, Patel A, Weingartl HM. 2011. Replication, pathogenicity, shedding, and transmission of Zaire Ebolavirus in pigs. J Infect Dis 204:200–208. doi: 10.1093/infdis/jir077. [DOI] [PubMed] [Google Scholar]
- 8.Weingartl HM, Embury-Hyatt C, Nfon C, Leung A, Smith G, Kobinger G. 2012. Transmission of Ebola virus from pigs to non-human primates. Sci Rep 2:811. doi: 10.1038/srep00811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gupta M, Mahanty S, Ahmed R, Rollin PE. 2001. Monocyte-derived human macrophages and peripheral blood mononuclear cells infected with Ebola virus secrete MIP-1 alpha and TNF-alpha and inhibit poly-IC-induced IFN-alpha in vitro. Virology 284:20–25. doi: 10.1006/viro.2001.0836. [DOI] [PubMed] [Google Scholar]
- 10.Hensley LE, Young HA, Jahrling PB, Geisbert TW. 2002. Proinflammatory response during Ebola virus infection of primate models: possible involvement of the tumor necrosis factor receptor superfamily. Immunol Lett 80:169–179. doi: 10.1016/S0165-2478(01)00327-3. [DOI] [PubMed] [Google Scholar]
- 11.Stroher U, West E, Bugany H, Klenk HD, Schnittler HJ, Feldmann H. 2001. Infection and activation of monocytes by Marburg and Ebola viruses. J Virol 75:11025–11033. doi: 10.1128/JVI.75.22.11025-11033.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baize S, Leroy EM, Georges AJ, Georges-Courbot MC, Capron M, Bedjabaga I, Lansoud-Soukate J, Mavoungou E. 2002. Inflammatory responses in Ebola virus-infected patients. Clin Exp Immunol 128:163–168. doi: 10.1046/j.1365-2249.2002.01800.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bray M, Geisbert TW. 2005. Ebola virus: the role of macrophages and dendritic cells in the pathogenesis of Ebola hemorrhagic fever. Int J Biochem Cell Biol 37:1560–1566. doi: 10.1016/j.biocel.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 14.Mahanty S, Bray M. 2004. Pathogenesis of filoviral haemorrhagic fevers. Lancet Infect Dis 4:487–498. doi: 10.1016/S1473-3099(04)01103-X. [DOI] [PubMed] [Google Scholar]
- 15.Feldmann H, Bugany H, Mahner F, Klenk HD, Drenckhahn D, Schnittler HJ. 1996. Filovirus-induced endothelial leakage triggered by infected monocytes/macrophages. J Virol 70:2208–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geisbert TW, Hensley LE, Larsen T, Young HA, Reed DS, Geisbert JB, Scott DP, Kagan E, Jahrling PB, Davis KJ. 2003. Pathogenesis of Ebola hemorrhagic fever in cynomolgus macaques—evidence that dendritic cells are early and sustained targets of infection. Am J Pathol 163:2347–2370. doi: 10.1016/S0002-9440(10)63591-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geisbert TW, Young HA, Jahrling PB, Davis KJ, Kagan E, Hensley LE. 2003. Mechanisms underlying coagulation abnormalities in Ebola hemorrhagic fever: overexpression of tissue factor in primate monocytes/macrophages is a key event. J Infect Dis 188:1618–1629. doi: 10.1086/379724. [DOI] [PubMed] [Google Scholar]
- 18.Takada A, Watanabe S, Ito H, Okazaki K, Kida H, Kawaoka Y. 2000. Downregulation of beta1 integrins by Ebola virus glycoprotein: implication for virus entry. Virology 278:20–26. doi: 10.1006/viro.2000.0601. [DOI] [PubMed] [Google Scholar]
- 19.Schornberg KL, Shoemaker CJ, Dube D, Abshire MY, Delos SE, Bouton AH, White JM. 2009. Alpha5beta1-integrin controls Ebolavirus entry by regulating endosomal cathepsins. Proc Natl Acad Sci U S A 106:8003–8008. doi: 10.1073/pnas.0807578106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kindrachuk J, Wahl-Jensen V, Safronetz D, Trost B, Hoenen T, Arsenault R, Feldmann F, Traynor D, Postnikova E, Kusalik A, Napper S, Blaney JE, Feldmann H, Jahrling PB. 2014. Ebola virus modulates transforming growth factor beta signaling and cellular markers of mesenchyme-like transition in hepatocytes. J Virol 88:9877–9892. doi: 10.1128/JVI.01410-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Angel P, Karin M. 1991. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta 1072:129–157. [DOI] [PubMed] [Google Scholar]
- 22.Halazonetis TD, Georgopoulos K, Greenberg ME, Leder P. 1988. c-Jun dimerizes with itself and with c-Fos, forming complexes of different DNA binding affinities. Cell 55:917–924. doi: 10.1016/0092-8674(88)90147-X. [DOI] [PubMed] [Google Scholar]
- 23.Tengku-Muhammad TS, Hughes TR, Foka P, Cryer A, Ramji DP. 2000. Cytokine-mediated differential regulation of macrophage activator protein-1 genes. Cytokine 12:720–726. doi: 10.1006/cyto.1999.0620. [DOI] [PubMed] [Google Scholar]
- 24.Monje P, Marinissen MJ, Gutkind JS. 2003. Phosphorylation of the carboxyl-terminal transactivation domain of c-Fos by extracellular signal-regulated kinase mediates the transcriptional activation of AP-1 and cellular transformation induced by platelet-derived growth factor. Mol Cell Biol 23:7030–7043. doi: 10.1128/MCB.23.19.7030-7043.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brenner DA, Ohara M, Angel P, Chojkier M, Karin M. 1989. Prolonged activation of Jun and collagenase genes by tumor necrosis factor-alpha. Nature 337:661–663. doi: 10.1038/337661a0. [DOI] [PubMed] [Google Scholar]
- 26.Goldgaber D, Harris HW, Hla T, Maciag T, Donnelly RJ, Jacobsen JS, Vitek MP, Gajdusek DC. 1989. Interleukin-1 regulates synthesis of amyloid beta-protein precursor mRNA in human endothelial cells. Proc Natl Acad Sci U S A 86:7606–7610. doi: 10.1073/pnas.86.19.7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muegge K, Williams TM, Kant J, Karin M, Chiu R, Schmidt A, Siebenlist U, Young HA, Durum SK. 1989. Interleukin-1 costimulatory activity on the interleukin-2 promoter via Ap-1. Science 246:249–251. doi: 10.1126/science.2799385. [DOI] [PubMed] [Google Scholar]
- 28.Shaulian E, Karin M. 2001. AP-1 in cell proliferation and survival. Oncogene 20:2390–2400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- 29.Shaulian E, Karin M. 2002. AP-1 as a regulator of cell life and death. Nat Cell Biol 4:E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 30.Parry GCN, Mackman N. 1995. Transcriptional regulation of tissue factor expression in human endothelial cells. Arterioscler Thromb Vasc Biol 15:612–621. doi: 10.1161/01.ATV.15.5.612. [DOI] [PubMed] [Google Scholar]
- 31.Takahra T, Smart DE, Oakley F, Mann DA. 2004. Induction of myofibroblast MMP-9 transcription in three-dimensional collagen I gel cultures: regulation by NF-kappa B, AP-1 and Sp1. Int J Biochem Cell Biol 36:353–363. doi: 10.1016/S1357-2725(03)00260-7. [DOI] [PubMed] [Google Scholar]
- 32.Eriksson M, Arminen L, Karjalainen-Lindsberg ML, Leppa S. 2005. AP-1 regulates alpha(2)beta(1) integrin expression by ERK-dependent signals during megakaryocytic differentiation of K562 cells. Exp Cell Res 304:175–186. doi: 10.1016/j.yexcr.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 33.Galvagni F, Orlandini M, Oliviero S. 2013. Role of the AP-1 transcription factor FOSL1 in endothelial cell adhesion and migration. Cell Adh Migr 7:408–411. doi: 10.4161/cam.25894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Angel P, Hattori K, Smeal T, Karin M. 1988. The Jun proto-oncogene is positively autoregulated by its product, Jun/Ap-1. Cell 55:875–885. doi: 10.1016/0092-8674(88)90143-2. [DOI] [PubMed] [Google Scholar]
- 35.Shabman RS, Jabado OJ, Mire CE, Stockwell TB, Edwards M, Mahajan M, Geisbert TW, Basler CF. 2014. Deep sequencing identifies noncanonical editing of Ebola and Marburg virus RNAs in infected cells. mBio 5:e02011. doi: 10.1128/mBio.02011-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. 2009. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ho Sui SJ, Fulton DL, Arenillas DJ, Kwon AT, Wasserman WW. 2007. oPOSSUM: integrated tools for analysis of regulatory motif over-representation. Nucleic Acids Res 35:W245–W252. doi: 10.1093/nar/gkm427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ho Sui SJ, Mortimer JR, Arenillas DJ, Brumm J, Walsh CJ, Kennedy BP, Wasserman WW. 2005. oPOSSUM: identification of over-represented transcription factor binding sites in co-expressed genes. Nucleic Acids Res 33:3154–3164. doi: 10.1093/nar/gki624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holzer M, Krahling V, Amman F, Barth E, Bernhart SH, Carmelo VAO, Collatz M, Doose G, Eggenhofer F, Ewald J, Fallmann J, Feldhahn LM, Fricke M, Gebauer J, Gruber AJ, Hufsky F, Indrischek H, Kanton S, Linde J, Mostajo N, Ochsenreiter R, Riege K, Rivarola-Duarte L, Sahyoun AH, Saunders SJ, Seemann SE, Tanzer A, Vogel B, Wehner S, Wolfinger MT, Backofen R, Gorodkin J, Grosse I, Hofacker I, Hoffmann S, Kaleta C, Stadler PF, Becker S, Marz M. 2016. Differential transcriptional responses to Ebola and Marburg virus infection in bat and human cells. Sci Rep 6:34589. doi: 10.1038/srep34589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sasaki T, Kojima H, Kishimoto R, Ikeda A, Kunimoto H, Nakajima K. 2006. Spatiotemporal regulation of c-Fos by ERK5 and the E3 ubiquitin ligase UBR1, and its biological role. Mol Cell 24:63–75. doi: 10.1016/j.molcel.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 41.Davis RJ. 2000. Signal transduction by the JNK group of MAP kinases. Cell 103:239–252. doi: 10.1016/S0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 42.Ng M, Ndungo E, Kaczmarek ME, Herbert AS, Binger T, Kuehne AI, Jangra RK, Hawkins JA, Gifford RJ, Biswas R, Demogines A, James RM, Yu M, Brummelkamp TR, Drosten C, Wang LF, Kuhn JH, Muller MA, Dye JM, Sawyer SL, Chandran K. 2015. Filovirus receptor NPC1 contributes to species-specific patterns of Ebolavirus susceptibility in bats. Elife 4:e11785. doi: 10.7554/eLife.11785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kash JC, Muhlberger E, Carter V, Grosch M, Perwitasari O, Proll SC, Thomas MJ, Weber F, Klenk HD, Katze MG. 2006. Global suppression of the host antiviral response by Ebola- and Marburgviruses: increased antagonism of the type I interferon response is associated with enhanced virulence. J Virol 80:3009–3020. doi: 10.1128/JVI.80.6.3009-3020.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ludwig S, Ehrhardt C, Neumeier ER, Kracht M, Rapp UR, Pleschka S. 2001. Influenza virus-induced AP-1-dependent gene expression requires activation of the JNK signaling pathway. J Biol Chem 276:10990–10998. doi: 10.1074/jbc.M009902200. [DOI] [PubMed] [Google Scholar]
- 45.Xie J, Pan H, Yoo S, Gao SJ. 2005. Kaposi's sarcoma-associated herpesvirus induction of AP-1 and interleukin 6 during primary infection mediated by multiple mitogen-activated protein kinase pathways. J Virol 79:15027–15037. doi: 10.1128/JVI.79.24.15027-15037.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kang SM, Lim S, Won SJ, Shin YJ, Lim YS, Ahn BY, Hwang SB. 2011. c-Fos regulates hepatitis C virus propagation. FEBS Lett 585:3236–3244. doi: 10.1016/j.febslet.2011.08.041. [DOI] [PubMed] [Google Scholar]
- 47.Hou CH, Lin FL, Hou SM, Liu JF. 2014. Cyr61 promotes epithelial-mesenchymal transition and tumor metastasis of osteosarcoma by Raf-1/MEK/ERK/Elk-1/TWIST-1 signaling pathway. Mol Cancer 13:236. doi: 10.1186/1476-4598-13-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Audet J, Kobinger GP. 2015. Immune evasion in Ebolavirus infections. Viral Immunol 28:10–18. doi: 10.1089/vim.2014.0066. [DOI] [PubMed] [Google Scholar]
- 49.Basler CF, Amarasinghe GK. 2009. Evasion of interferon responses by Ebola and Marburg viruses. J Interferon Cytokine Res 29:511–520. doi: 10.1089/jir.2009.0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pichlmair A, Lassnig C, Eberle CA, Gorna MW, Baumann CL, Burkard TR, Burckstummer T, Stefanovic A, Krieger S, Bennett KL, Rulicke T, Weber F, Colinge J, Muller M, Superti-Furga G. 2011. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat Immunol 12:624–630. doi: 10.1038/ni.2048. [DOI] [PubMed] [Google Scholar]
- 51.Leroy EM, Kumulungui B, Pourrut X, Rouquet P, Hassanin A, Yaba P, Delicat A, Paweska JT, Gonzalez JP, Swanepoel R. 2005. Fruit bats as reservoirs of Ebola virus. Nature 438:575–576. doi: 10.1038/438575a. [DOI] [PubMed] [Google Scholar]
- 52.Pourrut X, Souris M, Towner JS, Rollin PE, Nichol ST, Gonzalez JP, Leroy E. 2009. Large serological survey showing cocirculation of Ebola and Marburg viruses in Gabonese bat populations, and a high seroprevalence of both viruses in Rousettus aegyptiacus. BMC Infect Dis 9:159. doi: 10.1186/1471-2334-9-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Crameri G, Todd S, Grimley S, McEachern JA, Marsh GA, Smith C, Tachedjian M, De Jong C, Virtue ER, Yu M, Bulach D, Liu JP, Michalski WP, Middleton D, Field HE, Wang LF. 2009. Establishment, immortalisation and characterisation of pteropid bat cell lines. PLoS One 4:e8266. doi: 10.1371/journal.pone.0008266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am J Hyg 27:493–497. [Google Scholar]
- 55.Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mok L, Wynne JW, Ford K, Shiell B, Bacic A, Michalski WP. 2015. Proteomic analysis of Pteropus alecto kidney cells in response to the viral mimic, poly I:C. Proteome Sci 13:25. doi: 10.1186/s12953-015-0081-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wynne JW, Shiell BJ, Marsh GA, Boyd V, Harper JA, Heesom K, Monaghan P, Zhou P, Payne J, Klein R, Todd S, Mok L, Green D, Bingham J, Tachedjian M, Baker ML, Matthews D, Wang LF. 2014. Proteomics informed by transcriptomics reveals Hendra virus sensitizes bat cells to TRAIL-mediated apoptosis. Genome Biol 15:532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pfaffl MW, Horgan GW, Dempfle L. 2002. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res 30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trombley AR, Wachter L, Garrison J, Buckley-Beason VA, Jahrling J, Hensley LE, Schoepp RJ, Norwood DA, Goba A, Fair JN, Kulesh DA. 2010. Comprehensive panel of real-time TaqMan polymerase chain reaction assays for detection and absolute quantification of filoviruses, arenaviruses, and New World hantaviruses. Am J Trop Med Hyg 82:954–960. doi: 10.4269/ajtmh.2010.09-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.