

ABSTRACT
The inhibitors carbobenzoxy (Z)-d-Phe-l-Phe-Gly (fusion inhibitor peptide [FIP]) and 4-nitro-2-phenylacetyl amino-benzamide (AS-48) have similar efficacies in blocking membrane fusion and syncytium formation mediated by measles virus (MeV). Other homologues, such as Z-d-Phe, are less effective but may act through the same mechanism. In an attempt to map the site of action of these inhibitors, we generated mutant viruses that were resistant to the inhibitory effects of Z-d-Phe-l-Phe-Gly. These 10 mutations were localized to the heptad repeat B (HRB) region of the fusion protein, and no changes were observed in the viral hemagglutinin, which is the receptor attachment protein. Mutations were validated in a luciferase-based membrane fusion assay, using transfected fusion and hemagglutinin expression plasmids or with syncytium-based assays in Vero, Vero-SLAM, and Vero-Nectin 4 cell lines. The changes I452T, D458N, D458G/V459A, N462K, N462H, G464E, and I483R conferred resistance to both FIP and AS-48 without compromising membrane fusion. The inhibitors did not block hemagglutinin protein-mediated binding to the target cell. Edmonston vaccine/laboratory and IC323 wild-type strains were equally affected by the inhibitors. Escape mutations were mapped upon a three-dimensional (3D) structure modeled from the published crystal structure of parainfluenzavirus 5 fusion protein. The most effective mutations were situated in a region located near the base of the globular head and its junction with the alpha-helical stalk of the prefusion protein. We hypothesize that the fusion inhibitors could interfere with the structural changes that occur between the prefusion and postfusion conformations of the fusion protein.
IMPORTANCE Due to lapses in vaccination worldwide that have caused localized outbreaks, measles virus (MeV) has regained importance as a pathogen. Antiviral agents against measles virus are not commercially available but could be useful in conjunction with MeV eradication vaccine programs and as a safeguard in oncolytic viral therapy. Three decades ago, the small hydrophobic peptide Z-d-Phe-l-Phe-Gly (FIP) was shown to block MeV infections and syncytium formation in monkey kidney cell lines. The exact mechanism of its action has yet to be determined, but it does appear to have properties similar to those of another chemical inhibitor, AS-48, which appears to interfere with the conformational change in the viral F protein that is required to elicit membrane fusion. Escape mutations were used to map the site of action for FIP. Knowledge gained from these studies could help in the design of new inhibitors against morbilliviruses and provide additional knowledge concerning the mechanism of virus-mediated membrane fusion.
KEYWORDS: measles virus, fusion inhibitors, F protein, FIP, AS-48, Z-d-Phe-l-Phe-Gly, antiviral inhibitors, escape mutations, drug resistance, membrane fusion
INTRODUCTION
Measles virus (MeV) is a member of the genus Morbillivirus in the family Paramyxoviridae (1, 2). Recently, there has been a resurgence of measles in certain populations due to lapses in vaccination worldwide (3–8). Despite the availability of a very effective vaccine, MeV was responsible for almost 114,900 deaths in 2014 (WHO). In most patients, MeV causes the classical measles disease, which is characterized by a 10- to 14-day incubation period and a 2- to 3-day prodrome of fever, cough, coryza, conjunctivitis, and Koplik spots, followed around 4 days later by the characteristic maculopapular rash over the skin (2). There is no specific treatment for measles, although vitamin A is recommended by the WHO for populations where infant mortality due to measles is greater than 1% (9). This treatment is believed to enhance innate immunity and provide resistance against MeV (10). In healthy patients without any complications, natural recovery takes about 7 to 10 days following the appearance of the rash, and the individual often acquires lifelong immunity to the disease. Antivirals could be used to synergize with vaccination and prevent infections in locations where measles outbreaks occur (11). There has also been intense interest in using MeV as an oncolytic agent (12, 13), and antivirals could control potential infections in immune-suppressed individuals during therapy (14).
The negative-stranded RNA genome of MeV comprises 6 viral genes with 2 additional transcripts that specify V and C proteins, produced by RNA editing and via alternative start codon usage, respectively (2, 15). Two structural-membrane proteins are responsible for viral entry into cells. The hemagglutinin (H) protein recognizes and binds to the cellular receptors, whereas the fusion (F) protein mediates the merger of the viral envelope with the cellular membrane to enable virus entry. Clinical strains of MeV target cells of the immune system by their recognition and use of the signaling lymphocyte activation molecule SLAMF1/SLAM/CD150 as their receptor, whereas the vaccine strains use either SLAMF1 or the ubiquitous membrane cofactor protein MCP/CD46. Finally, both vaccine and wild-type (WT) strains of MeV can use the epithelial cell receptor Nectin-4/PVRL4, which is present on airway epithelial cells and adenocarcinomas of the lung, breast, colon, and ovary (16, 17).
Upon binding to its receptor, the H protein triggers a conformational change in the F protein, allowing it to fuse the viral and cellular membranes through a mechanism that is still not fully elucidated (18–23). Binding of H to its receptor elicits a conformational change in the attachment protein to reveal a trigger sequence in its stem region that interacts with the globular head of F. The F-binding domain in the stem region of morbillivirus H has been mapped to residues 110 to 118, and the F-triggering region corresponds to a larger, overlapping region consisting of residues 85 to 118 (24–27). Consistent with this is the observation that H protein lacking the globular head can still activate F protein (28, 29). The H protein trigger region is believed to interact with hydrophobic regions in the globular head of the F protein located near its Ig-like domain (30, 31). This trigger model is based upon the study of site-specific mutations in the stem and globular-head regions of F and H, respectively, but X-ray data on H-F complexes showing the molecular details of H interaction with F protein are still lacking.
Atomic structures for the postfusion forms of F proteins from human parainfluenza virus 3 (hPIV3), respiratory syncytial virus (RSV), and Newcastle disease virus (NDV) were reported first (32–38). Subsequently, the crystal structures of stabilized prefusion forms of F proteins from parainfluenza virus 5 (PIV5), RSV, Nipah virus (NiV), and Hendra virus (HeV) were also solved (39–44). These structures have provided detailed insight into the spatial organization of the F proteins as they mediate fusion between virus and host membranes. The F protein exists as a homotrimer and is synthesized as a precursor protein that is cleaved by a host furin-like protease to produce disulfide-bonded F1 and F2 subunits. The cleaved protein first forms a thermodynamically metastable conformation known as the prefusion protein (20–23). The amino terminus of the F1 subunit contains the fusion peptide, which is composed of hydrophobic amino acid residues. The fusion peptide is followed by the first (HRA) of two heptad repeats, which are alpha-helical domains featuring a hydrophobic amino acid in a 4-to-3 repetitive pattern. The second heptad repeat (HRB) is located just proximal to the C-terminal transmembrane region. HRA and HRB are separated by a spacer region that is approximately 250 amino acids long. In the metastable prefusion state, HRA and HRB do not interact, but the HRB region forms a short prefusion stalk. The trimeric F protein is thought to be in close proximity to the H protein tetramer (45). Upon receptor binding by the H protein, the F protein is activated and dissociates from the H complex, and the three HRA regions of the F trimer assemble into a triple-stranded coiled coil, propelling the fusion peptide toward the target cell membrane. This reorganization is followed by the formation of an antiparallel coiled coil or six-helix bundle (6HB) structure between the HRA and HRB domains, representing the postfusion state. The formation of the 6HB brings together the viral and cellular membranes and ultimately results in fusion of the two membranes.
The small hydrophobic fusion inhibitor peptide (FIP) (carbobenzoxy [Z]-d-Phe-l-Phe-Gly [ZfFG]) (Fig. 1) has previously been shown to block infections and syncytium formation by the Edmonston laboratory strain of MeV (MeV-Edm) in Vero, CV-1, and HeLa cells (46). The peptide also blocked hemolysis of African green monkey red blood cells mediated by MeV Edmonston-derived glycoproteins. Similar results had previously been demonstrated with Z-d-Phe-l-Phe-l(NO2)Arg (47–50). Past experiments were performed with the Edmonston strain of the virus, which uses a CD46 receptor that is expressed on most primate cells and monkey erythrocytes (51, 52). Plemper and colleagues developed small-molecule MeV entry inhibitors (4-nitro-2-phenylacetyl amino-benzamide [AS-48]) (Fig. 1) based on structural modeling of F (53–55) and have elaborated upon the theoretical mechanism of action of these inhibitors using drug-resistant mutants (56, 57). Despite FIP having been developed over 3 decades ago, the exact mechanism of its action has yet to be demonstrated, although some evidence that it prevents hemifusion by inhibiting outer lipid bilayer mixing has been reported (58–61). Regardless, the compound has been used to study various aspects of MeV biology (61–67). Membrane fusion inhibitors against RSV have also been developed and were reported to bind a central cavity located at the intersection of the globular-head and stalk domains of the prefusion F trimer and to stabilize the metastable conformation (68–73).
FIG 1.
Chemical structures of carbobenzoxy-d-phenylalanine (Z-d-Phe), carbobenzoxy-l-phenylalanine (Z-l-Phe), carbobenzoxy-d-phenylalanine-l-phenylalanine-glycine (Z-d-Phe-l-Phe-Gly [ZfFG]; FIP), and AS-48. These chemical compounds have been shown to specifically inhibit the membrane fusion properties of MeV.
In this study, we generated FIP-resistant mutants of MeV-Edm and characterized F proteins in the context of both vaccine/laboratory and wild-type IC323 (MeV-IC323) strains of MeV. Ten mutations were localized to the heptad repeat region (HRB) of the fusion protein, and no changes were observed in the viral hemagglutinin. The escape mutations also provided MeV resistance against AS-48. The MeV F mutations localized to a small region between the head and stalk of the prefusion conformation of F. We further discuss the hypothetical mechanism by which FIP could inhibit MeV fusion in terms of blocking the transition between pre- and postfusion conformations of the F protein using a model derived from preexisting paramyxovirus structures. Antifusion inhibitors are a valid approach for controlling viral infections, as evidenced by FDA approval of the HIV entry drug enfuvirtide (T-20) (74–76) and clinical trials with the RSV inhibitor GS-5806 (73, 77, 78). Another fusion inhibitor, Arbidol, is also being used in Russia and China as a prophylactic and therapeutic treatment for influenza virus infections (79).
RESULTS
FIP and AS-48 effectively inhibit MeV infection and plaque formation.
The small hydrophobic peptide FIP (Z-d-Phe-l-Phe-Gly) (Fig. 1) was previously shown to block infections and syncytium formation in Vero, CV-1, and HeLa cells by the Edmonston laboratory strain of MeV (46). The peptide also blocked hemolysis of African green monkey red blood cells mediated by this strain of MeV. Similar results were also demonstrated with Z-d-Phe-l-Phe-l-(NO2)Arg (49, 50). The carbobenzoxy (Z) and d-Phe moieties are critical to the activity of FIP, and Z-l-Phe-l-Phe-Gly was shown to be a less potent inhibitor (46). AS-48 (Fig. 1) is another small-molecule inhibitor that blocks membrane fusion and was developed through a structure-guided medicinal-chemistry approach (54, 55, 57). Previous evidence demonstrated that Z-d-Phe, the N-terminal component of FIP, also inhibited MeV membrane fusion, but less efficiently (46). Z-d-Phe has some of the chemical characteristics of AS-48, and consequently, Z-d-Phe and Z-l-Phe were included as less potent inhibitor controls in Fig. 1. Previous experiments with both FIP and AS-48 were performed with the Edmonston vaccine/laboratory strain of the virus, which uses CD46 receptor, expressed on the surfaces of most primate cells and monkey erythrocytes (46, 51). To determine whether FIP and AS-48 also inhibited replication and membrane fusion by the wild-type IC323 isolate of MeV, which uses SLAM rather than CD46 as a receptor (80, 81), we performed virus plaque inhibition studies with the fusion inhibitors Z-d-Phe, FIP, and AS-48 in Vero-SLAM cells using recombinant MeV IC323 containing an enhanced green fluorescent protein (eGFP) reporter gene. Results showing fluorescent viral plaques were quantified using a Kodak 4000MM imager (Fig. 2, top). Plaque reduction assays with inhibitor concentrations ranging between 0 and 250 μM were also assessed by counting the individual visible plaques, which proved to be more sensitive and quantitative (Fig. 2, bottom). Z-l-Phe was ineffective in preventing MeV plaque formation, which was only slightly reduced at a 250 μM concentration. Z-d-Phe had a 50% effective dose of 65 μM compared to 20 μM for FIP and 5 μM for AS-48. The hydrophobic nature of these inhibitors and their comparable chemical structures and similar effects upon virus-mediated cell fusion suggested the compounds could possibly work through a common mechanism.
FIG 2.
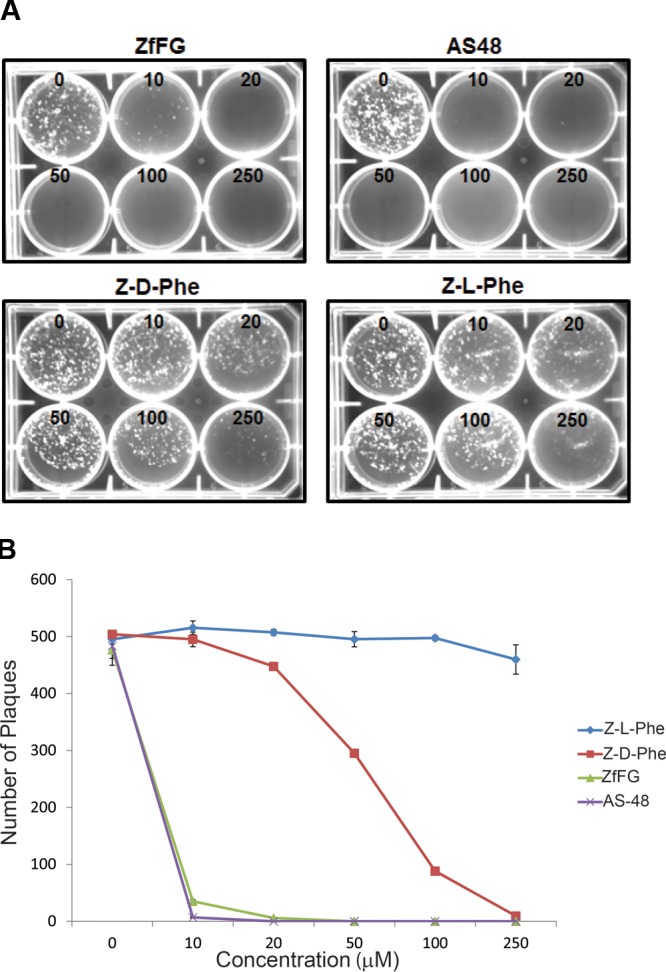
Replication of WT MeV IC323 containing eGFP (MeV-eGFP) within Vero-SLAM cells in the presence of varying concentrations of fusion inhibitors (FIP, AS-48, Z-d-Phe, and Z-l-Phe). (Top) Vero-SLAM cells in 6-well culture plates were infected with 500 PFU WT MeV-eGFP per well in the presence of 0, 10, 20, 50, 100, and 250 μM inhibitor. The infected cells were overlaid with 1% SeaPlaque agarose in DMEM containing 2% fetal calf serum and the specified inhibitor at the indicated concentrations (0, 10, 20, 50, 100, and 250 μM). Fluorescent plaques were visualized with a Kodak 4000MM imager. (Bottom) Viral plaques were quantified following the staining of infected cells with 0.005% neutral red at 96 h postinfection. Plaques visible to the eye were counted in triplicate experiments and averaged. This quantitation proved to be more sensitive than fluorescence imaging. Experimental error was calculated as SEM, and error bars are shown. The numbers of plaques were plotted against the concentrations (0, 10, 20, 50, 100, and 250 μM) of specific inhibitors (Z-l-Phe, Z-d-Phe, FIP, and AS-48).
MeV H can bind to its cellular receptor in the presence of FIP and AS-48.
FIP was originally reported for its ability to prevent membrane fusion and syncytium formation (46). Although FIP's sequence similarity with the fusion peptide sequence of the F protein was postulated, its interaction with either the F or H protein was not firmly established. Fusion is normally triggered when H recognizes its receptor, and its interaction with the F protein is modified to trigger fusion (18, 19, 23, 29, 82). In order to exclude the possibility that interaction between the fusion inhibitors and H prevented fusion, the ability of FIP or AS-48 to inhibit virus binding to MeV receptor-expressing cells was tested. MeV Edmonston was allowed to bind to Vero cells expressing CD46 receptor on their surfaces, and binding was measured by flow cytometry (fluorescence-activated cell sorting [FACS]) using a measles virus-specific antibody (Fig. 3A), as previously described (16). The virus binding assay was performed at 4°C for 90 min in the presence or absence of either FIP or AS-48. Both the FIP- and AS-48-treated samples exhibited the same shift in fluorescence as untreated MeV samples, indicating that virus binding was unaffected by the inhibitors (Fig. 3A). In addition, there was no difference in the ability of MeV-Edm to bind to Vero cells expressing either SLAM or PVRL4 in the presence of these fusion inhibitors (data not shown). The percentage of MeV-bound Vero cells compared to mock-treated Vero cells was quantitated using FCS Express software (De Novo Software, Glendale CA) and plotted as histograms (Fig. 3B). Inhibition of virus attachment to its cellular receptor is not the mechanism by which FIP or AS-48 prevents MeV-mediated membrane fusion.
FIG 3.

FIP does not prevent MeV (MV) from binding to Vero or CHO cells that express its known cellular receptors. (A) Vero cells that express CD46 receptor were inoculated with MeV Edmonston at an MOI of 10 in the presence of 100 μM FIP, 75 μM AS-48, or 0.5% DMSO for 1.5 h; washed; and incubated with anti-MeV hemagglutinin antibody or an isotype-matched control antibody (shaded). Cells incubated in the absence of virus, as a negative control, were also stained with anti-MeV hemagglutinin antibody (blue). Bound MeV-specific primary antibody was detected with Alexa Fluor 488-conjugated goat anti-mouse secondary antibody. The relative fluorescence intensity was measured on a Cyan ADP flow cytometer. (B) The percentage of MeV-bound Vero cells compared to mock-treated Vero cells was quantitated using FCS Express software. The data are means of the results from three independent experiments, and the error bars represent SEM. (C) CHO, CHO.CD46, CHO.SLAM, and CHO.PVRL4 cells were infected with IC323-eGFP WT MeV and Edmonston MeV-eGFP at an MOI of 5. After 1 h, the viral inoculum was removed and the cells were washed and treated with 50 μM FIP, 75 μM AS-48, or 0.5% DMSO (drug carrier) as indicated. Fluorescence images were captured 1 day postinfection in order to visualize the extent of virus replication and syncytium formation.
We next investigated whether these fusion inhibitors were capable of preventing MeV-Edm or MeV-IC323 from inducing syncytium formation in Chinese hamster ovary (CHO) cells expressing CD46, SLAM, or PVRL4 (Fig. 3C). Both FIP and AS-48 were able to block syncytium formation following infection with MeV-Edm or MeV-IC323, and this was independent of the cellular receptor used. Overall, these data suggest that the inhibitory activities of both FIP and AS-48 are directed against the MeV F protein and do not affect the ability of MeV H to bind to its cellular receptor. The data also show that the inhibitory effects of FIP and AS-48 are independent of the cellular receptor used for MeV entry.
FIP-resistant mutants of MeV cluster in the HRB region of the MeV F protein.
A previous paper suggested that a variety of changes in the protein sequence of the F protein of MeV-Edm may impart resistance to the inhibitor peptide Z-d-Phe-l-Phe-l-(NO2)Arg (83). However, the significance of these mutations was not validated. To identify amino acid residues in the MeV F protein that confer resistance to the fusion peptide inhibitor FIP, we repeatedly passaged MeV Edmonston in the presence of 50 μM FIP. Forty-eight individual mutant viruses were isolated and amplified by plaque purification. The F RNAs from these isolates were amplified by reverse transcription (RT)-PCR, cloned, and sequenced. Analyses of cloned sequences revealed 9 mutually exclusive mutations and 1 double mutation that occurred in the HRB region of MeV F: I452T, L454W, D458G, D458N, D458G/V459A, N462H, N462K, G464E, G464R, and I483R (Fig. 4B). Mutations were not found in other regions of the F gene, and no mutations were detected in the H gene sequence following 20 attempts to do so (results not shown). Of the 10 mutations, 7 resulted in changes from hydrophobic to polar amino acids. To validate the stability of each mutation in the virus genome, the mutant viruses were passaged and plaque purified three times through Vero cells in the presence of FIP, and the F gene was recloned and resequenced. Sequencing analyses showed that the mutations persisted after the three passages. However, removal of the FIP selection pressure caused reversion of the mutated amino acids to the original wild-type sequence after 5 passages, suggesting that the resistance mutations occurred at the expense of viral fitness. These data indicate that FIP inhibits the activity of MeV F by binding to a region of F involving HRB, either directly or indirectly. We hypothesize that FIP induces mutations that either prevent inhibitor binding or lower the activation threshold of F to facilitate a more efficient conformational change, leading to membrane fusion in the presence of the inhibitory peptide.
FIG 4.
FIP resistance mutations in MeV Edmonston F protein cluster within the HRB region. (A) Vero cells were infected with MeV-Edm at an MOI of 1. After 1 h, the viral inoculum was removed and the cells were washed and treated with 50 μM FIP or 0.5% DMSO as indicated. At 1 day postinfection, the cells were stained with Giemsa. Phase-contrast images were monitored to visualize the extent of syncytium formation. (B) Schematic representation of spontaneous mutations in MeV-Edm F in the presence of 50 to 100 μM FIP following 3 rounds of plaque purification. The disulfide bond (S—S) connects the F1 and F2 subunits. The boxes marked HRA and HRB indicate the positions of the corresponding heptad repeat regions. Other features of F include the fusion peptides (FP), the transmembrane region (TM), and the cytoplasmic tail (CPT). The FIP escape mutations are I452T, L454W, D458G, D458N, D458G/V459A, N462H, N462K, G464R, G464E, and I483R.
Characterization of MeV F inhibitor escape mutants using syncytium- and luciferase-based fusion assays.
The coding regions for the 10 MeV F mutant proteins and the MeV H protein were inserted into the pCAGGS expression vector. Each mutant was cotransfected into Vero cells together with MeV-Edm H in the presence or absence of either 50 μM FIP or 50 μM AS-48. The extent of syncytium formation following expression of these resistance mutants was compared following incubation with the inhibitors (Fig. 5A). Overall, the MeV F mutant proteins were able to produce syncytia in the presence of either of the MeV fusion inhibitors. The results suggested that AS-48 and FIP work in similar manners to inhibit MeV fusion. However, two of the MeV F mutants (D458G and G464R) expressed from plasmids failed to form syncytia in the presence or absence of FIP and AS-48. To investigate the abilities of the MeV F inhibitor resistance mutants to be processed and transported to the cell surface, whole-cell extracts were analyzed for F0 precursor protein cleavage and F1 expression on the cell surface. In extracts from cotransfected cells, the F0 form of the D458G and G464R MeV F mutants was not cleaved to the active F1 form (Fig. 5B). Since furin is the Golgi apparatus-resident protease that is responsible for proteolytic activation of F0, this suggests that these two particular mutants were not transported from the endoplasmic reticulum to the trans-Golgi network. The remaining F mutants appeared to be cleaved and transported to the cell surface. Biotinylation assays showed reduced surface expression of both the D458G and G464R mutants, while the remaining mutants were transported to the cell surface with varying efficiencies (Fig. 5C). With the exception of D458G and G464R mutants, FIP and AS-48 had similar effects upon syncytium formation in the wild type and remaining F variants.
FIG 5.

(A) Vero cells transfected with expression vectors expressing Edmonston H and Edmonston FIP-resistant F mutants induce syncytia in the presence of both FIP and AS-48. Vero cells were cotransfected with the expression plasmids, and after 5 h, the cells were treated with 50 μM FIP, 75 μM AS-48, or 0.5% DMSO. At 20 h posttransfection, the cells were stained with Giemsa to reveal syncytia, which were subsequently viewed by phase-contrast microscopy (×100 magnification). (B) HEK 293 cells were cotransfected with H and mutant F expression plasmids, and cells were harvested 24 h posttransfection. Cell lysate proteins were resolved by SDS-PAGE and subjected to immunoblot analysis with an antibody that recognizes the C terminus of MeV F. The multiple bands of the glycosylated F0 precursor (60 to 65 kDa) and the F1 subunit were detected. Actin was detected with a specific antibody as a loading control. (C) Surface proteins from transfected HEK 293 cells coexpressing H and mutant F proteins were labeled with biotin and precipitated using streptavidin-conjugated beads. The biotin-labeled surface proteins were resolved by SDS-PAGE, and F1 protein was detected by immunoblot analysis using the antibody against the MeV F protein and ECL detection. Protein intensities were determined using a Kodak 4000MM imager, compared to those of the wild-type Edmonston F1 protein, and expressed as a percentage. The values in parentheses are the standard deviations calculated from the results of three independent experiments. (D) Luciferase-based fusion assays between HEK293 cells expressing Edmonston H and wild-type F/FIP-resistant F proteins and Vero-SLAM cells. Vero-SLAM cells were cotransfected with plasmids encoding T7 polymerase and renilla luciferase under the control of the CMV promoter. At 20 h posttransfection, the cells were overlaid with HEK293 cells that had been cotransfected with expression plasmids encoding Edmonston H and firefly luciferase (under the control of the T7 promoter), together with WT or mutated Edmonston F expression plasmids. After 4 h, firefly luciferase activity was measured as RLU of bioluminescence. The negative control (Luc Vec) consisted of HEK293 cells transfected with the luciferase expression vector and Vero-SLAM cells transfected with the T7 polymerase vector. Nonnormalized bioluminescence values are presented as the averages of the results of three experiments, and the error bars represent SEM. (E) Dose response to FIP (0, 50, 100, and 200 μM) by F-resistant mutants using luciferase-based fusion assays. The fusion assays were performed as described above in the absence or presence of FIP, and bioluminescence was measured in RLU. FIP clearly inhibited membrane fusion mediated by the wild-type fusion protein (WT) but inhibited fusion activity from the mutant F proteins to varying degrees. Nonnormalized bioluminescence values are presented as the averages of the results of three independent experiments, and the error bars represent SEM. The I452T, L454W, and I483R mutants were more sensitive to FIP than the D458N, D458N/V459A, N462H, N462K, and G464E mutants. The D458N and N462K mutants were substantially more resistant to 200 μM FIP.
To quantify the membrane fusion activity of each F mutant, luciferase-based fusion assays were performed. The assays were used to evaluate membrane fusion between HEK293 cells expressing Edmonston H and wild-type F or FIP-resistant F proteins and Vero-SLAM cells. Vero-SLAM cells were cotransfected with plasmids encoding T7 polymerase and renilla luciferase under the control of the cytomegalovirus (CMV) promoter. At 20 h posttransfection, the cells were overlaid with HEK293 cells that had been cotransfected with expression plasmids encoding Edmonston H and firefly luciferase (under the control of the T7 promoter), together with WT or mutated Edmonston F expression plasmids. When cell fusion occurred between the two cell types, mRNA for luciferase was transcribed and translated to produce the reporter protein. After 4 h, bioluminescence from firefly luciferase was measured. The negative control (Luc Vec) consisted of HEK293 cells transfected with the luciferase expression vector and Vero-SLAM cells transfected with the T7 polymerase vector. Nonnormalized bioluminescence data, measured in relative light units (RLU), are presented as the means of the results of three independent experiments, and the error bars represent the standard errors of the mean (SEM) (Fig. 5D). Fusion activities elicited by WT and mutant F proteins were comparable, but the N462H, N462K, and G464E mutants consistently produced 10 to 20% higher levels of membrane fusion. It seems possible that these mutations could destabilize prefusion F and make the molecules more fusogenic. Next, the dose responses to FIP (0, 50, 100, and 200 μM) by F-resistant mutants were evaluated using luciferase-based fusion assays (Fig. 5E). The fusion assays were performed as described above in the absence or presence of FIP, and bioluminescence was measured in RLU. FIP clearly inhibited membrane fusion mediated by the wild-type fusion protein but inhibited fusion activity from the mutant F proteins to varying degrees. The I452T, L454W, and I483R mutants were more sensitive to FIP than the D458N, D458N/V459A, N462H, N462K, and G464E mutants. The D458N and N462K F proteins were completely resistant to 200 μM FIP, unlike the other mutants. It seems possible that FIP binds to and enhances the fusogenicity of these 2 mutants, and this scenario has been proposed for AS-48 and compound 3g (N-[3-cyanophenyl]-2-phenylacetamide) by another laboratory (23).
Comparison of membrane fusion mediated by F mutants in the presence and absence of FIP and AS-48 inhibitors using a luciferase-based fusion assay.
To quantify the membrane fusion activity of each F mutant in the presence of 50 μM FIP and 75 μM AS-48, luciferase-based fusion assays were performed. HEK293 cells were cotransfected with pCAGGS-EdmonstonH, pCAGGS-F, and pT7 promoter firefly luciferase expression plasmids. The transfected cells were allowed to fuse for 4 h with Vero cells expressing T7 polymerase in the presence or absence of MeV fusion inhibitors. For most of the F mutants, luciferase activity was evident whether FIP or AS-48 was present or not (Fig. 6A). In contrast, cells expressing parental wild-type MeV F exhibited an approximate 50% decrease in luciferase reporter activity when FIP and AS-48 were present (Fig. 6A). This inhibition occurred regardless of which cellular receptor (CD46, SLAM, or PVRL4) was expressed by the Vero cells (Fig. 6A, B, and C). Luciferase activity was never completely inhibited in the parental wild-type MeV F-expressing cells, despite the absence of visible syncytia in the cell lines by microscopy, yielding high background bioluminescence. It is possible that FIP and AS-48 may not prevent the formation of pores, allowing limited transfer of T7 luciferase reporter plasmid from 293T cells into Vero cells. Alternatively, some cell breakage may have occurred over the 4-h incubation period. The high background level seen when only the luciferase reporter vector was transfected into HEK293 cells (Fig. 5D) indicates that the sensitivity of the luciferase-based assay is somewhat limited. However, extensive fusion and widespread syncytium formation were visibly inhibited when the fusion inhibitors were present in cells expressing wild-type MeV F, but not the mutant F proteins (Fig. 5A). Again, as shown in Fig. 5E, the I452T and L454W mutants appeared more sensitive to the effects of FIP and AS-48 and the D458N, D458G, N462K, and G464E mutants were more resistant to the inhibitors.
FIG 6.
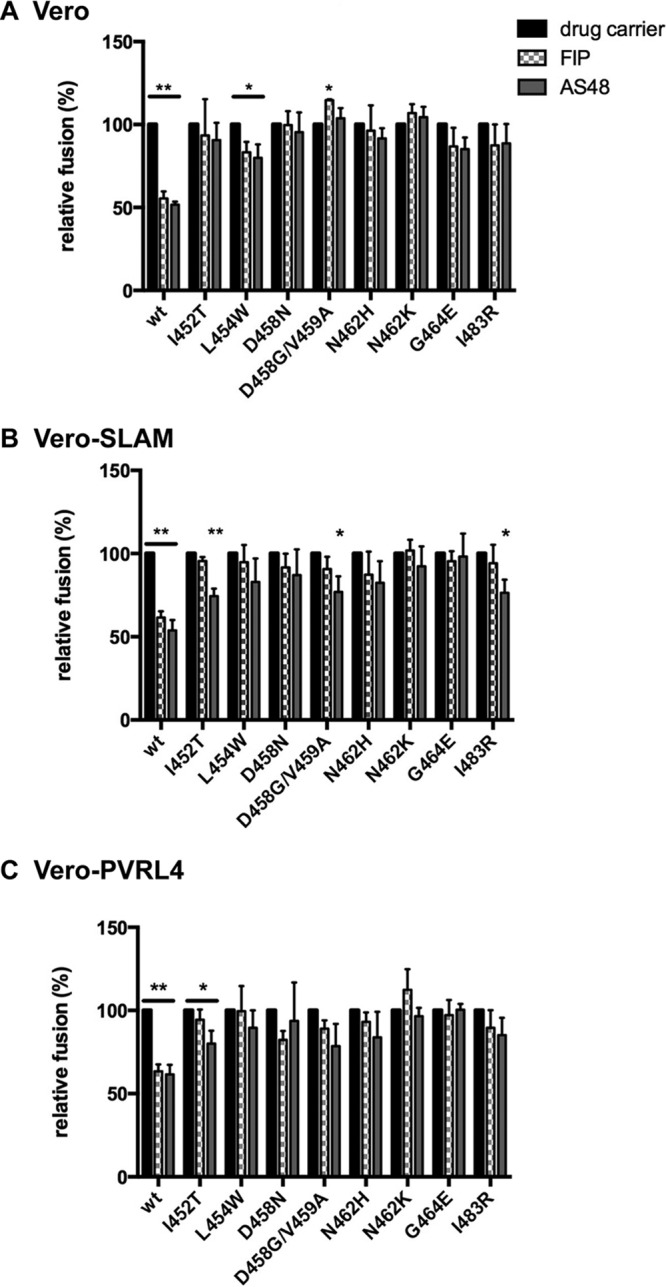
Quantitation of fusion in Vero (A), Vero-SLAM (B), and Vero-PVRL4 (C) cells expressing Edmonston H and FIP-resistant F proteins in the presence of FIP, AS-48, or 0.5% DMSO. Vero, Vero-SLAM, and Vero-PVRL4 cells were cotransfected with plasmids encoding T7 polymerase and renilla luciferase. At 20 h posttransfection, the cells were overlaid with HEK293 cells that had been cotransfected with expression plasmids encoding Edmonston H and firefly luciferase, together with WT or mutated Edmonston F. Fusion between the 2 cell lines was allowed to proceed in the presence of 50 μM FIP, 75 μM AS-48, or 0.5% DMSO. After 4 h, firefly luciferase bioluminescence was measured, and the effects of fusion inhibitors on the various F mutants were calculated as percentages of the bioluminescence in the absence of FIP and AS-48. The data are presented as the means of the results of three independent experiments, and the error bars represent SEM. Statistical probabilities (ANOVA P values) are indicated by asterisks (*, P < 0.05; **, P < 0.005).
FIP-resistant mutations are localized to a small region between the globular head and helical stalk of the prefusion conformation of F.
The mutation N462K was previously shown to confer resistance to AS-48, and a model of MeV F protein created by Prussia et al. indicated that changes in this region may destabilize a network of noncovalent interactions between HRB and the base of the globular head to favor a more efficient transition between the prefusion and postfusion conformations (57). We modeled the prefusion and postfusion structures of MeV F based on the published crystal structures of PIV5 F (36, 39). The FIP resistance mutations, shown in green and red, were mapped to the HRB-head junction regions in homology models of the prefusion conformation of MeV F protein (Fig. 7). In the prefusion conformation, the mutations conferring resistance to FIP localized to a small region present between the globular head and the linker region of the HRB stem. The I483R mutation was situated slightly farther down the stalk. An expanded ribbon structure of the MeV F prefusion model shows that the region containing the FIP resistance mutations (I452, L454, D458, V459, N462, and G464) lies below a central pocket in the globular head of the trimeric MeV F protein (Fig. 8). A similar hydrophobic cavity has recently been shown to exist in the prefusion crystal structure of the RSV F protein and has been implicated as a target and site of interaction for chemical inhibitors that inhibit the membrane fusion activity in these cocrystallization studies (73). The results reported in this paper do not prove a mechanism of action for FIP, but our resistance mutations parallel other studies performed with AS-48 and compound 3g, using measles and canine distemper viruses, respectively (57, 84). These authors suggest that resistance mutations, either directly or indirectly, prevent high-affinity binding of the small-molecule inhibitors to the pocket in order to stabilize prefusion F. Alternatively, they propose that the mutations could lower the activation threshold of prefusion F to facilitate a more efficient conformational change, leading to membrane fusion in the presence of the inhibitors (23).
FIG 7.
Homology space-filling models of the head and stalk regions of MeV F protein mutants predicted from the prefusion PIV5 F protein. The parental sequence for the MeV Edmonston F protein used in this study corresponded to GenBank accession no. 9626950, except for a glutamic acid at residue position 460 that was found in our isolate. The highest-scoring hits corresponded to PDB entries for paramyxovirus fusion proteins (1ZTM, 5EJB, 1G5G, and 2B9B), with sequence identities ranging from 28 to 35%. We used 2B9B (parainfluenza virus 5 F protein) (39) as a template to model the prefusion state of the MeV F protein. The HHpred-based alignments were used for the subsequent 3D modeling with MODELLER 9.9 (97). FIP-resistant MeV F amino acid mutations cluster in a small pocket between the head and stalk regions of the prefusion conformation. The predicted locations for mutations I452T, L454W, D458G, D458N, D458G/V459A, N462H, N462K, G464R, G464E, and I483R are indicated. Detailed views of the mutations in the HRB region of the prefusion state are shown, with green carbon and red oxygen atoms. The three chains of the helical stem are colored light gray, dark gray, and black, respectively. The space-filling model shows solvent-accessible surfaces on the globular head of the trimer. For the G464R mutations, the R464 side chain is mostly obscured and not solvent exposed.
FIG 8.
Homology model of the MeV F protein based on the PIV5 F protein in the prefusion state (PDB ID 2B9B). 3D modeling of the prefusion form of MeV F was performed with MODELLER 9.9 (97). The image was generated with the PyMOL molecular graphics system. The individual chains of the trimer are represented in ribbon format and colored red, green, and blue. Side chains that are substituted in the resistant viruses are indicated as van der Waals spheres in darker colors and occur at the junction between HRB and the globular head. Residues from the red chain that are frequently mutated are indicated with labels (I452, L454, D458, V459, N462, and G464).
DISCUSSION
Paramyxovirus infections constitute a large portion of biologically and economically important diseases of humans and livestock. Understanding the mechanism used by these viruses to gain entry into a host could lead to the development of novel entry inhibitors to prevent or treat active infections. FIP was developed as a specific fusion inhibitor over 3 decades ago and has been used extensively to study and block MeV entry. However, the underlying mechanism through which it works has not been reported. By analogy to recent studies with an inhibitor, AS-48, we provide evidence through resistance mutations that FIP targets a site on the F protein and interferes with the conformational change required to elicit membrane fusion.
In the present study, we investigated the mechanism of action of FIP through generation of spontaneous escape mutants that were resistant to the inhibitor. Our screen isolated 10 mutants, 8 of which were shown to be functional when expressed in conjunction with H in Vero cells expressing the known cellular receptors for MeV. Overall, both FIP and AS-48 were able to inhibit nonvariant strains of Edmonston and wild-type MeV IC323 at almost equal concentrations. All of the FIP-resistant mutations localized to a small region between the head and stalk regions of the prefusion conformation of MeV F. The same mutations also conferred resistance to AS-48.
As a prelude to the development of AS-48, Plemper et al. originally demonstrated that mutation of Val94 modulates F fusogenicity and also confers resistance to FIP (54, 66). A homology model based on a postfusion structure of paramyxovirus F described this residue as helping to define a small conserved hydrophobic cavity involving the HRC region at the C-terminal end of F2 (85). The cavity microdomain served as a basis for structure-informed drug identification (53–55). The lead candidates, OX-1 and AM-4, had 50% inhibitory concentrations (IC50s) of 50 μM and 260 nM, respectively, against MeV Edmonston and specifically inhibited virus entry and syncytium formation (54). The V94A mutation conferred resistance to OX-1, as was previously shown for FIP. AM-4 was subsequently subjected to synthetic optimization, which resulted in a first-generation lead, AS-48, with lower cell toxicity, increased stability, and potent inhibitory activity against a variety of wild-type MeV isolates (53, 55). The analogous properties and activity profiles between AS-48 and FIP prompted us to perform the studies outlined here.
Plemper and colleagues went on to isolate spontaneous escape mutants by growing Edmonston vaccine or Khartoum Sudan (KS) wild-type MeV in the presence of OX-1 and AM-4 and isolating resistant variants by plaque purification (56). The major resistance mutations were N462S, N462D, and N462K, which are found in the HRB region. Another mutation included A367T. After the prefusion structure of PIV5 became available (39), a homology model for MeV F was used to propose the following mechanism of action and the development of resistance mutations for AS-48 (57). The mutations N462S, N462D, N462K, and A367T were believed to function in a region comprised of HRB, HRB linker, and the globular head (domain I) that functions as a “conformational switch.” The changes to more hydrophilic amino acid residues at this site decreased the activation threshold to facilitate transition to the postfusion F trimer. AS-48 was proposed to interact with an intermediate structure along the fusion conformational pathway and to interfere with the rearrangements bringing HRB in proximity to HRA that were needed to form the postfusion 6HB. Binding of AS-48 to the intermediate would increase the energy barrier from the intermediate to the postfusion structure. In our recent experiments, escape mutations to FIP (I452T, L454W, D458G, D458N, D458G/V459A, N462H, N462K, G464E, G464R, and I483R) that could possibly lower the activation energy of fusion also occurred in HRB of MeV F. Russell and colleagues have similarly reported that mutating amino acid residues L447 and I449 in the fusion protein of PIV5 (equivalent to L457 and V459 in MeV, respectively) reduces the activation barrier of the metastable prefusion F, with aromatic substitutions promoting hyperactive fusion (86). We suggest that FIP employs a mechanism of action similar to that of AS-48 to inhibit the conformation change in MeV F and that escape mutations against each inhibitor act to destabilize the prefusion F and promote the conformational change leading to postfusion F and membrane fusion.
Interestingly, chemical fusion inhibitors of RSV were originally thought to bind to a late-stage fusion intermediate of RSV F and block the transition to the 6HB postfusion state (87, 88). However, recent work has demonstrated that escape mutations from all RSV entry inhibitors characterized so far are located at the intersection of the prefusion RSV R head and stalk domains (72). Similar to A-48-resistant MeV F, the resistant RSV F investigated in this study displayed enhanced fusion activity. Subsequently, cocrystal structures of several of these entry inhibitors (JNJ-2408068, JNJ-49153390, TMC-353121, and BMS-433771) in complex with prefusion F compounds showed that the compounds insert into the central cavity of the prefusion F trimer (73). The report suggests that RSV resistance to entry inhibitors is based on a combination of kinetic and primary resistances (89). These data suggest that these antagonists tether the fusion peptide to HRB and stabilize the prefusion conformation through aromatic stacking interactions, as well as additional electrostatic interactions. Some resistance mutations, although predicted to reduce viral fitness (73), were found to remain fully pathogenic in a mouse model of RSV disease (72), raising the concern that clinical use of RSV entry inhibitors may drive the development of preexisting resistance in circulating strains. In analogy to the RSV F cocrystal structure, we predict that crystallization of MeV F with AS-48 or FIP will likely reveal docking of these compounds into the central cavity of the globular head just above the HRB domain (Fig. 8).
Our studies with FIP have much in common with those performed with AS-38 and the RSV fusion inhibitors. Resistance mutations frequently occurred in the HRB region, but never in the HRA region. In our experiments, FIP escape mutations occurred exclusively in HRB (I452T, L454W, D458G, D458N, D458G/V459A, N462H, N462K, G464E, G464R, and I483R) and did not appear near the F peptide (V94M) or the cysteine-rich region (A362T), as was previously observed using AS-48. Removal of inhibitor selection allowed the FIP-resistant viruses to rapidly revert to the original phenotype. It is most likely that FIP binds to the prefusion form of F, as was confirmed recently for the AS-48 scaffold, using antibodies that are specific for prefusion F of MeV and canine distemper virus (CDV) (84). Future experiments with 186CA and 19GD6 monoclonal antibodies specific for prefusion MeV F and postfusion MeV, respectively (84), could be used to determine whether FIP also stabilizes the prefusion F and increases the activation energy required for transition to its fusogenic form. Alternatively, fast photochemical oxidative footprinting of solvent-exposed amino acid side chains coupled with high-resolution mass spectrometry could be used to follow the prefusion-to-postfusion kinetics, as was done with simian virus 5 (SV5) F (90). The effect of FIP on oxidative footprinting could be used to monitor the MeV F transition. Ultimately crystallization and structural determination of the prefusion form of MeV F in the presence of FIP will be the most definitive means to determine the mechanism of action and atomic interactions of the inhibitor. Another closely related possibility is that FIP and AS-48 inhibit the interaction of the trigger region on the stalk of H protein (residues 110 to 118) with hydrophobic acids in the IgG-like fold of domain II in the globular head of F (23). Again, this scenario can be verified only with structural cocrystallization studies of F, H, and inhibitor complexes However, complementary resistance mutations in the H protein were never observed in our studies, suggesting that F is the primary target for FIP and AS-48.
Inhibitors directed against MeV F may have some value for postexposure prophylactic anti-MeV therapy to limit outbreaks in conjunction with vaccination programs (11). Interestingly, successful postexposure prophylaxis against lethal morbillivirus disease was recently demonstrated with an orally bioavailable polymerase inhibitor in a CDV ferret model (91). Combination therapies of entry and polymerase inhibitors are conceivable and will likely lower the frequency of viral escape from inhibition. In addition, antivirals against MeV may be more valuable in preventing persistent infections that cause neurological disease, such as measles inclusion body encephalitis (MIBE) or subacute sclerosing panencephalitis (SSPE) (14). They could also be useful in controlling infections, such as giant-cell pneumonia, in individuals who are immune suppressed (92). Fusion inhibitors, such as GS-5806, have shown some success in early-phase clinical trials against RSV (77), but as yet, no truly effective antiviral or vaccine has been approved for the prevention or treatment of RSV infection (78). While the clinical impact of entry inhibitors against pneumoviruses and paramyxoviruses remains uncertain, available compounds unquestionably represent valuable tools that can aid in dissecting the mechanics of F protein-mediated membrane fusion.
MATERIALS AND METHODS
Reagents, cells, and virus.
The Edmonston strain of MeV was originally obtained from Erling Norrby (Karolinska Institute, Stockholm, Sweden) and was passaged on Vero cells. Edmonston strain and wild-type IC323 MeVs containing the eGFP reporter gene were supplied by R. Cattaneo. Vero cells and HEK293 cells were obtained from the American Type Culture Collection (Rockville, MD) and maintained at 37°C in Dulbecco's modified Eagle's medium (DMEM) (Wisent, St. Bruno, QC, Canada), 10% (vol/vol) fetal calf serum (Wisent), 10 μg/ml gentamicin (Invitrogen, Mississauga, ON, Canada), and 0.25 μg/ml amphotericin B (Fungizone; Invitrogen). Vero-SLAM cells were produced by transfecting pcDNA3.1-SLAM into Vero cells and selecting for G418 (Invitrogen)-resistant colonies. They were maintained in DMEM supplemented with 800 μg/ml G418. FIP was purchased from Sigma-Aldrich (Oakville, ON, Canada) and solubilized in either 80% (wt/vol) ethanol–phosphate-buffered saline (PBS) or dimethyl sulfoxide (DMSO) as a concentrated stock solution of 10 mM. AS-48, a measles fusion inhibitor, was obtained from R. Plemper and dissolved in DMSO as a concentrated stock solution of 50 mM.
Virus infection and titer determination.
Virus was diluted in Opti-MEM (Invitrogen) in the minimum volume necessary to cover the monolayer for infection. Vero cells were infected at 37°C for 1.5 h while being rocked at regular intervals. The titer of MeV was determined on Vero cells using a modified TCID50 protocol (93).
Production of FIP-resistant measles virus.
FIP-resistant MeV was produced by passaging the virus in the presence of FIP. Vero cells were infected at a multiplicity of infection (MOI) of 0.1 and overlaid with 1% (wt/vol) SeaPlaque agarose-DMEM in the presence of 50 or 100 μM FIP. Well-isolated single plaques were picked at 72 h postinfection (hpi). The virus was further amplified in 6-well plates in the presence of 100 μM FIP.
Cloning the fusion and hemagglutinin genes from FIP-resistant measles virus.
A single well of a 6-well plate of Vero cells was infected with FIP-resistant MeV in the presence of FIP. When approximately 90% of the cells had fused, total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's protocols. Reverse transcription reactions were carried out using Moloney leukemia virus reverse transcriptase (Invitrogen) following the manufacturer's protocols and using 0.5 μg of RNA and a random-hexamer primer. PCR was performed using 1 unit of Taq polymerase (Invitrogen), 0.1 unit of Pfu polymerase (Agilent Technologies, Cedar Creek, TX), and the following primers: F5′, CGGAATTCCATGGTAATGTCCATCATGGGTCTCAAGG; F3′, ACTGAACCTGAGGTCAGAGCGACCTTACATAGG; H5′, CGGAATTCCATGGTAATGTCACCACAACGAGACC; and H3′, CTGCAGAACCAGGGCATTGGCTATCTGCGATTGGTTCCATCTTCC.
A single 1.8-kb band was extracted from the agarose gel following gel electrophoresis using a QIA extraction kit (Qiagen, Mississauga, ON, Canada). The purified band was digested with BstXI and EcoRI for H and Bsu36I and EcoRI for F and ligated with a similarly digested pCAGGS vector (94). Amplified plasmid DNA was verified via restriction digestion and sequencing analysis (ACGT Corporation, Toronto, ON, Canada).
Luciferase fusion assay.
HEK293 cells (3 × 105 cells/12 wells) were transfected with 0.33 μg each of pCAGGS-H and pCAGGS-F and 1 μg of T7 luciferase reporter plasmid, which contains a firefly luciferase gene under the T7 promoter. In parallel, Vero cells (3 × 105 cells/well) were transfected with a plasmid that expresses T7 polymerase under the CMV promoter and a renilla luciferase expression plasmid. All transfections were carried out using Lipofectamine 2000 (Invitrogen) following the manufacturer's protocols. Twenty hours posttransfection, the HEK293 cells were detached by addition of cell dissociation buffer (Sigma) and added to the T7 polymerase-transfected Vero cells in the presence of varying amounts of FIP. The cell lysates were harvested after 4 h in cell culture lysis buffer (Promega, Madison, WI) at 4°C, and dual firefly and renilla luciferase activity was read using a Glomax luminometer (Promega).
Cell surface protein biotinylation and pulldown.
HEK293 cells (1 × 106 cells/6 wells) were transfected with 0.5 μg each of pCAGGS-H and pCAGGS-F; 17 h posttransfection, the cells were washed with PBS and resuspended, and surface proteins were biotinylated with 2 mM EZ-Link Sulfo-NHS-Biotin (Thermo Scientific, Rockford, IL) for 1 h at 4°C. Adding serum-free medium quenched the reaction. Following washes with PBS, the cells were lysed in 300 μl RIPA (radioimmunoprecipitation assay) buffer (50 mM Tris-HCl, pH 7.4, 1% [wt/vol] NP-40, 0.25% sodium deoxycholate, 150 mM sodium chloride, 1 mM EDTA) supplemented with protease inhibitors (Complete protease inhibitor cocktail [Roche, Mississauga, ON, Canada], 1 mM phenylmethylsulfonyl fluoride [PMSF], 1 mM sodium orthovanadate, 1 mM sodium fluoride). Neutravidin agarose resin (150 μl; Thermo Scientific) was added to the lysate, and the tubes were rocked at 4°C overnight. After adsorption, the resin was washed 3 times with RIPA buffer and resuspended in reducing sample buffer for immunoblotting. Densitometric analysis of the immunoblot was performed using the software accompanying the Kodak Image Station.
Western immunoblot analysis.
Sodium dodecyl sulfate-polyacrylamide (9%) gel electrophoresis (SDS-PAGE) was performed, and proteins were transferred onto a polyvinylidene fluoride membrane. The membrane was blocked with 5% (wt/vol) skim milk in PBS-Tween (0.5%) for 1 h and probed with antisera against the carboxy terminus of the F protein (95) at 1:1,000 dilution overnight. After washing, the membrane was probed with horseradish peroxidase-conjugated anti-rabbit antibody and developed using the ECL Plus reagent (GE Healthcare, Baie d'Urfe, QC, Canada) on a Kodak Image Station 4000MM (Mandel Scientific, Guelph, ON, Canada).
MeV binding assay.
To assess whether FIP prevented measles virus binding to the cell surface, Vero cells were incubated with 100 PFU/cell of MeV (Edmonston) for 90 min on ice in the presence or absence of 100 μM FIP. The cells were washed three times with PBS containing 1% (wt/vol) bovine serum albumin (BSA), 5 mM EDTA, and 0.1% (wt/vol) sodium azide and incubated with an anti-MeV hemagglutinin antibody (MAB8905; Millipore, Billerica, MA) on ice for 60 min. The cells were washed prior to incubation with an Alexa Fluor 488-conjugated anti-mouse antibody for 45 min on ice. The cells were washed again to remove any unbound antibodies, fixed in 1% (wt/vol) paraformaldehyde, and run on a Cyan ADP flow cytometer (Beckman Coulter). Data were processed using FCS Express software (De Novo Software, Glendale CA).
Homology modeling of F mutant structures.
HHpred (96) was used to search for profile hidden Markov models based on structures in the Protein Data Bank (PDB) that were compatible with the MeV F protein sequence. The wild-type sequence for the MeV-Edm F protein used in this study corresponded to GenBank accession no. U03657.1, except for a glutamic acid at residue position 460. The highest-scoring hits corresponded to PDB entries for paramyxovirus fusion proteins (1ZTM, 5EJB, 1G5G, and 2B9B), with sequence identities ranging from 28 to 35%. We used 2B9B (parainfluenza virus 5 F protein) (39) as a template to model the prefusion state and 1ZTM (paramyxovirus [hPIV3] F protein) (36) to model the postfusion state. The HHpred-based alignments were used for the subsequent 3D modeling with MODELLER 9.9 (97). Models for wild-type and mutant MeV F proteins were generated for residues 26 to 489 based on structure 2B9B and for residues 27 to 487 based on structure 1ZTM. Trimers were generated by including three chains (labeled A, B, and C) for both the input sequence and the template model. In order to preserve the 3-fold symmetry of both the pre- and postfusion conformations of the trimers, restraints were applied between all C-α atoms between the pairs of chains (A-B), (B-C), and (A-C). All MeV F mutant structures were generated independently from the template PDB files 2B9B and 1ZTM. In all cases, the mutant structures were highly similar to the wild-type structures with either the pre- or postfusion template. The figures were generated with the PyMOL molecular graphics system.
Microscopy.
Cells were viewed by phase-contrast and fluorescence microscopy using a Leica DMI4000B inverted microscope (Leica Microsystems, Concord, ON, Canada). A Zeiss LSM 510 microscope (Carl Zeiss Canada) was used to acquire images by oil immersion. In order to observe syncytia, cells were fixed and stained with Hema3 solution (Fisher Scientific) according to the manufacturer's instructions.
Data analysis.
Data are expressed as means ± SEM. Analysis of variance (ANOVA) was performed to identify statistically significant differences. P values below 0.05 were considered statistically significant. The data were processed using GraphPad (La Jolla, CA) Instat software.
Ethics statement.
The experiments reported in this article were performed at biological safety level 2 in accordance with the regulations set forth by the Public Health Agency of Canada and the Canada Food and Drug Inspection Agency. This work did not involve experimentation with animals or human beings.
ACKNOWLEDGMENTS
This work was supported by grants from the Canadian Institute for Health Research (MOP 10638, MOP 114949, and MOP 142775) and the Nova Scotia Health Research Foundation to C.D.R. In addition, the work was partly supported by an NIH/NIAID Public Health Service Grant (AI071002) awarded to R.K.P. C.D.R. is a Canada Research Chair (Tier I) in Vaccinology and Viral Therapeutics and received an equipment grant from the Canadian Foundation for Innovation. M.N.H. held a Natural Sciences and Engineering Research Council (NSERC) doctoral research award. R.S.N. was supported by a CIHR Banting Postdoctoral Fellowship. S.D. was supported by a trainee award from the Beatrice Hunter Cancer Research Institute with funds provided by Cancer Care Nova Scotia as part of the Terry Fox Strategic Health Research Training Program in Cancer Research at CIHR.
We thank Natalie Mishreky for help in preparing the figures and Angelita Alcos for technical help.
M.N.H., D.B., and C.D.R. conceived and designed experiments. M.N.H., S.D., R.S.N., D.B., G.S., and C.D.R. performed experiments. M.N.H., S.D., R.S.N., L.-T.L., R.K.P., G.G.P., and C.D.R. analyzed the data. K.M.B. analyzed and presented the data in Fig. 2. M.N.H., R.K.P., G.G.P., and C.D.R. wrote the paper.
REFERENCES
- 1.Moss WJ, Griffin DE. 2012. Measles. Lancet 379:153–164. doi: 10.1016/S0140-6736(10)62352-5. [DOI] [PubMed] [Google Scholar]
- 2.Griffin DE. 2013. Measles virus, p 1042–1069. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 1 Lippincott, Williams, & Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Dabbagh A, Gacic-Dobo M, Simons E, Featherstone D, Strebel P, Okwo-Bele JM, Hoekstra E, Chopra M, Uvicanin A, Cochi S. 2009. Global measles mortality, 2000-2008. MMWR Morb Mortal Wkly Rep 58:1321–1326.19959985 [Google Scholar]
- 4.Moss WJ. 2009. Measles control and the prospect of eradication. Curr Top Microbiol Immunol 330:173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perry RT, Dabbagh AJ, Gacic-Dobo M, Liu JL, Simons EA, Featherstone DA, Strebel PM, Okwo-Bele JM, Goodson JL. 2012. Progress in global measles control, 2000-2010. MMWR Morb Mortal Wkly Rep 61:73–78.22298303 [Google Scholar]
- 6.Perry RT, Gacic-Dobo M, Dabbagh A, Mulders MN, Strebel PM, Okwo-Bele JM, Rota PA, Goodson JL, Centers for Disease Control and Prevention. 2014. Global control and regional elimination of measles, 2000-2012. MMWR Morb Mortal Wkly Rep 63:103–107. [PMC free article] [PubMed] [Google Scholar]
- 7.Tanne JH. 2014. Rise in US measles cases is blamed on unimmunized travelers. BMJ 348:g3478. doi: 10.1136/bmj.g3478. [DOI] [PubMed] [Google Scholar]
- 8.Gastanaduy PA, Redd SB, Fiebelkorn AP, Rota JS, Rota PA, Bellini WJ, Seward JF, Wallace GS, Division of Viral Disease National Center for Immunization and Respiratory Diseases, CDC. 2014. Measles-United States, January 1-May 23, 2014. MMWR Morb Mortal Wkly Rep 63:496–499. [PMC free article] [PubMed] [Google Scholar]
- 9.D'Souza RM, D'Souza R. 2002. Vitamin A for preventing secondary infections in children with measles—a systematic review. J Trop Pediatr 48:72–77. doi: 10.1093/tropej/48.2.72. [DOI] [PubMed] [Google Scholar]
- 10.Trottier C, Colombo M, Mann KK, Miller WH Jr, Ward BJ. 2009. Retinoids inhibit measles virus through a type I IFN-dependent bystander effect. FASEB J 23:3203–3212. doi: 10.1096/fj.09-129288. [DOI] [PubMed] [Google Scholar]
- 11.Plemper RK, Hammond AL. 2014. Synergizing vaccinations with therapeutics for measles eradication. Expert Opin Drug Discov 9:201–214. doi: 10.1517/17460441.2014.867324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Russell SJ, Peng KW. 2009. Measles virus for cancer therapy. Curr Top Microbiol Immunol 330:213–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Msaouel P, Opyrchal M, Dispenzieri A, Peng KW, Federspiel MJ, Russell SJ, Galanis E. 22 February 2017. Clinical trials with oncolytic measles virus: current status and future prospects. Curr Cancer Drug Targets. doi: 10.2174/1568009617666170222125035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Welsch JC, Talekar A, Mathieu C, Pessi A, Moscona A, Horvat B, Porotto M. 2013. Fatal measles virus infection prevented by brain-penetrant fusion inhibitors. J Virol 87:13785–13794. doi: 10.1128/JVI.02436-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Griffin DE, Knipe DM, Lamb RA, Martin MA, Roizman B, Straus SE. 2007. Measles virus, p 1551–1586. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 16.Noyce RS, Bondre DG, Ha MN, Lin LT, Sisson G, Tsao MS, Richardson CD. 2011. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog 7:e1002240. doi: 10.1371/journal.ppat.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muhlebach MD, Mateo M, Sinn PL, Prufer S, Uhlig KM, Leonard VH, Navaratnarajah CK, Frenzke M, Wong XX, Sawatsky B, Ramachandran S, McCray PB Jr, Cichutek K, von Messling V, Lopez M, Cattaneo R. 2011. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 480:530–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Navaratnarajah CK, Oezguen N, Rupp L, Kay L, Leonard VH, Braun W, Cattaneo R. 2011. The heads of the measles virus attachment protein move to transmit the fusion-triggering signal. Nat Struct Mol Biol 18:128–134. doi: 10.1038/nsmb.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hashiguchi T, Ose T, Kubota M, Maita N, Kamishikiryo J, Maenaka K, Yanagi Y. 2011. Structure of the measles virus hemagglutinin bound to its cellular receptor SLAM. Nat Struct Mol Biol 18:135–141. doi: 10.1038/nsmb.1969. [DOI] [PubMed] [Google Scholar]
- 20.Plemper RK, Brindley MA, Iorio RM. 2011. Structural and mechanistic studies of measles virus illuminate paramyxovirus entry. PLoS Pathog 7:e1002058. doi: 10.1371/journal.ppat.1002058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jardetzky TS, Lamb RA. 2014. Activation of paramyxovirus membrane fusion and virus entry. Curr Opin Virol 5:24–33. doi: 10.1016/j.coviro.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bose S, Jardetzky TS, Lamb RA. 2015. Timing is everything: Fine-tuned molecular machines orchestrate paramyxovirus entry. Virology 479-480:518–531. doi: 10.1016/j.virol.2015.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plattet P, Alves L, Herren M, Aguilar HC. 2016. Measles virus fusion protein: structure, function and inhibition. Viruses 8:112. doi: 10.3390/v8040112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JK, Prussia A, Paal T, White LK, Snyder JP, Plemper RK. 2008. Functional interaction between paramyxovirus fusion and attachment proteins. J Biol Chem 283:16561–16572. doi: 10.1074/jbc.M801018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ader N, Brindley MA, Avila M, Origgi FC, Langedijk JP, Orvell C, Vandevelde M, Zurbriggen A, Plemper RK, Plattet P. 2012. Structural rearrangements of the central region of the morbillivirus attachment protein stalk domain trigger F protein refolding for membrane fusion. J Biol Chem 287:16324–16334. doi: 10.1074/jbc.M112.342493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Apte-Sengupta S, Navaratnarajah CK, Cattaneo R. 2013. Hydrophobic and charged residues in the central segment of the measles virus hemagglutinin stalk mediate transmission of the fusion-triggering signal. J Virol 87:10401–10404. doi: 10.1128/JVI.01547-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Navaratnarajah CK, Kumar S, Generous A, Apte-Sengupta S, Mateo M, Cattaneo R. 2014. The measles virus hemagglutinin stalk: structures and functions of the central fusion activation and membrane-proximal segments. J Virol 88:6158–6167. doi: 10.1128/JVI.02846-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bose S, Zokarkar A, Welch BD, Leser GP, Jardetzky TS, Lamb RA. 2012. Fusion activation by a headless parainfluenza virus 5 hemagglutinin-neuraminidase stalk suggests a modular mechanism for triggering. Proc Natl Acad Sci U S A 109:E2625–E2634. doi: 10.1073/pnas.1213813109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brindley MA, Suter R, Schestak I, Kiss G, Wright ER, Plemper RK. 2013. A stabilized headless measles virus attachment protein stalk efficiently triggers membrane fusion. J Virol 87:11693–11703. doi: 10.1128/JVI.01945-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Apte-Sengupta S, Negi S, Leonard VH, Oezguen N, Navaratnarajah CK, Braun W, Cattaneo R. 2012. Base of the measles virus fusion trimer head receives the signal that triggers membrane fusion. J Biol Chem 287:33026–33035. doi: 10.1074/jbc.M112.373308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Avila M, Khosravi M, Alves L, Ader-Ebert N, Bringolf F, Zurbriggen A, Plemper RK, Plattet P. 2015. Canine distemper virus envelope protein interactions modulated by hydrophobic residues in the fusion protein globular head. J Virol 89:1445–1451. doi: 10.1128/JVI.01828-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baker KA, Dutch RE, Lamb RA, Jardetzky TS. 1999. Structural basis for paramyxovirus-mediated membrane fusion. Mol Cell 3:309–319. doi: 10.1016/S1097-2765(00)80458-X. [DOI] [PubMed] [Google Scholar]
- 33.Zhao X, Singh M, Malashkevich VN, Kim PS. 2000. Structural characterization of the human respiratory syncytial virus fusion protein core. Proc Natl Acad Sci U S A 97:14172–14177. doi: 10.1073/pnas.260499197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen L, Gorman JJ, McKimm-Breschkin J, Lawrence LJ, Tulloch PA, Smith BJ, Colman PM, Lawrence MC. 2001. The structure of the fusion glycoprotein of Newcastle disease virus suggests a novel paradigm for the molecular mechanism of membrane fusion. Structure 9:255–266. doi: 10.1016/S0969-2126(01)00581-0. [DOI] [PubMed] [Google Scholar]
- 35.Yu M, Wang E, Liu Y, Cao D, Jin N, Zhang CW, Bartlam M, Rao Z, Tien P, Gao GF. 2002. Six-helix bundle assembly and characterization of heptad repeat regions from the F protein of Newcastle disease virus. J Gen Virol 83:623–629. doi: 10.1099/0022-1317-83-3-623. [DOI] [PubMed] [Google Scholar]
- 36.Yin HS, Paterson RG, Wen X, Lamb RA, Jardetzky TS. 2005. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc Natl Acad Sci U S A 102:9288–9293. doi: 10.1073/pnas.0503989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swanson K, Wen X, Leser GP, Paterson RG, Lamb RA, Jardetzky TS. 2010. Structure of the Newcastle disease virus F protein in the post-fusion conformation. Virology 402:372–379. doi: 10.1016/j.virol.2010.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McLellan JS, Yang Y, Graham BS, Kwong PD. 2011. Structure of respiratory syncytial virus fusion glycoprotein in the postfusion conformation reveals preservation of neutralizing epitopes. J Virol 85:7788–7796. doi: 10.1128/JVI.00555-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439:38–44. doi: 10.1038/nature04322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Welch BD, Liu Y, Kors CA, Leser GP, Jardetzky TS, Lamb RA. 2012. Structure of the cleavage-activated prefusion form of the parainfluenza virus 5 fusion protein. Proc Natl Acad Sci U S A 109:16672–16677. doi: 10.1073/pnas.1213802109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McLellan JS, Chen M, Joyce MG, Sastry M, Stewart-Jones GB, Yang Y, Zhang B, Chen L, Srivatsan S, Zheng A, Zhou T, Graepel KW, Kumar A, Moin S, Boyington JC, Chuang GY, Soto C, Baxa U, Bakker AQ, Spits H, Beaumont T, Zheng Z, Xia N, Ko SY, Todd JP, Rao S, Graham BS, Kwong PD. 2013. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 342:592–598. doi: 10.1126/science.1243283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McLellan JS, Chen M, Leung S, Graepel KW, Du X, Yang Y, Zhou T, Baxa U, Yasuda E, Beaumont T, Kumar A, Modjarrad K, Zheng Z, Zhao M, Xia N, Kwong PD, Graham BS. 2013. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 340:1113–1117. doi: 10.1126/science.1234914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu K, Chan YP, Bradel-Tretheway B, Akyol-Ataman Z, Zhu Y, Dutta S, Yan L, Feng Y, Wang LF, Skiniotis G, Lee B, Zhou ZH, Broder CC, Aguilar HC, Nikolov DB. 2015. Crystal structure of the pre-fusion Nipah virus fusion glycoprotein reveals a novel hexamer-of-trimers assembly. PLoS Pathog 11:e1005322. doi: 10.1371/journal.ppat.1005322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wong JJ, Paterson RG, Lamb RA, Jardetzky TS. 2016. Structure and stabilization of the Hendra virus F glycoprotein in its prefusion form. Proc Natl Acad Sci U S A 113:1056–1061. doi: 10.1073/pnas.1523303113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang A, Masante C, Buchholz UJ, Dutch RE. 2012. Human metapneumovirus (HMPV) binding and infection are mediated by interactions between the HMPV fusion protein and heparan sulfate. J Virol 86:3230–3243. doi: 10.1128/JVI.06706-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Richardson CD, Scheid A, Choppin PW. 1980. Specific inhibition of paramyxovirus and myxovirus replication by oligopeptides with amino acid sequences similar to those at the N-termini of the F1 or HA2 viral polypeptides. Virology 105:205–222. doi: 10.1016/0042-6822(80)90168-3. [DOI] [PubMed] [Google Scholar]
- 47.Miller FA, Dixon GJ, Arnett G, Dice JR, Rightsel WA, Schabel FM Jr, McLean IW Jr. 1968. Antiviral activity of carbobenzosy di- and tripeptides on measles virus. Appl Microbiol 16:1489–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nicolaides E, DeWald H, Lipnik M, Westland R, Posler J. 1968. Potential antiviral agents. Carbobenzoxy di- and tripeptides active against measles and herpes viruses. J Med Chem 11:74–79. [DOI] [PubMed] [Google Scholar]
- 49.Norrby E. 1971. The effect of a carbobenzoxy tripeptide on the biological activities of measles virus. Virology 44:599–608. doi: 10.1016/0042-6822(71)90374-6. [DOI] [PubMed] [Google Scholar]
- 50.Norrby E, Sievertsson H. 1974. Effect of certain polypeptides on the biological activities of measles virus. Antimicrob Agents Chemother 5:426–430. doi: 10.1128/AAC.5.4.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dorig RE, Marcil A, Chopra A, Richardson CD. 1993. The human CD46 molecule is a receptor for measles virus (Edmonston strain). Cell 75:295–305. doi: 10.1016/0092-8674(93)80071-L. [DOI] [PubMed] [Google Scholar]
- 52.Dorig RE, Marcil A, Richardson CD. 1994. CD46, a primate-specific receptor for measles virus. Trends Microbiol 2:312–318. doi: 10.1016/0966-842X(94)90447-2. [DOI] [PubMed] [Google Scholar]
- 53.Plemper RK, Doyle J, Sun A, Prussia A, Cheng LT, Rota PA, Liotta DC, Snyder JP, Compans RW. 2005. Design of a small-molecule entry inhibitor with activity against primary measles virus strains. Antimicrob Agents Chemother 49:3755–3761. doi: 10.1128/AAC.49.9.3755-3761.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Plemper RK, Erlandson KJ, Lakdawala AS, Sun A, Prussia A, Boonsombat J, Aki-Sener E, Yalcin I, Yildiz I, Temiz-Arpaci O, Tekiner B, Liotta DC, Snyder JP, Compans RW. 2004. A target site for template-based design of measles virus entry inhibitors. Proc Natl Acad Sci U S A 101:5628–5633. doi: 10.1073/pnas.0308520101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun A, Prussia A, Zhan W, Murray EE, Doyle J, Cheng LT, Yoon JJ, Radchenko EV, Palyulin VA, Compans RW, Liotta DC, Plemper RK, Snyder JP. 2006. Nonpeptide inhibitors of measles virus entry. J Med Chem 49:5080–5092. doi: 10.1021/jm0602559. [DOI] [PubMed] [Google Scholar]
- 56.Doyle J, Prussia A, White LK, Sun A, Liotta DC, Snyder JP, Compans RW, Plemper RK. 2006. Two domains that control prefusion stability and transport competence of the measles virus fusion protein. J Virol 80:1524–1536. doi: 10.1128/JVI.80.3.1524-1536.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Prussia AJ, Plemper RK, Snyder JP. 2008. Measles virus entry inhibitors: a structural proposal for mechanism of action and the development of resistance. Biochemistry 47:13573–13583. doi: 10.1021/bi801513p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Richardson CD, Choppin PW. 1983. Oligopeptides that specifically inhibit membrane fusion by paramyxoviruses: studies on the site of action. Virology 131:518–532. doi: 10.1016/0042-6822(83)90517-2. [DOI] [PubMed] [Google Scholar]
- 59.Epand RM, Epand RF, Richardson CD, Yeagle PL. 1993. Structural requirements for the inhibition of membrane fusion by carbobenzoxy-D-Phe-Phe-Gly. Biochim Biophys Acta 1152:128–134. doi: 10.1016/0005-2736(93)90239-V. [DOI] [PubMed] [Google Scholar]
- 60.Dentino AR, Westerman PW, Yeagle PL. 1995. A study of carbobenzoxy-D-phenylalanine-L-phenylalanine-glycine, an inhibitor of membrane fusion, in phospholipid bilayers with multinuclear magnetic resonance. Biochim Biophys Acta 1235:213–220. doi: 10.1016/0005-2736(95)80007-3. [DOI] [PubMed] [Google Scholar]
- 61.Weidmann A, Fischer C, Ohgimoto S, Ruth C, ter Meulen V, Schneider-Schaulies S. 2000. Measles virus-induced immunosuppression in vitro is independent of complex glycosylation of viral glycoproteins and of hemifusion. J Virol 74:7548–7553. doi: 10.1128/JVI.74.16.7548-7553.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Demotz S, Ammerlaan W, Fournier P, Muller CP, Barbey C. 1998. Processing of the DRB1*1103-restricted measles virus nucleoprotein determinant 185-199 in the endosomal compartment. Clin Exp Immunol 114:228–235. doi: 10.1046/j.1365-2249.1998.00716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Firsching R, Buchholz CJ, Schneider U, Cattaneo R, ter Meulen V, Schneider-Schaulies J. 1999. Measles virus spread by cell-cell contacts: uncoupling of contact-mediated receptor (CD46) downregulation from virus uptake. J Virol 73:5265–5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Klagge IM, ter Meulen V, Schneider-Schaulies S. 2000. Measles virus-induced promotion of dendritic cell maturation by soluble mediators does not overcome the immunosuppressive activity of viral glycoproteins on the cell surface. Eur J Immunol 30:2741–2750. doi:. [DOI] [PubMed] [Google Scholar]
- 65.Weidmann A, Maisner A, Garten W, Seufert M, ter Meulen V, Schneider-Schaulies S. 2000. Proteolytic cleavage of the fusion protein but not membrane fusion is required for measles virus-induced immunosuppression in vitro. J Virol 74:1985–1993. doi: 10.1128/JVI.74.4.1985-1993.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Plemper RK, Compans RW. 2003. Mutations in the putative HR-C region of the measles virus F2 glycoprotein modulate syncytium formation. J Virol 77:4181–4190. doi: 10.1128/JVI.77.7.4181-4190.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Makhortova NR, Askovich P, Patterson CE, Gechman LA, Gerard NP, Rall GF. 2007. Neurokinin-1 enables measles virus trans-synaptic spread in neurons. Virology 362:235–244. doi: 10.1016/j.virol.2007.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Andries K, Moeremans M, Gevers T, Willebrords R, Sommen C, Lacrampe J, Janssens F, Wyde PR. 2003. Substituted benzimidazoles with nanomolar activity against respiratory syncytial virus. Antiviral Res 60:209–219. doi: 10.1016/j.antiviral.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 69.Cianci C, Genovesi EV, Lamb L, Medina I, Yang Z, Zadjura L, Yang H, D'Arienzo C, Sin N, Yu KL, Combrink K, Li Z, Colonno R, Meanwell N, Clark J, Krystal M. 2004. Oral efficacy of a respiratory syncytial virus inhibitor in rodent models of infection. Antimicrob Agents Chemother 48:2448–2454. doi: 10.1128/AAC.48.7.2448-2454.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cianci C, Yu KL, Combrink K, Sin N, Pearce B, Wang A, Civiello R, Voss S, Luo G, Kadow K, Genovesi EV, Venables B, Gulgeze H, Trehan A, James J, Lamb L, Medina I, Roach J, Yang Z, Zadjura L, Colonno R, Clark J, Meanwell N, Krystal M. 2004. Orally active fusion inhibitor of respiratory syncytial virus. Antimicrob Agents Chemother 48:413–422. doi: 10.1128/AAC.48.2.413-422.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bonfanti JF, Meyer C, Doublet F, Fortin J, Muller P, Queguiner L, Gevers T, Janssens P, Szel H, Willebrords R, Timmerman P, Wuyts K, van Remoortere P, Janssens F, Wigerinck P, Andries K. 2008. Selection of a respiratory syncytial virus fusion inhibitor clinical candidate. 2. Discovery of a morpholinopropylaminobenzimidazole derivative (TMC353121). J Med Chem 51:875–896. [DOI] [PubMed] [Google Scholar]
- 72.Yan D, Lee S, Thakkar VD, Luo M, Moore ML, Plemper RK. 2014. Cross-resistance mechanism of respiratory syncytial virus against structurally diverse entry inhibitors. Proc Natl Acad Sci U S A 111:E3441–E3449. doi: 10.1073/pnas.1405198111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Battles MB, Langedijk JP, Furmanova-Hollenstein P, Chaiwatpongsakorn S, Costello HM, Kwanten L, Vranckx L, Vink P, Jaensch S, Jonckers TH, Koul A, Arnoult E, Peeples ME, Roymans D, McLellan JS. 2016. Molecular mechanism of respiratory syncytial virus fusion inhibitors. Nat Chem Biol 12:87–93. doi: 10.1038/nchembio.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wild C, Greenwell T, Matthews T. 1993. A synthetic peptide from HIV-1 gp41 is a potent inhibitor of virus-mediated cell-cell fusion. AIDS Res Hum Retroviruses 9:1051–1053. doi: 10.1089/aid.1993.9.1051. [DOI] [PubMed] [Google Scholar]
- 75.Zhang D, Li W, Jiang S. 2015. Peptide fusion inhibitors targeting the HIV-1 gp41: a patent review (2009-2014). Expert Opin Ther Pat 25:159–173. doi: 10.1517/13543776.2014.987752. [DOI] [PubMed] [Google Scholar]
- 76.Yi HA, Fochtman BC, Rizzo RC, Jacobs A. 2016. Inhibition of HIV entry by targeting the envelope transmembrane subunit gp41. Curr HIV Res 14:283–294. doi: 10.2174/1570162X14999160224103908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.DeVincenzo JP, Whitley RJ, Mackman RL, Scaglioni-Weinlich C, Harrison L, Farrell E, McBride S, Lambkin-Williams R, Jordan R, Xin Y, Ramanathan S, O'Riordan T, Lewis SA, Li X, Toback SL, Lin SL, Chien JW. 2014. Oral GS-5806 activity in a respiratory syncytial virus challenge study. N Engl J Med 371:711–722. doi: 10.1056/NEJMoa1401184. [DOI] [PubMed] [Google Scholar]
- 78.Heylen E, Neyts J, Jochmans D. 2017. Drug candidates and model systems in respiratory syncytial virus antiviral drug discovery. Biochem Pharmacol 127:1–12. doi: 10.1016/j.bcp.2016.09.014. [DOI] [PubMed] [Google Scholar]
- 79.Kadam RU, Wilson IA. 2017. Structural basis of influenza virus fusion inhibition by the antiviral drug Arbidol. Proc Natl Acad Sci U S A 114:206–214. doi: 10.1073/pnas.1617020114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hsu EC, Iorio C, Sarangi F, Khine AA, Richardson CD. 2001. CDw150(SLAM) is a receptor for a lymphotropic strain of measles virus and may account for the immunosuppressive properties of this virus. Virology 279:9–21. doi: 10.1006/viro.2000.0711. [DOI] [PubMed] [Google Scholar]
- 81.Tatsuo H, Ono N, Tanaka K, Yanagi Y. 2000. SLAM (CDw150) is a cellular receptor for measles virus. Nature 406:893–897. doi: 10.1038/35022579. [DOI] [PubMed] [Google Scholar]
- 82.Navaratnarajah CK, Negi S, Braun W, Cattaneo R. 2012. Membrane fusion triggering: three modules with different structure and function in the upper half of the measles virus attachment protein stalk. J Biol Chem 287:38543–38551. doi: 10.1074/jbc.M112.410563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hull JD, Krah DL, Choppin PW. 1987. Resistance of a measles virus mutant to fusion inhibitory oligopeptides is not associated with mutations in the fusion peptide. Virology 159:368–372. doi: 10.1016/0042-6822(87)90475-2. [DOI] [PubMed] [Google Scholar]
- 84.Ader N, Brindley M, Avila M, Orvell C, Horvat B, Hiltensperger G, Schneider-Schaulies J, Vandevelde M, Zurbriggen A, Plemper RK, Plattet P. 2013. Mechanism for active membrane fusion triggering by morbillivirus attachment protein. J Virol 87:314–326. doi: 10.1128/JVI.01826-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Plemper RK, Lakdawala AS, Gernert KM, Snyder JP, Compans RW. 2003. Structural features of paramyxovirus F protein required for fusion initiation. Biochemistry 42:6645–6655. doi: 10.1021/bi034385k. [DOI] [PubMed] [Google Scholar]
- 86.Russell CJ, Kantor KL, Jardetzky TS, Lamb RA. 2003. A dual-functional paramyxovirus F protein regulatory switch segment: activation and membrane fusion. J Cell Biol 163:363–374. doi: 10.1083/jcb.200305130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cianci C, Langley DR, Dischino DD, Sun Y, Yu KL, Stanley A, Roach J, Li Z, Dalterio R, Colonno R, Meanwell NA, Krystal M. 2004. Targeting a binding pocket within the trimer-of-hairpins: small-molecule inhibition of viral fusion. Proc Natl Acad Sci U S A 101:15046–15051. doi: 10.1073/pnas.0406696101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Douglas JL, Panis ML, Ho E, Lin KY, Krawczyk SH, Grant DM, Cai R, Swaminathan S, Chen X, Cihlar T. 2005. Small molecules VP-14637 and JNJ-2408068 inhibit respiratory syncytial virus fusion by similar mechanisms. Antimicrob Agents Chemother 49:2460–2466. doi: 10.1128/AAC.49.6.2460-2466.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cox R, Plemper RK. 2016. Structure-guided design of small-molecule therapeutics against RSV disease. Expert Opin Drug Discov 21:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Poor TA, Jones LM, Sood A, Leser GP, Plasencia MD, Rempel DL, Jardetzky TS, Woods RJ, Gross ML, Lamb RA. 2014. Probing the paramyxovirus fusion (F) protein-refolding event from pre- to postfusion by oxidative footprinting. Proc Natl Acad Sci U S A 111:E2596–E2605. doi: 10.1073/pnas.1408983111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Krumm SA, Yan D, Hovingh ES, Evers TJ, Enkirch T, Reddy GP, Sun A, Saindane MT, Arrendale RF, Painter G, Liotta DC, Natchus MG, von Messling V, Plemper RK. 2014. An orally available, small-molecule polymerase inhibitor shows efficacy against a lethal morbillivirus infection in a large animal model. Sci Transl Med 6:232ra52. doi: 10.1126/scitranslmed.3008517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wyplosz B, Lafarge M, Escaut L, Stern JB. 2013. Fatal measles pneumonitis during Hodgkin's lymphoma. BMJ Case Rep 2013:bcr2013200252. doi: 10.1136/bcr-2013-200252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hierholzer JC, Killington RA. 1996. Chapter 2. Virus isolation and quantitation, p 25–46. In Mahy BWJ, Kangro H (ed), Virology methods manual. Academic Press, London, United Kingdom. [Google Scholar]
- 94.Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199. doi: 10.1016/0378-1119(91)90434-D. [DOI] [PubMed] [Google Scholar]
- 95.Vialard J, Lalumiere M, Vernet T, Briedis D, Alkhatib G, Henning D, Levin D, Richardson C. 1990. Synthesis of the membrane fusion and hemagglutinin proteins of measles virus, using a novel baculovirus vector containing the beta-galactosidase gene. J Virol 64:37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Soding J, Biegert A, Lupas AN. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sali A, Blundell TL. 1993. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]