

ABSTRACT
An effective AIDS vaccine should elicit strong humoral and cellular immune responses while maintaining low levels of CD4+ T-cell activation to avoid the generation of target cells for viral infection. The present study investigated two prime-boost regimens, both starting vaccination with single-cycle immunodeficiency virus, followed by two mucosal boosts with either recombinant adenovirus (rAd) or fowlpox virus (rFWPV) expressing SIVmac239 or SIVmac251 gag/pol and env genes, respectively. Finally, vectors were switched and systemically administered to the reciprocal group of animals. Only mucosal rFWPV immunizations followed by systemic rAd boost significantly protected animals against a repeated low-dose intrarectal challenge with pathogenic SIVmac251, resulting in a vaccine efficacy (i.e., risk reduction per exposure) of 68%. Delayed viral acquisition was associated with higher levels of activated CD8+ T cells and Gag-specific gamma interferon (IFN-γ)-secreting CD8+ cells, low virus-specific CD4+ T-cell responses, and low Env antibody titers. In contrast, the systemic rFWPV boost induced strong virus-specific CD4+ T-cell activity. rAd and rFWPV also induced differential patterns of the innate immune responses, thereby possibly shaping the specific immunity. Plasma CXCL10 levels after final immunization correlated directly with virus-specific CD4+ T-cell responses and inversely with the number of exposures to infection. Also, the percentage of activated CD69+ CD8+ T cells correlated with the number of exposures to infection. Differential stimulation of the immune response likely provided the basis for the diverging levels of protection afforded by the vaccine regimen.
IMPORTANCE A failed phase II AIDS vaccine trial led to the hypothesis that CD4+ T-cell activation can abrogate any potentially protective effects delivered by vaccination or promote acquisition of the virus because CD4+ T helper cells, required for an effective immune response, also represent the target cells for viral infection. We compared two vaccination protocols that elicited similar levels of Gag-specific immune responses in rhesus macaques. Only the animal group that had a low level of virus-specific CD4+ T cells in combination with high levels of activated CD8+ T cells was significantly protected from infection. Notably, protection was achieved despite the lack of appreciable Env antibody titers. Moreover, we show that both the vector and the route of immunization affected the level of CD4+ T-cell responses. Thus, mucosal immunization with FWPV-based vaccines should be considered a potent prime in prime-boost vaccination protocols.
KEYWORDS: AIDS vaccine, SCIV, recombinant fowlpox virus, recombinant adenovirus, repeated low-dose challenge, SIV, innate immunity, simian immunodeficiency virus
INTRODUCTION
In AIDS vaccine development, different immunization protocols have been applied in nonhuman primate models, using a broad panel of viral and bacterial recombinant vectors that resulted in different levels of protection (1–3). To improve vaccine efficacy, the most promising immunogens were administered by prime-boost regimens (4, 5). In addition, several studies showed that the route of vaccine delivery is also important because it can significantly influence the magnitude and duration of antibody and cytotoxic T lymphocyte (CTL) responses (6, 7). Given that human immunodeficiency virus (HIV) transmission occurs mainly through the rectogenital mucosa and that vaccines administered systemically do not generally elicit an effective immune response at these sites, there is great interest in developing vaccines that induce mucosal immunity (8). Oral and intranasal (i.n.) antigen administration have been shown to induce both systemic and mucosal immune responses at distant effector sites (9, 10). Importantly, it has recently been suggested that activated CD4+ T cells, which are required for the induction of immunity, can abrogate all protective vaccine effects because they represent perfect target cells for viral entry and replication. The magnitude of vaccine-elicited T helper cell response thus represents a critical factor in AIDS vaccine development (11–14). So far, however, only a few studies have described the deleterious effects of strong CD4+ T-cell activation on vaccine efficacy (11, 13–15).
Adenovirus (Ad) and poxvirus vectors are the most extensively studied vehicles for the construction of recombinant immunogens (16–18). In a previous study (19), the single-cycle immunodeficiency virus (SCIV) was successfully used in a heterologous prime-boost protocol for priming, followed by atraumatic tonsillar administration of non-replication-competent adenoviral vector (rAd) expressing the SIVmac239 gag/pol and env genes. This regimen induced a robust simian immunodeficiency virus (SIV)-specific immune response and significantly reduced viral RNA levels in macaques after challenge with the neutralization-resistant SIVmac239 strain (19). Although adenovirus-vectored vaccines raise potent cellular immune responses, there are concerns about their safety because they seem to facilitate HIV infection instead of impeding it (12, 14, 20, 21). Fowlpox virus recombinants (rFWPV) have been used as vaccines in several preclinical trials in mice, rabbits, and macaques to evaluate and compare immunogenicity and efficacy against SIV, simian-human immunodeficiency virus (SHIV), or HIV (22–31). Intranasal priming with rFWPV followed by intramuscular (i.m.) boosting was shown to induce long-lasting systemic and mucosal T-cell responses (32).
Based on these findings, we assessed the efficacy of the combination of Ad- and FWPV-vectored vaccines administered in reverse order by two different routes. We investigated two prime-boost regimens using SCIV for priming, followed by two mucosal boosters either with Ad-SIVgag/pol (Adgp) plus Ad-SIVenv (Adenv) (vaccine group 1 [V1]) or with rFWPV expressing SIV gag/pol and env genes (FWPVgp and FWPVenv) (vaccine group 2 [V2]). A final crossover booster immunization was performed by systemic administration of rFWPV in V1 and rAd in V2. With this strategy, we intended not only to minimize possible negative effects of vector-specific immune responses, but also to examine which vector order provided the best efficacy. Vaccine efficacy was determined by a stringent, repeated, low-dose mucosal challenge protocol with pathogenic SIVmac. Acquisition of SIV was significantly delayed in one arm of the study, and immune correlates of protection were identified.
RESULTS
Vectors used for immunization expressed transgenes correctly.
Our vaccine strategy comprised three different viral vectors and combined mucosal and systemic immunizations. Two different vaccination regimens were compared using identical priming with SCIV, followed by two booster immunizations with either rFWPV or rAd carrying SIV gag/pol and env genes, both given mucosally, and a final systemic crossover immunization as outlined in Fig. 1. Our repeated low-dose challenge study provided 75% power to detect a vaccine efficacy of 50%, assuming a per-challenge infection probability without vaccine of 0.5.
FIG 1.

Timeline, immunization regimens, and vaccine doses.
SCIV and the SIV transgenes carried by the rAd vectors were correctly expressed in vitro, as reported previously (19, 33, 34). Expression of the SIV transgenes by rFWPV was analyzed by Western blotting (WB) and immunofluorescence (IF) assay in replication-restricted human MRC-5 cells, Vero cells, and replication-permissive chicken embryo fibroblasts (CEFs). The WB analysis of cell lysates prepared from cells infected with FWPVgp or FWPVenv showed bands at either 27 or 120 kDa (Fig. 2A), corresponding to the SIV Gag and Env proteins, respectively. IF analysis (Fig. 2B) confirmed the recognition of the nondenatured form of the proteins and their cytoplasmic localization, suggesting that in vitro findings should translate into in vivo infection of nonhuman primates and humans. No SIV-specific bands were seen after wild-type FWPV (FWPVwt) infection (data not shown).
FIG 2.
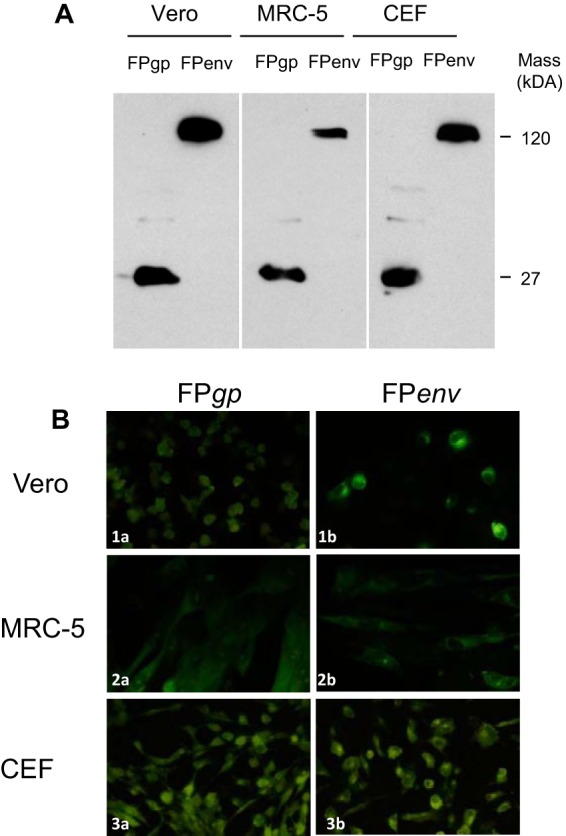
Transgene expression of rFWPVgp and rFWPVenv in replication-permissive avian CEFs and replication-restrictive mammalian Vero and MRC-5 cells. (A) Western blotting was used to reveal the presence of 27- and 120-kDa proteins in infected cells using a SIV-positive monkey serum pool as a primary polyclonal antibody. (B) Using immunofluorescence staining, specific signals were detectable in all cell lines after infection with FWPVgp (FPgp) (1a, 2a, and 3a) and FWPVenv (FPenv) recombinants (1b, 2b, and 3b), using the same polyclonal antibodies as for WB. FITC-conjugated rabbit anti-monkey IgG was used as secondary antibody. For both WB and IF assay, cells infected with wild-type FWPV were used as a negative control (data not shown).
The SIVmac251 10/09 stock and its parental SIVmac251 stock show considerable env sequence diversity.
To compare our SIVmac251 10/09 challenge stock with other commonly used SIVmac251 stocks and to evaluate its suitability for vaccine studies, single-genome amplification (SGA) sequencing analysis of the full-length env gene was performed, and an env phylogeny was constructed (Fig. 3). The SIVmac251 10/09 stock and its parental SIVmac251 stock (Aubertin), which has been used for numerous nonhuman primate vaccine studies in Europe over the last few decades, were analyzed and compared with other previously published sequences of SIVmac251 (35). The maximum env gene sequence diversity in the SIVmac251 Aubertin stock was 2.0%, whereas in the SIVmac251 10/09 stock it was 2.1%, with one highly divergent sequence (Fig. 3A). In particular, these two SIVmac251 stocks share a homogeneous cluster at the top of the tree (Fig. 3B). Overall, the stock shares many of the features of the 251-RD virus stock and is one of the more heterogeneous stocks of SIVmac251 currently used in the United States (35).
FIG 3.

Neighbor-joining trees of SIVmac251 challenge stocks. A phylogenetic tree was constructed from single-genome amplification-derived full-length env sequences of the SIVmac251 stocks 10/09 used here and its parental stock (Aubertin) combined with sequences of frequently utilized SIVmac251 stocks from the United States (A) and the 10/09 and Aubertin sequences by themselves (B). The branch colors indicate the stock from which each sequence was derived. The scale bars indicate genetic distances between sequences.
Vaccine group 2 exhibited delayed acquisition of SIV.
To assess vaccine efficacy, all 18 animals were challenged intrarectally (i.r.) with low doses (120 50% tissue culture infective doses [TCID50]) of the highly pathogenic SIVmac251 10/09 stock at weekly intervals, starting 8 weeks after the final immunization (week 42). Prior to each challenge, blood samples were collected, and the viral load was determined. Challenges were suspended when viral RNA exceeded 100 copies/ml plasma.
The numbers of SIV exposures leading to infection varied from one to eight, except for one animal that remained uninfected after eight exposures (Table 1). In particular, four of six animals from the control group (66%) became infected after the first exposure and four of six animals from V1 (66%) became infected after the second virus exposure. Conversely, only one of six macaques became infected after two exposures (16.6%) in V2, whereas five of six (83.4%) required at least 5 exposures for infection, including the uninfected animal (Table 1). No influence upon viral acquisition was observed for Mamu-A1*001:01 carriership (data not shown). In summary, a significant delay in viral acquisition was observed in V2, which needed more than 35 inoculations in total, compared to V1 and controls, which required 16 and 15 inoculations to infect all the animals, respectively (Table 1 and Fig. 4A) (omnibus log rank, P = 0.036, 2 df; bivariate comparisons with 1 df, V1 versus V2, P = 0.0092, and controls versus V2, P = 0.025). Relative to the controls, V2 showed a reduction of infection risk per exposure of 68% (hazard ratio [HR] = 0.315; 95% confidence interval [CI], 0.083 to 1.195; P = 0.083) compared to only 12% for V1 (HR = 0.875; 95% CI, 0.252 to 3.030; P = 0.817). Comparison of viral acquisition between the controls and 10 unvaccinated animals from another experiment infected with the same virus stock and dose by the same route showed that our challenge protocol yielded reproducible results (36). Inclusion of these historical controls in the calculations resulted in a reduction of the observed infection risk per exposure, relative to controls, of 75% for V2 (HR = 0.248; 95% CI, 0.087 to 0.712; P = 0.012).
TABLE 1.
Genetic background of vaccinees and controls and numbers of challenges needed to cause infection with SIVmac251
| Group | Animal ID | MHC class Ia | No. of exposures to infection | Total no. of exposures |
|---|---|---|---|---|
| Controls, vaccine 1 | 2249 | A1*001 | 4 | |
| 2290 | 1 | |||
| 2294 | A1*001 | 1 | ||
| Controls, vaccine 2 | 2302 | 7 | ||
| 2305 | 1 | |||
| 2325 | A1*001, B*017:01 | 1 | 15 | |
| Vaccine 1 | 2317 | 2 | ||
| 2328 | 2 | |||
| 2332 | 2 | |||
| 2337 | 3 | |||
| 13923 | A1*001 | 5 | ||
| 13930 | A1*001 | 2 | 16 | |
| Vaccine 2 | 2275 | 8 | ||
| 2295 | >8b | |||
| 2324 | A1*001, B*017:01 | 6 | ||
| 2338 | A1*001, B*017:01 | 5 | ||
| 13929 | A1*001 | 6 | ||
| 13934 | A1*001 | 2 | >35 |
Major histocompatibility complex genotypes associated with slow disease progression.
Not infected after 8 exposures.
FIG 4.

Challenge outcome as evaluated by infection rate and plasma viral load. (A) Kaplan-Meier plot showing the percentages of uninfected macaques after weekly intrarectal exposure to 120 TCID50 of SIVmac251 (omnibus log rank, P = 0.036, 2 df; bivariate comparisons with 1 df, controls versus V1, P = 0.23, V1 versus V2, P = 0.0092, controls versus V2, P = 0.025). (B) Plasma viral RNA copies (geometric mean) of the infected macaques from V1, V2, and controls normalized to weeks postinfection. (C) Plasma viral RNA copies of V1, V2, and controls until 20 weeks postinfection, expressed as AUC and shown as a box-and-whiskers plot, indicating minimum, mean, maximum, and 25th and 75th percentiles. P values are shown (Mann-Whitney test).
Both vaccine groups showed lower peak plasma viral loads than the controls.
Peak viremia at week 2 postinfection (p.i.) was significantly lower in both vaccine groups than in controls (Mann-Whitney test; P ≤ 0.041) (Fig. 4B). In addition, the overall viral load (weeks 0 to 20 p.i.) was also significantly lower in both vaccine groups than in controls (Fig. 4C) (area under the concentration-time curve [AUC] weeks 0 to 20 p.i., V1 versus control [Mann-Whitney test], P = 0.002; V2 versus control, P = 0.041). Notably, two animals from V2 had already cleared the virus from the peripheral blood by week 4 after infection (data not shown). In all the groups, CD4+ T-cell levels dropped during acute infection but were best conserved in V2 (data not shown). However, after stratification for MhcMamu-A1*001 carriership, the two vaccine groups did not differ in terms of viral load or CD4+ T-cell counts.
Systemic rFWPV immunization elicited higher SIV-specific neutralizing antibodies.
Gag- and Env-binding antibodies were determined by enzyme-linked immunosorbent assay (ELISA) (Fig. 5A and B). The two vaccine groups had similar anti-Gag p27 antibody levels, reaching the highest titers after the first and third boosters (Fig. 5A). Anti-Env gp130 antibodies were low or almost completely lacking in V1 and V2 until the third booster, but in contrast to V2, they increased in V1 after systemic rFWPV immunization (Fig. 5B). In particular, 2 weeks after the third booster (week 36) and 1 week before the first challenge (week 41), V1 had significantly higher anti-Env antibody titers than V2 (Mann-Whitney test; P = 0.002). In addition, at week 41, SIV neutralizing antibody (NAb) titers were significantly higher in V1 than in V2 (Fig. 5C) (Mann-Whitney test; P = 0.01).
FIG 5.

Humoral immune responses elicited by vaccination. Longitudinal SIV-specific serum antibody levels against SIVp27 (A) and SIVgp130 (B) were determined by ELISA. The data are shown as means and standard errors of the mean (SEM) of six animals per group. Immunization time points are indicated by the arrows (weeks 0, 4, 12, 20, and 34). Anti-SIV Env antibodies measured 2 weeks after the last immunization and 1 week before the first challenge differed significantly between the groups (Mann-Whitney test; *, P = 0.002). (C) Neutralizing antibody titers were determined 1 week before the first challenge (week 41) in a TZM-bl assay against SIVmac32H and are shown as reciprocal log10 dilutions. The horizontal line marks the mean of each group. The P value is shown (Mann-Whitney test). (D) IgA antibody levels in blood against SIVp27 and gp130 at week 36 determined by ELISA. The horizontal lines mark the means of the groups. (E) Binding antibodies against undisrupted fowlpox virus (FP) particles were determined after mucosal and intramuscular immunization. A cutoff of 0.4 defined positivity. The red diamonds represent the positively reacting animals; the blue circles represent the negative ones; the horizontal lines are the means.
IgA antibody levels against Gag and Env proteins in blood did not differ significantly between the two vaccine groups 2 weeks post-final immunization (Fig. 5D) and were undetectable in rectal swabs (data not shown). Notably, the animal with the highest plasma IgA level in V1 was infected by the second challenge, whereas the one in V2 remained uninfected, thus not revealing an influence of systemic IgA antibody levels on protection.
Very low levels of FWPV-specific antibodies were induced by the oral immunization route.
While vector-specific immunity to Ad proteins upon oral and systemic immunization was analyzed previously (19), corresponding data following FWPV immunization are lacking. Therefore, ELISAs were performed with sera of those animals that received either FWPVwt or rFWPV (Fig. 5E). After the two oral immunizations, only one of nine FWPV-immunized macaques (11%) was positive (week 22). Vector-neutralizing activity could not be detected after oral immunization (data not shown). Conversely, 2 weeks after the single i.m. immunization with rFWPV (V1; week 36), six of nine animals (66.7%) were anti-FWPV antibody positive, and at week 38, all the macaques immunized i.m. with FWPV had elicited antibodies against the vector. FWPV oral immunization induced almost no vector-specific antibodies, in contrast to the FWPV systemic immunization.
A systemic booster with rFWPV elicited stronger CD4+ cellular immune responses than the systemic rAd booster.
To compare the potencies of the two vaccine regimens to induce T-cell responses, gamma interferon (IFN-γ) enzyme-linked immunosorbent spot (ELISpot) assays, carboxyfluorescein succinimidyl ester (CFSE) (Invitrogen) proliferation assays, and intracellular cytokine staining (ICS) were performed. After the second SCIV immunization at week 6 p.i., SIV Gag-specific IFN-γ ELISpot responses were mainly detected in V2 (Fig. 6A, middle and left). After the first and second booster immunizations, stimulation with SIV Gag-specific peptides revealed no significant differences between the two vaccine groups (Fig. 6A, right, week 22). The systemic immunization boosted Gag-specific IFN-γ responses in both vaccine groups, but higher numbers of spot-forming cells (SFCs) were found in three of six animals in V2 (Fig. 6A, right, week 36).
FIG 6.

Cellular immune responses throughout immunization. (A and B) IFN-γ ELISpot responses in blood of vaccinees during immunization are presented as SFCs per 106 PBMCs after stimulation with a SIVGag 15-mer peptide pool (A) and SIV AT-2 (B). Individual values are shown. The four- and five-digit numbers are monkey designations. The arrows indicate immunization with either SCIV (S), adenovirus (A), or fowlpox virus (F). The asterisks mark monkeys with MHC class I genotype Mamu-A1*001:01. (Right) Values for week 22 and week 36 after initial immunization are shown for group comparison. (C and D) Proliferative CD8+ and CD4+ T-cell responses in blood of vaccinees against SIV AT-2 determined by a CFSE proliferation assay shown as individual percentages of responding CD8+ (C) and CD4+ (D) T cells after initial immunization. Monkeys are indicated by four- or five-digit numbers. (C and D, right) Comparisons between values for week 22 and week 36 after initial immunization. At week 36, data from only 5 animals were available for V1. (E) Cell-mediated responses measured by ELISpot assay upon SIV AT-2 stimulation are CD4+ T-cell restricted. To identify which T-cell subsets were activated through SIV AT-2 stimulation in the IFN-γ ELISpot assay, we used PBMCs from long-term SIV-infected macaques known to be strongly reactive against SIV AT-2. IFN-γ ELISpots were performed with PBMCs without depletion (SIV AT-2 stimulation w/o depletion) and after depletion (SIV AT-2 stimulation w depletion) of CD4+ (n = 4) and CD8+ (n = 2) cells and the corresponding negative controls (microvesicle w/o depletion and microvesicle w depletion). (F) The ratio of the ELISpot results after stimulation with SIV AT-2 and Gag peptides was calculated for each individual monkey, and the kinetics of the ratio (median) of the two vaccine groups are displayed. The asterisks indicate significance between the two vaccine groups (Mann-Whitney test; P < 0.05). (G) Percentages of Gag-specific CD8+ T cells among total CD8+ CD3+ cells expressing either TNF-α or IFN-γ determined by ICS at week 36 in V1 (blue) and V2 (red). Polyfunctional CD8+ cells expressing IFN-γ plus IL-2, IFN-γ plus TNF-α, and IFN-γ plus IL-2 plus TNF-α observed only in animal 2295 (V2) are shown cumulatively. IFN-γ- plus TNF-α-expressing cells were detected in animal 13929 (V2).
The two immunizations with SCIV elicited weak responses against aldrithiol-2-inactivated SIV (SIV AT-2) in 5 of the 12 vaccinees (Fig. 6B, left and middle, week 6). A booster effect in IFN-γ ELISpot responses following stimulation with SIV AT-2 was observed in four of six animals in V2 after the first oral FWPV administration (Fig. 6B, middle, week 14). This effect was delayed and weaker in V1 (Fig. 6B, left, week 22). The systemic crossover immunization with rFWPV elicited significantly higher SIV AT-2 responses in V1 than systemic rAd immunization in V2 (Fig. 6B, right, week 36).
Similar results were obtained when the CFSE proliferation assay was performed with SIV AT-2 as the stimulus (Fig. 6C and D). While the proliferation rates of SIV-specific CD8+ and CD4+ cells did not differ significantly between the two vaccine groups at week 22, the rate of proliferating CD4+ T cells was significantly higher in V1 than in V2 after the third booster (week 36; Mann-Whitney test; P = 0.017) (Fig. 6D, right).
Depletion studies were performed to investigate which cell types responded to the different stimuli when employing an ELISpot assay. The results revealed that the IFN-γ ELISpot response to SIV AT-2 stimulation was restricted to CD4+ cells (Fig. 6E). We therefore regard SIV AT-2-induced ELISpot responses as being CD4+ T cell restricted. Furthermore, the proportions of activated T helper cells (CD4+ HLA-DR+) were also higher in V1 than in V2 2 weeks post-final vaccination (Mann-Whitney test; P = 0.026) (data not shown). In contrast, blood lymphocytes of the two vaccine groups did not differ significantly in the percentages of CXCR3-expressing CD4+ T cells, CD8+ T cells, and CD20+ B cells or of potential Th17 cells (CD4+ CD95+ CD196+) (data not shown).
Potential differences between the vectors regarding differential stimulation of CD4+ and CD8+ T cells are depicted in Fig. 6F. For this, we calculated the ratio between the CD4+ cell-dependent SIV AT-2 and the predominantly CD8+ T-cell-mediated Gag peptide ELISpot response (37) (Fig. 6A and B). Figure 6F shows that at the second boost the CD4+ T-cell ELISpot response dominated in V2 (ratios of >1) while the two T-cell subsets were equally stimulated in V1. Notably, the ratios were inverted after the final boost, when the vaccine constructs were switched, resulting in a significant difference between the SFC ratios in the two groups (P = 0.026) (Fig. 6F). Similar results were obtained when proliferation ratios after SIV-AT2 stimulation of CD4+ and CD8+ T cells were calculated (data not shown).
Thus, after the final booster, CD4+ T-cell responses detected by the SIV AT-2 IFN-γ ELISpot and CFSE proliferation assays were significantly stronger in V1 than in V2. Notably, when testing responses by ICS, hardly any reactive CD4+ cells were detected (data not shown).
Overall, the analysis of proliferating CD8+ cells revealed no significant difference between the two vaccine groups after the final booster, even though V1 showed a stronger response than V2 (Fig. 6C). Furthermore, Gag-CM9 tetramer staining of isolated mononuclear cells from blood and various organs (colon, cervix, and lung surface) of Mamu-A1*001-positive monkeys also revealed no significant differences between the two vaccine regimens after the second and third boost immunizations (data not shown). Likewise, the total SIV-Gag-specific CD8+ T-cell magnitudes determined by ICS at week 36 did not differ between groups. However, the virus-specific CD8+ cells in V1 were mainly monofunctional, secreting tumor necrosis factor alpha (TNF-α) (four of six animals), whereas in V2 the response was dominated by IFN-γ-producing cells in four of six animals (Fig. 6G, left). Notably, two vaccinees from V2, one infected after 6 exposures and the only uninfected animal (2295), displayed appreciable percentages of polyfunctional CD8+ cells (Fig. 6G, right).
Innate immune responses differed between V1 and V2 after the third booster immunization.
To investigate whether the two vaccine regimens influenced innate immunity, the RNA levels of genes of innate immunity in whole blood were examined at different time points, namely, before the onset of immunization (data not shown), 24 and 48 h after the second and third boosters (weeks 20 and 34), and on the day of the first SIV exposure (week 42). MX1 and BST2 were chosen as type I IFN-responsive genes (Fig. 7A to C) and FAM26F and CXCL10 as IFN-γ-responsive genes (Fig. 7D to F).
FIG 7.

Innate immune responses 24 and 48 h after second (week 20) and final (week 34) booster immunizations and on the day of first challenge. C1, controls for V1; C2, controls for V2. Data for V1 are shown in blue and those for V2 in red. Significant P values discriminating V1 and V2 are shown (Mann-Whitney test). Means and standard errors of the mean are depicted for each group. (A to C) Relative RNA levels of MX1, BST2, FAM26F, and CXCL10 in PBMCs at 24 and 48 h after the second booster (A) and the third booster (B) immunization and on the day of first SIV exposure (C). (D) Plasma IFN-α was assessed by a panspecific IFN-α ELISA at 24 and 48 h after the third booster immunization. The cutoff value (7 pg/ml) is shown by the dashed line. (E) Plasma IFN-γ at 24 and 48 h after the third booster immunization was determined by macaque-specific ELISA. Plasma IFN-γ levels of all control samples and of V1 obtained at 48 h after the final immunization were below the detection limit of the assay (7 pg/ml). (F) Plasma CXCL10 was determined by a macaque-specific ELISA at 24 and 48 h after the third booster immunization (week 34).
The second booster induced no significant changes in relative RNA levels of the examined genes at both 24 and 48 h postimmunization (Fig. 7A), whereas significant changes were observed after the third booster (Fig. 7B). In particular, after 24 h, RNA levels of BST2, FAM26F, and CXCL10 were significantly higher in V2 than in V1 (BST2 [Mann-Whitney test], P = 0.026; FAM26F, P = 0.005; CXCL10, P = 0.002) (Fig. 7B). RNA levels declined at 48 h postimmunization. The control animals showed a reaction pattern similar to that of the vaccinees, which suggests that innate responses were also mediated by the vector itself and/or by the route of immunization.
On the day of the first SIV exposure (week 42) (Fig. 7C), RNA levels of FAM26F and CXCL10 had returned to preinfection levels, whereas MX1 and BST2 RNA levels were elevated (preinfection data not shown). BST2 levels did not differ between the vaccine groups before the first challenge. However, MX1 RNA levels were significantly higher in V2 than in V1 (Mann-Whitney test; P = 0.030) (Fig. 7C, left).
Plasma IFN-α, IFN-γ, and CXCL10 were quantified by ELISA. Just before the third booster, plasma IFN-α and IFN-γ levels were undetectable (data not shown), whereas CXCL10 levels varied between 30 and 125 pg/ml (mean, 60 pg/ml) but did not differ significantly between the groups (data not shown). At 24 h post-final immunization (week 34), V2 had numerically higher plasma IFN-α levels than V1 (Fig. 7D), and IFN-α correlated with the RNA levels of the two responsive genes, MX1 and BST2 (MX1, Spearman rs = 0.73, and permutation, P = 0.0007; BST2, rs = 0.71, P = 0.0009 [data not shown]).
IFN-γ levels were significantly higher in V2 than in V1 (Mann-Whitney test; P = 0.008) (Fig. 7E) and correlated with the relative RNA levels of FAM26F and CXCL10 (FAM26F, rs = 0.67, P < 0.0001 [36]; CXCL10, rs = 0.69, P = 0.015 [data not shown]). Surprisingly, in contrast to IFN-γ, plasma CXCL10 levels were significantly higher in V1 than in V2 (Mann-Whitney test; P = 0.026 at 24 h and P = 0.017 at 48 h) (Fig. 7F). The finding that V1 had high plasma CXCL10 but low plasma IFN-γ levels and low transcript levels of the responsive genes CXCL10 and FAM26F can be explained by assuming that strong and rapid IFN-γ secretion had occurred in V1 during the 24 h postimmunization. As a result, protein levels of CXCL10 were still peaking in V1 at 24 h postimmunization whereas plasma IFN-γ, as well as RNA levels of CXCL10 and FAM26F, were already declining.
Plasma CXCL10 correlated directly with CD4+ T-cell response and inversely with vaccine efficacy.
We investigated the potential impacts of innate immune responses on vaccine-induced cellular immunity and vaccine efficacy. Plasma CXCL10 levels 24 and 48 h after the third boost immunization (week 34) correlated significantly with the CD4+ T-cell-dependent SIV AT-2 ELISpot responses measured at week 36 (24 h, rs = 0.87, P = 0.0002; 48 h, rs = 0.94, P < 0.0001) (Fig. 8A) and week 41 (24 h, rs = 0.77, P = 0.0034) (data not shown), as well as with activated T helper cells (CD4+ HLA-DR+, 24 h, rs = 0.74, P = 0.0078; 48 h, rs = 0.64, P = 0.026) (Fig. 8B). Plasma CXCL10 also correlated with the CD4+ T-cell proliferative response after SIV AT-2 stimulation at week 36 (24 h, rs = 0.60, P = 0.035; 48 h, rs = 0.62, P = 0.043) (data not shown), indicating that innate responses measured within 48 h after the third booster translated into cellular immune responses. Importantly, plasma CXCL10 levels at 48 h post-final immunization also correlated inversely with the number of exposures to infection (rs = −0.64; P = 0.026) (Fig. 8C). A trend was found toward a correlation of SIV AT-2 ELISpot responses with the number of exposures required for infection (rs = −0.55; P = 0.067) (Fig. 8D).
FIG 8.

Innate and adaptive immune correlates. The minimal number of exposures required for infection was assumed to be 9 for the uninfected monkey. (A) Plasma CXCL10 levels at 24 and 48 h post-final immunization (week 34) correlated significantly with SIV AT-2 ELISpot responses measured at week 36. P values and Spearman's rank correlation coefficients are shown. (B) Plasma CXCL10 levels at 24 and 48 h post-final immunization correlated significantly with CD4+ HLA-DR+ cells detected at week 36. P values and Spearman's rank correlation coefficients are shown. (C) Plasma CXCL10 levels at 48 h post-final immunization (week 34) correlated inversely with the number of exposures required to infect the immunized monkeys. The P value and Spearman's rank correlation coefficient are shown. (D) Cell-mediated responses (number of SFCs) measured by ELISpot assay upon SIV AT-2 stimulation 2 weeks after the final booster (week 36) plotted against the number of exposures required for infection. The P value and Spearman's rank correlation coefficient are shown. (E) The percentages of IFN-γ-secreting CD8+ T cells among CD3+ CD8+ T cells were determined by intracellular-cytokine staining after stimulation with Gag peptide pools and stratified according to the number of exposures to infection. The percentage of IFN-γ-positive cells was summed up for each animal. The arithmetic means and P value are shown (Mann-Whitney rank sum test). Note that no animal was infected by the fourth challenge. (F) Percentages of CD69+ cells among CD8+ T cells in V1 and V2 at weeks 36 and 41 post-first immunization. The medians are indicated by horizontal lines. (G) Mean percentages of CD69+ CD8+ T cells from weeks 36 and 41 in immunized macaques determined by flow cytometry correlated significantly with the number of exposures required for infection (Spearman rank correlation).
Vaccine efficacy correlated with virus-specific and activated CD8+ T cells before challenge.
When analyzing virus-specific CD8+ cells by ICS 2 weeks post-final boost, a higher level of IFN-γ-producing cells was associated with delayed acquisition (Fig. 8E). Notably, this T-cell subset was almost lacking in V1 (Fig. 6G).
Another potential correlate of protection that was unrelated to virus-specific cellular responses was the proportion of CD8+ CD69+ cells in peripheral blood mononuclear cells (PBMCs). As inferred from the expression of the early activation marker CD69, V2 had higher proportions of activated CD8+ T cells 48 h after the final immunization (data not shown) and at week 41, shortly before the first inoculation (Fig. 8F). The proportion of CD8+ CD69+ T cells at week 41 (rs = 0.58; P = 0.011) (data not shown), as well as the individual means of their levels at weeks 36 and 41 (rs = 0.74; P = 0.005), correlated with the number of exposures required for infection (Fig. 8G).
Interestingly, one macaque of V2 (13934) that presented with low SIV AT-2 ELISpot and low SIV AT-2 CD4+ proliferative responses (Fig. 6B and D, middle), but also with the lowest levels of CD8+ CD69+ cells, became infected after only 2 exposures to SIV. If this animal was removed from the analysis, the CD4+ T-cell-dependent SIV AT-2 ELISpot responses and the proliferative CD4+ cell responses at week 36 correlated inversely with the number of exposures required for infection (SIV AT-2 ELISpot, rs = −0.71, P = 0.014; SIV AT-2 CD4+ proliferative response, rs = −0.87, P = 0.001) (data not shown).
DISCUSSION
The efficacy of a vaccine is influenced by the type of antigen, the immunization regimen, the route of delivery, and the experimental challenge model. In our vaccination study, we compared two viral vectors and mucosal versus systemic vaccine delivery after identical priming. FWPV recombinants were compared with those of Ad. Although immunization with rAd can evoke potent immune responses, it has raised safety concerns because of the frequent preexisting Ad5 immunity in humans (38). FWPV recombinants were chosen because FWPV-based vaccine vectors are regarded as safe (39). They are similar to the canarypox virus (CNPV)-based ALVAC vaccine (40, 41) that was already used, with moderate success, for prophylaxis against HIV-1 in humans (42). Notably, poxvirus-derived vectors can elicit differential innate and adaptive immune responses (43, 44), providing a rationale for testing different recombinant poxviruses in preclinical studies. Reports have shown, e.g., that HIV-1 env gene expression lasted longer in FWPV- than in canarypox virus-infected cells (41), and the gene expression patterns in spleens of mice following infection grouped CNPV and modified vaccinia Ankara (MVA) together but allocated FWPV to a separate cluster (43). However, the gene expression pattern of FWPV was more closely related to CNPV and MVA than to lumpy skin disease virus (LSDV) (43). Vaccination experiments performed in mice indicated that FWPV recruited a significantly smaller portion of the unfavorable CD11b− CD103+ but elevated levels of favorable CD11b+ CD103− dendritic cells to lung tissue than MVA or vaccinia virus (44). FWPV-based vaccines, administered in combination with a DNA prime, have been reported to induce neutralizing antibodies (45, 46) and strong CD4+ and CD8+ T-cell responses (23–25, 30, 47, 48). Therefore, they have been considered for vaccination against cancer in humans (47, 49, 50) and against a variety of viral infections in different species (31, 32, 45, 46). Both vaccination regimes used in our study resulted in significantly reduced peak viremia and reduced set point viral load after infection compared to controls. However, only V2 animals that were boosted mucosally with rFWPV followed by a systemic immunization with rAd showed a significant delay in acquisition of the virus, corresponding to a vaccine efficacy of 68%. Notably, we used a stringent challenge protocol that led to the productive infection of 66% of the control macaques after the first challenge. With this protocol, potentially protective effects that may arise through repeated low-dose challenges can probably be neglected (51, 52). In addition, SGA sequence data revealed that the challenge virus was heterogeneous, showing a maximal env diversity of 2.1%, similar to that of the original 251 stock.
After priming with SCIV and two rounds of mucosal immunizations, SIV-specific immune responses were moderate and did not differ significantly between the two vaccine groups. Adverse reactions did not become obvious, and antibodies against the FWPV proteins were not elicited in the vast majority of animals.
Upon systemic immunization, the immune response increased substantially, and differences between the two vaccine groups became evident. The main differences noted were specific to the titer of anti-SIV NAbs, the induction of SIV-specific CD4+ cells, the functionality of SIV-specific CD8+ T-cell responses, and the proportion of CD8+ CD69+ T cells in blood.
After crossover immunization, the levels of anti-Gag antibodies were similar in the two groups, but while V1 animals developed anti-Env NAbs, this was not the case in V2. Thus, protection did not seem to be antibody dependent. The low level of anti-Env antibodies after systemic rAd immunization in V2 was unexpected because oral Adenv immunization seemed to have efficiently primed for these antibodies following systemic rFWPV administration. That the presence of NAbs per se is not necessarily associated with protection is a well-known fact (53).
After the final booster, the two groups also differed significantly in the magnitudes of their CD4+ T-cell-dependent SIV AT-2-ELISpot and lymphoproliferative responses. The systemic booster with rFWPV in V1 also appeared to trigger a strong IFN-γ and a potential TNF-α response, which resulted in strong synthesis/secretion of CXCL10 that in turn probably translated into increased virus-specific cellular CD4+ T-cell responses. This might have induced a larger number of SIV-specific memory T cells than in V2. Moreover, plasma CXCL10 after the final vaccination not only correlated with CD4+ cellular responses, but was also inversely related to the number of exposures to infection. Increased SIV replication through the presence of activated (13) or infiltrating (54) CD4+ T cells on the day of challenge has been reported previously. A detrimental effect of vaccine-induced CD4+ T cells on vaccine efficacy has also been suggested (11, 14). While in our study, in contrast to results reported by Fouts et al. (11), antibodies probably did not play a major role in protection, high IFN-γ ELISpot responses were associated with attenuation of protection in all three vaccine studies, including ours. We extended these findings by narrowing the responses to virus-specific CD4+ T cells and related them to IFN-γ/CXCL10 secretion after vaccination. The strong CD4+ T-cell activation probably abrogated the protective responses mediated by CD8+ T cells and even NAbs in V1. The virus-specific memory CD4+ T lymphocytes not only might have served as target cells, but also could have proliferated upon sensing of the virus, thereby providing increased numbers of highly susceptible target cells at the portal of entry and fueling viral replication instead of inhibiting it. However, polyfunctional CD4+ cells detected by ICS may or may not contribute to vaccine efficacy (55–57). Notably, in our experiment, hardly any reactive CD4+ T cells were detected by ICS, whereas SIV-specific CD4+ T cells were readily measured in the proliferation assays and by IFN-γ ELISpot. An explanation for the discrepancy may be that the assays differ in sensitivity and that, because of the varying stimulation times, different CD4+ T-cell subsets were activated. Furthermore, we might have missed the peak in cytokine production that can occur in CD4+ T cells 1 week after rFWPV boosting (30).
The causal relationship between CXCL10 secretion and the expansion and differentiation of CD4+ cells remains elusive. CXCl10 is a pleiotropic chemokine that can promote chemotactic activity of CXCR3+ cells toward inflamed tissue and can induce apoptosis and regulate cell growth and proliferation in infectious, inflammatory diseases and cancer. It acts via the CXCR3 receptor and is secreted by numerous cell types, like leukocytes, monocytes, neutrophils, eosinophils, fibroblasts, and stromal cells (58, 59). It has been reported that CXCL10 can polarize CD4+ T cells into Th1/Th17 cells by increased transcription of T-bet and RORγT in mice (60). Furthermore, evidence suggests that subsets of Th17 cells are highly susceptible to infection by HIV (61, 62) or promote HIV-1 persistence (63). Elevated preinfection CXCL10 levels have been associated with advanced disease progression upon HIV/SIV infection (64). In this study, we found no correlation between plasma CXCL10 and levels of potential Th17 cells (CD4+ CD95+ CCR6+) in blood. Instead, we found a correlation between plasma CXCL10 and activated T helper cells (CD4+ HLA-DR+). With regard to the elusive causal relationships, plasma CXCL10 concentrations after vaccination may be no more than an early biomarker associated with vaccine efficacy in this study. Further investigations to identify T-cell subsets that influence HIV acquisition in the context of vaccination should include a fine-tuned analysis of the distinct functional subsets of CD4+ T cells, especially Th17 and CXCR3-expressing T cells in the different immune compartments.
Concerning the CD8+ T-cell-mediated immune response after the final booster, the two vaccine groups did not differ significantly, either in their cellular CD8+ T-cell immune responses or in the percentages of CM9-specific CD8+ T cells or in their magnitude as detected by ICS. However, the responses probably differed functionally, because Gag-specific CD8+ T cells in V1 were found to primarily secrete TNF-α, whereas in V2, mainly monofunctional cells secreting IFN-γ were detected by ICS after the final booster. These Gag-specific CD8+ IFN-γ-secreting T cells probably contributed to vaccine efficacy. Polyfunctional CD8+ T-cell subsets were detected in the animal that remained uninfected. In addition, CD8+ T-cell activation determined by the percentage of CD8+ CD69+ cells before first challenge was also significantly associated with the relative resistance to SIV infection and thus to vaccine efficacy. Thus, protection was associated with functional differences in the CD8+ T lymphocyte responses and when reactive CD4+ cells were present at relatively low levels. This setting seems to be driven by the vaccine vector.
Finally, our results show that systemic rFWPV immunization not only resulted in enhanced CXCL10 secretion compared to rAd vaccination, but the kinetics of the induction of immune responses also differed. Since V1 already had high plasma CXCL10 levels at 24 h postvaccination, IFN-γ secretion must have occurred very rapidly within 24 h postimmunization, followed by a fast drop. Conversely, in V2, plasma IFN-γ probably peaked later and potentially at lower levels than in V1, resulting in lower levels of plasma CXCL10 at 24 and 48 h post-final immunization but higher IFN-γ and higher transcript levels of the responding genes FAM26F and CXCL10. In summary, systemic rFWPV immunization probably elicited a faster innate immune response than systemic rAd immunization. Our observations are supported by a report that transgenes are already expressed in vivo 6 h after rFWPV vaccination (65). Moreover, similar to our results, other studies showed that Ad5 in general evokes relatively low innate immune responses compared to other adenoviruses (66, 67), probably because Ad5 migrates into the nucleus relatively quickly (68). A comparison between Ad5 and FWPV has not been performed to date.
In summary, our vaccination regimen provided significant protection in spite of a stringent challenge protocol. Different patterns of immunity were shaped in response to the vector and route of immunization. Delayed acquisition of SIV was associated with activated CD8+ cells, IFN-γ-expressing CD8+ T cells, and low virus-specific CD4+ T-cell responses in the absence of Env antibodies. Strong CD4+ T-cell activity obviously mitigated any protective effects mediated by Gag-specific T cells and by NAbs. Mucosal immunization with rFWPV provided immunological memory that resulted in strong CD8+ activation after systemic booster immunization with rAd while avoiding excessive levels of virus-specific CD4+ T cells. To our knowledge, this is one of the first studies showing that a nonreplicating HIV vaccine candidate that induced almost no anti-Env antibodies, but CD8+ T-cell responses, delayed viral acquisition, suggesting early containment of infection by local CTLs (69).
Mucosal immunization with FWPV-based vaccines should therefore be considered a potent prime in prime-boost vaccination protocols. Moreover, our results also confirm that systemic immunization with FWPV-vectored vaccines can elicit NAbs against the transgene, and thus, the vector appears to be a promising candidate when induction of NAbs and CTL responses are desirable and the level of T helper cell activation is irrelevant. It will, however, remain a major challenge for an effective AIDS vaccine to induce strong cellular and humoral immune responses in a setting of low memory CD4+ T-cell responses. Mucosal, but not systemic, priming with an FWPV-vectored vaccine may represent an option in this endeavor.
MATERIALS AND METHODS
Vaccine preparation.
SCIVs were produced by transient cotransfection of 293T cells with SX2Äfrxn, X2-Lys3, and pHIT-G plasmids as described previously (19, 33). Briefly, after 12 h, Dulbecco modified minimal essential medium (DMEM) was replaced by AIMV medium (ThermoFisher Scientific), and culture supernatants were harvested 2 and 3 days after transfection, pooled, and concentrated by low-speed centrifugation. The pellets were resuspended in phosphate-buffered saline (PBS), and the virus infectious titer was 9.05 × 107 LacZ-forming units/ml when determined on S-MAGI cells. The absence of replication-competent SIV in the SCIV stock was confirmed by inoculating CEM-SEAP cells cultured for 80 days.
Adenoviral recombinants Ad-Sgp-syn (34) and Ad-Senv-co expressing the gag/pol and env genes of SIVmac239 (19), here termed Adgp and Adenv, respectively, were purified by CsCl gradient centrifugation. Based upon optical density (OD), titers of 8.8 × 1012 and 1.55 × 1013 particles/ml were obtained and used to calculate the number of particles used for immunization. The Adenv preparation was also analyzed for its gene-transducing units. Briefly, 293T cells were infected with 100 μl of serially diluted virus for 2 days, fixed with 80% ethanol, and incubated for 1 h with 100 μl of a 1:400 dilution of mouse anti-SIVgp120 KK68 monoclonal antibody (MAb) (NIH AIDS Reagent Program). After washing, the cells were treated with anti-mouse IgG horseradish peroxidase (HRP) conjugate and stained with 3-amino-9-ethyl carbazole, and plaques were counted. A titer of 5 × 1012 particles/ml was calculated.
FWPVgp and FWPVenv expressing the gag/pol and env genes of SIVmac251, respectively, were originally kindly provided by D. Panicali (Therion Biologics Corp.) (70) and verified for correct transgene expression by WB and IF assay. The recombinant viruses were amplified on specific-pathogen-free CEFs (Charles River Laboratories), purified on a discontinuous sucrose density gradient, and titrated as described previously (40). Briefly, the infected cells were harvested and ultracentrifuged at 30,000 × g for 2 h at 4°C, and the pellets, resuspended in 1 mM Tris, 150 mM NaCl, 1 mM EDTA, pH 7.4, were sonicated to release the intracellular virus. The supernatant was overlaid onto a discontinuous 30% to 45% (wt/wt) sucrose gradient in the same buffer. After ultracentrifugation at 38,000 × g for 1 h, the viral band at the interface was recovered, diluted with 1 mM Tris-HCl, pH 9, and pelleted at 67,000 × g for 1 h. The purified virus was resuspended in PBS and sonicated, and aliquots were stored at −80°C until use.
Transgene expression by the viral recombinants.
The level of expression of SIV proteins by SCIV was determined by WB. Detection of the expression of SIV proteins encoded by Adgp and Adenv was performed as described previously (19, 34).
SIV Gag/Pol and Env protein expression by FWPVgp and FWPVenv was examined by WB after infection (10 PFU/cell) of Vero cells, MRC-5 cells, and CEFs using a 1:100 dilution of a SIV-positive monkey serum pool as a primary antibody (kindly supplied by J. Heeney, Department of Veterinary Medicine, University of Cambridge, Cambridge, United Kingdom), followed by rabbit anti-monkey IgG conjugated with HRP (1:2,000 dilution; Sigma-Aldrich). Bands were visualized using an enhanced-chemiluminescence (ECL) system (EuroClone). Protein expression by rFWPV was also determined by IF assay in the same cells described previously (71). In detail, after infection with 3 PFU/cell, cells were grown for 4 h and incubated with a 1:100 dilution of a SIV-positive serum pool. A 1:30 dilution of fluorescein isothiocyanate (FITC)-conjugated rabbit anti-monkey IgG (Sigma-Aldrich) was used as a secondary antibody. For both WB and IF assay, cells infected with FWPVwt were used as a negative control.
Animals.
Eighteen colony-bred adult rhesus monkeys (Macaca mulatta) of Indian origin (14 males and 4 females) were housed at the German Primate Center under standard conditions, in accordance with the German Animal Welfare Act and the European Union guidelines on the use of nonhuman primates in biomedical research. The animal experiment was approved by an ethics committee authorized by the Lower Saxony State Office for Consumer Protection and Food Safety and performed under project license AZ 33.14-42502-04-072/08, issued by the same authority. All the animals were tuberculosis free and antibody negative for SIV, simian retrovirus, FWPV, and T-cell leukemia virus. Physical examinations, bleedings, and immunizations were carried out under ketamine anesthesia (10 mg/kg body weight). For bronchoalveolar lavage (BAL), collection of intestinal biopsy specimens and cervical cytobrush samples, and virus inoculation, a deeper anesthesia consisting of a mixture of 5% ketamine, 1% xylazine, and 0.1% atropine was used (72). The animals were humanely euthanized by an overdose of pentobarbital sodium (Narcoren) under anesthesia. They were euthanized either without clinical symptoms at the preset endpoint of the experiment or because of mild suffering, defined by a scoring system of termination criteria that was approved by the external ethics committee and that corresponded to IACUC endpoint guidelines.
Immunization and challenge protocol.
The 18 rhesus monkeys were divided into two vaccine groups (V1 and V2; six animals each) and two control groups (control 1 and control 2; three animals each). All the animals were characterized with regard to major histocompatibility complex (MHC) class I Mamu-A1*001:01, B*008:01, and B*017:01 alleles, as described previously (73, 74). The nine Mamu-A1*001:01-positive animals (5 males and 4 females) were distributed to the four different groups as shown in Table 1. Two females were assigned to each of the two vaccine groups. The timeline, vaccine regimens, and vaccine doses are outlined in Fig. 1. Both vaccine groups received two priming immunizations with SCIV subcutaneously (s.c.), followed by two orally administered mucosal boosters with Adgp and Adenv (V1) or FWPVgp and FWPVenv (V2). For the final booster, the vectors were switched and applied i.m.: V1 received FWPVgp plus FWPVenv, whereas V2 was immunized with Adgp plus Adenv. Three animals of the control group received Ad-GFP expressing the green fluorescent protein (control 1), and three received FWPVwt (control 2) orally, followed by a crossover immunization given i.m. The oral spray administration has been previously described in detail (19, 75).
Eight weeks after final immunization (i.e., week 42 after the first immunization), repeated low-dose i.r. challenges were performed with SIVmac251 stock 10/09 (120 TCID50/dose/animal). This SIV stock was prepared by an additional in vitro passage of an early preparation of SIVmac251 (76), kindly provided by A. M. Aubertin (Université Louis Pasteur, Strasbourg, France). Additional details regarding the virus and the challenge procedure have been reported previously (14). Viral exposures were performed at weekly intervals, and the plasma viral load was determined. Challenges were suspended 1 week after the viral RNA copies exceeded the predefined cutoff: 100 copies/ml of plasma. To determine the number of viral lineages in our virus challenge stock, SGA was performed as described previously (35). An env phylogenetic tree was constructed by the neighbor-joining method to compare our virus stock with other stocks commonly used as SIV challenge stocks in vaccine studies (35).
Sample collection, mononuclear cell purification, and flow cytometry.
Blood samples were obtained by puncture of the femoral vein at predefined time points. PBMCs were Ficoll purified and used immediately for the immune assays. The purification of mononuclear cells from colonic biopsy specimens and BAL fluid was carried out as previously described (77).
To collect cervical cells, female monkeys were positioned in ventral recumbency with their hips elevated. A pediatric speculum was inserted into the vagina to locate the ectocervix. The tip of a flocked nylon swab (microRheologics; 501CS01) was gently inserted without touching the vaginal mucosa and rotated briefly. After removing the swab, the tip was cut off, transferred into a 5-ml tube containing PBS, and kept on ice until further processing. For each animal, four swabs were taken and inserted into four tubes. After vortexing the tubes at the highest speed for 2 s, the swabs were removed and the samples were pooled. To maximize cell yield, the swabs were placed in a sputum liquefying solution (Copan), vortexed at 80% of maximum speed until the mucus dissolved (20 to 30 s), and washed once with PBS. The cell pellet was resuspended in PBS, and lymphocytes and epithelial cells were counted.
Phenotypic analyses of leukocytes were performed by polychromatic flow cytometry as described previously (78, 79) using an LSRII flow cytometer (BD Biosciences). FlowJo version 8.7 (Tree Star) was used for the analysis of the list mode data files. The following antibody-fluorochrome combinations were used: CD8 (RPA-T8; Pacific Orange; Caltag), CD20 (2H7; eFluor 625NC; eBioscience), CD45 (M34-6D6; FITC; Miltenyi), CD69 (TP1.55.3; ECD; Beckman Coulter), CD3 (SP34.2; AF700), CD4 (L200; V450), CD28 and CD95 (DX2; phycoerythrin [PE]-Cy7), CD183 (IC6/CXCR3; PE), CD196 (11A9; peridinin chlorophyll protein [PerCP]-Cy5.5), HLA-DR (L243; allophycocyanin [APC]-Cy7) (all from BD-Biosciences unless otherwise stated).
Serological assays.
To determine the SIV-specific humoral immune response in blood, a standard ELISA was performed using SIV p27 (EVA643) and gp130 (EVA670) recombinant proteins, kindly provided by the Centre for AIDS Reagents (National Institute for Biological Standards and Control [NIBSC], United Kingdom). Antibodies against the SIV proteins were quantified using 1:200-diluted plasma by measuring the optical density at 450 nm (80). Anti-SIV IgA antibody titers were also determined by limiting-dilution assay (79) 2 weeks after the final immunization.
To detect anti-FWPV antibodies, an ELISA was performed using the purified virus as a plate-bound antigen. Briefly, 96-well MaxiSorp microtiter plates (Nunc) were coated with FWPVwt (2 × 105 PFU/well) in 0.05 M carbonate-bicarbonate buffer, pH 9.6, and incubated overnight at 4°C. The sera were then added at 1:100 dilution, and binding was revealed by adding a 1:10,000 dilution of rabbit anti-monkey IgG conjugated with HRP (1:2,000 dilution; Sigma-Aldrich) and tetramethylbenzidine (TMB) substrate (Sigma). The cutoff for positivity was an OD of 0.4, i.e., twice the mean of the background of all the sera. The individual background ODs ranged from 0.09 to 0.36 (see Results).
Neutralization assays.
SIV-neutralizing antibodies were quantified by a luciferase reporter gene assay (Bright Glo Luciferase Assay; Promega) according to the manufacturer's instructions. The test was performed in duplicate, using the TZM-bl cell line. Briefly, heat-inactivated serum samples were diluted serially in DMEM (PAN Biotech), supplemented with 10% fetal calf serum (FCS) (PAA) and 1% P/S (100 U/ml penicillin and 100 mg/ml streptomycin; PAN Biotech). The replication-competent SIVmac32H stock was added to the cells at a dilution sufficient to cause a luciferase signal at least 100-fold higher than that of virus-negative cells. After incubation for 1 h at 37°C, 1 × 104 cells/well were added in DMEM containing DEAE dextran (Sigma) at a final concentration of 15 μg/ml. After 48 h at 37°C, the cells were washed, lysed, and transferred into a 96-well plate (Corning), and the luciferase activity was determined in a PlateChameleonV reader (Hiddex). Positive results are reported as the reciprocal serum dilution that resulted in a 95% reduction of relative light units compared to the virus control in the absence of serum. The monoclonal SIV-neutralizing KK9 antibody (Centre for AIDS Reagents, NIBSC, United Kingdom) was used as a positive control.
To determine neutralizing activity against the FWPV vector, FWPVwt (102 PFU/petri dish) was preincubated with an equal volume of heat-inactivated, serially diluted sera of vaccinated animals for 1 h at 37°C, using preimmune serum as a negative control. The cells were overlaid with 5 ml DMEM containing 0.7% low-electroendosmosis agarose (SeaKem; FMC BioProducts). After adding an additional agarose layer containing 1.5% neutral red (Gibco), the plaques were counted on day 4 postinfection. The neutralizing activity was determined as the percentage of plaque reduction versus the control, where the FWPVwt was incubated without serum.
ELISpot assay and tetramer and intracellular-cytokine staining.
To determine the SIV-specific T-cell response in the vaccinees, an IFN-γ ELISpot assay was carried out as described previously (81). Virus-specific stimulation was performed with either SIV Gag peptides (EVA7066.1-16, Centre for AIDS Reagents, NIBSC, United Kingdom) or aldrithiol-2-inactivated whole SIVmac251 particles (SIV AT-2) (ARP1018.1, kindly provided by Jeff Lifson, National Cancer Institute [NCI], Frederick, MD, USA, via the Centre for AIDS Reagents, NIBSC, United Kingdom). A pool of six 20-mer peptides derived from the hepatitis C virus NS3 gene was included to assess the background peptide stimulation (82). Microvesicles (ARP1018.2) were used as controls for viral particle stimulation. The numbers of virus-specific SFCs were calculated as previously reported (81, 84). To analyze whether SIV AT-2-specific responses were mediated by CD4+ or CD8+ subsets, PBMCs from long-term SIV-infected monkeys presenting with strong ELISpot responses were used. PBMCs were depleted of CD4+ or CD8+ cells using the Magnetic Activated Cell Sorting (MACS) system (Milteny Biotec). Next, the respective fractions were stimulated with SIV AT-2 or microvesicles and analyzed by an IFN-γ ELISpot assay as outlined above.
Mononuclear cells isolated from blood, BAL fluid, colon biopsy specimens, and cervical brushes of Mamu-A1*001:01-positive macaques were analyzed for SIVgag-specific tetramer-binding CD8+ cells as described previously (85).
For intracellular-cytokine staining, 0.5 × 106 to 1.5 × 106 Ficoll-purified PBMCs were stimulated with SIV Gag peptides (see above) with a final concentration of 2 μg/ml/peptide at 37°C in a 5% CO2 atmosphere for 1 h. Staphyloccocus enterotoxin B (SEB) (Sigma)-stimulated cells served as positive controls and those cultured with unrelated peptides (see “ELISpot assay and tetramer and intracellular-cytokine staining” above) and plain culture medium as negative controls. All the cultures were supplemented with anti-CD28 (clone CD28.2; BD Biosciences Pharmingen) as a costimulus. Next, brefeldin A (Sigma) was added to the cultures at 10 μg/ml. After a further 5 h of incubation, the cells were centrifuged and stained with optimal dilutions of fluorochrome-conjugated MAbs for the cell surface markers CD3 (SP34-2; AF 700) and CD4 (L200; PerCP-Cy5.5) from BD Biosciences and CD8 (3B5; Pacific Orange) from Caltag. After washing with PBS-0.5% bovine serum albumin (BSA), the cells were fixed with 4% formaldehyde and stored at 4°C overnight. The next day, the cells were permeabilized with a saponin buffer (PBS-BSA-0.5% saponin [Sigma]), followed by incubation with a mixture of antibodies against three different cytokines, i.e., anti-interleukin 2 (IL-2) (MQ1-17H12; FITC), anti-TNF-α (MAb11; PE), and anti-IFN-γ (B27; APC), all used in optimal concentrations and obtained from BD Biosciences. Samples were analyzed as described above. From the lymphocyte population gated via low forward scatter and side scatter, first duplets and then dead cells were excluded, followed by identification of CD3+ T cells. The T cells were further divided into CD4+ and CD8+ subsets and analyzed for expression of the respective cytokines.
CFSE proliferation assay.
A CFSE assay was applied to determine T-cell proliferation. Labeling of PBMCs with CFSE at 1 μM was performed as previously described (83). Next, the cells were transferred to a 96-well round-bottom plate at 2 × 105 per well and stimulated in triplicate with either 2 μg/ml SIV-AT2, microvesicles, or SEB (Sigma). After 6 days, the triplicates were pooled, and T cells were stained with CD3 (SP34-2; AF700; BD Biosciences), CD4 (L200; V450; BD Biosciences), CD8 (DK25; PE; Dako), CD45RA (2H4; ECD; Beckman Coulter), CD28 (L293; PerCP-Cy5.5; BD Bioscience), and CD95 (DX2; APC; BD Biosciences). The stained cells were processed on an LSRII flow cytometer (BD Bioscience) using FACSDiva software (BD Bioscience) and analyzed with FlowJo (Treestar). Proliferating CD4+ and CD8+ T cells were defined as CFSE− CD45RA−.
Real-time quantitative reverse transcription-PCR (qRT-PCR).
Total cellular RNA was isolated from whole blood stored in PAX Gene blood RNA tubes (BD Diagnostics) with a PAX Gene RNA kit (Qiagen). The RNA quality of most samples was determined using an RNA 6000 Nano kit (Agilent Technologies) and a 2100 Bioanalyzer (Agilent Technologies). RNA integrity numbers were ≥8.0. The absence of genomic DNA was randomly verified by PCR amplification of samples without cDNA synthesis. As a positive control, cDNA from one unrelated control animal was used. Relative RNA levels were quantified by real-time PCR using GAPDH as the reference gene. Primer sequencing for the amplification of GAPDH, CXCL10, MX1, BST-2, and FAM26F, as well as reverse transcription and PCRs, was performed as reported previously (36, 86). Each sample was tested in triplicate using the ABI Prism 7500 system (PE Applied Biosystems). Results were analyzed with sequence detection software (Applied Biosystems). Relative RNA copy numbers (rE) were calculated as follows: rE = 100 × 2ΔCt.
Quantification of plasma IFN-α, IFN-γ, and IP-10.
Plasma IFN-α and IFN-γ were quantified by ELISA using panspecific antibodies for IFN-α (Mabtech) and rhesus macaque-specific antibodies for IFN-γ (Mabtech), following the instructions of the manufacturer. Briefly, plasma was added to high-binding microtiter plates (Greiner Bio-One GmbH) that were previously coated with the respective capture antibodies. Bound interferons from plasma samples were quantified by adding biotinylated detection antibody, streptavidin-HRP conjugate, and TMB substrate (Sigma). CXCL10 was quantified using a cross-reacting human CXCL10/IP-10 ELISA kit (R&D Systems) according to the manufacturer's specifications. Absorbance was measured at 405-nm wavelength with a 550 microplate reader (Bio-Rad Laboratories).
Determination of viral load by qRT-PCR.
The viral load was determined by quantifying SIV RNA copies using TaqMan-based real-time PCR (87). Viral RNA was isolated from frozen plasma samples following the MagAttract Virus Mini M48 protocol (Qiagen). Amplified RNA was expressed as SIV RNA copies/ml of plasma. The detection limit was 40 RNA copies/ml of plasma.
Statistical analyses.
The power of our repeated low-dose challenge study design to detect differences in vaccine efficacy between animal groups was determined using a Web-based calculator that emerged from a study by Hudgens and Gilbert (88). Power was evaluated assuming a significance level of 0.05, a maximum of eight challenges per animal, and a per-challenge probability of infection without vaccine of 0.5.
Cumulative-incidence analysis, including log rank tests for group comparisons, was performed with Sigma Stat 3.5 (Systat Software GmbH), treating challenges as time units. HRs with 95% confidence intervals were calculated employing a Cox regression model as implemented in procedure SURVEYPHREG of SAS version 9.4 (SAS Institute, Cary, NC, USA). Vaccine efficacy (VE) was calculated as follows: VE = (1 − HR) × 100. All other statistical analyses were performed with GraphPad Prism version 5 (GraphPad Software). Differences between groups were evaluated using a two-tailed Mann-Whitney U test. Relationships between parameters were quantified by means of Spearman's rank correlation coefficient (rs) and assessed for statistical significance using a permutation test. P values of <0.05 were considered significant.
ACKNOWLEDGMENTS
We thank K. Eckelmann, J. Hampe, and S. Heine for their excellent technical support; T. Eggers, G. Marschhausen, and P. Müller for animal care; K. Listemann, K. Töpfer, and B. Neumann for support in flow cytometry; and S. Petersen for proofreading the manuscript. We thank Young-C. Sung for providing rAd/GFP. The stimulation reagents for ELISA and the ELISpot assay and the KK9 antibody were kindly provided through the European Union Program EVA/MRC Centralized Facility for AIDS Reagents, NIBSC, United Kingdom (grant numbers QLK2-CT-1999-00609 and GP828102). SIV AT-2 was supplied by Jeff Lifson through the European Union Program EVA Centre for AIDS Reagents, NIBSC, United Kingdom (AVIP contract number LSHP-CT-2004-503487) and NIH AIDS Reagent Program for provision of KK68 antibody. We thank J. Heeney for provision of sera from SIV-infected monkeys. The FWPVgp and FWPVenv recombinants expressing the gag/pol and env genes of SIVmac251, respectively, were kindly provided by D. Panicali (Therion Biologics Corp., Cambridge, MA).
This project has been funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract no. HHSN261200800001E.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
REFERENCES
- 1.Barouch DH, Alter G, Broge T, Linde C, Ackerman ME, Brown EP, Borducchi EN, Smith KM, Nkolola JP, Liu J, Shields J, Parenteau L, Whitney JB, Abbink P, Ng‘ang'a DM, Seaman MS, Lavine CL, Perry JR, Li W, Colantonio AD, Lewis MG, Chen B, Wenschuh H, Reimer U, Piatak M, Lifson JD, Handley SA, Virgin HW, Koutsoukos M, Lorin C, Voss G, Weijtens M, Pau MG, Schuitemaker H. 2015. Protective efficacy of adenovirus/protein vaccines against SIV challenges in rhesus monkeys. Science 349:320–324. doi: 10.1126/science.aab3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roederer M, Keele BF, Schmidt SD, Mason RD, Welles HC, Fischer W, Labranche C, Foulds KE, Louder MK, Yang ZY, Todd JP, Buzby AP, Mach LV, Shen L, Seaton KE, Ward BM, Bailer RT, Gottardo R, Gu W, Ferrari G, Alam SM, Denny TN, Montefiori DC, Tomaras GD, Korber BT, Nason MC, Seder RA, Koup RA, Letvin NL, Rao SS, Nabel GJ, Mascola JR. 2014. Immunological and virological mechanisms of vaccine-mediated protection against SIV and HIV. Nature 505:502–508. doi: 10.1038/nature12893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sui Y, Gordon S, Franchini G, Berzofsky JA. 2013. Nonhuman primate models for HIV/AIDS vaccine development. Curr Protoc Immunol 102:Unit 12.14. doi: 10.1002/0471142735.im1214s102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kardani K, Bolhassani A, Shahbazi S. 2016. Prime-boost vaccine strategy against viral infections: mechanisms and benefits. Vaccine 34:413–423. doi: 10.1016/j.vaccine.2015.11.062. [DOI] [PubMed] [Google Scholar]
- 5.Musich T, Robert-Guroff M. 2016. New developments in an old strategy: heterologous vector primes and envelope protein boosts in HIV vaccine design. Expert Rev Vaccines 15:1015–1027. doi: 10.1586/14760584.2016.1158108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lehner T. 2003. Innate and adaptive mucosal immunity in protection against HIV infection. Vaccine 21(Suppl 2):S68–S76. doi: 10.1016/S0264-410X(03)00204-4. [DOI] [PubMed] [Google Scholar]
- 7.Lehner T, Wang Y, Cranage M, Bergmeier LA, Mitchell E, Tao L, Hall G, Dennis M, Cook N, Brookes R, Klavinskis L, Jones I, Doyle C, Ward R. 1996. Protective mucosal immunity elicited by targeted iliac lymph node immunization with a subunit SIV envelope and core vaccine in macaques. Nat Med 2:767–775. doi: 10.1038/nm0796-767. [DOI] [PubMed] [Google Scholar]
- 8.Aldovini A. 2016. Mucosal vaccination for prevention of HIV infection and AIDS. Curr HIV Res 14:247–259. doi: 10.2174/1570162X14999160224103025. [DOI] [PubMed] [Google Scholar]
- 9.Ogra PL, Faden H, Welliver RC. 2001. Vaccination strategies for mucosal immune responses. Clin Microbiol Rev 14:430–445. doi: 10.1128/CMR.14.2.430-445.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poles J, Alvarez Y, Hioe CE. 2014. Induction of intestinal immunity by mucosal vaccines as a means of controlling HIV infection. AIDS Res Hum Retroviruses 30:1027–1040. doi: 10.1089/aid.2014.0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fouts TR, Bagley K, Prado IJ, Bobb KL, Schwartz JA, Xu R, Zagursky RJ, Egan MA, Eldridge JH, LaBranche CC, Montefiori DC, Le Buanec H, Zagury D, Pal R, Pavlakis GN, Felber BK, Franchini G, Gordon S, Vaccari M, Lewis GK, DeVico AL, Gallo RC. 2015. Balance of cellular and humoral immunity determines the level of protection by HIV vaccines in rhesus macaque models of HIV infection. Proc Natl Acad Sci U S A 112:E992–E999. doi: 10.1073/pnas.1423669112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lewis GK, DeVico AL, Gallo RC. 2014. Antibody persistence and T-cell balance: two key factors confronting HIV vaccine development. Proc Natl Acad Sci U S A 111:15614–15621. doi: 10.1073/pnas.1413550111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Staprans SI, Barry AP, Silvestri G, Safrit JT, Kozyr N, Sumpter B, Nguyen H, McClure H, Montefiori D, Cohen JI, Feinberg MB. 2004. Enhanced SIV replication and accelerated progression to AIDS in macaques primed to mount a CD4 T cell response to the SIV envelope protein. Proc Natl Acad Sci U S A 101:13026–13031. doi: 10.1073/pnas.0404739101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tenbusch M, Ignatius R, Temchura V, Nabi G, Tippler B, Stewart-Jones G, Salazar AM, Sauermann U, Stahl-Hennig C, Überla K. 2012. Risk of immunodeficiency virus infection may increase with vaccine-induced immune response. J Virol 86:10533–10539. doi: 10.1128/JVI.00796-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qureshi H, Ma ZM, Huang Y, Hodge G, Thomas MA, DiPasquale J, DeSilva V, Fritts L, Bett AJ, Casimiro DR, Shiver JW, Robert-Guroff M, Robertson MN, McChesney MB, Gilbert PB, Miller CJ. 2012. Low-dose penile SIVmac251 exposure of rhesus macaques infected with adenovirus type 5 (Ad5) and then immunized with a replication-defective Ad5-based SIV gag/pol/nef vaccine recapitulates the results of the phase IIb step trial of a similar HIV-1 vaccine. J Virol 86:2239–2250. doi: 10.1128/JVI.06175-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Afkhami S, Yao Y, Xing Z. 2016. Methods and clinical development of adenovirus-vectored vaccines against mucosal pathogens. Mol Ther Methods Clin Dev 3:16030. doi: 10.1038/mtm.2016.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ertl HC. 2016. Viral vectors as vaccine carriers. Curr Opin Virol 21:1–8. doi: 10.1016/j.coviro.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 18.Ewer KJ, Lambe T, Rollier CS, Spencer AJ, Hill AV, Dorrell L. 2016. Viral vectors as vaccine platforms: from immunogenicity to impact. Curr Opin Immunol 41:47–54. doi: 10.1016/j.coi.2016.05.014. [DOI] [PubMed] [Google Scholar]
- 19.Stahl-Hennig C, Kuate S, Franz M, Suh YS, Stoiber H, Sauermann U, Tenner-Racz K, Norley S, Park KS, Sung YC, Steinman R, Racz P, Überla K. 2007. Atraumatic oral spray immunization with replication-deficient viral vector vaccines. J Virol 81:13180–13190. doi: 10.1128/JVI.01400-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buchbinder SP, Mehrotra DV, Duerr A, Fitzgerald DW, Mogg R, Li D, Gilbert PB, Lama JR, Marmor M, Del Rio C, McElrath MJ, Casimiro DR, Gottesdiener KM, Chodakewitz JA, Corey L, Robertson MN, Step Study Protocol Team. 2008. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet 372:1881–1893. doi: 10.1016/S0140-6736(08)61591-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gray GE, Allen M, Moodie Z, Churchyard G, Bekker LG, Nchabeleng M, Mlisana K, Metch B, de Bruyn G, Latka MH, Roux S, Mathebula M, Naicker N, Ducar C, Carter DK, Puren A, Eaton N, McElrath MJ, Robertson M, Corey L, Kublin JG, HVTN 503/Phambili Study Team. 2011. Safety and efficacy of the HVTN 503/Phambili study of a clade-B-based HIV-1 vaccine in South Africa: a double-blind, randomised, placebo-controlled test-of-concept phase 2b study. Lancet Infect Dis 11:507–515. doi: 10.1016/S1473-3099(11)70098-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coupar BE, Purcell DF, Thomson SA, Ramshaw IA, Kent SJ, Boyle DB. 2006. Fowlpox virus vaccines for HIV and SHIV clinical and pre-clinical trials. Vaccine 24:1378–1388. doi: 10.1016/j.vaccine.2005.09.044. [DOI] [PubMed] [Google Scholar]
- 23.De Rose R, Batten CJ, Smith MZ, Fernandez CS, Peut V, Thomson S, Ramshaw IA, Coupar BE, Boyle DB, Venturi V, Davenport MP, Kent SJ. 2007. Comparative efficacy of subtype AE simian-human immunodeficiency virus priming and boosting vaccines in pigtail macaques. J Virol 81:292–300. doi: 10.1128/JVI.01727-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Rose R, Chea S, Dale CJ, Reece J, Fernandez CS, Wilson KM, Thomson S, Ramshaw IA, Coupar BE, Boyle DB, Sullivan MT, Kent SJ. 2005. Subtype AE HIV-1 DNA and recombinant Fowlpoxvirus vaccines encoding five shared HIV-1 genes: safety and T cell immunogenicity in macaques. Vaccine 23:1949–1956. doi: 10.1016/j.vaccine.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 25.Nacsa J, Radaelli A, Edghill-Smith Y, Venzon D, Tsai WP, Morghen CDG, Panicali D, Tartaglia J, Franchini G.. 2004. Avipox-based simian immunodeficiency virus (SIV) vaccines elicit a high frequency of SIV-specific CD4+ and CD8+ T-cell responses in vaccinia-experienced SIVmac251-infected macaques. Vaccine 22:597–606. doi: 10.1016/j.vaccine.2003.08.028. [DOI] [PubMed] [Google Scholar]
- 26.Radaelli A, Bonduelle O, Beggio P, Mahe B, Pozzi E, Elli V, Paganini M, Zanotto C, De Giuli Morghen C, Combadiere B. 2007. Prime-boost immunization with DNA, recombinant fowlpox virus and VLP(SHIV) elicit both neutralizing antibodies and IFNgamma-producing T cells against the HIV-envelope protein in mice that control env-bearing tumour cells. Vaccine 25:2128–2138. doi: 10.1016/j.vaccine.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 27.Radaelli A, Nacsa J, Tsai WP, Edghill-Smith Y, Zanotto C, Elli V, Venzon D, Tryniszewska E, Markham P, Mazzara GP, Panicali D, De Giuli Morghen C, Franchini G. 2003. Prior DNA immunization enhances immune response to dominant and subdominant viral epitopes induced by a fowlpox-based SIVmac vaccine in long-term slow-progressor macaques infected with SIVmac251. Virology 312:181–195. doi: 10.1016/S0042-6822(03)00184-3. [DOI] [PubMed] [Google Scholar]
- 28.Radaelli A, Zanotto C, Perletti G, Elli V, Vicenzi E, Poli G, De Giuli Morghen C. 2003. Comparative analysis of immune responses and cytokine profiles elicited in rabbits by the combined use of recombinant fowlpox viruses, plasmids and virus-like particles in prime-boost vaccination protocols against SHIV. Vaccine 21:2052–2064. doi: 10.1016/S0264-410X(02)00773-9. [DOI] [PubMed] [Google Scholar]
- 29.Santra S, Sun Y, Parvani JG, Philippon V, Wyand MS, Manson K, Gomez-Yafal A, Mazzara G, Panicali D, Markham PD, Montefiori DC, Letvin NL. 2007. Heterologous prime/boost immunization of rhesus monkeys by using diverse poxvirus vectors. J Virol 81:8563–8570. doi: 10.1128/JVI.00744-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stratov I, Dale CJ, Kent SJ. 2005. Phenotypic and kinetic analysis of effective simian-human immunodeficiency virus-specific T cell responses in DNA- and fowlpox virus-vaccinated macaques. Virology 337:222–234. doi: 10.1016/j.virol.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 31.Zanotto C, Elli V, Basavecchia V, Brivio A, Paganini M, Pinna D, Vicenzi E, De Giuli Morghen C, Radaelli A. 2003. Evaluation in rabbits of different anti-SHIV vaccine strategies based on DNA/fowlpox priming and virus-like particle boosting. FEMS Immunol Med Microbiol 35:59–65. doi: 10.1111/j.1574-695X.2003.tb00649.x. [DOI] [PubMed] [Google Scholar]
- 32.Ranasinghe C, Medveczky JC, Woltring D, Gao K, Thomson S, Coupar BE, Boyle DB, Ramsay AJ, Ramshaw IA. 2006. Evaluation of fowlpox-vaccinia virus prime-boost vaccine strategies for high-level mucosal and systemic immunity against HIV-1. Vaccine 24:5881–5895. doi: 10.1016/j.vaccine.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 33.Kuate S, Stahl-Hennig C, ten Haaft P, Heeney J, Uberla K. 2003. Single-cycle immunodeficiency viruses provide strategies for uncoupling in vivo expression levels from viral replicative capacity and for mimicking live-attenuated SIV vaccines. Virology 313:653–662. doi: 10.1016/S0042-6822(03)00388-X. [DOI] [PubMed] [Google Scholar]
- 34.Kuate S, Stefanou D, Hoffmann D, Wildner O, Überla K. 2004. Production of lentiviral vectors by transient expression of minimal packaging genes from recombinant adenoviruses. J Gene Med 6:1197–1205. doi: 10.1002/jgm.623. [DOI] [PubMed] [Google Scholar]
- 35.Del Prete GQ, Scarlotta M, Newman L, Reid C, Parodi LM, Roser JD, Oswald K, Marx PA, Miller CJ, Desrosiers RC, Barouch DH, Pal R, Piatak M Jr, Chertova E, Giavedoni LD, O'Connor DH, Lifson JD, Keele BF. 2013. Comparative characterization of transfection- and infection-derived simian immunodeficiency virus challenge stocks for in vivo nonhuman primate studies. J Virol 87:4584–4595. doi: 10.1128/JVI.03507-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Javed A, Leuchte N, Salinas G, Opitz L, Stahl-Hennig C, Sopper S, Sauermann U. 2016. Pre-infection transcript levels of FAM26F in peripheral blood mononuclear cells inform about overall plasma viral load in acute and post-acute phase after simian immunodeficiency virus infection. J Gen Virol 97:3400–3412. doi: 10.1099/jgv.0.000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Egan MA, Chong SY, Megati S, Montefiori DC, Rose NF, Boyer JD, Sidhu MK, Quiroz J, Rosati M, Schadeck EB, Pavlakis GN, Weiner DB, Rose JK, Israel ZR, Udem SA, Eldridge JH. 2005. Priming with plasmid DNAs expressing interleukin-12 and simian immunodeficiency virus gag enhances the immunogenicity and efficacy of an experimental AIDS vaccine based on recombinant vesicular stomatitis virus. AIDS Res Hum Retroviruses 21:629–643. doi: 10.1089/aid.2005.21.629. [DOI] [PubMed] [Google Scholar]
- 38.Fauci AS, Marovich MA, Dieffenbach CW, Hunter E, Buchbinder SP.. 2014. Immunology. Immune activation with HIV vaccines. Science 344:49–51. doi: 10.1126/science.1250672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schat KA, Erb HN. 2014. Lack of evidence that avian oncogenic viruses are infectious for humans: a review. Avian Dis 58:345–358. doi: 10.1637/10847-041514-Review.1. [DOI] [PubMed] [Google Scholar]
- 40.Pacchioni S, Volonte L, Zanotto C, Pozzi E, De Giuli Morghen C, Radaelli A. 2010. Canarypox and fowlpox viruses as recombinant vaccine vectors: an ultrastructural comparative analysis. Arch Virol 155:915–924. doi: 10.1007/s00705-010-0663-7. [DOI] [PubMed] [Google Scholar]
- 41.Zanotto C, Pozzi E, Pacchioni S, Volonte L, De Giuli Morghen C, Radaelli A. 2010. Canarypox and fowlpox viruses as recombinant vaccine vectors: a biological and immunological comparison. Antiviral Res 88:53–63. doi: 10.1016/j.antiviral.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 42.Pitisuttithum P, Rerks-Ngarm S, Bussaratid V, Dhitavat J, Maekanantawat W, Pungpak S, Suntharasamai P, Vanijanonta S, Nitayapan S, Kaewkungwal J, Benenson M, Morgan P, O'Connell RJ, Berenberg J, Gurunathan S, Francis DP, Paris R, Chiu J, Stablein D, Michael NL, Excler JL, Robb ML, Kim JH. 2011. Safety and reactogenicity of canarypox ALVAC-HIV (vCP1521) and HIV-1 gp120 AIDSVAX B/E vaccination in an efficacy trial in Thailand. PLoS One 6:e27837. doi: 10.1371/journal.pone.0027837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Offerman K, Deffur A, Carulei O, Wilkinson R, Douglass N, Williamson AL. 2015. Six host-range restricted poxviruses from three genera induce distinct gene expression profiles in an in vivo mouse model. BMC Genomics 16:510. doi: 10.1186/s12864-015-1659-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trivedi S, Jackson RJ, Ranasinghe C. 2014. Different HIV pox viral vector-based vaccines and adjuvants can induce unique antigen presenting cells that modulate CD8 T cell avidity. Virology 468-470:479–489. doi: 10.1016/j.virol.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 45.Bissa M, Quaglino E, Zanotto C, Illiano E, Rolih V, Pacchioni S, Cavallo F, De Giuli Morghen C, Radaelli A. 2016. Protection of mice against the highly pathogenic VVIHD-J by DNA and fowlpox recombinant vaccines, administered by electroporation and intranasal routes, correlates with serum neutralizing activity. Antiviral Res 134:182–191. doi: 10.1016/j.antiviral.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li J, Yang T, Xu Q, Sun E, Feng Y, Lv S, Zhang Q, Wang H, Wu D. 2015. DNA vaccine prime and recombinant FPV vaccine boost: an important candidate immunization strategy to control bluetongue virus type 1. Appl Microbiol Biotechnol 99:8643–8652. doi: 10.1007/s00253-015-6697-8. [DOI] [PubMed] [Google Scholar]
- 47.Bissa M, Illiano E, Pacchioni S, Paolini F, Zanotto C, De Giuli Morghen C, Massa S, Franconi R, Radaelli A, Venuti A. 2015. A prime/boost strategy using DNA/fowlpox recombinants expressing the genetically attenuated E6 protein as a putative vaccine against HPV-16-associated cancers. J Transl Med 13:80. doi: 10.1186/s12967-015-0437-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robinson HL, Montefiori DC, Johnson RP, Manson KH, Kalish ML, Lifson JD, Rizvi TA, Lu S, Hu SL, Mazzara GP, Panicali DL, Herndon JG, Glickman R, Candido MA, Lydy SL, Wyand MS, McClure HM. 1999. Neutralizing antibody-independent containment of immunodeficiency virus challenges by DNA priming and recombinant pox virus booster immunizations. Nat Med 5:526–534. doi: 10.1038/8406. [DOI] [PubMed] [Google Scholar]
- 49.Hoffmann PR, Panigada M, Soprana E, Terry F, Bandar IS, Napolitano A, Rose AH, Hoffmann FW, Ndhlovu LC, Belcaid M, Moise L, De Groot AS, Carbone M, Gaudino G, Matsui T, Siccardi A, Bertino P. 2015. Preclinical development of HIvax: human survivin highly immunogenic vaccines. Hum Vaccin Immunother 11:1585–1595. doi: 10.1080/21645515.2015.1050572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Singh P, Pal SK, Alex A, Agarwal N. 2015. Development of PROSTVAC immunotherapy in prostate cancer. Future Oncol 11:2137–2148. doi: 10.2217/fon.15.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Manrique M, Kozlowski PA, Cobo-Molinos A, Wang SW, Wilson RL, Martinez-Viedma MDP, Montefiori DC, Carville A, Aldovini A. 2014. Resistance to infection, early and persistent suppression of simian immunodeficiency virus SIVmac251 viremia, and significant reduction of tissue viral burden after mucosal vaccination in female rhesus macaques. J Virol 88:212–224. doi: 10.1128/JVI.02523-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Selinger C, Strbo N, Gonzalez L, Aicher L, Weiss JM, Law GL, Palermo RE, Vaccari M, Franchini G, Podack ER, Katze MG. 2014. Multiple low-dose challenges in a rhesus macaque AIDS vaccine trial result in an evolving host response that affects protective outcome. Clin Vaccine Immunol 21:1650–1660. doi: 10.1128/CVI.00455-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burton SL, Kilgore KM, Smith SA, Reddy S, Hunter E, Robinson HL, Silvestri G, Amara RR, Derdeyn CA. 2015. Breakthrough of SIV strain smE660 challenge in SIV strain mac239-vaccinated rhesus macaques despite potent autologous neutralizing antibody responses. Proc Natl Acad Sci U S A 112:10780–10785. doi: 10.1073/pnas.1509731112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Y, Abel K, Lantz K, Krieg AM, McChesney MB, Miller CJ. 2005. The Toll-like receptor 7 (TLR7) agonist, imiquimod, and the TLR9 agonist, CpG ODN, induce antiviral cytokines and chemokines but do not prevent vaginal transmission of simian immunodeficiency virus when applied intravaginally to rhesus macaques. J Virol 79:14355–14370. doi: 10.1128/JVI.79.22.14355-14370.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Janes HE, Cohen KW, Frahm N, De Rosa SC, Sanchez B, Hural J, Magaret CA, Karuna S, Bentley C, Gottardo R, Finak G, Grove D, Shen M, Graham BS, Koup RA, Mulligan MJ, Koblin B, Buchbinder SP, Keefer MC, Adams E, Anude C, Corey L, Sobieszczyk M, Hammer SM, Gilbert PB, McElrath MJ. 2017. Higher T cell responses induced by DNA/rAd5 HIV-1 preventive vaccine are associated with lower HIV-1 infection risk in an efficacy trial. J Infect Dis 215:1376–1385. doi: 10.1093/infdis/jix086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin L, Finak G, Ushey K, Seshadri C, Hawn TR, Frahm N, Scriba TJ, Mahomed H, Hanekom W, Bart PA, Pantaleo G, Tomaras GD, Rerks-Ngarm S, Kaewkungwal J, Nitayaphan S, Pitisuttithum P, Michael NL, Kim JH, Robb ML, O'Connell RJ, Karasavvas N, Gilbert PC, De Rosa S, McElrath MJ, Gottardo R. 2015. COMPASS identifies T-cell subsets correlated with clinical outcomes. Nat Biotechnol 33:610–616. doi: 10.1038/nbt.3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mooij P, Balla-Jhagjhoorsingh SS, Koopman G, Beenhakker N, van Haaften P, Baak I, Nieuwenhuis IG, Kondova I, Wagner R, Wolf H, Gomez CE, Najera JL, Jimenez V, Esteban M, Heeney JL.. 2008. Differential CD4+ versus CD8+ T-cell responses elicited by different poxvirus-based human immunodeficiency virus type 1 vaccine candidates provide comparable efficacies in primates. J Virol 82:2975–2988. doi: 10.1128/JVI.02216-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Karin N, Wildbaum G, Thelen M. 2016. Biased signaling pathways via CXCR3 control the development and function of CD4+ T cell subsets. J Leukoc Biol 99:857–862. doi: 10.1189/jlb.2MR0915-441R. [DOI] [PubMed] [Google Scholar]
- 59.Van Raemdonck K, Van den Steen PE, Liekens S, Van Damme J, Struyf S. 2015. CXCR3 ligands in disease and therapy. Cytokine Growth Factor Rev 26:311–327. doi: 10.1016/j.cytogfr.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 60.Zohar Y, Wildbaum G, Novak R, Salzman AL, Thelen M, Alon R, Barsheshet Y, Karp CL, Karin N. 2014. CXCL11-dependent induction of FOXP3-negative regulatory T cells suppresses autoimmune encephalomyelitis. J Clin Invest 124:2009–2022. doi: 10.1172/JCI71951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alvarez Y, Tuen M, Shen G, Nawaz F, Arthos J, Wolff MJ, Poles MA, Hioe CE. 2013. Preferential HIV infection of CCR6+ Th17 cells is associated with higher levels of virus receptor expression and lack of CCR5 ligands. J Virol 87:10843–10854. doi: 10.1128/JVI.01838-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bernier A, Cleret-Buhot A, Zhang Y, Goulet JP, Monteiro P, Gosselin A, DaFonseca S, Wacleche VS, Jenabian MA, Routy JP, Tremblay C, Ancuta P. 2013. Transcriptional profiling reveals molecular signatures associated with HIV permissiveness in Th1Th17 cells and identifies peroxisome proliferator-activated receptor gamma as an intrinsic negative regulator of viral replication. Retrovirology 10:160. doi: 10.1186/1742-4690-10-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun H, Kim D, Li X, Kiselinova M, Ouyang Z, Vandekerckhove L, Shang H, Rosenberg ES, Yu XG, Lichterfeld M. 2015. Th1/17 polarization of CD4 T cells supports HIV-1 persistence during antiretroviral therapy. J Virol 89:11284–11293. doi: 10.1128/JVI.01595-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ploquin MJ, Madec Y, Casrouge A, Huot N, Passaes C, Lecuroux C, Essat A, Boufassa F, Jacquelin B, Jochems SP, Petitjean G, Angin M, Gartner K, Garcia-Tellez T, Noel N, Booiman T, Boeser-Nunnink BD, Roques P, Saez-Cirion A, Vaslin B, Dereudre-Bosquet N, Barre-Sinoussi F, Ghislain M, Rouzioux C, Lambotte O, Albert ML, Goujard C, Kootstra N, Meyer L, Muller-Trutwin MC. 2016. Elevated basal pre-infection CXCL10 in plasma and in the small intestine after infection are associated with more rapid HIV/SIV disease onset. PLoS Pathog 12:e1005774. doi: 10.1371/journal.ppat.1005774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Townsend DG, Trivedi S, Jackson RJ, Ranasinghe C. 2017. Recombinant fowlpox virus vector-based vaccines: expression kinetics, dissemination and safety profile following intranasal delivery. J Gen Virol 98:496–505. doi: 10.1099/jgv.0.000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Quinn KM, Zak DE, Costa A, Yamamoto A, Kastenmuller K, Hill BJ, Lynn GM, Darrah PA, Lindsay RW, Wang L, Cheng C, Nicosia A, Folgori A, Colloca S, Cortese R, Gostick E, Price DA, Gall JG, Roederer M, Aderem A, Seder RA. 2015. Antigen expression determines adenoviral vaccine potency independent of IFN and STING signaling. J Clin Invest 125:1129–1146. doi: 10.1172/JCI78280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Teigler JE, Iampietro MJ, Barouch DH. 2012. Vaccination with adenovirus serotypes 35, 26, and 48 elicits higher levels of innate cytokine responses than adenovirus serotype 5 in rhesus monkeys. J Virol 86:9590–9598. doi: 10.1128/JVI.00740-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Teigler JE, Kagan JC, Barouch DH. 2014. Late endosomal trafficking of alternative serotype adenovirus vaccine vectors augments antiviral innate immunity. J Virol 88:10354–10363. doi: 10.1128/JVI.00936-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu H, Andersson AM, Ragonnaud E, Boilesen D, Tolver A, Jensen BAH, Blanchard JL, Nicosia A, Folgori A, Colloca S, Cortese R, Thomsen AR, Christensen JP, Veazey RS, Holst PJ. 2017. Mucosal vaccination with heterologous viral vectored vaccine targeting subdominant SIV accessory antigens strongly inhibits early viral replication. EBioMedicine 18:204–215. doi: 10.1016/j.ebiom.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jenkins S, Gritz L, Fedor CH, O'Neill EM, Cohen LK, Panicali DL. 1991. Formation of lentivirus particles by mammalian cells infected with recombinant fowlpox virus. AIDS Res Hum Retroviruses 7:991–998. doi: 10.1089/aid.1991.7.991. [DOI] [PubMed] [Google Scholar]
- 71.Zanotto C, Pozzi E, Pacchioni S, Bissa M, De Giuli Morghen C, Radaelli A. 2011. Construction and characterisation of a recombinant fowlpox virus that expresses the human papilloma virus L1 protein. J Transl Med 9:190. doi: 10.1186/1479-5876-9-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grunwald T, Tenbusch M, Schulte R, Raue K, Wolf H, Hannaman D, de Swart RL, Überla K, Stahl-Hennig C. 2014. Novel vaccine regimen elicits strong airway immune responses and control of respiratory syncytial virus in nonhuman primates. J Virol 88:3997–4007. doi: 10.1128/JVI.02736-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mühl T, Krawczak M, Ten Haaft P, Hunsmann G, Sauermann U. 2002. MHC class I alleles influence set-point viral load and survival time in simian immunodeficiency virus-infected rhesus monkeys. J Immunol 169:3438–3446. doi: 10.4049/jimmunol.169.6.3438. [DOI] [PubMed] [Google Scholar]
- 74.Sauermann U, Siddiqui R, Suh YS, Platzer M, Leuchte N, Meyer H, Mätz-Rensing K, Stoiber H, Nürnberg P, Hunsmann G, Stahl-Hennig C, Krawczak M. 2008. MHC class I haplotypes associated with survival time in simian immunodeficiency virus (SIV)-infected rhesus macaques. Genes Immun 9:69–80. doi: 10.1038/sj.gene.6364448. [DOI] [PubMed] [Google Scholar]
- 75.Stolte-Leeb N, Bieler K, Kostler J, Heeney J, Haaft PT, Suh YS, Hunsmann G, Stahl-Hennig C, Wagner R. 2008. Better protective effects in rhesus macaques by combining systemic and mucosal application of a dual component vector vaccine after rectal SHIV89.6P challenge compared to systemic vaccination alone. Viral Immunol 21:235–246. doi: 10.1089/vim.2007.0103. [DOI] [PubMed] [Google Scholar]
- 76.Neildez O, Le Grand R, Caufour P, Vaslin B, Cheret A, Matheux F, Theodoro F, Roques P, Dormont D. 1998. Selective quasispecies transmission after systemic or mucosal exposure of macaques to simian immunodeficiency virus. Virology 243:12–20. doi: 10.1006/viro.1997.9026. [DOI] [PubMed] [Google Scholar]
- 77.Schultheiss T, Stolte-Leeb N, Sopper S, Stahl-Hennig C. 2011. Flow cytometric characterization of the lymphocyte composition in a variety of mucosal tissues in healthy rhesus macaques. J Med Primatol 40:41–51. doi: 10.1111/j.1600-0684.2010.00446.x. [DOI] [PubMed] [Google Scholar]
- 78.Bischoff T, Stahl-Hennig C, Matz-Rensing K, Koutsilieri E, Sopper S. 2011. Definition of leukocyte subsets in primate central nervous system by polychromatic flow cytometry. Cytometry A 79:436–445. doi: 10.1002/cyto.a.21046. [DOI] [PubMed] [Google Scholar]
- 79.Schulte R, Suh YS, Sauermann U, Ochieng W, Sopper S, Kim KS, Ahn SS, Park KS, Stolte-Leeb N, Hunsmann G, Sung YC, Stahl-Hennig C. 2009. Mucosal prior to systemic application of recombinant adenovirus boosting is more immunogenic than systemic application twice but confers similar protection against SIV-challenge in DNA vaccine-primed macaques. Virology 383:300–309. doi: 10.1016/j.virol.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 80.Stolte-Leeb N, Sauermann U, Norley S, Fagrouch Z, Heeney J, Franz M, Hunsmann G, Stahl-Hennig C.. 2006. Sustained conservation of CD4+ T cells in multiprotein triple modality-immunized rhesus macaques after intrarectal challenge with simian immunodeficiency virus. Viral Immunol 19:448–457. doi: 10.1089/vim.2006.19.448. [DOI] [PubMed] [Google Scholar]
- 81.Suh YS, Park KS, Sauermann U, Franz M, Norley S, Wilfingseder D, Stoiber H, Fagrouch Z, Heeney J, Hunsmann G, Stahl-Hennig C, Sung YC. 2006. Reduction of viral loads by multigenic DNA priming and adenovirus boosting in the SIVmac-macaque model. Vaccine 24:1811–1820. doi: 10.1016/j.vaccine.2005.10.026. [DOI] [PubMed] [Google Scholar]
- 82.Stahl-Hennig C, Kuate S, Franz M, Suh YS, Stoiber H, Sauermann U, Tenner-Racz K, Norley S, Park KS, Sung YC, Steinman R, Racz P, Uberla K. 2007. Atraumatic oral spray immunization with replication-deficient viral vector vaccines. J Virol 81:13180–13190. doi: 10.1128/JVI.01400-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stahl-Hennig C, Eisenblätter M, Jasny E, Rzehak T, Tenner-Racz K, Trumpfheller C, Salazar AM, Überla K, Nieto K, Kleinschmidt J, Schulte R, Gissmann L, Muller M, Sacher A, Racz P, Steinman RM, Uguccioni M, Ignatius R. 2009. Synthetic double-stranded RNAs are adjuvants for the induction of T helper 1 and humoral immune responses to human papillomavirus in rhesus macaques. PLoS Pathog 5:e1000373. doi: 10.1371/journal.ppat.1000373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stolte-Leeb N, Loddo R, Antimisiaris S, Schultheiss T, Sauermann U, Franz M, Mourtas S, Parsy C, Storer R, La Colla P, Stahl-Hennig C. 2011. Topical nonnucleoside reverse transcriptase inhibitor MC 1220 partially prevents vaginal RT-SHIV infection of macaques. AIDS Res Hum Retroviruses 27:933–943. doi: 10.1089/aid.2010.0339. [DOI] [PubMed] [Google Scholar]
- 85.Tenbusch M, Ignatius R, Nchinda G, Trumpfheller C, Salazar AM, Töpfer K, Sauermann U, Wagner R, Hannaman D, Tenner-Racz K, Racz P, Stahl-Hennig C, Überla K. 2012. Immunogenicity of DNA vaccines encoding simian immunodeficiency virus antigen targeted to dendritic cells in rhesus macaques. PLoS One 7:e39038. doi: 10.1371/journal.pone.0039038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mussil B, Javed A, Töpfer K, Sauermann U, Sopper S. 2015. Increased BST2 expression during simian immunodeficiency virus infection is not a determinant of disease progression in rhesus monkeys. Retrovirology 12:92. doi: 10.1186/s12977-015-0219-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Javed A, Leuchte N, Neumann B, Sopper S, Sauermann U.. 2015. Noncytolytic CD8+ cell mediated antiviral response represents a strong element in the immune response of simian immunodeficiency virus-infected long-term non-progressing rhesus macaques. PLoS One 10:e0142086. doi: 10.1371/journal.pone.0142086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hudgens MG, Gilbert PB. 2009. Assessing vaccine effects in repeated low-dose challenge experiments. Biometrics 65:1223–1232. doi: 10.1111/j.1541-0420.2009.01189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]