

ABSTRACT
B cell subsets with phenotypes characteristic of naive, non-isotype-switched, memory (Bmem) cells and antibody-secreting cells (ASC) accumulate in various models of central nervous system (CNS) inflammation, including viral encephalomyelitis. During neurotropic coronavirus JHMV infection, infiltration of protective ASC occurs after T cell-mediated viral control and is preceded by accumulation of non-isotype-switched IgD+ and IgM+ B cells. However, the contribution of peripheral activation events in cervical lymph nodes (CLN) to driving humoral immune responses in the infected CNS is poorly defined. CD19, a signaling component of the B cell receptor complex, is one of multiple regulators driving B cell differentiation and germinal center (GC) formation by lowering the threshold of antigen-driven activation. JHMV-infected CD19−/− mice were thus used to determine how CD19 affects CNS recruitment of B cell subsets. Early polyclonal ASC expansion, GC formation, and virus-specific ASC were all significantly impaired in CLN of CD19−/− mice compared to wild-type (WT) mice, consistent with lower and unsustained virus-specific serum antibody (Ab). ASC were also significantly reduced in the CNS, resulting in increased infectious virus during persistence. Nevertheless, CD19 deficiency did not affect early CNS IgD+ B cell accumulation. The results support the notion that CD19-independent factors drive early B cell mobilization and recruitment to the infected CNS, while delayed accumulation of virus-specific, isotype-switched ASC requires CD19-dependent GC formation in CLN. CD19 is thus essential for both sustained serum Ab and protective local Ab within the CNS following JHMV encephalomyelitis.
IMPORTANCE CD19 activation is known to promote GC formation and to sustain serum Ab responses following antigen immunization and viral infections. However, the contribution of CD19 in the context of CNS infections has not been evaluated. This study demonstrates that antiviral protective ASC in the CNS are dependent on CD19 activation and peripheral GC formation, while accumulation of early-recruited IgD+ B cells is CD19 independent. This indicates that IgD+ B cells commonly found early in the CNS do not give rise to local ASC differentiation and that only antigen-primed, peripheral GC-derived ASC infiltrate the CNS, thereby limiting potentially harmful nonspecific Ab secretion. Expanding our understanding of activation signals driving CNS migration of distinct B cell subsets during neuroinflammatory insults is critical for preventing and managing acute encephalitic infections, as well as preempting reactivation of persistent viruses during immune-suppressive therapies targeting B cells in multiple sclerosis (MS), such as rituximab and ocrelizumab.
KEYWORDS: B cell responses, CD19, central nervous system infections, coronavirus, neuroimmunology, viral encephalomyelitis
INTRODUCTION
Infections of the central nervous system (CNS) commonly require a humoral immune component for effective long-term control (1–4). However, little is known about the signals driving B cell activation and differentiation during neurotropic infections or how peripheral factors affect local immunity in the CNS. Following CNS infection with a glial-tropic sublethal variant of mouse hepatitis virus (MHV), designated JHMV-v2.2-1, both T and B cell priming occurs in draining cervical lymph nodes (CLN) (5–7). While virus-specific CD4 and CD8 T cells are essential to clear infectious virus, virus-specific antibody (Ab)-secreting cells (ASC) and sustained neutralizing IgG Ab are required for long-term control of persisting virus, which is detectable only by the ongoing presence of low levels of viral RNA (5, 8, 9). Virus-specific ASC in CLN are CD4 dependent (10) and reach peak frequencies at ∼14 days postinfection (p.i.) (7, 11), coincident with germinal center (GC) formation (12). However, their accumulation in the CNS is not robust until day 21 p.i., when more mature GC, characterized by dark and light zones, become evident (12). Irrespective of the temporal and spatial ASC organization in CLN, chemokine guidance mediated via CXCR3 ligands, predominantly CXCL10, is essential for protective ASC accumulation in the CNS (11, 13).
The extent to which peripheral mature GC formation imprints ASC migration to the CNS is still unclear despite the temporal correlation between GC formation and ASC accumulation in the CNS. Key lymphoid chemokines organizing B cell compartmentalization in follicles and GCs are CXCL12 and CXCL13, as well as CCL19 and CCL21, which act through their cognate receptors, CXCR4, CXCR5, and CCR7, respectively (9, 14). Specifically, CXCL13 contributes to GC formation by recruiting activated B and follicular helper CD4+ T (TFH) cells expressing CXCR5 (15–17). CXCL13 is also associated with lymphoid neogenesis in nonlymphoid tissue, such as joints during rheumatoid arthritis (18) and CNS meninges during some neuroinflammatory diseases, including multiple sclerosis (MS) and Lyme disease (19–23). However, despite impaired GC formation, CXCL13−/− mice infected with glial-tropic JHMV mounted effective peripheral ASC and serum Ab responses (24). Importantly, the specific deficit in ASC and memory (Bmem) cells in the CNS did not affect virus control during persistence. Moreover, initial non-isotype-switched B cell recruitment into the CNS was not affected (24), similar to studies in Sindbis virus infection and experimental allergic encephalomyelitis (EAE) (25).
These results called into question the nature of activation signals required, not only for effective ASC responses in the CNS, but also to mobilize early-activated IgD+ cells, whose function and specificity are unknown, to the CNS. One critical signaling component promoting B cell receptor (BCR)-mediated activation and GC formation under conditions of low antigen (Ag) dose and/or limited proinflammatory stimuli is the BCR coreceptor CD19 (26). Together with CD21, also known as complement receptor type 2 (CR2), and CD81, CD19 forms a multimeric signal transduction complex on mature B cells (27–29). Activation and attachment of complement C3d, a cleavage product of complement C3, to Ag forms a covalent complex, which can lower the threshold for B cell activation by coligation of the BCR and coreceptor complex (28, 30, 31). The C3d-Ag complex also acts directly on activated B cells in follicles and the GC to enhance their survival (28, 32). Complement-mediated retention of Ag on follicular dendritic cells further promotes clonal selection of activated B cells in GCs. While many studies assessing the role of CD19 have focused on CD19−/− mice immunized with protein or inactivated virus, reports describing responses to replicating viruses are limited (26, 31). The latter revealed that CD19 is required for T cell-dependent responses, such lymphocytic choriomeningitis virus (LCMV) infection, but this requirement is overcome by infections associated with high viral doses, exemplified by vesicular stomatitis virus (VSV) (26). While a direct role of CD19 was not determined following peripheral herpes simplex virus 1 (HSV-1) infection, efficient humoral responses were dependent on complement C3 and CD21 (33). Overall, both Ag and viral studies support a costimulatory role for CD19 in inducing long-term B cell memory, with CD19 playing a more prominent role in GC formation under limiting Ag conditions (26, 28, 31, 34).
The studies mentioned above focused largely on ASC and Ab responses to peripheral infections, leaving the impact of CD19-dependent activation on B cell migration to inflamed tissue unclear. CNS inflammation is associated with accumulation of B cells in various differentiation and activation stages, independent of the insult, suggesting a common denominator driving their initial activation (12, 35). Using the JHMV-induced encephalomyelitis model, this study set out to identify a role of CD19 in driving early-activated B cells into the CNS, as well as promoting antiviral ASC. Coincident with severely impaired GC formation, virus-specific ASC were significantly reduced in CLN and barely detectable in the CNS compared to wild-type (WT) mice. While infected CD19−/− mice mounted virus-specific serum Abs, they were not sustained. Surprisingly, early polyclonal ASC expansion prior to GC formation was also significantly reduced. However, the absence of BCR-CD19 coligation did not appear to affect early CNS B cell accumulation. The results further demonstrate that elevated persisting virus in the CNS, as a result of impaired humoral immunity, can still be effectively controlled by reemerging T cell activity without causing increased clinical disease.
RESULTS
CD19 deficiency abrogates peripheral GC formation following CNS infection.
CLN are the predominant sites of both T cell and ASC expansion following JHMV infection, consistent with CLN as the draining sites for CNS-derived Ag (5, 36–38). Temporal analysis of GC formation in CLN following JHMV infection using GL7 to identify GC B cells previously showed formation of GL7+ foci as early as day 7 p.i., more defined structures by day 14 p.i., and segregation into light and dark zones by day 21 p.i (12). To assess whether accumulation of ASC within the CNS is dependent on CD19 signaling and GC formation in the periphery, JHMV-infected CD19−/− and WT mice were initially compared for GC formation in CLN. In WT CLN, defined structures of GL7+ B220+ B cells started to form at day 14 p.i. and were robust by day 21 p.i. (Fig. 1A), confirming previous results (12). In contrast, in the absence of CD19, GL7+ cells were very sparse and scattered at day 14 p.i. Although GC were evident in some follicles by day 21 p.i., they were small and disorganized compared to those in WT mice (Fig. 1A). GL7+ B cells were scattered within the follicle (Fig. 1A, arrows) without forming structures resembling GC architecture. Quantitative evaluation of the GL7+ area by immunofluorescence highlighted significantly reduced reactivity in CD19−/− compared to WT mice, especially at day 21 p.i., when the numbers of pixels per frame were 18-fold reduced (Fig. 1B). Impaired GC formation could not be attributed to disrupted lymphoid organization, as B cell follicle formation and lymphoid architecture were intact in naive CD19−/− mice (39).
FIG 1.

Infected CD19−/− mice exhibit diminished GC in CLN. WT and CD19−/− mice were infected with sublethal doses of JHMV, and CLN were harvested at the indicated times for histology, flow cytometric analysis, and gene expression. (A) Representative images of fluorescence immunohistochemistry performed at days 14 (d14) and 21 p.i. Sections were stained for T cells (CD3; blue), B cells (B220; red), and activated GC B cells (GL7; green). The arrows indicate GL7+ B cells. The images were taken at ×40 magnification. (B) Percentages of pixels per image frame resulting from GL7+ staining in CLN follicles. The bars represent the mean percentages plus SEM of GL7+ pixels per frame from 2 to 3 frames per mouse from 2 to 6 mice per time point. (C) Flow cytometry of CLN suspensions stained for B220, GL7, and CD95 and gated as indicated. The bar graph shows percentages of GL7+ CD95+ cells among the total B cells over time. The data represent the mean and SEM of 2 to 4 individual mice per time point per group from 4 separate experiments. (D) CLN tissue was assessed for aicda RNA transcript levels by RT PCR over time. The data represent the means plus SEM of transcript levels relative to gapdh mRNA of individual mice from 2 separate experiments, each comprising 3 to 5 individual mice per time point and group. Statistically significant differences between WT and CD19−/− mice are denoted by asterisks: *, P < 0.05; ***, P < 0.001.
The extent of impaired GC formation was further confirmed by flow cytometry using the B220+ GL7+ CD95+ phenotype to identify GC B cells (Fig. 1C). The population of GL7+ CD95+ B cells in CLN of both naive WT and CD19−/− mice was below 0.5%, consistent with no or sparse GC activity. In WT mice, GL7+ CD95+ B cells started to emerge at day 5 and continued to increase to ∼3% by day 14.p.i., consistent with anatomical GC formation. The frequency of GC phenotype B cells was maintained at ∼3 to 4% through days 21 to 28 p.i. In contrast, GL7+ CD95+ B cells were only slightly elevated to <1% in CD19−/− mice and remained barely detectable throughout the infection (Fig. 1C). Functionally, GC B cells are characterized by upregulation of activation-induced cytidine deaminase (AICDA), an enzyme required for somatic hypermutation and class switch recombination to increase Ab diversity and affinity. As B cell maturation can occur in the absence of GC (24, 25, 40), we also assessed transcript levels of the gene encoding AICDA (aicda). In CLN of infected WT mice, the progressive increase of aicda mRNA levels from days 7 to 21 p.i. correlated with GC formation and maturation (Fig. 1D). While CD19−/− mice exhibited modestly increased aicda mRNA levels in CLN between days 7 and 21 p.i., these levels did not significantly differ from those in naive CD19−/− mice until day 21 p.i. (Fig. 1D). These results demonstrate a retarded and diminished capacity to initiate GC reactions in JHMV-infected CD19−/− relative to WT mice. Nevertheless, the relative population of GL7+ CD95+ B cells in CD19−/− CLN reached only ∼15% of WT levels at day 21 p.i., while aicda mRNA levels reached ∼40% of WT levels, suggesting that CD19−/− B cells exhibit modest maturation capacity despite severely impaired GC formation.
To support the notion that deficient GC formation is a result of poor B cell activation in the absence of CD19 rather than extrinsic factors related to GC formation, we assessed transcript levels for several chemokines and cytokines regulating B cell migration and differentiation (Fig. 2). Compared to infected WT mice, CD19−/− mice exhibited no significant changes in relative levels or kinetics of mRNAs encoding CXCL13, CCL19, or CCL21, lymphoid chemokines regulating B cell migration within follicles (14) (Fig. 2). The CXCL13 chemokine receptor CXCR5 is upregulated on B cells migrating to and forming GCs (15, 41). It is also highly expressed by TFH cells, which are essential for GC formation and maintenance and B cell differentiation and survival by producing interleukin 21 (IL-21). Although cxcr5 transcripts were elevated in CLN of CD19−/− mice throughout infection, differences reached statistical significance only at day 21 p.i. (Fig. 2). Il21 mRNA levels were not significantly altered in the absence of CD19 (Fig. 2), consistent with similar TFH cell frequencies characterized by their PD-1+ CXCR5+ phenotype using flow cytometry in both WT and CD19−/− mice (data not shown). Furthermore, transcript levels of the Tnfsf13b gene, which encodes the survival factor B cell activation factor (BAFF), were also similar in the two groups, suggesting no deficits in retaining B cell viability. We also assessed upregulation of transcripts encoding complement protein C3. C3-Ag adducts not only lower the threshold for B cell activation by coligation of the CD21/CD19/CD81 BCR coreceptor, but also enhance B cell survival in follicles and GC (32). CNS infection indeed resulted in significant upregulation of c3 mRNA in both groups at days 7 to 14 p.i., and it remained elevated to day 21 p.i. Overall, these results suggested that the absence of CD19 did not impair cues for B cell localization to and trafficking between anatomical structures of the CLN. The deficit in GC formation more likely resides in deficient CD19 engagement either directly or indirectly via the complement C3d binding receptor CD21 (27, 32).
FIG 2.

CD19 deficiency does not impair expression of factors promoting GC formation within CLN. CLN harvested from infected WT and CD19−/− mice at the indicated times p.i. were analyzed for mRNA expression levels of Cxcl13, Ccl19, Ccl21, Cxcr5, Il-21, Tnfsf13b (BAFF), and complement C3. The data represent the means and SEM of transcript levels relative to gapdh mRNA of individual mice from 2 separate experiments, each comprising 3 to 5 individual mice per time point and group. Statistically significant differences between WT and CD19−/− mice, determined by unpaired t test, are denoted by asterisks: *, P < 0.05.
Impaired GC formation is associated with induction of, but reduced and rapid decline in, virus-specific humoral responses in the periphery.
Impaired GC formation in CLN was also a hallmark of CXCL13−/− mice infected with JHMV. However, differentiation of B cells into virus-specific IgG ASC was reduced by only ∼25% in the CLN and 50% in the CNS (24). Modestly impaired, but not abrogated, virus-specific serum Ab in the absence of CXCL13 confirmed that formation of mature GC in CLN is not an absolute requirement for development of peripheral or CNS humoral immunity. We therefore assessed how severely impaired GC formation in CD19−/− affects JHMV-specific serum Ab throughout infection. Virus-specific IgM levels were below detection at day 7 p.i. in CD19−/− mice but reached levels similar to those in WT mice by day 14 p.i. (Fig. 3A). However, while WT mice sustained virus-specific IgM levels out to day 28 p.i., they were progressively lost after day 14 p.i. in CD19−/− mice. Virus-specific IgG2a was induced in both mouse groups by day 7 p.i. and increased by day 14 p.i. (Fig. 3A). However, the levels were lower in the absence of CD19, reaching ∼3 log10 relative to ∼4 to 4.5 log10 units in WT mice. Furthermore, while virus-specific serum IgG2a increased slightly in WT mice by day 21 p.i. and then stabilized, it had already started to decline by day 28 p.i. in CD19−/− mice. Importantly, although neutralizing serum Ab was initially reduced in infected CD19−/− relative to WT mice at day 7 p.i., it reached levels found in WT mice by day 14 p.i. However, distinct from the gradual increase of neutralizing Ab throughout infection in WT mice, it significantly declined by day 28 p.i. in CD19−/− mice (Fig. 3A). Thus, despite impaired GC formation in the absence of CD19, virus-specific serum Ab responses were induced, albeit with delayed kinetics, at lower levels, and with shorter longevity. The inability of serum Ab to be maintained supported a critical role for CD19 in the generation of both IgM and isotype-switched long-lived ASC (31).
FIG 3.

Reduced and unsustained virus-specific and neutralizing serum Ab coincides with decreased virus-specific ASC in the absence of CD19. (A) Virus-specific IgM, IgG, and neutralizing Ab in sera of uninfected (day 0 p.i.) and infected mice at the indicated times p.i. (B and C) CLN (B) and bone marrow (C) were analyzed for virus-specific IgG ASC by ELISPOT assay. Representative wells are shown for CLN at day 14 and BM at day 21 p.i. The graphs show frequencies of virus-specific IgG ASC spots and spot diameters. The data represent the means and SEM of individual mice from 3 separate experiments, each comprising 2 to 4 individual mice per time point and group. Statistically significant differences between WT and CD19−/− mice, determined by unpaired t test, are indicated by asterisks: *, P < 0.05; **, P < 0.01; ***, P < 0.001. Significant differences between time points within the same group are also indicated: #, P < 0.05.
The induction, but not maintenance, of virus-specific serum Ab prompted us to investigate the kinetics of virus-specific IgG ASC accumulation in CLN and bone marrow (BM) by enzyme-linked immunosorbent spot (ELISPOT) analysis (Fig. 3B and C). Virus-specific IgG ASC emerged at day 7 in CLN of WT mice, reached peak frequencies at day 14 p.i., and declined by day 21 p.i., consistent with previous data (7, 11, 42). Although virus-specific IgG ASC followed similar kinetics in CD19−/− mice, peak frequencies were reduced by ∼70% compared to WT mice and dropped to barely detectable levels by day 21 p.i. Furthermore, in contrast to the modest increase in virus-specific ASC in BM by day 21 p.i., virus-specific IgG ASC remained at baseline levels in BM of CD19−/− mice (Fig. 3C). Impaired ASC maturation was supported by assessment of spot size as an indicator for the ASC Ab secretion rate (Fig. 3B and C). On average, ASC derived from WT CLN and BM at days 14 and 21 p.i., respectively, formed more intense spots with an increased diameter compared to CD19−/− counterparts, suggesting higher rates of IgG secretion. Sparse GC formation following JHMV infection of CD19−/− mice thus coincides with reduced virus-specific ASC maturation and longevity. These results indicated CD19 is essential to drive activation and differentiation of high-affinity virus-specific ASC competent to egress CLN and migrate to survival niches in BM (43).
CD19 deficiency severely diminishes peripheral ASC expansion following CNS infection.
Impaired virus-specific ASC and serum responses as early as day 7 p.i. were consistent with a defect in early activation, potentially due to the absence of complement-mediated costimulation through CD21. Moreover, the peak of virus-specific ASC at day 14 p.i. in CLN following JHMV infection was preceded by a peak in polyclonal ASC expansion at day 7 p.i. (11, 12). To assess whether CD19 deficiency specifically affected virus-specific ASC, overall ASC expansion in CLN was monitored by flow cytometry using CD138 as a phenotypic marker (Fig. 4A and B). The frequency of CD138+ B cells was reduced by ∼70% relative to WT mice at day 7 p.i. prior to expansion of virus-specific ASC and remained low through days 14 to 28 p.i. (Fig. 4). Similar numbers of B220+ B cells in CLN of both groups during infection indicated no overall deficiencies in the B cell compartment or recruitment to CLN following infection (data not shown). These data were consistent with a polyclonal defect in early B cell activation in CD19−/− mice following JHMV infection preceding impaired generation of virus-specific ASC. Taken together, these results demonstrate a diminished capacity to initiate GC reactions, resulting in impaired differentiation of long-lived ASC in the absence of CD19 in the periphery.
FIG 4.
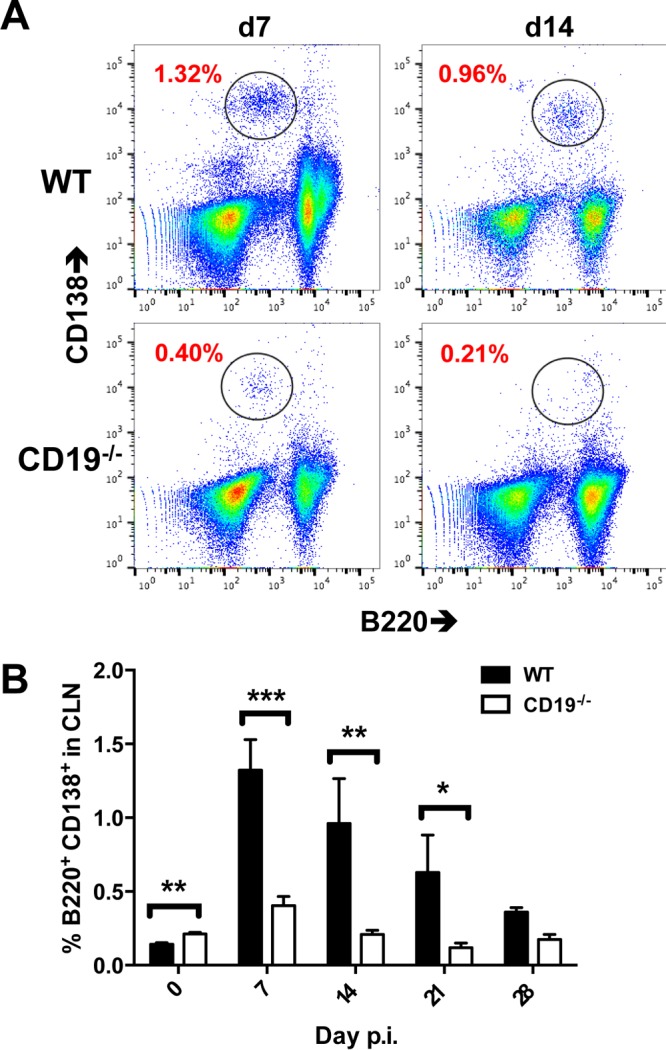
CD138+ ASC are decreased in CLN of CD19−/− mice following infection. Pooled CLN cells from infected WT and CD19−/− mice (n = 2 or 3 mice per time point per group from 3 separate experiments) were stained for B220 and the ASC marker CD138. (A) Representative density plots depicting CD138+ and B220+ B cells at 7 and 14 days p.i. (gated on live cells). The numbers represent percentages of CD138+ B220+ ASC among total cells. (B) Mean and SEM percentages of CD138+ B220+ ASC within total CLN cells over time. Statistically significant differences between WT and CD19−/− mice are denoted by asterisks: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
CD19 deficiency severely diminishes CNS humoral responses.
ASC expanding prior to GC formation in CLN appear not to have egress or migration capacity, as ASC do not emerge in the CNS until 14 days p.i. and prominently increase only thereafter (7, 11, 24, 42). This observation suggests that accumulation of ASC in the CNS is dependent on differentiation in GCs. Moreover, unlike CXCR4-driven migration of ASC to BM, ASC accumulation in the CNS is dependent on CXCR3 expression (11). We therefore assessed whether the few ASC primed in CLN of CD19−/− mice nevertheless traffic to the infected CNS, as observed in CXCL13−/− mice (21). Measurement of virus-specific IgG ASC by ELISPOT assay revealed similar frequencies at day 14 p.i. when brains or spinal cords of WT and CD19−/− mice, respectively, were compared (Fig. 5A). However, while frequencies in WT mice increased by day 21 p.i. and remained steady at day 28 p.i., they never increased in the CNS of CD19−/− mice. Overall, virus-specific ASC frequencies were significantly higher in spinal cords than in brains in WT mice at days 21 and 28 p.i., consistent with their preferential accumulation at the site of persistence and elevated inflammation (44). The differences between ASC frequencies were therefore especially pronounced in spinal cords, amounting to an almost 10-fold disparity, whereas differences were only 3- to 4-fold in the brain (Fig. 5A). Similar to CLN, average spot sizes of virus-specific CD19−/− ASC were also reduced at days 21 and 28 p.i. compared to WT ASC, reflecting poor affinity maturation and differentiation. The paucity in virus-specific ASC in the brain was supported by direct analysis of supernatants from brain homogenates for virus-specific IgG2a Ab, the most abundant and protective Ab isotype during JHMV infection (7, 45, 46). Compared to the progressive increase of virus-specific IgG2a from days 14 to 28 p.i. in WT mice, the levels were barely above background in CD19−/− mice (Fig. 5B).
FIG 5.

Virus-specific ASC and Ab are severely decreased in the CNS of CD19−/− mice. (A) Brains and spinal cords of infected WT and CD19−/− were analyzed for virus-specific IgG ASC by ELISPOT assay. Representative wells from day 28 p.i. are shown. The graphs show the frequencies of virus-specific IgG ASC spots, as well as spot diameters. Statistically significant differences between WT and CD19−/− mice (individual brains and pooled spinal cords from 2 to 6 mice per group per time point from 4 separate experiments), determined by unpaired t test, are denoted by asterisks: *, P < 0.05; **, P < 0.01; ***, P < 0.001. (B) Virus-specific IgG2a in brain supernatants. Individual titers from 2 to 6 mice per group per time point from 2 separate experiments are shown. Statistically significant differences between WT and CD19−/− mice, determined by unpaired t test, are denoted by asterisks: *, P < 0.05; ***, P < 0.001.
To ascertain that CD19 deficiency affected overall ASC CNS migration independent of specificity, we monitored accumulation of ASC via expression of mRNAs encoding IgG and IgM heavy chains, which are highly expressed in ASC compared to non-Ab-secreting B cells (8). Both brains and spinal cords of infected WT mice exhibited a significant increase in IgG heavy chain mRNA by 14 days p.i., which was further elevated at days 21 through 28 p.i. (Fig. 6A). As anticipated, CD19−/− mice exhibited significantly decreased transcript levels throughout days 14 to 28 p.i. (Fig. 6A). Although a slight trend toward elevated transcript levels was noted in spinal cords, the levels at day 28 p.i. did not even reach those of WT mice at day 14 p.i. To delineate potential factors abrogating accumulation of ASC within the CNS of CD19−/− mice, we used flow cytometry to assess CXCR3 expression on CD138+ ASC in the CLN (Fig. 6B). CXCR3 is upregulated by gamma interferon (IFN-γ) and is essential in driving ASC migration to and entry into the CNS parenchyma (11). CD19−/− mice expressed significantly reduced frequencies of CXCR3+ ASC in the CLN throughout infection, with differences most prominent at day 14 p.i. (Fig. 6B). However, the mean fluorescent intensities (MFI) of CXCR3+ were similar in WT and CD19−/− ASC (Fig. 6C). These data suggest that there is no inherent deficit in the ability of CD19−/− ASC to express CXCR3. Similarly to CXCR3, CXCR4 is upregulated during ASC differentiation (11, 47). Interactions with its ligand, CXCL12, are important in mediating ASC homing to, and retention within, the BM (43, 47). CD19−/− mice also demonstrated a significant reduction in the frequency of ASC expressing CXCR4 in the CLN, beginning at day 14 p.i. and continuing throughout the infection (data not shown). As was the case with CXCR3, the MFI of CXCR4 on ASC did not differ between WT and CD19−/− mice (data not shown).
FIG 6.

Total CXCR3+ ASC are reduced in CLN of CD19−/− mice. (A) Brains (BR) and spinal cords (SC) harvested from infected WT and CD19−/− mice at the indicated times p.i. were analyzed for expression of IgG heavy chain (Ighg) mRNA. The data represent the means and SEM of transcript levels relative to gapdh mRNA of individual mice from 2 separate experiments, each comprising 2 to 6 individual mice per time point and group. (B) CLN cells pooled from infected mice were stained for CD138 and CXCR3. Representative histograms gated on B220+ CD138+ ASC at 21 days p.i. are shown for WT and CD19−/− mice; the graph depicts mean and SEM percentages of CXCR3+ cells within ASC (n = 2 to 4 individual mice per group per time point from 3 separate experiments). Statistically significant differences between WT and CD19−/− mice are denoted by asterisks: *, P < 0.05; **, P < 0.01; ***, P < 0.001. (C) MFI of CXCR3+ ASC from the cells depicted in panel B.
We further assessed how CD19 deficiency affects overall B cell recruitment using CD45hi and B220 as expression markers of infiltrating B cells. There were no differences in total CNS cell yields or frequencies of CD45hi infiltrating bone marrow-derived cells. CD8+ and CD4+ T cells were also similar between the groups (data not shown), confirming no overall deficits in leukocyte trafficking to the CNS. Although an ∼40% reduction in the frequency of B220+ B cells in the CNS was noted at 7 days p.i., frequencies were similar to those in WT mice by days 14 and 21 (Fig. 7). Moreover, ∼60% of B220+ B cells were IgD+ in both groups at days 7 and 14 p.i., indicating that a majority of these B cells were undifferentiated. By day 21 p.i., the proportions decreased similarly to ∼25%. These results suggest that CD19 has a minor impact on migration of IgD+ early-activated B cells into the CNS following JHMV infection but specifically affects overall ASC accumulation at day 14 p.i. and thereafter.
FIG 7.
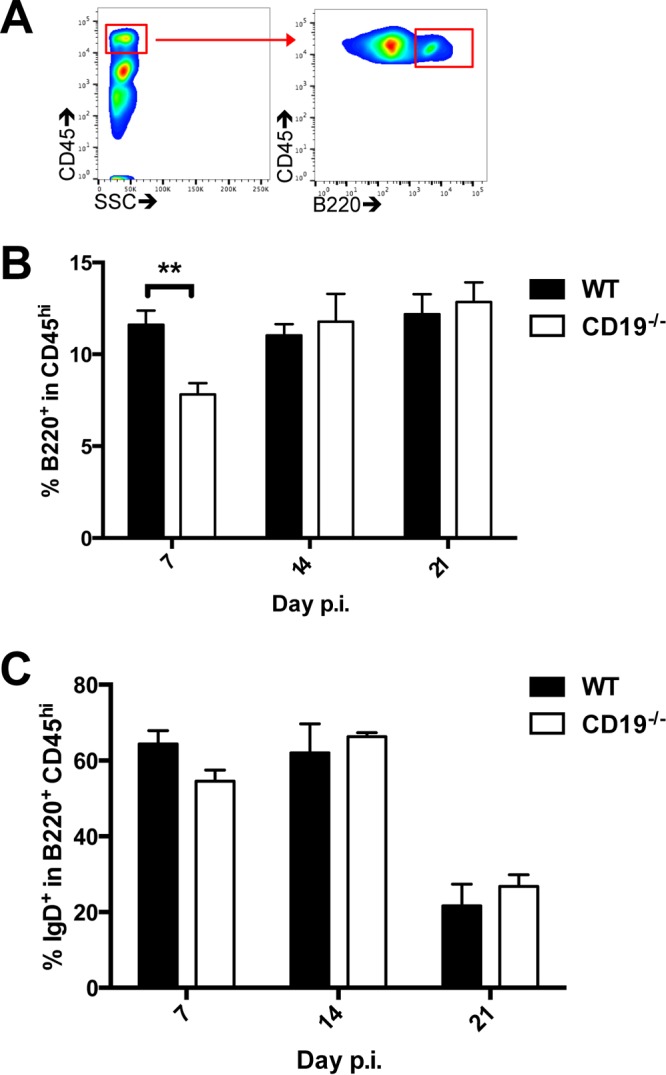
CD19 deficiency does not impair recruitment of IgD+ B cells into the CNS. Brains from infected WT and CD19−/− mice were analyzed for accumulation of total CD45hi cells, as well as undifferentiated IgD+ B220+ B cells, by flow cytometry. (A) Representative density plots showing CD45 staining and the side scatter (SSC) pattern and gating strategy for B220+ cells within infiltrating CD45hi cells. (B and C) Percentages of B220+ cells within the CD45hi infiltrating population (B) and percentages of IgD+ cells among CD45hi B220+ B cells (C) over time. The data represent the means and SEM of individual mice from 2 separate experiments, each comprising 2 to 4 individual mice per time point and group. Statistically significant differences between WT and CD19−/− mice are denoted by asterisks: **, P < 0.01.
The deficit in CNS humoral immunity is associated with increased infectious virus balanced by reactivation of T cell responses.
The severe deficit in virus-specific ASC and antiviral IgG2a in the CNS of JHMV-infected mice predicted loss of virus control during the persistent phase of infection, which requires local ASC (7, 45, 46, 48). The infectious virus load in the CNS of CD19−/− mice was similar to and even lower than that in WT mice at days 7 and 14 p.i. (Fig. 8A), consistent with T cell-mediated control. However, whereas WT mice reduced infectious virus to below detection by day 21 p.i., it remained detectable between days 14 and 28 p.i. in the absence of CD19 (Fig. 8A). The inability to maintain effective virus control was confirmed by elevated, and even increasing, transcript levels of viral RNA encoding nucleocapsid protein in both brain and spinal cord at day 21 compared to day 28 p.i. (Fig. 8B), although the increase was modest. Importantly however, mice also did not show signs of increased clinical disease (data not shown), distinct from mice totally deficient in virus-specific Ab production, in which vastly increasing virus was associated with worsening clinical disease (46, 48, 49). Although T cells did not increase in the CNS during persistence in CD19−/− mice (data not shown), we assessed reemergence of T cell effector activity to explain partial ongoing viral control during persistence. IFN-γ is the most critical cytokine essential for JHMV control (5, 50, 51). IFN-γ transcripts peaked to similar levels at day 7 p.i. in both the brains and spinal cords of CD19−/− and WT mice, confirming effective T cell stimulation by virus Ag presentation (Fig. 8C). IFN-γ mRNA levels dropped by days 10 and 14 p.i., coincident with viral control, and were similarly low in both groups at day 21 p.i., implying minimal in vivo stimulation and antiviral T cell activity. However, by day 28 p.i., IFN-γ mRNA levels were significantly elevated in both brains and spinal cords of CD19−/− mice. These results indicated that an elevated persisting viral load triggers retained or reemerging T cells to produce IFN-γ and exert antiviral activity.
FIG 8.

Impaired virus control associated with sparse local humoral immunity reinvigorates T-cell effector function. (A) Infectious virus within the brains of WT and CD19−/− mice (n = 2 to 6 mice per group per time point from 4 separate experiments) determined by plaque assay. The dashed line indicates the limit of detection. (B and C) Levels of viral RNA encoding nucleocapsid protein (B) and IFN-γ (C) in brains (BR) and spinal cords (SC) determined by real-time PCR. The data represent the means and SEM of transcript levels relative to gapdh mRNA of individual mice from 2 or 3 separate experiments, each comprising 2 to 6 individual mice per time point and group. Statistically significant differences between WT and CD19−/− mice, determined by unpaired t test, are denoted by asterisks: *, P < 0.05; ***, P < 0.001.
To provide evidence for reemergence of other T cell functions, as well as IFN-γ-dependent activities, both brains and spinal cords were temporally screened for expression of factors associated with increased T cell function (Fig. 9). IL-21 not only enhances B cell differentiation in CLN, but also enhances and prolongs CD8 T cell function within the CNS (10). While IL-21 was similarly regulated in the CNS of WT and CD19−/− mice out to day 14 p.i., it was sustained and even increased in both brains and spinal cords of CD19−/− compared to WT mice during persistence, reaching statistically significant differences by day 28 p.i. Surprisingly, transcript levels of the lymphoid chemokine CXCL13, which regulates migration of IL-21-expressing CXCR5+ T cells, was also notably elevated at day 28 p.i. in the CNS of CD19−/− mice. Increased functional IFN-γ activity in the CNS of CD19−/− mice in vivo was supported by elevated IFN-γ-inducible Tnfsf13b and cxcl9 mRNAs relative to WT mice, specifically at day 28 p.i. In contrast, C3 mRNA as an activation marker peaked in the brain at day 7 and was only marginally increased at day 28 p.i. in the absence of CD19. Surprisingly, spinal cords revealed increasing C3 mRNA levels throughout persistence in both groups. Lastly, IL-10 mRNA, expressed most prominently by CD4 T cells during JHMV infection (52), peaked in both groups and organs at day 7 p.i. and declined during persistence but was sustained at higher levels in CD19−/− mice, indicative of elevated regulatory T cell activity (53, 54). Assessment of the CCR7 binding lymphoid chemokines CCL19 and CCL21, known to be expressed in the inflamed CNS (55), revealed surprisingly distinct patterns between WT and CD19−/− mice. Compared to WT mice, CCL19 mRNA was consistently elevated in brains of CD19−/− mice throughout infection, whereas it was mainly increased at day 28 in spinal cords. Unexpectedly, CCL21 mRNA levels were vastly increased at basal levels in the CNS of CD19−/− mice but remained unaltered by infection, similar to naive WT mice. Overall the data imply that retriggering of T cell activity by elevated virus replication in settings of impaired humoral immunity potentially contributes to steady-state viral control during JHMV persistence.
FIG 9.

CD19 deficiency leads to reemergence of T cell-associated functions. Brains and spinal cords harvested from uninfected (day 0 p.i.) and infected WT and CD19−/− mice at the indicated times p.i. were analyzed for mRNA encoding cytokines and chemokines associated with regulation of humoral responses. The data represent the means and SEM of transcript levels relative to gapdh mRNA of individual mice from 2 or 3 separate experiments, each comprising 2 to 6 individual mice per time point and group. Statistically significant differences between WT and CD19−/− mice, determined by unpaired t test, are denoted by asterisks: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
DISCUSSION
A variety of human viral CNS infections, including those caused by measles, mumps, rubella, polio, varicella zoster, and herpes simplex viruses and Japanese encephalitis virus (JEV), are characterized by intrathecal Ab in the cerebrospinal fluid (CSF), consistent with local ASC (2, 56–59). Overall Ab detection is more transient in cases of acute encephalitis but persists during chronic disease, such as measles virus-associated subacute sclerosing panencephalitis (60, 61). Although the role of intrathecal humoral responses in humans is difficult to assess, they correlate with protective functions during JEV encephalitis (62), CNS retrovirus infection (63–65), and human T cell lymphotropic virus type 1 (HTLV-1)-associated myelopathy (65). Experimental CNS infections in rodents support an essential local protective role of ASC, as evidenced by infections established by Sindbis, rabies, and neurotropic coronaviruses (3, 11, 46, 66–70). Sustained Ab output by local ASC provides a potent nonlytic mechanism of immune control potentially beneficial for a variety of neurotropic infections that are prone to persist. However, little is known about peripheral activation signals and the necessity for GC formation in regulating B cell migration and accumulation in the CNS.
Expression of the lymphoid chemokines CXCL13 and CCL19/21 within the CNS has been associated with formation of ectopic follicle-like structures harboring multiple B cell differentiation phenotypes giving rise to de novo local Ab production (71–73). Upregulation of B cell survival factors (8, 74, 75) is thought to contribute to clonal B cell expansion and local humoral responses distinct from those occurring systemically. However, follicle-like structures have not been observed in experimental viral encephalomyelitis (12, 76), suggesting activation and GC formation in draining lymphoid organs imprints activated B cells for migration to the inflamed site. As CD19 is a key signaling component initiating B cell activation, as well as driving GC formation, we assessed the relevance of CD19 in humoral responses to neurotropic JHMV infection. As anticipated, based on sparse peripheral replication and consequently Ag load (77), GC formation was significantly impaired in the absence of CD19. Despite induction of virus-specific serum IgG and neutralizing Ab, both IgM and IgG responses were overall reduced and not sustained. Reduced anti-virus serum Ab, lower virus-specific ASC, and very scant accumulation of IgG ASC in BM are consistent with less differentiation and longevity of isotype-switched CD19−/− ASC than of WT ASC.
Contrasting with the scant migration to BM, virus-specific ASC trafficking to the CNS was initially similar to that in WT mice. However, their ongoing accumulation was significantly impaired after day 14 p.i. The more prominent defect in the spinal cord reflects the overall enhanced ASC accumulation in the spinal cord relative to the brain associated with increased and sustained expression of ASC-promoting factors (Fig. 9), as shown previously in WT mice (44). Initial recruitment of virus-specific ASC to the CNS, but not BM, supports the notion that CXCR3-driven recruitment to the inflamed site overrides CXCR4-driven recruitment to BM (42). Similar CXCR3 expression levels on CD19−/− and WT ASC in CLN, irrespective of their low numbers, implies that defective progressive accumulation of ASC is due to their less differentiated, short-lived phenotype as a result of impaired GC formation rather than an inherent defect in migration or ASC-promoting factors in the CNS of CD19−/− mice.
JHMV infection of CXCL13−/− mice also revealed impaired GC formation with reduced frequencies of GL7+ B cells (24). Nevertheless, distinct from CD19−/− mice, virus-specific serum Ab responses were not significantly impaired through day 21 p.i., consistent with similar ASC in BM and only slightly reduced ASC in CLN. Although ASC in the CNS were reduced by 50% in CXCL13−/− mice, they were sufficient to control persisting viral RNA. Potential differences may reside in the lymphoid structure. While CXCL13−/− mice are devoid of most lymph nodes, they retain cervical lymph nodes (41). However, the absence of B cell follicles and disrupted lymphoid architecture likely alters typical migration patterns. In contrast, CD19−/− mice exhibit normal lymphoid architecture with intact B cell follicles (34), which may put more restraints on CD4 T cell-B cell interactions. In any case, the absence of CD19 leads to more severely impaired generation of protective ASC during JHMV infection than in CXCL13−/− mice. An early defect in B cell activation was already noted by reduced overall expansion of ASC prior to detection of virus-specific ASC. While the early increase in ASC was previously attributed to IFN-α/β-induced bystander activation, similar to influenza virus and West Nile virus (WNV) (78, 79), our results imply a role for CD19 coreceptor activation via CD21 coligation by virus/C3d adducts. JHMV infection resulted in a robust increase of C3 mRNA in both CLN and the CNS. Viral Ag/C3d-mediated activation of the BCR coreceptor can thus reduce the threshold for B cell activation in the periphery, as well as the CNS, where astrocytes, microglia, and potentially neurons and oligodendrocytes, are likely sources of C3 (80–82). Surprisingly however, early-activated IgD+ B cells recruited to the CNS were not altered by CD19 deficiency, suggesting BCR-independent signals drive their trafficking to the CNS. In vitro stimulation to assess virus specificity in this population provided no evidence for these early B cells as precursors of ASC (83). The nature of signals driving IgD+ B cells to the CNS during infection, as well as their role, thus remains to be determined but likely involves Toll-like receptor (TLR)-driven signals (84, 85).
The biological impact of significantly impaired ASC within the CNS was manifested by the inability of CD19−/− mice to control infectious virus during persistence. Surprisingly, the elevated viral load triggered T cell reactivation, as evidenced by reemerging IFN-γ mRNA in both brains and spinal cords. Biological IFN-γ function was directly reflected by coincidently increased expression of IFN-γ-inducible CXCL9 and CXCL10 mRNAs (8). Although retention of both CD4 and CD8 T cells in the CNS during persistence makes them likely sources of IFN-γ, de novo recruitment of T cells cannot be excluded. In this context, it is interesting that IL-21 mRNA levels were also increased, suggesting IL-21 contributes to reinvigorating local CD8 T cell effector function (10). Together, these data suggest that persisting viral replication in the absence of protective ASC is kept in check by reemerging T cell activity. The significantly higher constitutive levels of the CCR7 binding chemokine CCL21 in both the brain and spinal cord, but not CLN, of CD19−/− mice are also of interest. Elevated CCL21 mRNA was also noted in CXCL13−/− and CXCR3−/− mice (11, 24). Following infection with the JHMV heterologous MHV-A59 strain, CCR7 ligands produced by CNS stromal cells were crucial to support recruitment and local reactivation of antiviral CD8+ T cells (86). These studies indicated that CNS stromal cells generate confined microenvironments that control T cell immunity and protect the host from lethal neuroinflammatory disease. Surprisingly CD19−/− mice did not exhibit increased clinical symptoms or disability at day 28.p.i., suggesting CNS pathology manifested in demyelination and axonal damage was not significantly worsened. Possible explanations reside in CCR7-mediated restraint of T cell function to perivascular spaces and or elevated protective IL-10. In summary, the effect of CD19 deficiency on JHMV-induced humoral responses was overall similar to reduced GC formation in peripheral-infection models associated with limited viral antigen (26, 31). Moreover, the results are the first to demonstrate that protective ASC in the CNS are dependent on CD19 activation. In contrast, CD19-independent accumulation of early-recruited IgD+ B cells in the CNS suggests that independent activation mechanisms drive the emergence of early versus more differentiated, isotype-switched B cells accumulating in the CNS as GCs are formed in the periphery. A better understanding of distinct B cell subsets in the CNS is essential to preempt reactivation of persistent viruses in the CNS during immune-suppressive therapies (87, 88), as well as to combat susceptibility to acute encephalitic arboviral infections.
MATERIALS AND METHODS
Mice, virus infection, and virus titer.
C57BL/6 mice were purchased from the National Cancer Institute (Frederick, MD). B6.129P2(C)-Cd19tm1(cre)Cgn/J mice (89) were purchased from the Jackson Laboratory (Bar Harbor, ME), and homozygous mice were utilized as CD19−/− mice. Female and male mice were housed at the Cleveland Clinic Lerner Research Institute under pathogen-free conditions. All animal procedures were executed in accordance with guidelines approved by the Cleveland Clinic Lerner Research Institute Institutional Animal Care and Use Committee. Mice 6 to 7 weeks of age were infected by intracranial injection with 1,000 PFU of the glial-tropic JHMV variant designated v2.2-1 (90). The infected animals were evaluated daily for clinical signs, utilizing the following scale: 0, healthy; 1, hunched back and ruffled fur; 2, inability to correct to upright position or partial hind limb paralysis; 3, complete hind limb paralysis and wasting; 4, moribund or deceased. Virus titers within the CNS were determined in clarified supernatants via plaque assay with the murine delayed brain tumor (DBT) astrocytoma, as previously described (90). Plaques were quantified following a 48-h incubation at 37°C.
Quantitative real-time PCR gene expression analysis.
Spinal cords and brains harvested from individual mice were snap-frozen, treated with 1 ml TRIzol (Invitrogen, Grand Island, NY), and homogenized using a TissueLyser and stainless steel beads (Qiagen, Valenica, CA). RNA was extracted according to the manufacturer's instructions. DNA contamination was eliminated via DNase I treatment for 30 min at 37°C (DNA-free kit; Ambion, Austin, TX). cDNA was synthesized from RNA using Moloney murine leukemia virus (MMLV) reverse transcriptase (Invitrogen) and a 1:1 mixture of oligo(dT) primers and random primers (Promega, Madison, WI). Quantitative real-time (RT) PCR was performed using either SYBR green master mix or Applied Biosystems gene expression assays with universal TaqMan master mix on a 7500 fast real-time PCR system (Applied Biosystems, Foster City, CA). The primers used for transcripts encoding glyceraldehyde 3-phosphate dehydrogenase (GAPDH), tumor necrosis factor (TNF), a proliferation-inducing ligand (APRIL; TNFSF13), IL-21, CXCL9, CXCL10, viral nucleocapsid, and C3 (5′-AAGCATCAACACACCCAACA-3′ and 5′-CTTGAGCTCCATTCGTGACA-3′) were used in conjunction with SYBR green master mix as described previously (8, 24, 91). GAPDH, activation-induced cytidine deaminase (AID), IgG, IFN-γ, CXCR5, CCL19, CCL21, CXCL13, BAFF (TNFSF13B), and IL-10 mRNA levels were determined using TaqMan primers (Applied Biosystems). Transcript levels were calculated relative to the GAPDH housekeeping gene using the following formula: 2[CT(GAPDH) − CT(target gene)] × 1,000.
Mononuclear cell isolation and flow cytometric analysis.
Cells were isolated from the CNS as described previously (24, 92). Briefly, brains harvested from phosphate-buffered saline (PBS)-perfused mice were mechanically homogenized in Dulbecco's PBS using ice-cold Tenbroeck grinders. The resulting suspension was centrifuged at 450 × g for 7 min at 4°C, the supernatants were stored at −80°C for subsequent analysis, and the cells were resuspended in RPMI medium. The cells were adjusted to 30% Percoll (Pharmacia, Piscataway, NJ), underlaid with 1 ml 70% Percol,l and collected from the 30%-70% Percoll interface following centrifugation at 850 × g for 30 min at 4°C. After washing, the cells were resuspended in fluorescence-activated cell sorter (FACS) buffer and incubated with a mixture of mouse, goat, and horse sera (1:1:1) and rat anti-mouse FcγIII/II MAb (2.4G2: BD Bioscience, San Diego, CA) for 20 min on ice. The cells were then stained with specific monoclonal Ab (MAb) to determine expression of cell surface markers for CD45 (30-F11), CD4 (L3T4), B220 (RA3-6B2), CD138 (281-2), CD95 (Jo2), CXCR4 (2B11), T cell and B cell activation antigen (GL7) (all from BD Pharmingen), CD8 (53-6.7) and PD-1 (RMP1-30) (both from eBioscience), and CXCR3 (R&D Systems). Cell surface expression of CXCR5 was determined via staining with biotin rat anti-mouse CXCR5 MAb and streptavidin phycoerythrin (both from BD Bioscience). The cells were then washed with FACS buffer, fixed with 2% paraformaldehyde, and analyzed on a BD LSR II flow cytometer. The resulting data were analyzed with FlowJo software (Tree Star Inc., Ashland, OR).
Serum, brain supernatant, and neutralizing Ab quantification.
JHMV-specific IgG and IgM in serum and brain supernatant were quantified by enzyme-linked immunosorbent assay (ELISA) using plates coated with serum-free supernatant from infected DBT cells and biotinylated goat anti-mouse IgG2a and IgM as detection Abs, as described previously (7, 91). Ab titers are expressed as the log of the highest dilution with an optical density value exceeding 3 standard deviations above the mean background. Titers for brain supernatants are expressed using the following formula: (absorbance at 450 nm/0.1) × dilution factor × total volume of clarified CNS supernatant. Neutralizing serum Ab was measured by incubation of serial 2-fold dilutions of heat-inactivated serum from individual mice with 50 PFU JHMV in 96-well plates for 90 min at 37°C. DBT cells were added, and the plates were incubated at 37°C for 48 h. The neutralization titers represent the log of the highest average serum dilution that inhibited cytopathic effect.
ELISPOT.
ASC were determined by ELISPOT assay as described previously (12, 24). Briefly, sterile, white 96-well filter plates with 0.45-μm-pore-size hydrophobic polyvinylidene difluoride (PVDF) membranes (Merck Millipore, Billerica, MA) were stripped with 70% ethanol for 2 min, washed with 0.1 M sodium bicarbonate buffer, and coated with either polyclonal goat anti-mouse Ig (Life Technologies, Eugene, OR) at a concentration of 2 mg/ml or undiluted virus supernatant for approximately 16 h at 4°C. The wells were washed with washing buffer (0.05% Tween 20 in 1× PBS) and blocked with 5% fetal calf serum (FCS) in RPMI medium for 2 h at 37°C. The blocking buffer was replaced with cell suspensions at serial dilutions in triplicate in RPMI medium. The plates were incubated at 37°C for 4 h and then washed thoroughly to remove the cells. After addition of biotinylated rabbit anti-mouse IgG (Southern Biotech, Birmingham, AL) at a concentration of 0.5 μg/ml, the plates were incubated for ∼16 h at 4°C, washed with washing buffer, and incubated with streptavidin horseradish peroxidase for 1 h at 25°C. Following washes with washing buffer and 1× PBS, diaminobenzidine (DAB) solution was added to visualize spots. Once spots had developed sufficiently, the reaction was stopped by flushing the wells with H2O, and the plates were left to dry in the dark. The plates were scanned, and the spots were quantified via Immunospot (Cellular Technologies Ltd., Shaker Heights, OH). The threshold criterion for spot size was defined as between 0.0009 and 0.2257 mm2.
Immunohistochemistry.
CLN from PBS-perfused mice were snap-frozen in Tissue-Tek OCT compound (Sakura Finetex, Torrance, CA) and sectioned at 10 μm using a Thermo Shandon cryostat. Slide-mounted tissue sections were fixed with ice-cold 70% acetone for 5 min, blocked with 5% bovine serum albumin and 10% goat serum for 1 h, and stained with rat anti-mouse B220 MAb (BD Biosciences), rabbit anti-mouse CD3 polyclonal Ab (Abcam, Cambridge, MA), and rat anti-mouse fluorescein isothiocyanate (FITC)-conjugated GL7 MAb (BD Biosciences) overnight at 4°C. Sections were then incubated with secondary Ab using Alexa Fluor 594 goat anti-rat (Life Technologies, Grand Island, NY) and Cy5 goat anti-rabbit (Life Technologies, Grand Island, NY) Ab for 1 h at room temperature. The sections were mounted with Vectashield Hard Set mounting medium with DAPI (4,6-diamidino-2-phenylindole) (Vector Laboratories, Burlingame, CA) and examined using a Leica TCS SP5 II confocal microscope (Leica Microsystems, Exton, PA).
Statistical analysis.
All the results are expressed as the means and standard errors of the mean (SEM). The data were plotted, and statistical significance was determined utilizing GraphPad Prism 6 software. Statistically significant differences are indicated.
ACKNOWLEDGMENTS
This work was supported by U.S. National Institutes of Health grant NS086299. The funding source had no involvement in the study design, writing of the manuscript, decision to submit, or collection, analysis, and interpretation of data.
We sincerely thank Mi-Hyun Hwang for intracranial virus administration.
REFERENCES
- 1.Sedgwick JD, Dörries R. 1991. The immune system response to viral infection of the CNS. Semin Neurosci 3:93–100. doi: 10.1016/1044-5765(91)90003-7. [DOI] [Google Scholar]
- 2.Phares TW, Stohlman SA, Bergmann CC. 2013. Intrathecal humoral immunity to encephalitic RNA viruses. Viruses 5:732–752. doi: 10.3390/v5020732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Griffin D, Levine B, Tyor W, Ubol S, Despres P. 1997. The role of antibody in recovery from alphavirus encephalitis. Immunol Rev 159:155–161. doi: 10.1111/j.1600-065X.1997.tb01013.x. [DOI] [PubMed] [Google Scholar]
- 4.Griffin DE. 2010. Recovery from viral encephalomyelitis: immune-mediated noncytolytic virus clearance from neurons. Immunol Res 47:123–133. doi: 10.1007/s12026-009-8143-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bergmann CC, Lane TE, Stohlman SA. 2006. Coronavirus infection of the central nervous system: host-virus stand-off. Nat Rev Microbiol 4:121–132. doi: 10.1038/nrmicro1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marten NW, Stohlman SA, Zhou J, Bergmann CC. 2003. Kinetics of virus-specific CD8+-T-cell expansion and trafficking following central nervous system infection. J Virol 77:2775–2778. doi: 10.1128/JVI.77.4.2775-2778.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tschen S-I, Bergmann CC, Ramakrishna C, Morales S, Atkinson R, Stohlman SA. 2002. Recruitment kinetics and composition of antibody-secreting cells within the central nervous system following viral encephalomyelitis. J Immunol 168:2922–2929. doi: 10.4049/jimmunol.168.6.2922. [DOI] [PubMed] [Google Scholar]
- 8.Phares TW, Marques CP, Stohlman SA, Hinton DR, Bergmann CC. 2011. Factors supporting intrathecal humoral responses following viral encephalomyelitis. J Virol 85:2589–2598. doi: 10.1128/JVI.02260-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okada T, Cyster JG. 2006. B cell migration and interactions in the early phase of antibody responses. Curr Opin Immunol 18:278–285. doi: 10.1016/j.coi.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 10.Phares TW, DiSano KD, Hinton DR, Hwang M, Zajac AJ, Stohlman SA, Bergmann CC. 2013. IL-21 optimizes T cell and humoral responses in the central nervous system during viral encephalitis. J Neuroimmunol 263:43–54. doi: 10.1016/j.jneuroim.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marques CP, Kapil P, Hinton DR, Hindinger C, Nutt SL, Ransohoff RM, Phares TW, Stohlman SA, Bergmann CC. 2011. CXCR3-dependent plasma blast migration to the central nervous system during viral encephalomyelitis. J Virol 85:6136–6147. doi: 10.1128/JVI.00202-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DiSano KD, Stohlman SA, Bergmann CC. 2017. Activated GL7+ B cells are maintained within the inflamed CNS in the absence of follicle formation during viral encephalomyelitis. Brain Behav Immun 60:71–83. doi: 10.1016/j.bbi.2016.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Phares TW, Stohlman SA, Hinton DR, Bergmann CC. 2013. Astrocyte-derived CXCL10 drives accumulation of antibody-secreting cells in the central nervous system during viral encephalomyelitis. J Virol 87:3382–3392. doi: 10.1128/JVI.03307-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cyster JG. 2005. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu Rev Immunol 23:127–159. doi: 10.1146/annurev.immunol.23.021704.115628. [DOI] [PubMed] [Google Scholar]
- 15.Victora GD, Nussenzweig MC. 2012. Germinal centers. Annu Rev Immunol 30:429–457. doi: 10.1146/annurev-immunol-020711-075032. [DOI] [PubMed] [Google Scholar]
- 16.Zotos D, Tarlinton DM. 2012. Determining germinal centre B cell fate. Trends Immunol 33:281–288. doi: 10.1016/j.it.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 17.Crotty S. 2012. The 1-1-1 fallacy. Immunol Rev 247:133–142. doi: 10.1111/j.1600-065X.2012.01117.x. [DOI] [PubMed] [Google Scholar]
- 18.Wengner AM, Höpken UE, Petrow PK, Hartmann S, Schurigt U, Bräuer R, Lipp M. 2007. CXCR5-and CCR7-dependent lymphoid neogenesis in a murine model of chronic antigen-induced arthritis. Arthritis Rheum 56:3271–3283. doi: 10.1002/art.22939. [DOI] [PubMed] [Google Scholar]
- 19.Krumbholz M, Theil D, Cepok S, Hemmer B, Kivisäkk P, Ransohoff RM, Hofbauer M, Farina C, Derfuss T, Hartle C. 2006. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up-regulation is differentially linked to CNS immune cell recruitment. Brain 129:200–211. doi: 10.1093/brain/awh680. [DOI] [PubMed] [Google Scholar]
- 20.Haas J, Bekeredjian-Ding I, Milkova M, Balint B, Schwarz A, Korporal M, Jarius S, Fritz B, Lorenz H-M, Wildemann B. 2011. B cells undergo unique compartmentalized redistribution in multiple sclerosis. J Autoimmun 37:289–299. doi: 10.1016/j.jaut.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 21.Finch DK, Ettinger R, Karnell JL, Herbst R, Sleeman MA. 2013. Effects of CXCL13 inhibition on lymphoid follicles in models of autoimmune disease. Eur J Clin Invest 43:501–509. doi: 10.1111/eci.12063. [DOI] [PubMed] [Google Scholar]
- 22.Kowarik MC, Cepok S, Sellner J, Grummel V, Weber MS, Korn T, Berthele A, Hemmer B. 2012. CXCL13 is the major determinant for B cell recruitment to the CSF during neuroinflammation. J Neuroinflammation 9:93. doi: 10.1186/1742-2094-9-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rupprecht TA, Plate A, Adam M, Wick M, Kastenbauer S, Schmidt C, Klein M, Pfister H-W, Koedel U. 2009. The chemokine CXCL13 is a key regulator of B cell recruitment to the cerebrospinal fluid in acute Lyme neuroborreliosis. J Neuroinflammation 6:42. doi: 10.1186/1742-2094-6-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Phares TW, DiSano KD, Stohlman SA, Segal BM, Bergmann CC. 2016. CXCL13 promotes isotype-switched B cell accumulation to the central nervous system during viral encephalomyelitis. Brain Behav Immun 54:128–139. doi: 10.1016/j.bbi.2016.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rainey-Barger EK, Rumble JM, Lalor SJ, Esen N, Segal BM, Irani DN. 2011. The lymphoid chemokine, CXCL13, is dispensable for the initial recruitment of B cells to the acutely inflamed central nervous system. Brain Behav Immun 25:922–931. doi: 10.1016/j.bbi.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fehr T, Rickert RC, Odermatt B, Roes J, Rajewsky K, Hengartner H, Zinkernagel RM. 1998. Antiviral protection and germinal center formation, but impaired B cell memory in the absence of CD19. J Exp Med 188:145–155. doi: 10.1084/jem.188.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carter RH, Wang Y, Brooks S. 2002. Role of CD19 signal transduction in B cell biology. Immunol Res 26:45–54. doi: 10.1385/IR:26:1-3:045. [DOI] [PubMed] [Google Scholar]
- 28.Rickert RC. 2005. Regulation of B lymphocyte activation by complement C3 and the B cell coreceptor complex. Curr Opin Immunol 17:237–243. doi: 10.1016/j.coi.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 29.Del Nagro CJ, Otero DC, Anzelon AN, Omori SA, Kolla RV, Rickert RC. 2005. CD 19 function in central and peripheral B-cell development. Immunol Res 31:119–131. doi: 10.1385/IR:31:2:119. [DOI] [PubMed] [Google Scholar]
- 30.Baiu DC, Prechl J, Tchorbanov A, Molina HD, Erdei A, Sulica A, Capel PJ, Hazenbos WL. 1999. Modulation of the humoral immune response by antibody-mediated antigen targeting to complement receptors and Fc receptors. J Immunol 162:3125–3130. [PubMed] [Google Scholar]
- 31.Ochsenbein AF, Pinschewer DD, Odermatt B, Carroll MC, Hengartner H, Zinkernagel RM. 1999. Protective T cell-independent antiviral antibody responses are dependent on complement. J Exp Med 190:1165–1174. doi: 10.1084/jem.190.8.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fischer MB, Goerg S, Shen L, Prodeus AP, Goodnow CC, Kelsoe G, Carroll MC. 1998. Dependence of germinal center B cells on expression of CD21/CD35 for survival. Science 280:582–585. doi: 10.1126/science.280.5363.582. [DOI] [PubMed] [Google Scholar]
- 33.Verschoor A, Brockman MA, Knipe DM, Carroll MC. 2001. Cutting edge: myeloid complement C3 enhances the humoral response to peripheral viral infection. J Immunol 167:2446–2451. doi: 10.4049/jimmunol.167.5.2446. [DOI] [PubMed] [Google Scholar]
- 34.Rickert RC, Rajewsky K, Roes J. 1995. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature 376:352. doi: 10.1038/376352a0. [DOI] [PubMed] [Google Scholar]
- 35.Meinl E, Krumbholz M, Hohlfeld R. 2006. B lineage cells in the inflammatory central nervous system environment: migration, maintenance, local antibody production, and therapeutic modulation. Ann Neurol 59:880–892. doi: 10.1002/ana.20890. [DOI] [PubMed] [Google Scholar]
- 36.Zhao J, Zhao J, Perlman S. 2014. Virus-specific regulatory T cells ameliorate encephalitis by repressing effector T cell functions from priming to effector stages. PLoS Pathog 10:e1004279. doi: 10.1371/journal.ppat.1004279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stern JN, Yaari G, Vander Heiden JA, Church G, Donahue WF, Hintzen RQ, Huttner AJ, Laman JD, Nagra RM, Nylander A. 2014. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci Transl Med 6:248ra107. doi: 10.1126/scitranslmed.3008879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Kevin SL, Harris TH, Kipnis J. 2015. Structural and functional features of central nervous system lymphatics. Nature 523:337. doi: 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Engel P, Zhou L-J, Ord DC, Sato S, Koller B, Tedder TF. 1995. Abnormal B lymphocyte development, activation, and differentiation in mice that lack or overexpress the CD19 signal transduction molecule. Immunity 3:39–50. doi: 10.1016/1074-7613(95)90157-4. [DOI] [PubMed] [Google Scholar]
- 40.Berkowska MA, Driessen GJ, Bikos V, Grosserichter-Wagener C, Stamatopoulos K, Cerutti A, He B, Biermann K, Lange JF, van der Burg M, van Dongen JJM, van Zelm MC. 2011. Human memory B cells originate from three distinct germinal center-dependent and-independent maturation pathways. Blood 118:2150–2158. doi: 10.1182/blood-2011-04-345579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ansel KM, Ngo VN, Hyman PL, Luther SA, Förster R, Sedgwick JD, Browning JL, Lipp M, Cyster JG. 2000. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature 406:309–314. doi: 10.1038/35018581. [DOI] [PubMed] [Google Scholar]
- 42.Tschen SI, Stohlman SA, Ramakrishna C, Hinton DR, Atkinson RD, Bergmann CC. 2006. CNS viral infection diverts homing of antibody-secreting cells from lymphoid organs to the CNS. Eur J Immunol 36:603–612. doi: 10.1002/eji.200535123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Radbruch A, Muehlinghaus G, Luger EO, Inamine A, Smith KG, Dörner T, Hiepe F. 2006. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat Rev Immunol 6:741–750. doi: 10.1038/nri1886. [DOI] [PubMed] [Google Scholar]
- 44.Phares TW, DiSano KD, Stohlman SA, Bergmann CC. 2014. Progression from IgD+ IgM+ to isotype-switched B cells is site specific during coronavirus-induced encephalomyelitis. J Virol 88:8853–8867. doi: 10.1128/JVI.00861-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith AL, Barthold S, De Souza M, Bottomly K. 1991. The role of gamma interferon in infection of susceptible mice with murine coronavirus, MHV-JHM. Arch Virol 121:89–100. doi: 10.1007/BF01316746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramakrishna C, Bergmann CC, Atkinson R, Stohlman SA. 2003. Control of central nervous system viral persistence by neutralizing antibody. J Virol 77:4670–4678. doi: 10.1128/JVI.77.8.4670-4678.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muehlinghaus G, Cigliano L, Huehn S, Peddinghaus A, Leyendeckers H, Hauser AE, Hiepe F, Radbruch A, Arce S, Manz RA. 2005. Regulation of CXCR3 and CXCR4 expression during terminal differentiation of memory B cells into plasma cells. Blood 105:3965–3971. doi: 10.1182/blood-2004-08-2992. [DOI] [PubMed] [Google Scholar]
- 48.Ramakrishna C, Stohlman SA, Atkinson RD, Shlomchik MJ, Bergmann CC. 2002. Mechanisms of central nervous system viral persistence: the critical role of antibody and B cells. J Immunol 168:1204–1211. doi: 10.4049/jimmunol.168.3.1204. [DOI] [PubMed] [Google Scholar]
- 49.Lin MT, Hinton DR, Marten NW, Bergmann CC, Stohlman SA. 1999. Antibody prevents virus reactivation within the central nervous system. J Immunol 162:7358–7368. [PubMed] [Google Scholar]
- 50.Parra B, Hinton DR, Marten NW, Bergmann CC, Lin MT, Yang CS, Stohlman SA. 1999. IFN-γ is required for viral clearance from central nervous system oligodendroglia. J Immunol 162:1641–1647. [PubMed] [Google Scholar]
- 51.Bergmann CC, Parra B, Hinton DR, Chandran R, Morrison M, Stohlman SA. 2003. Perforin-mediated effector function within the central nervous system requires IFN-γ-mediated MHC up-regulation. J Immunol 170:3204–3213. doi: 10.4049/jimmunol.170.6.3204. [DOI] [PubMed] [Google Scholar]
- 52.Puntambekar SS, Bergmann CC, Savarin C, Karp CL, Phares TW, Parra GI, Hinton DR, Stohlman SA. 2011. Shifting hierarchies of interleukin-10-producing T cell populations in the central nervous system during acute and persistent viral encephalomyelitis. J Virol 85:6702–6713. doi: 10.1128/JVI.00200-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perlman S, Zhao J. 2017. Roles of regulatory T cells and IL-10 in virus-induced demyelination. J Neuroimmunol 308:6–11. doi: 10.1016/j.jneuroim.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Puntambekar SS, Hinton DR, Yin X, Savarin C, Bergmann CC, Trapp BD, Stohlman SA. 2015. Interleukin-10 is a critical regulator of white matter lesion containment following viral induced demyelination. Glia 63:2106–2120. doi: 10.1002/glia.22880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lalor SJ, Segal BM. 2010. Lymphoid chemokines in the CNS. J Neuroimmunol 224:56–61. doi: 10.1016/j.jneuroim.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sköldenberg B, Kalimo K, Forsgren M, Halonen P. 1981. Herpes simplex encephalitis: a serological follow-up study. Acta Neurol Scand 63:273–285. [PubMed] [Google Scholar]
- 57.Schultze D, Weder B, Cassinotti P, Vitek L, Krausse K, Fierz W. 2004. Diagnostic significance of intrathecally produced herpes simplex and varicella-zoster virus-specific antibodies in central nervous system infections. Swiss Med Wkly 134:700. [DOI] [PubMed] [Google Scholar]
- 58.Narayan K, Dail D, Li L, Cadavid D, Amrute S, Fitzgerald-Bocarsly P, Pachner AR. 2005. The nervous system as ectopic germinal center: CXCL13 and IgG in Lyme neuroborreliosis. Ann Neurol 57:813–823. doi: 10.1002/ana.20486. [DOI] [PubMed] [Google Scholar]
- 59.Burke DS, Nisalak A, Lorsomrudee W, Ussery MA, Laorpongse T. 1985. Virus-specific antibody-producing cells in blood and cerebrospinal fluid in acute Japanese encephalitis. J Med Virol 17:283–292. doi: 10.1002/jmv.1890170310. [DOI] [PubMed] [Google Scholar]
- 60.Vandvik B, Weil ML, Grandien M, Norrby E. 1978. Progressive rubella virus panencephalitis: synthesis of oligoclonal virus-specific IgG antibodies and homogeneous free light chains in the central nervous system. Acta Neurol Scand 57:53–64. [DOI] [PubMed] [Google Scholar]
- 61.Vandvik B, Norrby E. 1973. Oligoclonal IgG antibody response in the central nervous system to different measles virus antigens in subacute sclerosing panencephalitis. Proc Natl Acad Sci U S A 70:1060–1063. doi: 10.1073/pnas.70.4.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thakare JP, Gore MM, Risbud AR, Banerjee K, Ghosh SN. 1991. Detection of virus specific IgG subclasses in Japanese encephalitis patients. Indian J Med Res 93:271–276. [PubMed] [Google Scholar]
- 63.Kaiser R, Dorries R, Luer W, Poser S, Pohle HD, Felgenhauer K, ter Meulen V. 1989. Analysis of oligoclonal antibody bands against individual HIV structural proteins in the CSF of patients infected with HIV. J Neurol 236:157–160. doi: 10.1007/BF00314332. [DOI] [PubMed] [Google Scholar]
- 64.Ryzhova E, Aye P, Harvey T, Cao W, Lackner A, Gonzalez-Scarano F. 2009. Intrathecal humoral responses are inversely associated with the frequency of simian immunodeficiency virus macrophage-tropic variants in the central nervous system. J Virol 83:8282–8288. doi: 10.1128/JVI.00235-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Puccioni-Sohler M, Rios M, Bianco C, Zhu SW, Oliveira C, Novis SA, Pombo-de-Oliveira MS. 1999. An inverse correlation of HTLV-I viral load in CSF and intrathecal synthesis of HTLV-I antibodies in TSP/HAM. Neurology 53:1335–1339. doi: 10.1212/WNL.53.6.1335. [DOI] [PubMed] [Google Scholar]
- 66.Hooper DC, Phares TW, Fabis MJ, Roy A. 2009. The production of antibody by invading B cells is required for the clearance of rabies virus from the central nervous system. PLoS Negl Trop Dis 3:e535. doi: 10.1371/journal.pntd.0000535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fragkoudis R, Ballany CM, Boyd A, Fazakerley JK. 2008. In Semliki Forest virus encephalitis, antibody rapidly clears infectious virus and is required to eliminate viral material from the brain, but is not required to generate lesions of demyelination. J Gen Virol 89:2565–2568. doi: 10.1099/vir.0.2008/002238-0. [DOI] [PubMed] [Google Scholar]
- 68.Levine B, Hardwick JM, Trapp BD, Crawford TO, Bollinger RC, Griffin DE. 1991. Antibody-mediated clearance of alphavirus infection from neurons. Science 254:856–860. doi: 10.1126/science.1658936. [DOI] [PubMed] [Google Scholar]
- 69.Pachner AR, Brady J, Narayan K. 2007. Antibody-secreting cells in the central nervous system in an animal model of MS: phenotype, association with disability, and in vitro production of antibody. J Neuroimmunol 190:112–120. doi: 10.1016/j.jneuroim.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 70.Lee H, Sunden Y, Ochiai K, Umemura T. 2011. Experimental intracerebral vaccination protects mouse from a neurotropic virus by attracting antibody secreting cells to the CNS. Immunol Lett 139:102–109. doi: 10.1016/j.imlet.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 71.Corcione A, Casazza S, Ferretti E, Giunti D, Zappia E, Pistorio A, Gambini C, Mancardi GL, Uccelli A, Pistoia V. 2004. Recapitulation of B cell differentiation in the central nervous system of patients with multiple sclerosis. Proc Natl Acad Sci U S A 101:11064–11069. doi: 10.1073/pnas.0402455101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Owens GP, Bennett JL, Gilden DH, Burgoon MP. 2006. The B cell response in multiple sclerosis. Neurol Res 28:236–244. doi: 10.1179/016164106X98099. [DOI] [PubMed] [Google Scholar]
- 73.Owens GP, Ritchie AM, Burgoon MP, Williamson RA, Corboy JR, Gilden DH. 2003. Single-cell repertoire analysis demonstrates that clonal expansion is a prominent feature of the B cell response in multiple sclerosis cerebrospinal fluid. J Immunol 171:2725–2733. doi: 10.4049/jimmunol.171.5.2725. [DOI] [PubMed] [Google Scholar]
- 74.Krumbholz M, Theil D, Derfuss T, Rosenwald A, Schrader F, Monoranu CM, Kalled SL, Hess DM, Serafini B, Aloisi F, Wekerle H, Hohlfeld R, Meinl E. 2005. BAFF is produced by astrocytes and up-regulated in multiple sclerosis lesions and primary central nervous system lymphoma. J Exp Med 201:195–200. doi: 10.1084/jem.20041674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thangarajh M, Masterman T, Hillert J, Moerk S, Jonsson R. 2007. A proliferation-inducing ligand (APRIL) is expressed by astrocytes and is increased in multiple sclerosis. Scand J Immunol 65:92–98. doi: 10.1111/j.1365-3083.2006.01867.x. [DOI] [PubMed] [Google Scholar]
- 76.Metcalf TU, Baxter VK, Nilaratanakul V, Griffin DE. 2013. Recruitment and retention of B cells in the central nervous system in response to alphavirus encephalomyelitis. J Virol 87:2420–2429. doi: 10.1128/JVI.01769-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cervantes-Barragán L, Kalinke U, Züst R, König M, Reizis B, López-Macías C, Thiel V, Ludewig B. 2009. Type I IFN-mediated protection of macrophages and dendritic cells secures control of murine coronavirus infection. J Immunol 182:1099–1106. doi: 10.4049/jimmunol.182.2.1099. [DOI] [PubMed] [Google Scholar]
- 78.Coro ES, Chang WW, Baumgarth N. 2006. Type I IFN receptor signals directly stimulate local B cells early following influenza virus infection. J Immunol 176:4343–4351. doi: 10.4049/jimmunol.176.7.4343. [DOI] [PubMed] [Google Scholar]
- 79.Purtha WE, Chachu KA, Virgin HW, Diamond MS. 2008. Early B-cell activation after West Nile virus infection requires alpha/beta interferon but not antigen receptor signaling. J Virol 82:10964–10974. doi: 10.1128/JVI.01646-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Veerhuis R, Janssen I, De Groot CJ, Van Muiswinkel FL, Hack CE, Eikelenboom P. 1999. Cytokines associated with amyloid plaques in Alzheimer's disease brain stimulate human glial and neuronal cell cultures to secrete early complement proteins, but not C1-inhibitor. Exp Neurol 160:289–299. doi: 10.1006/exnr.1999.7199. [DOI] [PubMed] [Google Scholar]
- 81.Hosokawa M, Klegeris A, Maguire J, McGeer PL. 2003. Expression of complement messenger RNAs and proteins by human oligodendroglial cells. Glia 42:417–423. doi: 10.1002/glia.10234. [DOI] [PubMed] [Google Scholar]
- 82.Veerhuis R, Nielsen HM, Tenner AJ. 2011. Complement in the brain. Mol Immunol 48:1592–1603. doi: 10.1016/j.molimm.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.DiSano KD, Stohlman SA, Bergmann CC. 2017. An optimized method for enumerating CNS derived memory B cells during viral-induced inflammation. J Neurosci Methods 285:58–68. doi: 10.1016/j.jneumeth.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Baccala R, Hoebe K, Kono DH, Beutler B, Theofilopoulos AN. 2007. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat Med 13:543. doi: 10.1038/nm1590. [DOI] [PubMed] [Google Scholar]
- 85.Das A, Heesters BA, Bialas A, O'Flynn J, Rifkin IR, Ochando J, Mittereder N, Carlesso G, Herbst R, Carroll MC. 2017. Follicular dendritic cell activation by TLR ligands promotes autoreactive B cell responses. Immunity 46:106–119. doi: 10.1016/j.immuni.2016.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cupovic J, Onder L, Gil-Cruz C, Weiler E, Caviezel-Firner S, Perez-Shibayama C, Rülicke T, Bechmann I, Ludewig B. 2016. Central nervous system stromal cells control local CD8+ T cell responses during virus-induced neuroinflammation. Immunity 44:622–633. doi: 10.1016/j.immuni.2015.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wingerchuk DM, Carter JL. 2014. Multiple sclerosis: current and emerging disease-modifying therapies and treatment strategies. Mayo Clin Proc 89:225–240. doi: 10.1016/j.mayocp.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 88.Chalkias S, Dang X, Bord E, Stein MC, Kinkel RP, Sloane JA, Donnelly M, Ionete C, Houtchens MK, Buckle GJ. 2014. JC virus reactivation during prolonged natalizumab monotherapy for multiple sclerosis. Ann Neurol 75:925–934. doi: 10.1002/ana.24148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rickert RC, Roes J, Rajewsky K. 1997. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res 25:1317–1318. doi: 10.1093/nar/25.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fleming J, Trousdale M, El-Zaatari F, Stohlman S, Weiner L. 1986. Pathogenicity of antigenic variants of murine coronavirus JHM selected with monoclonal antibodies. J Virol 58:869–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Phares TW, Stohlman SA, Hinton DR, Atkinson R, Bergmann CC. 2010. Enhanced antiviral T cell function in the absence of B7-H1 is insufficient to prevent persistence but exacerbates axonal bystander damage during viral encephalomyelitis. J Immunol 185:5607–5618. doi: 10.4049/jimmunol.1001984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bergmann CC, Altman JD, Hinton D, Stohlman SA. 1999. Inverted immunodominance and impaired cytolytic function of CD8+ T cells during viral persistence in the central nervous system. J Immunol 163:3379–3387. [PubMed] [Google Scholar]