

ABSTRACT
The neuroimmune dialogue between peripheral neurons and Langerhans cells (LCs) within mucosal epithelia protects against incoming pathogens. LCs rapidly internalize human immunodeficiency virus type 1 (HIV-1) upon its sexual transmission and then trans-infect CD4+ T cells. We recently found that the neuropeptide calcitonin gene-related peptide (CGRP), secreted mucosally from peripheral neurons, inhibits LC-mediated HIV-1 trans-infection. In this study, we investigated the mechanism of CGRP-induced inhibition, focusing on HIV-1 degradation in LCs and its interplay with trans-infection. We first show that HIV-1 degradation occurs in endolysosomes in untreated LCs, and functionally blocking such degradation with lysosomotropic agents results in increased trans-infection. We demonstrate that CGRP acts via its cognate receptor and at a viral postentry step to induce faster HIV-1 degradation, but without affecting the kinetics of endolysosomal degradation. We reveal that unexpectedly, CGRP shifts HIV-1 degradation from endolysosomes toward the proteasome, providing the first evidence for functional HIV-1 proteasomal degradation in LCs. Such efficient proteasomal degradation significantly inhibits the first phase of trans-infection, and proteasomal, but not endolysosomal, inhibitors abrogate CGRP-induced inhibition. Together, our results establish that CGRP controls the HIV-1 degradation mode in LCs. The presence of endogenous CGRP within innervated mucosal tissues, especially during the sexual response, to which CGRP contributes, suggests that HIV-1 proteasomal degradation predominates in vivo. Hence, proteasomal, rather than endolysosomal, HIV-1 degradation in LCs should be enhanced clinically to effectively restrict HIV-1 trans-infection.
IMPORTANCE During sexual transmission, HIV-1 is internalized and degraded in LCs, the resident antigen-presenting cells in mucosal epithelia. Yet during trans-infection, infectious virions escaping degradation are transferred to CD4+ T cells, the principal HIV-1 targets. We previously found that the neuroimmune dialogue between LCs and peripheral neurons, innervating mucosal epithelia, significantly inhibits trans-infection via the action of the secreted neuropeptide CGRP on LCs. In this study, we investigated whether CGRP-induced inhibition of trans-infection is linked to CGRP-controlled HIV-1 degradation in LCs. We show that in untreated LCs, HIV-1 is functionally degraded in endolysosomes. In sharp contrast, we reveal that in CGRP-treated LCs, HIV-1 is diverted toward and degraded via another cytosolic protein degradative pathway, namely, the proteasome. These results establish that CGRP regulates HIV-1 degradation in LCs. As CGRP contributes to the sexual response and present within mucosal epithelia, HIV-1 proteasomal degradation in LCs might predominate in vivo and should be enhanced clinically.
KEYWORDS: calcitonin gene-related peptide (CGRP), degradation, human immunodeficiency virus type 1 (HIV-1), Langerhans cells (LCs), proteasome, trans-infection
INTRODUCTION
HIV-1 invades the body principally during unprotected sexual intercourse and is rapidly internalized by resident Langerhans cells (LCs) in the male (1, 2) and female (3) genital epithelia. Depending on the LC preparation/isolation method (4) and HIV-1 load (5), and especially ex vivo (1–3), infectious virions might be retained by LCs within tetraspanin-rich intracellular compartments (6) and then transferred to CD4+ T cells during the first phase of a process termed trans-infection (7). Although HIV-1 productive infection of LCs is limited (8, 9), virions produced de novo are additionally transferred later, during the second phase of trans-infection (7).
The endogenous factors controlling HIV-1 trans-infection in vivo remain ill defined. Peripheral neurons, which innervate all mucosal epithelia, are in direct contact with LCs and affect their function via the secreted neuropeptide calcitonin gene-related peptide (CGRP) (10, 11). Such neuroimmune dialogue might play an important role during mucosal HIV-1 transmission but is routinely ignored. Hence, we recently investigated the role of CGRP during HIV-1 trans-infection and reported that CGRP modulates the early mucosal molecular/cellular interactions between LCs and HIV-1, resulting in strong inhibition of HIV-1 trans-infection (12). Such inhibition is further reinforced by a CGRP-mediated autocrine/paracrine feedback mechanism that enhances the anti-HIV-1 activity of CGRP (13).
The first phase of HIV-1 trans-infection is mediated by virions escaping degradation in LCs (7). Indeed, HIV-1 binding to the LC-specific C-type lectin langerin (14) induces viral endocytosis and endolysosomal degradation in vitro. This paradigm is based on previous studies that reported colocalization of HIV-1 with Birbeck granules and/or lysosomes in LCs at the ultrastructural level (5, 9, 15). To date, however, functional blocking of HIV-1 degradation in LCs with appropriate inhibitors has not been reported, nor has its potential outcome on trans-infection. As CGRP markedly decreases HIV-1 trans-infection (12), we investigated in this study whether CGRP affects HIV-1 degradation in LCs to inhibit subsequent trans-infection. Our results confirm that HIV-1 degradation in untreated LCs indeed takes place via the endolysosomal machinery, and functionally blocking this degradation mode using lysosomotropic agents results in increased trans-infection. We further report that CGRP operates at a postentry step to inhibit trans-infection by inducing more efficient viral degradation. Unexpectedly, CGRP does not modulate the kinetics of HIV-1 endolysosomal degradation. Instead, CGRP diverts HIV-1 away from this cellular pathway to induce faster viral degradation via the proteasome. In turn, proteasomal, but not endolysosomal, inhibitors abrogate CGRP-induced inhibition of trans-infection.
RESULTS
CGRP mediates faster and more efficient HIV-1 degradation in LCs.
To investigate whether CGRP affects HIV-1 degradation, monocyte-derived LCs (MDLCs) were left untreated or treated for 24 h with 100 nM CGRP, i.e., the effective concentration that we previously reported to significantly inhibit trans-infection from MDLCs to CD4+ T cells (12, 13). The cells were then pulsed with HIV-1 for 30 min at room temperature to facilitate efficient binding, shifted to 37°C to initiate viral endocytosis and degradation, and chased for different time periods (i.e., 0, 4, and 24 h at 37°C). Total HIV-1 was then measured in the cell lysates using p24 enzyme-linked immunosorbent assay (ELISA). In both untreated and CGRP-treated MDLCs, HIV-1 was progressively degraded, with greater degradation rates between 0 and 4 h than between 4 and 24 h (Fig. 1A), suggesting that most virions are degraded during the first hours following internalization. At 4 h and 24 h, total HIV-1 was significantly lower in CGRP-treated than in untreated MDLCs (Fig. 1A). Importantly, viral degradation showed faster kinetics after CGRP treatment, with percentages of decrease in total HIV-1 content of 92.0 versus 81.4 (0 to 4 h) and 59.8 versus 40.9 (4 to 24 h) for CGRP-treated versus untreated MDLCs, respectively (Fig. 1A).
FIG 1.

CGRP induces efficient HIV-1 degradation in MDLCs. MDLCs were left untreated or were pretreated for 24 h at 37°C with 100 nM CGRP. (A) The cells were pulsed with HIV-1JR-CSF at room temperature for 30 min, washed, shifted to 37°C, chased for 0, 4, or 24 h, and lysed, and total HIV-1 was measured in the cell lysates using p24 ELISA. Shown are means ± SEMs (using MDLCs from 4 different donors) for percentages of total HIV-1 normalized against values at 0 h. *, P = 0.0229 (4 h); **, P = 0.0049 (24 h) (CGRP-treated versus untreated cells). (B) The cells were incubated with HIV-1ADA directly at 37°C for 4 h, washed, treated immediately or 24 h later with trypsin for 10 min to remove surface-bound virus, and lysed, and intracellular HIV-1 was measured in the cell lysates using p24 ELISA. Shown are means ± SEMs (n = 3) for concentrations of intracellular HIV-1 p24. ***, P = 0.0006 (4 h); **, P = 0.0083 (24 h) (CGRP-treated versus untreated cells). (C) The cells were incubated at 4°C with the mouse langerin MAb DCGM4, washed, and further incubated with a biotin-conjugated anti-mouse IgG Ab. The cells were then chased for 0, 5, and 30 min at either 4°C (dashed lines) or 37°C (solid lines), cooled, incubated with FITC-conjugated streptavidin at 4°C, and examined by flow cytometry. Shown are means ± SEMs (n = 3) for percentages of langerin internalization, calculated as the ratios of langerin-positive cells at each time point over that at 0 min and normalized against untreated cells. ***, P < 0.0001 (untreated); *, P = 0.0174 (CGRP treated) (4°C versus 37°C, respectively, at 30 min).
To confirm that the decrease in HIV-1 content was due to viral degradation, untreated or CGRP-treated MDLCs were pulsed with HIV-1 directly for 4 h at 37°C to increase the viral input and facilitate continuous degradation. The cells were then washed and treated with trypsin to remove surface-bound virus, either immediately or 24 h after the viral pulse. Intracellular HIV-1 was then measured in the cell lysates using p24 ELISA. Directly after the 4-h pulse, intracellular HIV-1 content was significantly lower in CGRP-treated than in untreated MDLCs (Fig. 1B). Intracellular HIV-1 content continued to significantly decline at 24 h and at a higher rate, with percentages of decrease in intracellular HIV-1 content of 75.4 versus 68.0 (4 to 24 h) for CGRP-treated versus untreated MDLCs, respectively (Fig. 1B).
To verify that the decrease in total and intracellular HIV-1 contents was not merely a result of potential CGRP-induced inhibition of langerin-mediated HIV-1 endocytosis, we measured langerin internalization (16). Untreated or CGRP-treated MDLCs were labeled at 4°C with the langerin mouse monoclonal antibody (MAb) DCGM4, followed by a biotin-conjugated anti-mouse Ab. The cells were then chased for different times (i.e., 0, 5, and 30 min) at either 4°C or 37°C, and the remaining levels of noninternalized cell surface langerin were evaluated using fluorescent streptavidin and flow cytometry. In untreated MDLCs, langerin was rapidly internalized at 37°C but not at 4°C, as previously reported (16). In line with our previous observations (12), CGRP induced upregulation in langerin cell surface expression (Fig. 1C, 0 min). In such CGRP-treated MDLCs, langerin was also rapidly internalized at 37°C but not at 4°C, with a >90% decrease at 30 min (Fig. 1C).
Taken together, these results show that HIV-1 is degraded more efficiently in MDLCs following CGRP treatment.
CGRP directs HIV-1 for proteasomal degradation in LCs to inhibit trans-infection.
HIV-1 was previously reported to colocalize within lysosomes in LCs at the ultrastructural level, suggesting that viral degradation takes place via the endolysosomal machinery. Therefore, we speculated based on our results described above that CGRP might modify the kinetics of endolysosomal HIV-1 degradation to induce faster viral degradation. To test this assumption, we included different inhibitors during the viral pulse period in order to functionally block viral degradation, which was not yet reported in LCs, and then calculated the corresponding degradation indexes (i.e., the ratios between the intracellular HIV-1 contents in the presence over absence of these inhibitors). We first used chloroquine, a lysosomotropic weak base that becomes trapped within endolysosomes in a protonated form and hence prevents their acidification. In untreated MDLCs, the HIV-1 degradation index was increased in the presence of chloroquine, indicating that HIV-1 degradation indeed occurred via the endolysosomal machinery (Fig. 2A). Surprisingly, chloroquine had no effect on the HIV-1 degradation index in CGRP-treated MDLCs (mean ± standard error of the mean [SEM] of 0.96 ± 0.09 [Fig. 2A]), demonstrating that CGRP-induced efficient HIV-1 degradation does not occur via endolysosomes or modifications of endolysosomal degradation kinetics but rather occurs via another mechanism.
FIG 2.

CGRP acts at a viral postentry step and directs HIV-1 for proteasomal degradation to inhibit trans-infection in MDLCs. (A) Untreated or CGRP-treated (24 h; 100 nM) MDLCs were pulsed with HIV-1ADA at 37°C for 4 h in the absence or presence of either chloroquine (50 μM) or MG132 (10 μM), washed, treated with trypsin for 10 min to remove surface-bound virus, and lysed, and intracellular HIV-1 was measured in the cell lysates using p24 ELISA. Shown are means ± SEMs (n = 3) for HIV-1 degradation indexes, calculated as the ratios of intracellular HIV-1 in the presence over absence of inhibitors. *, P = 0.0127 (untreated) and 0.0434 (CGRP treated) (MG132 versus chloroquine, respectively). (B to D) Untreated or CGRP-treated (24 h, 100 nM) MDLCs were pulsed with HIV-1JR-CSF for 4 h at 37°C in the absence (B) or presence of the indicated concentrations of NH4Cl and lactacystin (C) or dynasore (D). The cells were then cocultured with autologous CD4+ T cells for a week. In panel B, the cells were collected, stained for surface CD1a (upper and middle portions) or CD3/CD4 (lower portion) followed by intracellular p24, and examined by flow cytometry. Shown are representative fluorescence-activated cell sorting (FACS) plots, with regular numbers indicating the means ± SEMs (n = 3 for each panel) for percentages of p24+ cells out of FSChigh SSChigh CD1a+ MDLCs (upper portion), FSClow SSClow CD1a− T cells (middle portion), or SSClow CD3+ CD4+ T cells (lower portion). P = 0.0083, 0.0005, and 0.0302 for CGRP-treated versus untreated CD1a+ MDLCs, CD1a− T cells, or CD4+ T cells, respectively. Numbers in italics show means ± SEMs for mean fluorescence intensity (MFI) of CD4 expression (lower portion). FSC, forward scatter; SSC, side scatter. (C and D) HIV-1 replication was measured in the coculture supernatants by p24 ELISA. Shown are means ± SEMs for HIV-1 trans-infection percentages, normalized against untreated cells serving as the 100% set point, using MDLCs and T cells of 5 (C) or 3 (D) different donors.
Endolysosomal and proteasomal degradation are the two major pathways for intracellular protein degradation. Hence, we further investigated whether HIV-1 might be degraded via the proteasome in CGRP-treated LCs, using the proteasome inhibitor MG132. In untreated MDLCs, MG132 had no effect on the HIV-1 degradation index (Fig. 2A), demonstrating a lack of HIV-1 proteasomal degradation in these cells. Unexpectedly and in sharp contrast, MG132 significantly increased the HIV-1 degradation index in CGRP-treated MDLCs (1.30 ± 0.08 [Fig. 2A]), showing that CGRP exerts its effect by diverting viral degradation toward this pathway.
Next, we studied the impact of endolysosomal versus CGRP-induced proteasomal HIV-1 degradation on trans-infection. In principle, the first phase of trans-infection is mediated by virions escaping degradation (7), and therefore, more efficient HIV-1 degradation should decrease virion availability and ensuing trans-infection. To first set suitable conditions to measure HIV-1 degradation and subsequent trans-infection, untreated or CGRP-treated MDLCs were pulsed with HIV-1 for 4 h at 37°C at a multiplicity of infection (MOI) of 0.2, also used in the experiments described above, which facilitates efficient viral trans-infection (7). The cells were then cocultured with autologous CD4+ T cells, and a week later HIV-1 content within the cocultured cells was examined by flow cytometry. CGRP significantly decreased the percentage of HIV-1 p24+ cells out of the gated FSChigh SSChigh CD1a+ MDLCs (Fig. 2B, upper portion). Such CGRP-induced reduction in intracellular HIV-1 content within MDLCs resulted in decreased trans-infection, which was evidenced by a lower percentage of HIV-1 p24+ cells out of the gated FSClow SSClow CD1a− (Fig. 2B, middle portion) or SSClow CD3+ CD4+ (Fig. 2B, lower portion) cocultured T cells, confirming the suitability of our experimental settings. Importantly, CD4 surface expression was similar on T cells cocultured with either untreated or CGRP-treated MDLCs (Fig. 2B, lower portion), showing that CGRP-induced inhibition of trans-infection was not due to reduced CD4 expression.
To block the rapid process of HIV-1 degradation during the trans-infection assay described above, we included two other inhibitors during the viral pulse period: either NH4Cl, which impairs endolysosomal acidification similarly to chloroquine, or lactacystin, which inhibits proteasomal activity. HIV-1 trans-infection, which results in viral replication in CD4+ T cells, was measured within the coculture supernatants a week later using p24 ELISA. As for the HIV-1 degradation index described above, trans-infection in untreated MDLCs was significantly increased in a dose-dependent manner by the endolysosomal acidification inhibitor NH4Cl but was unaffected by lactacystin (Fig. 2C). In line with our previous observations (12, 13), CGRP significantly inhibited HIV-1 trans-infection, by approximately 65% (Fig. 2C). In contrast to the case with untreated MDLCs, CGRP-induced inhibition was significantly abrogated in a dose-dependent manner only by the proteasomal inhibitor lactacystin and was unaffected by NH4Cl (Fig. 2C). These results show that HIV-1 is degraded by unique mechanisms in CGRP-treated versus untreated MDLCs, and blocking each respective degradation pathway results in increased trans-infection.
To further identify whether CGRP directs HIV-1 to the proteasome from the plasma membrane or after viral internalization, we included during the HIV-1 pulse period the dynamin inhibitor dynasore, which prevents the scission of clathrin-coated vesicles. HIV-1 trans-infection was significantly inhibited, by approximately 60%, when MDLCs were treated with dynasore alone (Fig. 2D), demonstrating that clathrin-mediated endocytosis is required for subsequent trans-infection. The lack of complete inhibition of trans-infection following dynasore treatment points to the involvement of other HIV-1 uptake mechanisms, e.g., langerin/caveolin-mediated internalization (15). trans-Infection was significantly inhibited as before by CGRP alone but was unaffected by subsequent addition of dynasore during the viral pulse period (Fig. 2D). Hence, as there was no defect in langerin internalization in CGRP-treated MDLCs (Fig. 1C), the lack of any additive/abrogation effects when cells were treated with CGRP followed by dynasore shows that CGRP directs HIV-1 to the proteasome at a clathrin-mediated postentry step without interfering with caveolin-mediated viral uptake.
Overall, these results show that HIV-1 trans-infection is mediated by virions escaping degradation. Such degradation takes place by distinct mechanisms, namely, via endolysosomes in untreated MDLCs versus the proteasome in CGRP-treated MDLCs. The latter is induced at a postentry step, is more efficient, and results in significant restriction of trans-infection.
CGRP stimulates HIV-1 proteasomal degradation during the first phase of trans-infection.
LC-mediated HIV-1 trans-infection is a dynamic process characterized by two separate temporal phases: following HIV-1 internalization and vesicular trafficking, the first phase is mediated by virions escaping degradation and is completed at 48 h postinfection (p.i.); following LC productive infection, the second phase is mediated by de novo-produced virions and occurs at 48 to 96 h p.i. (7). As both internalized virions and newly produced viral proteins might be degraded in LCs, we next investigated at which trans-infection phase CGRP induces HIV-1 proteasomal degradation.
MUTZ3 cell-derived LCs (MULCs) were previously reported to mediate only the first phase of HIV-1 trans-infection due to a lack of CCR5 surface expression that prevents HIV-1 fusion and productive infection (7). In contrast, two other studies documented low levels of surface CCR5 on MULCs (17, 18). In our study, 20.1% ± 2.1% of nondifferentiated parental MUTZ3 cells were positive for surface CCR5 (Fig. 3A). Following 7 days of differentiation, only 6.6% ± 1.3% of MULCs expressed surface CCR5, which further significantly decreased to 1.9% ± 0.8% after an additional 3 days of differentiation (Fig. 3A). Therefore, MULCs differentiated for 10 days were used in subsequent studies to ensure low/no second-phase trans-infection.
FIG 3.
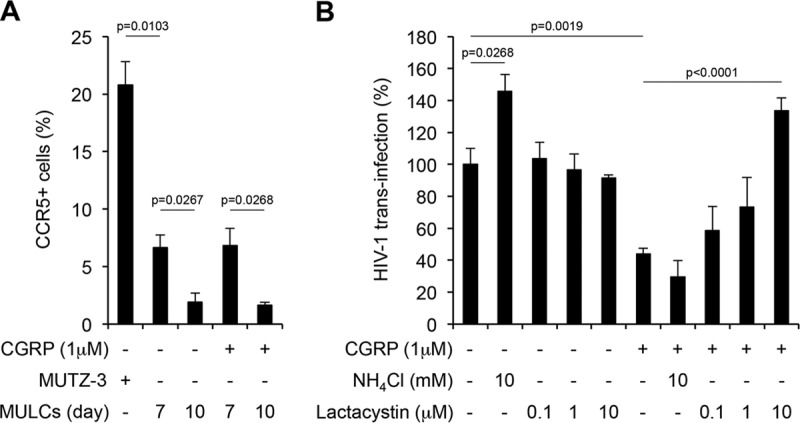
CGRP stimulates HIV-1 proteasomal degradation to restrict first-phase trans-infection in MULCs. (A) Parental MUTZ3 cells, or untreated and CGRP-treated (24 h, 1 μM) MULCs at different days during their differentiation process, were stained for surface CCR5 and examined by flow cytometry. Shown are means ± SEMs for percentages of CCR5+ cells from 3 experiments. (B) Untreated or CGRP-treated (24 h, 1 μM) MULCs were pulsed with HIV-1JR-CSF for 4 h at 37°C in the presence of the indicated concentrations of NH4Cl and lactacystin. The cells were then cocultured with CD4+ T cells for a week, and HIV-1 replication was measured in the coculture supernatants by p24 ELISA. Shown are means ± SEMs for HIV-1 trans-infection percentages, normalized against untreated cells serving as the 100% set point, using T cells from 4 different donors.
We previously reported that CGRP at 100 nM significantly inhibits MULC-mediated trans-infection, but to a lower extent than for MDLC-mediated trans-infection (12). We therefore treated MULCs with a higher CGRP concentration, 1 μM, which resulted in increased inhibition of trans-infection (Fig. 3B), similar to that measured in MDLCs (Fig. 2C). This concentration of CGRP had no effect on the low CCR5 surface levels in MULCs (Fig. 3A). As with MDLCs described above, lactacystin abrogated in a dose-dependent manner only the inhibition of HIV-1 trans-infection in CGRP-treated MULCs, while NH4Cl significantly increased only trans-infection in untreated MULCs (Fig. 3B). These results show that HIV-1 degradation restricts first-phase trans-infection and occurs via the proteasome in CGRP-treated MULCs versus endolysosomes in untreated MULCs.
We also monitored the first and second phases of trans-infection separately but within the same cellular system, as previously described (7). Hence, untreated or CGRP-treated MDLCs were pulsed with HIV-1 for 4 h at 37°C and washed. To measure first-phase trans-infection, CD4high CCR5high green fluorescent protein (GFP) reporter CD4+ T cells were then added immediately after HIV-1 pulse and cocultured with MDLCs for 48 h. To measure second-phase trans-infection, MDLCS were first cultured in medium alone for 48 h, followed by addition of GFP reporter CD4+ T cells at this later time point and coculture with MDLCs for additional 48 h (i.e., 96 h p.i.). In these settings (see the scheme of the experimental approach in Fig. 4A), the CCR5 antagonist maraviroc significantly inhibited, as expected, only the second phase of trans-infection (data not shown). In untreated MDLCs, both phases of trans-infection were insensitive to lactacystin (Fig. 4B and C), demonstrating a lack of HIV-1 proteasomal degradation. CGRP significantly inhibited both phases of trans-infection, but lactacystin abrogated only CGRP-induced inhibition during the first phase (Fig. 4B and C). First-phase trans-infection was also evaluated in the presence of the CGRP receptor antagonist CGRP8-37, added 15 min before addition of CGRP. In these experiments, lactacystin and CGRP8-37 alone had no effect on trans-infection from untreated MDLCs (Fig. 4D). CGRP significantly inhibited first-phase trans-infection as before, and such CGRP-induced inhibition was significantly abrogated by both lactacystin and CGRP8-37, alone or in combination (Fig. 4D). These results demonstrate that CGRP switches HIV-1 degradation mode by operating via its cognate CGRP receptor. Finally, while proteasomal activity in MDLCs, which was measured by binding of a fluorescent proteasome probe, was inhibited as expected by lactacystin, it was unaffected by CGRP (Fig. 4E; similar results were obtained with MULCs [data not shown]).
FIG 4.

CGRP induces HIV-1 proteasomal degradation to restrict only first-phase trans-infection in MDLCs, without affecting proteasome activity. (A) Schematic representation of the experimental approach to measure separately first-phase versus second-phase trans-infection using the same untreated/CGRP-treated MDLCs. (B to D) Untreated or CGRP-treated (24 h, 100 nM) MDLCs were pulsed with HIV-1ADA for 4 h at 37°C in the absence or presence of lactacystin (25 μM). In panel D, the CGRP receptor antagonist CGRP8-37 (1 μM) was added 15 min before CGRP. CD4high CCR5high GFP reporter T cells were then added immediately (first phase [B to D]) or 48 h (second phase [B and C]) after HIV-1 pulse and cocultured with MDLCs for an additional 48 h. Shown are FACS plots (B) of a representative experiment, with numbers indicating the percentages of GFP+ cells after the first or second trans-infection phase (top and bottom rows, respectively). Graphs in panels C and D show means ± SEMs for HIV-1 trans-infection percentages after first or second phase, normalized against untreated cells serving as the 100% set point and using MDLCs of 5 (C) or 3 (D) different donors. (E) Untreated or CGRP-treated (24 h, 100 nM) MDLCs were incubated for 1 h with either medium alone or containing lactacystin (20 μM), washed, and resuspended for 2 h in medium alone or containing the fluorescent proteasomal probe Me4BodipyFL-Ahx3Leu3VS (200 nM). The cells were then fixed and examined by flow cytometry. Shown is a representative histogram overlay for 3 different donors. Numbers indicate MFIs of the proteasome-bound probe in untreated MDLCs incubated with medium alone (black line) or medium containing lactacystin (dashed line) followed by probe, as well as in CGRP-treated MDLCs incubated with medium alone followed by probe (gray filled histogram). Untreated MDLCs incubated without probe/lactacystin served as a negative control (dotted line).
These results show that CGRP-induced proteasomal degradation of HIV-1 is completed during and restricts only the first phase of trans-infection. Moreover, CGRP directly delivers HIV-1 from endolysosome to the proteasome, rather than affecting proteasomal activity.
DISCUSSION
Overall, we show here for the first time that HIV-1 can be degraded in LCs via the proteasome. Hence, the current paradigm that HIV-1 is degraded in LCs only via endolysosomes should be refined to consider also the possibility of HIV-1 proteasomal degradation in LCs under specific conditions, especially in vivo. Our results show that in untreated LCs, functional HIV-1 degradation indeed occurs via the endolysosomal pathway, in agreement with morphological observations showing the presence of HIV-1 virions within Birbeck granules/lysosomes in such cells (5, 9, 15). Moreover, a recent study suggested that following langerin-mediated internalization and fusion of HIV-1 within Birbeck granules, an autophagy-activating scaffold assembles with HIV-1 capsids to target the virus into autophagosomes and subsequently lysosomes (9). In sharp contrast, we reveal herein that in the presence of CGRP, HIV-1 is not degraded in LCs by this pathway. Instead, functional HIV-1 degrading takes place only via the cytosolic proteasomal machinery. This process is faster and more efficient than that in endolysosomes in untreated LCs, resulting in significant CGRP-mediated restriction of HIV-1 trans-infection. Importantly, functional blocking of the respective HIV-1 degradation mode in untreated or CGRP-treated LCs results in increased trans-infection, formally linking infectious virions escaping degradation to the first phase of trans-infection.
In CD4+ T cells, virions entering via endocytosis are degraded via both endolysosomes and the proteasome, as inhibitors of both pathways increase HIV-1 infectivity in CD4+ T cells and reporter cell lines (19–22). Importantly, both pathways operate simultaneously in T cells, as the combination of endolysosomal and proteasomal inhibitors results in additive and/or synergistic effects (22). In contrast, these two degradative pathways are mutually exclusive and functionally independent in LCs, as HIV-1 degradation occurs either in endolysosomes in untreated cells or the proteasome in CGRP-treated cells, as we show herein.
We further show that CGRP operates at a viral postentry step and without affecting proteasome activity. Therefore, we speculate that CGRP mediates direct delivery of HIV-1 from endolysosomes into the cytosol, by potentially affecting langerin-mediated intracellular signaling. For example, leukocyte-specific protein 1 (LSP1) associates with langerin to direct HIV-1 for autophagy-mediated lysosomal degradation in LCs (9). Yet CGRP has no significant effect on LSP1 expression in MDLCs (data not shown), suggesting that LSP1 has little or no role in CGRP-induced HIV-1 proteasomal degradation in LCs. In addition, CGRP could increase the expression and/or function of a Birbeck granule/endosomal translocon to facilitate HIV-1 exit into the cytosol for subsequent proteasomal degradation. Recently, Sec61 was identified as essential for endosome-to-cytosol antigen translocation during cross presentation (23). Yet our preliminary results show no effect of CGRP on Sec61 gene expression (data not shown). Further studies are now needed to identify the cellular factors that might prevent autophagy and/or induce translocation of HIV-1, thus potentially maintaining virions within the cytosol and accessible for proteasomal degradation.
CGRP-mediated HIV-1 proteasomal degradation in LCs occurs and subsequently controls only first-phase trans-infection. In contrast, the inhibitory effect of CGRP also on second-phase trans-infection probably results from CCR5 downregulation, as we previously reported (13), which would decrease LC productive infection and formation of de novo virions during this later phase. Interestingly, the increase in langerin surface expression in CGRP-treated MDLCs that we previously reported (12) and confirmed in this study did not result in higher HIV-1 binding, as measured after pulsing MDLCs with HIV-1 for 4 h at 4°C and evaluating HIV-1 surface levels (data not shown). We therefore speculate that CGRP-induced langerin might contain unfavorable modifications in its oligomeric status, which would affect HIV-1 binding capacity (7, 24) but not langerin internalization. While a previous study reported that langerin is involved in both phases of trans-infection (7), it would be difficult to decipher the exact contribution of langerin alone to these distinct trans-infection phases in CGRP-treated MDLCs. Yet as CGRP also diverted HIV-1 degradation toward the proteasomal pathway in MULCs but had no effect on langerin expression levels in these cells (data not shown), we believe that the CGRP-induced switch in HIV-1 degradation mode is independent of the increase in langerin expression per se in MDLCs.
CGRP-mediated HIV-1 proteasomal degradation, described herein, might be relevant during the sexual transmission of HIV-1 in vivo (see Fig. 5 for a schematic summary of our findings). CGRP is a potent vasodilator (25) and plays a functional role during the human sexual response, by contributing to penile erection (26) and controlling vaginal blood flow (27). Moreover, sexual intercourse might generate mechanical and/or thermal stimuli with appropriate thresholds to activate peripheral neurons (28), inducing, in turn, mucosal secretion of CGRP. Hence, the presence of CGRP within innervated mucosal tissues implies that proteasome-mediated HIV-1 degradation in LCs probably predominates in vivo and is responsible for efficient restriction of HIV-1 trans-infection. Such mucosal sensory neuroimmune cross talk between LCs and CGRP-secreting peripheral neurons therefore has important consequences during HIV-1 sexual transmission and the ensuing LC-mediated immune responses. Targeting LCs via the CGRP receptor and/or developing strategies to increase HIV-1 proteasomal, not endolysosomal, degradation in LCs might turn out to be clinically useful in the fight against HIV-1.
FIG 5.

Summary of HIV-1 degradation pathways in LCs. (Left) In untreated LCs in vitro, HIV-1 binding to langerin induces viral internalization (1) and trafficking along the endolysosomal pathway, resulting in viral degradation in endolysosomes (2). Infectious virions escaping degradation subsequently trans-infect CD4+ T cells (3) and induce productive T-cell infection (4). (Right) Within mucosal tissues in vivo, CGRP secreted from peripheral neurons (5), either as part of the sexual response or following potential mechanical/thermal stimuli (6), binds its receptor expressed by LCs. This interaction directs HIV-1 from endolysosomes toward the proteasome (7). As proteasomal HIV-1 degradation is faster and more efficient, fewer infectious virions are available to trans-infect CD4+ T cells, resulting in significant CGRP-induced inhibition of first-phase trans-infection (8) and ensuing T-cell productive infection (9).
MATERIALS AND METHODS
Cells.
Peripheral blood mononuclear cells (PBMCs) from healthy HIV-1-seronegative individuals were separated from whole blood by a standard Ficoll gradient. CD14+ monocytes and CD4+ T cells were obtained from PBMCs by negative magnetic selection (Stemcell Technologies, Grenoble, France) according to the manufacturer's instructions and maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 U/ml of penicillin, and 100 μg/ml of streptomycin (Gibco Invitrogen, Carlsbad, CA). MUTZ3 cells were obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ) and maintained according to the supplier's guidelines in minimum essential medium alpha (αMEM) containing ribonucleosides and deoxyribonucleosides, supplemented with 20% fetal bovine serum, 2 mM glutamine, 100 U/ml of penicillin, and 100 μg/ml of streptomycin (Gibco). MDLCs and MULCs were prepared as previously described (29, 30). Briefly, 1 × 106 monocytes or 0.2 × 106 MUTZ3 cells were seeded in 1 ml of their respective complete media in 12-well plates and supplemented with 100 ng/ml of granulocyte-macrophage colony-stimulating factor (GM-CSF), 10 ng/ml of interleukin 4 (IL-4), and either 10 ng/ml of transforming growth factor β1 (TGF-β1) for MDLCs or 2.5 ng/ml of tumor necrosis factor alpha (TNF-α) for MULCs (R&D Systems, Minneapolis, MN). On days 2 and 4 of culture, 0.5 ml of complete medium supplemented with the same cytokines at the same concentrations was added to each well. MDLCs were used on day 7 and MULCs on day 10 following differentiation. A CD4high CCR5high GFP reporter T-cell line was a kind gift from O. Kutsch (The University of Alabama at Birmingham) and was maintained as described previously (31).
Virus.
HIV-1 stocks were prepared by transfecting 293T cells with plasmids of the molecular clones JR-CSF and ADA (R5 tropism, clade B) and titrated over GHOST (3) X4/R5 GFP reporter cells according to the supplier's recommendation's (AIDS Research and Reference Reagent Program, NIAID, NIH). HIV-1 was used at MOIs of 0.2 for MDLCs and 0.7 for MULCs.
HIV-1 degradation and trans-infection.
MDLCs (1 × 105) or MULCs (0.25 × 105) were seeded in 96-well round-bottom plates (200 μl/well) and treated for 24 h at 37°C with 100 nM or 1 μM CGRP (Sigma, St. Louis, MO), respectively. For total HIV-1 content, cells were pulsed with HIV-1 for 30 min at room temperature, washed, shifted to 37°C, and chased at different time points. For intracellular HIV-1 content, cells were pulsed with HIV-1 for 4 h at 37°C, washed, and incubated with trypsin-EDTA (Gibco) for 10 min at 37°C to remove surface-bound virus either immediately after viral pulse or 24 h later. When desired, the inhibitors chloroquine and MG132 (Sigma) were added 10 min before HIV-1 and were maintained throughout the pulse period. The cells were then lysed with NP-40, and HIV-1 contents were measured in the cell lysates using a p24 ELISA (Innotest, Ghent, Belgium). The HIV-1 degradation index was calculated as intracellular HIV-1 content with inhibitor/intracellular HIV-1 content without inhibitor. For trans-infection of CD4+ T cells, LCs were pulsed with HIV-1 for 4 h at 37°C. When desired, the inhibitors NH4Cl, lactacystin, and dynasore (Sigma) were added 10 min before HIV-1 and were maintained throughout the pulse period. Cells were then incubated with CD4+ T cells (3 × 105, autologous for MDLCs) at 37°C. HIV-1 was measured by p24 ELISA in the coculture supernatants or by flow cytometry a week later. In other experiments, the CGRP receptor antagonist CGRP8-37 (1 μM; Sigma) was added 15 min before CGRP, and immediately (first phase) or 48 h (second phase) following HIV-1 pulse of MDLCs (0.5 × 105), CD4high CCR5high GFP reporter T cells (0.5 × 105) were added and cocultured for additional 48 h, as described previously (7). The cells were then fixed with 4% paraformaldehyde (PFA), and HIV-1 infection levels in the cell cocultures were monitored by analysis of GFP expression using flow cytometry.
Langerin internalization and flow cytometry.
MDLCs (1 × 105) were treated with CGRP as described above. The cells were subsequently incubated for 30 min at 4°C with the mouse anti-human langerin DCGM4 MAb (20 μg/ml; Beckman Coulter, Marseille, France) diluted in phosphate-buffered saline (PBS; final volume of 50 μl/well), washed, and further incubated for 30 min at 4°C with a biotin-conjugated horse anti-mouse IgG Ab (10 μg/ml; Vector Laboratories, Burlingame, CA). The cells were then resuspended in RPMI 1640 medium, chased for 0, 5, and 30 min at either 37°C or 4°C, washed with precooled RPMI 1640 medium, incubated for 15 min at 4°C with fluorescein isothiocyanate (FITC)-conjugated streptavidin (5 μg/ml; BD Pharmingen, San Jose, CA), fixed with 4% PFA, and examined by flow cytometry. Langerin internalization was calculated as the percentage of langerin-positive cells at each time point over that at 0 min. In other experiments, surface staining was performed for 30 min at 4°C with 10 μl of mouse anti-human CD1a-phycoerythrin (PE), CCR5-PE, CD3-allophycocyanin (APC), and CD4-FITC MAbs (Pharmingen), diluted in PBS as described above. When desired, cells were fixed with 4% PFA, permeabilized with PBS–0.1% saponin, and stained intracellularly for HIV-1 p24 using KC57-FITC or KC57-PE (1:160 dilution; Beckman) for 15 min at room temperature. Matched isotype control MAbs served as negative controls. Fluorescence profiles were recorded using a Guava easyCyte flow cytometer and analyzed with InCyte software (Merck Millipore, Guyancourt, France).
Proteasomal activity.
Proteasomal activity was evaluated following 2 h of incubation at 37°C of untreated or CGRP-treated MDLCs or MULCs (1 × 105) with the fluorescent probe Me4BodipyFL-Ahx3Leu3VS (200 nM; BostonBiochem, Cambridge, MA) (32). Preincubation with lactacystin for 1 h before the probe served as a control. Fluorescence in the green channel of fixed cells was monitored by flow cytometry, as described above.
Statistical analysis.
Statistical significance was analyzed by the two-tailed Student t test.
ACKNOWLEDGMENTS
We thank J. Y. Pasquier and A. Weibel for their assistance.
This study was supported by a grant from the Agence Nationale de Recherche sur le Sida et les Hépatites (ANRS).
We declare no competing financial interests.
REFERENCES
- 1.Ganor Y, Zhou Z, Tudor D, Schmitt A, Vacher-Lavenu MC, Gibault L, Thiounn N, Tomasini J, Wolf JP, Bomsel M. 2010. Within 1 h, HIV-1 uses viral synapses to enter efficiently the inner, but not outer, foreskin mucosa and engages Langerhans-T cell conjugates. Mucosal Immunol 3:506–522. doi: 10.1038/mi.2010.32. [DOI] [PubMed] [Google Scholar]
- 2.Zhou Z, Barry de Longchamps N, Schmitt A, Zerbib M, Vacher-Lavenu MC, Bomsel M, Ganor Y. 2011. HIV-1 efficient entry in inner foreskin is mediated by elevated CCL5/RANTES that recruits T cells and fuels conjugate formation with Langerhans cells. PLoS Pathog 7:e1002100. doi: 10.1371/journal.ppat.1002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hladik F, Sakchalathorn P, Ballweber L, Lentz G, Fialkow M, Eschenbach D, McElrath MJ. 2007. Initial events in establishing vaginal entry and infection by human immunodeficiency virus type-1. Immunity 26:257–270. doi: 10.1016/j.immuni.2007.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Botting RA, Rana H, Bertram KM, Rhodes JW, Baharlou H, Nasr N, Cunningham AL, Harman AN. 3 January 2017. Langerhans cells and sexual transmission of HIV and HSV. Rev Med Virol doi: 10.1002/rmv.1923. [DOI] [PubMed] [Google Scholar]
- 5.de Witte L, Nabatov A, Pion M, Fluitsma D, de Jong MA, de Gruijl T, Piguet V, van Kooyk Y, Geijtenbeek TB. 2007. Langerin is a natural barrier to HIV-1 transmission by Langerhans cells. Nat Med 13:367–371. doi: 10.1038/nm1541. [DOI] [PubMed] [Google Scholar]
- 6.Fahrbach KM, Barry SM, Ayehunie S, Lamore S, Klausner M, Hope TJ. 2007. Activated CD34-derived Langerhans cells mediate transinfection with human immunodeficiency virus. J Virol 81:6858–6868. doi: 10.1128/JVI.02472-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nasr N, Lai J, Botting RA, Mercier SK, Harman AN, Kim M, Turville S, Center RJ, Domagala T, Gorry PR, Olbourne N, Cunningham AL. 2014. Inhibition of two temporal phases of HIV-1 transfer from primary Langerhans cells to T cells: the role of langerin. J Immunol 193:2554–2564. doi: 10.4049/jimmunol.1400630. [DOI] [PubMed] [Google Scholar]
- 8.Czubala MA, Finsterbusch K, Ivory MO, Mitchell JP, Ahmed Z, Shimauchi T, Karoo RO, Coulman SA, Gateley C, Birchall JC, Blanchet FP, Piguet V. 2016. TGFbeta induces a SAMHD1-independent post-entry restriction to HIV-1 infection of human epithelial Langerhans cells. J Invest Dermatol 136:1981–1989. doi: 10.1016/j.jid.2016.05.123. [DOI] [PubMed] [Google Scholar]
- 9.Ribeiro CM, Sarrami-Forooshani R, Setiawan LC, Zijlstra-Willems EM, van Hamme JL, Tigchelaar W, van der Wel NN, Kootstra NA, Gringhuis SI, Geijtenbeek TB. 2016. Receptor usage dictates HIV-1 restriction by human TRIM5alpha in dendritic cell subsets. Nature 540:448–452. doi: 10.1038/nature20567. [DOI] [PubMed] [Google Scholar]
- 10.Ding W, Stohl LL, Wagner JA, Granstein RD. 2008. Calcitonin gene-related peptide biases Langerhans cells toward Th2-type immunity. J Immunol 181:6020–6026. doi: 10.4049/jimmunol.181.9.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hosoi J, Murphy GF, Egan CL, Lerner EA, Grabbe S, Asahina A, Granstein RD. 1993. Regulation of Langerhans cell function by nerves containing calcitonin gene-related peptide. Nature 363:159–163. doi: 10.1038/363159a0. [DOI] [PubMed] [Google Scholar]
- 12.Ganor Y, Drillet-Dangeard AS, Lopalco L, Tudor D, Tambussi G, Delongchamps NB, Zerbib M, Bomsel M. 2013. Calcitonin gene-related peptide inhibits Langerhans cell-mediated HIV-1 transmission. J Exp Med 210:2161–2170. doi: 10.1084/jem.20122349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ganor Y, Drillet-Dangeard AS, Bomsel M. 2015. Calcitonin gene-related peptide inhibits human immunodeficiency type 1 transmission by Langerhans cells via an autocrine/paracrine feedback mechanism. Acta Physiol (Oxf) 213:432–441. doi: 10.1111/apha.12366. [DOI] [PubMed] [Google Scholar]
- 14.Turville SG, Cameron PU, Handley A, Lin G, Pohlmann S, Doms RW, Cunningham AL. 2002. Diversity of receptors binding HIV on dendritic cell subsets. Nat Immunol 3:975–983. doi: 10.1038/ni841. [DOI] [PubMed] [Google Scholar]
- 15.van den Berg LM, Ribeiro CM, Zijlstra-Willems EM, de Witte L, Fluitsma D, Tigchelaar W, Everts V, Geijtenbeek TB. 2014. Caveolin-1 mediated uptake via langerin restricts HIV-1 infection in human Langerhans cells. Retrovirology 11:123. doi: 10.1186/s12977-014-0123-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valladeau J, Duvert-Frances V, Pin JJ, Dezutter-Dambuyant C, Vincent C, Massacrier C, Vincent J, Yoneda K, Banchereau J, Caux C, Davoust J, Saeland S. 1999. The monoclonal antibody DCGM4 recognizes Langerin, a protein specific of Langerhans cells, and is rapidly internalized from the cell surface. Eur J Immunol 29:2695–2704. [DOI] [PubMed] [Google Scholar]
- 17.de Jong MA, de Witte L, Santegoets SJ, Fluitsma D, Taylor ME, de Gruijl TD, Geijtenbeek TB. 2010. Mutz-3-derived Langerhans cells are a model to study HIV-1 transmission and potential inhibitors. J Leukoc Biol 87:637–643. doi: 10.1189/jlb.0809577. [DOI] [PubMed] [Google Scholar]
- 18.Ouwehand K, Spiekstra SW, Reinders J, Scheper RJ, de Gruijl TD, Gibbs S. 2010. Comparison of a novel CXCL12/CCL5 dependent migration assay with CXCL8 secretion and CD86 expression for distinguishing sensitizers from non-sensitizers using MUTZ-3 Langerhans cells. Toxicol In Vitro 24:578–585. doi: 10.1016/j.tiv.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 19.Fredericksen BL, Wei BL, Yao J, Luo T, Garcia JV. 2002. Inhibition of endosomal/lysosomal degradation increases the infectivity of human immunodeficiency virus. J Virol 76:11440–11446. doi: 10.1128/JVI.76.22.11440-11446.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schaeffer E, Soros VB, Greene WC. 2004. Compensatory link between fusion and endocytosis of human immunodeficiency virus type 1 in human CD4 T lymphocytes. J Virol 78:1375–1383. doi: 10.1128/JVI.78.3.1375-1383.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwartz O, Marechal V, Friguet B, Arenzana-Seisdedos F, Heard JM. 1998. Antiviral activity of the proteasome on incoming human immunodeficiency virus type 1. J Virol 72:3845–3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei BL, Denton PW, O'Neill E, Luo T, Foster JL, Garcia JV. 2005. Inhibition of lysosome and proteasome function enhances human immunodeficiency virus type 1 infection. J Virol 79:5705–5712. doi: 10.1128/JVI.79.9.5705-5712.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zehner M, Marschall AL, Bos E, Schloetel JG, Kreer C, Fehrenschild D, Limmer A, Ossendorp F, Lang T, Koster AJ, Dubel S, Burgdorf S. 2015. The translocon protein Sec61 mediates antigen transport from endosomes in the cytosol for cross-presentation to CD8(+) T cells. Immunity 42:850–863. doi: 10.1016/j.immuni.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 24.Chabrol E, Nurisso A, Daina A, Vassal-Stermann E, Thepaut M, Girard E, Vives RR, Fieschi F. 2012. Glycosaminoglycans are interactants of Langerin: comparison with gp120 highlights an unexpected calcium-independent binding mode. PLoS One 7:e50722. doi: 10.1371/journal.pone.0050722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brain SD, Williams TJ, Tippins JR, Morris HR, MacIntyre I. 1985. Calcitonin gene-related peptide is a potent vasodilator. Nature 313:54–56. doi: 10.1038/313054a0. [DOI] [PubMed] [Google Scholar]
- 26.Stief CG, Wetterauer U, Schaebsdau FH, Jonas U. 1991. Calcitonin-gene-related peptide: a possible role in human penile erection and its therapeutic application in impotent patients. J Urol 146:1010–1014. doi: 10.1016/S0022-5347(17)37989-2. [DOI] [PubMed] [Google Scholar]
- 27.Uckert S, Albrecht K, Kuczyk MA, Hedlund P, Oelke M. 2013. Phosphodiesterase type 1, calcitonin gene-related peptide and vasoactive intestinal polypeptide are involved in the control of human vaginal arterial vessels. Eur J Obstet Gynecol Reprod Biol 169:283–286. doi: 10.1016/j.ejogrb.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 28.Lumpkin EA, Caterina MJ. 2007. Mechanisms of sensory transduction in the skin. Nature 445:858–865. doi: 10.1038/nature05662. [DOI] [PubMed] [Google Scholar]
- 29.Geissmann F, Prost C, Monnet JP, Dy M, Brousse N, Hermine O. 1998. Transforming growth factor beta1, in the presence of granulocyte/macrophage colony-stimulating factor and interleukin 4, induces differentiation of human peripheral blood monocytes into dendritic Langerhans cells. J Exp Med 187:961–966. doi: 10.1084/jem.187.6.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Masterson AJ, Sombroek CC, De Gruijl TD, Graus YM, van der Vliet HJ, Lougheed SM, van den Eertwegh AJ, Pinedo HM, Scheper RJ. 2002. MUTZ-3, a human cell line model for the cytokine-induced differentiation of dendritic cells from CD34+ precursors. Blood 100:701–703. doi: 10.1182/blood.V100.2.701. [DOI] [PubMed] [Google Scholar]
- 31.Jones J, Whitford W, Wagner F, Kutsch O. 2007. Optimization of HIV-1 infectivity assays. Biotechniques 43:589–590, 592, 594. [DOI] [PubMed] [Google Scholar]
- 32.de Jong A, Schuurman KG, Rodenko B, Ovaa H, Berkers CR. 2012. Fluorescence-based proteasome activity profiling. Methods Mol Biol 803:183–204. doi: 10.1007/978-1-61779-364-6_13. [DOI] [PubMed] [Google Scholar]