

ABSTRACT
Nipah virus is an emerging, highly pathogenic, zoonotic virus of the Paramyxoviridae family. Human transmission occurs by close contact with infected animals, the consumption of contaminated food, or, occasionally, via other infected individuals. Currently, we lack therapeutic or prophylactic treatments for Nipah virus. To develop these agents we must now improve our understanding of the host-virus interactions that underpin a productive infection. This aim led us to perform the present work, in which we identified 101 human-Nipah virus protein-protein interactions (PPIs), most of which (88) are novel. This data set provides a comprehensive view of the host complexes that are manipulated by viral proteins. Host targets include the PRP19 complex and the microRNA (miRNA) processing machinery. Furthermore, we explored the biologic consequences of the interaction with the PRP19 complex and found that the Nipah virus W protein is capable of altering p53 control and gene expression. We anticipate that these data will help in guiding the development of novel interventional strategies to counter this emerging viral threat.
IMPORTANCE Nipah virus is a recently discovered virus that infects a wide range of mammals, including humans. Since its discovery there have been yearly outbreaks, and in some of them the mortality rate has reached 100% of the confirmed cases. However, the study of Nipah virus has been largely neglected, and currently we lack treatments for this infection. To develop these agents we must now improve our understanding of the host-virus interactions that underpin a productive infection. In the present work, we identified 101 human-Nipah virus protein-protein interactions using an affinity purification approach coupled with mass spectrometry. Additionally, we explored the cellular consequences of some of these interactions. Globally, this data set offers a comprehensive and detailed view of the host machinery's contribution to the Nipah virus's life cycle. Furthermore, our data present a large number of putative drug targets that could be exploited for the treatment of this infection.
KEYWORDS: MS, Nipah, protein-protein interaction, virus-host interactions, virology, mass spectrometry, paramyxovirus
INTRODUCTION
Nipah virus is an emerging, highly pathogenic, zoonotic virus from the Paramyxoviridae family (1, 2). The virus was first detected in humans in 1998 in Malaysia (3), and since then fatal cases have been reported yearly. The first outbreaks in Malaysia and Singapore were associated with severe febrile encephalitis with a case fatality rate of 38%. More recent outbreaks in Bangladesh and India are linked with respiratory disease and manifest an even higher case fatality rate that occasionally reaches a staggering 100% (4). Transmission to humans occurs mostly by close contact with infected animals or by consuming contaminated food (5). However, incidents of human-to-human transmission have also been reported (6), with these being the major route of infection for the Bangladeshi strains (7). Currently, there are neither therapeutic nor prophylactic treatments for Nipah virus. Despite the low number of annual cases, the virus's broad host range and increased pathogenesis warrants attention given the major health threat that this virus could pose. However, disappointingly, the study of Nipah virus has been largely neglected.
The Nipah virus comprises a 6-gene, 18.2-kb, negative-sense single-stranded RNA (ssRNA) genome, which encodes 9 proteins: nucleoprotein (N), phosphoprotein (P), the interferon antagonists W and V, the viral C protein, a matrix protein (M), viral fusion and glycoproteins (F and G, respectively), and a large polymerase (L). Host attachment is achieved by the viral G protein that binds to the host cell surface receptor ephrin B2 or B3 (8, 9). The cellular and viral membranes fuse next, and the capsid disassembles to deliver the viral genome into the cell. Once in the cytoplasm, the viral messenger and genomic RNAs are synthesized and translated to generate viral proteins. Successful infection requires evasion of the alpha/beta interferon (IFN-α/β) response (a key component of the innate immune response to virus infection) (10, 11). Four Nipah virus-encoded proteins (P, V, W, and C, all encoded by the P gene) participate in overcoming the innate immune response (12, 13). The generation of these proteins relies on varying the reading frame or using an alternate reading frame when transcribing P. In order to vary the reading frame, the fidelity of transcription is interrupted. Specifically, the polymerase stutters at an AG patch of sequence, which results in the insertion of supplementary G residues into the nascent mRNA. While the unedited P mRNA encodes the P protein, insertion of one or two extra G residues shifts the frame to produce the V and W proteins; the C protein is synthesized from an alternate open reading frame (14). A large proportion of the P, V, and W protein sequences are identical (406 amino acids of 709, 456, and 449 amino acids, respectively) and, consequently, share some functions. All three proteins inhibit the Jak-STAT signaling pathway by binding to STAT1 and preventing its phosphorylation in response to IFN (15–18). In addition, the V protein inhibits MDA5 signaling (which is conserved across paramyxoviruses) (19–21), while the W protein blocks Toll-like receptor (TLR) and Rig-I-like receptor (RLR) signaling (downstream of IFN regulatory factor 3 activation) (22). The different functions of the V and W proteins can be attributed to their sequence differences and distinct subcellular localizations. While V is mostly cytosolic, W is conveyed to the nucleus by virtue of a nuclear localization signal (NLS) at its unique carboxy terminus (Ct). In addition to the proteins encoded by the P gene, the M proteins have also been reported to be involved in antagonizing the innate immune system by targeting TRIM6, IKK, and unanchored polyubiquitin chains (23).
Identifying and understanding host-virus interactions are fundamental to our comprehension of the mechanism of infection and how this can be used to inform the development of vaccines or antivirals. Previously, a variety of technologies have been used to identify host-virus protein-protein interactions (PPIs). These include tandem affinity purification coupled with mass spectrometry (TAP-MS) (24, 25), two-hybrid system screens (26), or protein-fragment complementation assays (27). The TAP-MS approach, particularly for a biosafety level 4 (BSL4) pathogen, such as Nipah virus, offers significant advantages. First, TAP-MS is a sensitive and unbiased method with which to identify PPIs. Second, the expression, purification, and identification of PPIs for individual viral proteins constitutes a risk-free and laboratory-friendly approach. Third, the resultant data set should provide a comprehensive analysis of the complexes that are formed and the pathways that are triggered during the viral life cycle. However, TAP-MS of noncellular genes expressed from a plasmid-driven system does not fully recapitulate the viral context and might lead to an erroneous identification of PPI.
In the present work, we aimed to identify the human proteins associated with Nipah virus proteins using a TAP-MS approach. We identified 101 PPIs, 88 of which were previously unreported. We uncovered a number of host complexes targeted by the virus, including interactions with the PRP19 complex and microRNA (miRNA) processing machinery. Furthermore, we explored the biologic consequences of these PPIs and found that the Nipah virus W protein is capable of altering p53 control and gene expression. Globally, this data set offers a comprehensive and detailed view of the host machinery's contribution to the Nipah virus's life cycle. Furthermore, our data present a large number of putative drug targets that could be exploited for the treatment of this life-threatening infection. Nonetheless, before drawing any conclusions on the functional relevance of any of these interactions, results must be validated in a viral context.
RESULTS
Purification, identification, and analyses of human-Nipah virus PPIs.
To identify human proteins associated with Nipah virus, we cloned the sequence of each known Nipah virus protein using a mammalian expression vector. Each protein was fused to a TAP tag (comprised of 2× Strep-tag II and 1× Flag) (28) at either its amino (Nt) or carboxy (Ct) terminus. Independent tags at either terminus were used to minimize the possibility of missing a PPI due to tag masking. This strategy also allowed us to identify which protein end, if any, was prone to the interaction. Enhanced yellow fluorescent protein (EYFP; Aequorea victoria), red fluorescent protein (RFP; Discosoma sp.), and an empty vector were included as negative controls. Clones were transfected into HEK-293T cells with tagged proteins purified 48 h posttransfection using, in tandem, the Streptavidin (Strep) and Flag affinity labels. Final eluates then were analyzed by SDS-PAGE for quality control and by MS to identify interaction partners (Fig. 1A). After careful evaluation, the L protein-derived samples were not submitted for MS analysis due to insufficient protein. Fusion of the C protein to EYFP was used to improve purification yields prior to PPI identification by MS. See Materials and Methods for a complete list of the constructs used and the number of replicates analyzed by MS. For each bait, only proteins identified in two or more replicates were considered to be potential interactors. Further, protein hits identified in any of the control experiments (using EYFP, RFP, or empty plasmid) were discarded. To further eliminate artifacts, we compared our hits to those recorded in the CRAPome database (29), which lists affinity purification contaminants. Specifically, the experimentally obtained ratio for a protein hit (i.e., the number of times a protein was identified with a particular bait/replicate number [for that bait]) had to exceed that recorded in the CRAPome database by (at least) 5-fold. The abundance of the interaction was then estimated using the exponentially modified protein abundance index (emPAI). To compare protein abundance across experiments, emPAIs for each protein were divided by the median emPAI value for the sample (emPAI/med). Since some hits were obtained in 2, 3, 4, or even 5 replicates, to accurately evaluate their abundance we calculated the average logarithm of the emPAI/med [ave log(emPAI/med)]. As expected, the higher the number of times a protein was found, the higher its ave log(emPAI/median) value (Fig. 1B; the unique PPI identified in 5 replicates is not represented in the bar graph). These data suggested a robust protein identification and selection protocol. A total of 101 PPIs were identified between human and Nipah virus proteins (Table 1), of which 88 were previously unreported. As we had placed the TAP tag at the N and C termini of the bait constructs, we could analyze which position, if any, favored the detection of the interactor proteins. Overall, 64.4% of the interactors were identified irrespective of tag position, while 18.8% and 16.8% of the PPIs were identified exclusively when the affinity tag as either N or C terminal, respectively (Fig. 1C). For some Nipah virus proteins the tag position was irrelevant in terms of identifying partner proteins (e.g., for the P, V, and W proteins) (Fig. 1C). For other Nipah proteins (C and, in particular, G), the location of the affinity label greatly influenced the host protein-bait interaction. Interestingly, three of these proteins (F, G, and C) associate with cellular membranes. It is worth noting that, for proteins detected in two replicates, the ave log(emPAI/med) values were higher when identification was tag position dependent (Fig. 1B).
FIG 1.

Affinity purification of Nipah virus proteins and analyses of MS data. (A) Flowchart of the TAP-MS approach used to identify Nipah virus-human interactions. hpt, hours posttransfection. (B) The ave log(emPAI/med) values of the 101 PPIs were grouped according to the number of times (replicates) they were found (2, 3, or 4). The single PPI found in 5 replicates was excluded from the representation. For each group, values for the number of interactors (top) and the averaged log(emPAI/med) values are shown. Those proteins found in 2 replicates were color coded based on the TAP tag position in the bait: blue, Nt; green, Ct; black, either Nt or Ct. The averaged log(emPAI/med) values of tag position-dependent (tpd) and -independent (tpi) interactions are also indicated. (C) Influence of tag position on identification of the PPI. The number of interactors for each bait (Nipah virus proteins N, P, V, W, C, M, F, and G) are shown. Bars are color coded according to the location of the tag. Gray indicates a host protein identified using a bait tagged at either terminus; blue indicates a protein that bound bait tagged at its N terminus (Nt) exclusively; green indicates the opposite, an interaction between host protein and bait tagged exclusively at the C terminus (Ct). The pie chart above the bar graph indicates the percentage of interactors identified with Nt (blue)-, Ct (green)-, or Nt and Ct (gray)-tagged baits. (D and E) Heat maps representing enriched cellular components (D) and biological processes (E) deduced from the human proteins captured with the Nipah virus baits. ER, endoplasmic reticulum. (F) The percentage of PPIs identified in the TAP-MS experiment that have been shown to be related (in the literature) to viral infection. Significance (Fisher test) was calculated using a random list of 101 proteins as a reference set.
TABLE 1.
Proteins identified by the TAP-MS
| Bait | UniProt accession no. | Gene name | Protein name | No. of replicatesa | Found ratiob | No. of replicates inc: |
Avg log(emPAI/med)d | Referencee | |
|---|---|---|---|---|---|---|---|---|---|
| Nt | Ctc | ||||||||
| W | O00505 | KPNA3 | Importin-alpha 3 | 3 | 0.75 | 1 | 2 | 1.59 | 22, 37 |
| P | O75934 | SPF27 | Pre-mRNA-splicing factor spf27 | 4 | 1.00 | 2 | 2 | 1.43 | |
| P | Q9UMS4 | PRP19 | Pre-mRNA-processing factor 19 | 4 | 1.00 | 2 | 2 | 1.37 | |
| M | Q92688 | ANP32B | Acidic leucine-rich nuclear phosphoprotein 32 | 2 | 0.50 | 1 | 1 | 1.28 | 38 |
| W | O00629 | KPNA4 | Importin subunit alpha-4 | 4 | 1.00 | 2 | 2 | 1.26 | 22, 37 |
| W | B2R7W3 | BCAS2 | Pre-mRNA-splicing factor spf27 | 4 | 1.00 | 2 | 2 | 1.23 | |
| W | Q9UMS4 | PRP19 | Pre-mRNA-processing factor 19 | 4 | 1.00 | 2 | 2 | 1.19 | |
| P | Q99459 | CDC5L | Cell division cycle 5-like protein | 4 | 1.00 | 2 | 2 | 1.18 | |
| V | B2R7W3 | BCAS2 | Pre-mRNA-splicing factor spf27 | 3 | 0.75 | 1 | 2 | 1.08 | |
| V | P52630 | STAT2 | Signal transducer and activator of transcription 2 | 2 | 0.50 | 1 | 1 | 0.98 | 16, 37 |
| V | Q9UMS4 | PRP19 | Pre-mRNA-processing factor 19 | 4 | 1.00 | 2 | 2 | 0.94 | |
| P | O43660 | PLRG1 | Pleiotropic regulator 1 | 4 | 1.00 | 2 | 2 | 0.89 | |
| C | P40855 | PEX19 | Peroxisomal biogenesis factor 19a | 2 | 0.50 | 2 | 0 | 0.83 | |
| F | Q53H37 | CALM5 | Calmodulin-like protein 5 | 2 | 0.40 | 0 | 2 | 0.80 | |
| C | P35613 | BSG | Basigin/cd147emmpirin | 3 | 0.75 | 2 | 1 | 0.80 | |
| V | O43660 | PLRG1 | Pleiotropic regulator 1 | 2 | 0.50 | 0 | 2 | 0.79 | |
| F | Q9Y5M8 | SRPRB | Signal recognition particle receptor subunit beta | 3 | 0.60 | 1 | 1 | 0.78 | |
| N | O75190 | DNAJB6 | DnaJ subfamily b member 6 | 2 | 0.33 | 1 | 1 | 0.76 | |
| C | Q9UHG3 | PCYOX1 | Prenylcysteine lyase | 2 | 0.50 | 2 | 0 | 0.73 | |
| M | P49458 | SRP9 | Signal recognition particle 9-kDa protein | 2 | 0.50 | 1 | 1 | 0.67 | |
| M | A0A024R2I7 | RAD18 | Postreplication repair protein hrad18p | 4 | 1.00 | 2 | 2 | 0.67 | 32 |
| W | O43660 | PLRG1 | Pleiotropic regulator 1 | 3 | 0.75 | 1 | 2 | 0.66 | |
| F | I2G9F8 | HLA-C | Major histocompatibility complex class I-c | 3 | 0.60 | 2 | 1 | 0.62 | 39* |
| F | P11441 | UBL4A | Ubiquitin-like protein 4a | 4 | 0.80 | 2 | 1 | 0.59 | |
| P | P51116 | FXR2 | Fragile x mental retardation syndrome-related protein | 4 | 1.00 | 2 | 2 | 0.55 | |
| V | P42771 | CDKN2A | P16-ink4 | 2 | 0.50 | 1 | 1 | 0.48 | |
| N | D3DR22 | HSD17B12 | Hydroxysteroid (17-beta) dehydrogenase 12 | 2 | 0.33 | 2 | 0 | 0.48 | |
| C | O00161 | SNAP23 | Snap-23 | 2 | 0.50 | 2 | 0 | 0.47 | |
| M | O15355 | PPM1G | Protein phosphatase 1g | 4 | 1.00 | 2 | 2 | 0.47 | |
| V | Q99459 | CDC5L | Cell division cycle 5-like protein | 3 | 0.75 | 1 | 2 | 0.36 | |
| G | P07203 | GPX1 | Glutathione peroxidase | 2 | 0.40 | 1 | 1 | 0.33 | |
| N | M4QFU4 | HLA-B | Major histocompatibility complex class I antigen | 2 | 0.33 | 0 | 2 | 0.33 | 39* |
| M | Q9UPY3 | DICER1 | Hypothetical helicase k12h4.8-like protein | 2 | 0.50 | 0 | 2 | 0.30 | |
| M | Q7Z6Z7 | HUWE1 | E3 ubiquitin-protein ligase huwe1 | 2 | 0.50 | 0 | 2 | 0.28 | |
| M | Q8WVZ9 | KBTBD7 | Kelch repeat and btb domain-containing protein 7 | 2 | 0.50 | 1 | 1 | 0.27 | |
| W | Q99459 | CDC5L | Cell division cycle 5-like protein | 3 | 0.75 | 1 | 2 | 0.26 | |
| F | A0A024RBE6 | NAP1L1 | Nucleosome assembly protein 1-like 1, cra_b | 2 | 0.40 | 0 | 2 | 0.25 | |
| M | Q15633 | TARBP2 | RISC-loading complex subunit tarbp2 b | 3 | 0.75 | 1 | 2 | 0.23 | |
| C | Q9P0T7 | TMEM9 | Transmembrane protein 9 | 2 | 0.50 | 0 | 2 | 0.23 | |
| N | O00217 | NDUFS8 | NADH dehydrogenase iron-sulfur protein 8 | 2 | 0.33 | 2 | 0 | 0.22 | |
| G | Q15768 | EFNB3 | Ephrin-b3 | 2 | 0.40 | 2 | 0 | 0.19 | 32 |
| C | Q9H9H4 | VPS37B | Vacuolar protein sorting-associated protein 37b | 4 | 1.00 | 2 | 2 | 0.19 | |
| V | Q13126 | MTAP | Methylthioadenosine phosphorylase | 3 | 0.75 | 1 | 2 | 0.18 | |
| F | Q7L5D6 | GET4 | Cgi-20 protein | 2 | 0.40 | 2 | 0 | 0.14 | |
| F | A4D0U5 | TES | Testin 1 | 4 | 0.80 | 2 | 2 | 0.14 | |
| G | Q9UK22 | FBX2 | F-box protein Fbx2 | 5 | 1.00 | 2 | 2 | 0.13 | |
| N | Q9Y4R8 | TELO2 | Kiaa0683 protein | 2 | 0.33 | 1 | 1 | 0.12 | |
| F | Q3SY69 | ALDH1L2 | Aldehyde dehydrogenase 1 family, member l2 | 2 | 0.40 | 1 | 1 | 0.09 | |
| M | Q9UL15 | BAG5 | Bag family molecular chaperone regulator 5 | 2 | 0.50 | 1 | 1 | 0.06 | |
| P | O43709 | WBSCR22 | Uncharacterized methyltransferase wbscr22 | 2 | 0.50 | 1 | 1 | 0.05 | |
| G | P27544 | CERS1 | Ceramide synthase 1 | 2 | 0.40 | 2 | 0 | 0.05 | |
| G | O60613 | SEP15 | Selenoprotein 1 precursor | 2 | 0.40 | 2 | 0 | 0.05 | |
| C | Q9BRK5 | SDF4 | Calcium-binding protein 1 precursor | 2 | 0.50 | 0 | 2 | 0.03 | |
| F | P51648 | ALDH3A2 | Fatty aldehyde dehydrogenase 2 | 3 | 0.60 | 1 | 1 | 0.03 | |
| F | Q96AY3 | FKBP10 | Fk506-binding protein | 2 | 0.40 | 2 | 0 | 0.01 | |
| N | Q8NBQ5 | HSD17B11 | Unnamed protein product | 3 | 0.50 | 1 | 2 | 0.01 | |
| P | P52630 | STAT2 | Signal transducer and activator of transcription 2 | 2 | 0.50 | 1 | 1 | 0.01 | 16, 37 |
| F | P02786 | TFRC | Transferrin receptor protein 1 | 3 | 0.60 | 2 | 1 | 0.00 | 40** |
| G | P52799 | EFNB2 | Ephrin-b2 | 2 | 0.40 | 2 | 0 | −0.01 | 8, 41 |
| M | Q6PEV8 | FAM199X | Fam199x | 2 | 0.50 | 0 | 2 | −0.02 | |
| M | Q9Y4B6 | VPRBP | Vprbp 1 | 3 | 0.75 | 1 | 2 | −0.04 | 42 |
| G | Q9Y3A6 | TMED5 | Transmembrane emp24 protein transport | 3 | 0.60 | 0 | 2 | −0.06 | |
| F | P46459 | NSF | Vesicle-fusing ATPase | 2 | 0.40 | 2 | 0 | −0.09 | |
| C | P51648 | ALDH3A2 | Fatty aldehyde dehydrogenase 2 | 2 | 0.50 | 1 | 1 | −0.09 | |
| C | Q9NV96 | TMEM30A | Cell cycle control protein 50a | 2 | 0.50 | 0 | 2 | −0.10 | |
| F | Q9UNL2 | SSR3 | Translocon-associated protein subunit gamma | 3 | 0.60 | 1 | 1 | −0.10 | |
| W | Q14765 | STAT4 | Signal transducer and activator of transcription 4 | 3 | 0.75 | 2 | 2 | −0.10 | |
| P | P56182 | RRP1 | Ribosomal RNA processing protein 1 | 2 | 0.50 | 1 | 1 | −0.11 | |
| M | Q9BTT0 | ANP32E | Acidic leucine-rich nuclear phosphoprotein 32 | 2 | 0.50 | 1 | 1 | −0.12 | |
| C | P25490 | YY1 | Transcriptional repressor protein yy1 | 2 | 0.50 | 0 | 2 | −0.13 | |
| N | Q9H078 | CLPB | Caseinolytic peptidase b | 3 | 0.50 | 2 | 1 | −0.14 | |
| N | Q8WVC6 | DCAKD | Dephospho-coenzyme A kinase domain containing | 3 | 0.50 | 3 | 0 | −0.15 | |
| N | O75306 | NDUFS2 | NADH dehydrogenase-ubiquinone Fe-S protein 2 | 3 | 0.50 | 1 | 2 | −0.15 | |
| F | Q7L099 | RUFY3 | Protein Rufy3 | 2 | 0.40 | 0 | 2 | −0.16 | |
| G | O15173 | PGRC2 | Membrane-associated progesterone receptor component 2 | 3 | 0.60 | 2 | 0 | −0.16 | |
| F | Q15165 | PON2 | Paraoxonase 2 | 2 | 0.40 | 2 | 0 | −0.16 | |
| C | Q9H4A5 | GOLPH3L | Golgi phosphoprotein 3-like | 2 | 0.50 | 2 | 0 | −0.16 | |
| M | Q9GZU8 | FAM192A | Protein fam192a | 3 | 0.75 | 2 | 1 | −0.16 | |
| M | A0A0C4DGV5Z | RANB2 | Zis1 | 4 | 1.00 | 2 | 2 | −0.20 | |
| N | P10155 | TROVE2 | Ro ribonucleoprotein | 4 | 0.67 | 3 | 1 | −0.21 | |
| M | Q7Z4V5 | HDGFRP2 | Hepatoma-derived growth factor-related protein 2 | 2 | 0.50 | 1 | 1 | −0.22 | |
| M | Q96JK2 | DCAF5 | Ddb1- and Cul4-associated factor 5 | 2 | 0.50 | 0 | 2 | −0.22 | |
| C | Q9ULX6 | AKAP8L | A-kinase anchor protein 8-like | 2 | 0.50 | 1 | 1 | −0.25 | |
| M | Q96EY7 | PTCD3 | Pentatricopeptide repeat domain-containing protein 3 | 4 | 1.00 | 2 | 2 | −0.25 | |
| C | O95071 | UBR5 | E3 ubiquitin-protein ligase Ubr5 | 2 | 0.50 | 0 | 2 | −0.25 | |
| N | Q8IWV7 | UBR1 | E3 ubiquitin-protein ligase Ubr1 | 4 | 0.67 | 1 | 3 | −0.26 | |
| M | O00203 | AP3B1 | Ap-3 complex beta3a subunit | 2 | 0.50 | 1 | 1 | −0.29 | 42 |
| M | O43164 | PJA2 | Praja ring finger 2 | 3 | 0.75 | 1 | 2 | −0.30 | |
| F | Q8IXB1 | DNAJC10 | Hypothetical protein | 2 | 0.40 | 1 | 1 | −0.32 | |
| C | Q8TEB1 | DCAF11 | Gl014 | 3 | 0.75 | 2 | 1 | −0.32 | |
| C | O43933 | PEX1 | Peroxisome biogenesis factor 1 | 2 | 0.50 | 0 | 2 | −0.36 | |
| G | Q9NRX5 | SERINC1 | Kiaa1253 protein | 2 | 0.40 | 2 | 0 | −0.38 | |
| N | Q96AC1 | FERMT2 | Mitogen-inducible gene mig-2 | 3 | 0.50 | 0 | 3 | −0.39 | |
| C | Q13557 | CAMK2D | Calcium/calmodulin-dependent protein kinase 2 delta | 2 | 0.50 | 0 | 2 | −0.39 | |
| M | P30414 | NKTR | NK-tumor recognition protein | 2 | 0.50 | 1 | 1 | −0.50 | |
| N | A0A024RCG7 | ARMCX2 | Armadillo repeat containing 6 | 3 | 0.50 | 2 | 1 | −0.54 | |
| N | I0B0K5 | FLG | Truncated profilaggrin | 2 | 0.33 | 1 | 1 | −0.56 | |
| M | P07199 | CENPB | Cenp-b | 2 | 0.50 | 0 | 2 | −0.62 | |
| N | Q5VYK3 | ECM29 | Proteasome-associated protein Ecm29 | 2 | 0.33 | 1 | 1 | −0.67 | |
| F | Q5T5U3 | ARHGAP21 | Rho-GTPase activating protein 10 | 2 | 0.40 | 1 | 1 | −0.94 | |
| C | Q92621 | NUP205 | Nuclear pore complex protein nup205 | 3 | 0.75 | 2 | 1 | −0.97 | |
Number of replicates in which the protein were found.
Number of replicates in which the protein was found/total number of replicates performed with the indicated bait.
Number of replicates in which the protein was found with the Nt or Ct TAP tag.
Proteins were sorted based on the average log(emPAI/med).
*, PPI reported with other Nipah virus protein; **, PPI reported with the equivalent Hendra virus protein.
We next analyzed the functional categories of host proteins associated with each Nipah virus bait. Based on the current literature, certain results were anticipated. These included an association of the F protein with the endoplasmic reticulum (30) and the G protein with the Ephrin receptor pathway (8). The nuclear localization of W (18), the shuttling of protein C between the cytoplasm and nucleus (31), and the association of M with the ubiquitin machinery (23, 32) all were consistent with previous data (Fig. 1D and E). Conversely, some other highly enriched categories were unexpected. These included an association of the PRP19 complex with the P, V, and W proteins and the enrichment of RNA interference (RNAi)-related proteins in the M-interactor list. Furthermore, a careful review of the literature revealed that 36.4% of the hits had previously been reported as viral protein interactors (Fig. 1F). When a random list of 101 proteins was queried, only 8% were described as viral interactors (Fisher's test enrichment log2 P value of −17.14). The identification of known functions of Nipah virus proteins using gene ontology (GO) term analyses and the elevated percentage of hits previously associated with viral functions greatly increase our confidence in this data set.
One of the main objectives of our work was to identify potential drug targets for the treatment of Nipah virus. With this in mind, we searched for drugs that could target any of the proteins obtained in our TAP-MS analyses. To this end, we utilized the DrugBank database (a bioinformatics and cheminformatics resource that combines drug data [i.e., chemical, pharmacological, and pharmaceutical] with comprehensive drug target information [i.e., sequence, structure, and pathway]) (33, 34). In our search we identified 11 drugs (including FDA-approved compounds) that could target the Nipah virus's cellular interactors. These data are summarized in Table 2.
TABLE 2.
List of drugs acting on potential Nipah virus therapeutic targets
| Target | DrugBank ID | Drug group | Protein acted on |
|---|---|---|---|
| CAMK2D | DB06616 | Approved | N |
| DB08699 | Exptl | ||
| HLA-C | DB02740 | Exptl | F |
| ALDH1L2 | DB00116 | Approved | F |
| ALDH3A2 | DB00157 | Nutraceutical | F |
| TFRC | DB05260 | Approved | F |
| DB06784 | Approved | ||
| DB01592 | Approved | ||
| NDUS2 | DB00157 | Nutraceutical | N |
| DB00997 | Approved | ||
| MTAP | DB00173 | Approved | V |
| DB02158 | Exptl | ||
| DB02281 | Exptl | ||
| DB02282 | Exptl | ||
| DB02933 | Exptl |
To visualize these host-virus interactions, we plotted the 101 PPIs as a network schematic (Fig. 2), with inclusion of the PPIs for cellular proteins derived from the IntACT (https://www.ebi.ac.uk/intact/) (35) and String (http://string-db.org/) (36) databases (Fig. 2, dotted lines). Collectively, the network contains 93 nodes (corresponding to human and Nipah virus proteins) and 126 interactions (edges). Many of the known PPIs (involving the viral G, F, M, W, and V proteins) were identified in our assays (denoted by the double black lines) (8, 9, 21, 22, 32, 37–42) and support the quality of our approach. Surprisingly, we also detected an interaction between the W protein and STAT4, although a previously reported interaction with STAT1 went undetected (18). To confirm the potentially new interaction, we performed a pulldown experiment using V, W, VG121E, and WG121E protein lysates (the mutants cannot bind STAT1 [43]) with the resolved proteins probed for STAT1 and STAT4 expression. EYFP was included in the assay as a negative control. The experiments revealed strong interactions between the wild-type V and W proteins and both STATs 1 and 4 (Fig. 3). Conversely, neither VG121E nor WG121E bound either STAT.
FIG 2.

Network representation of TAP-MS-identified Nipah virus-human PPIs. A total of 126 interactions (edges) and 93 proteins (nodes) are represented. Newly identified PPIs are indicated by solid black lines; previously described PPIs are those highlighted with a double solid line. Gray dotted lines indicate human-human PPIs obtained from the String and intACT databases. Nipah virus proteins are shown in light red. Cellular proteins are shown in either blue (Nt) or green (Ct) depending on the tag position (as described previously). Protein names (obtained from UniProt) are provided within each node.
FIG 3.
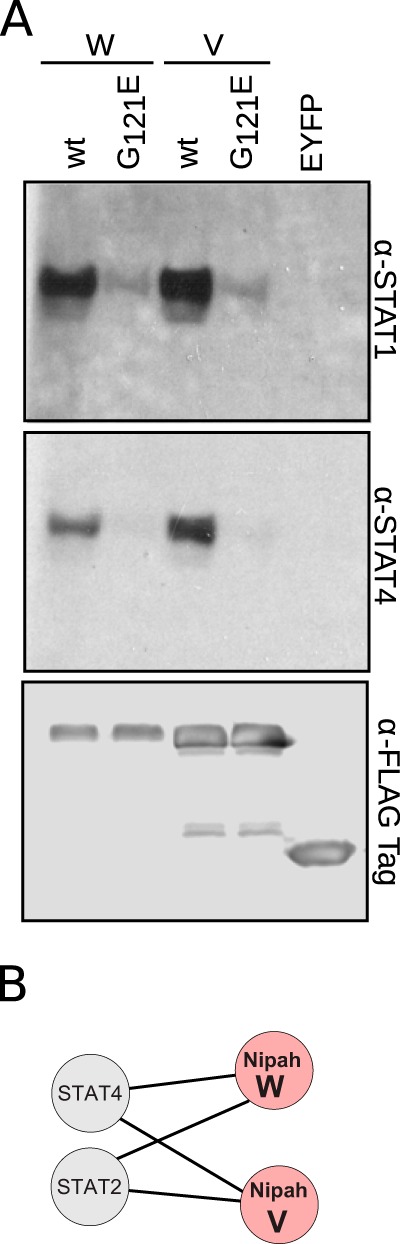
Nipah virus proteins W and V interact with STAT4. (A) TAP of tagged V, W, VG121E, WG121E, and EYFP. Postpurification, samples were immunoblotted for STAT1, STAT4, and the FLAG tag. (B) Network representation of the Nipah virus V and W interactions with STAT family members, as obtained in the TAP-MS experiments. Shown is the TAP of tagged V, W, VG121E, WG121E, and EYFP after purification samples were probed against STAT1, STAT4, and FLAG.
Interaction of the Nipah virus with the PRP19 complex.
The network map helped to identify robust associations between the P, V, and W proteins and the PRP19 complex (Fig. 2 and 4A and B). The PRP19 complex (also known as PRPF19 or the NeeTeen complex [NTC]) participates in key cellular processes (for a full review of PRP19 complex functions, see reference 44) that include splicing (45), transcription elongation (46), and genome maintenance (47). Additionally, the PRP19 complex has been identified as a negative regulator of p53 (48, 49). We first sought to validate the PRP19-viral PPI identified in the TAP-MS experiments. Using the TAP tag, we pulled down both the V and W proteins (and EYFP) and then performed immunoblots for select PRP19 complex constituents (namely, CDC5L, PRP19, PLRG1, and BCAS2) (Fig. 4C). Mutants (G121E) of the Nipah virus V and W proteins were also included in this assay. Subsequent results confirmed the TAP-MS data, with an association between the viral proteins and the PRP19 complex detected, even in the presence of mutants deficient in STAT1 binding (VG121E and WG121E).
FIG 4.
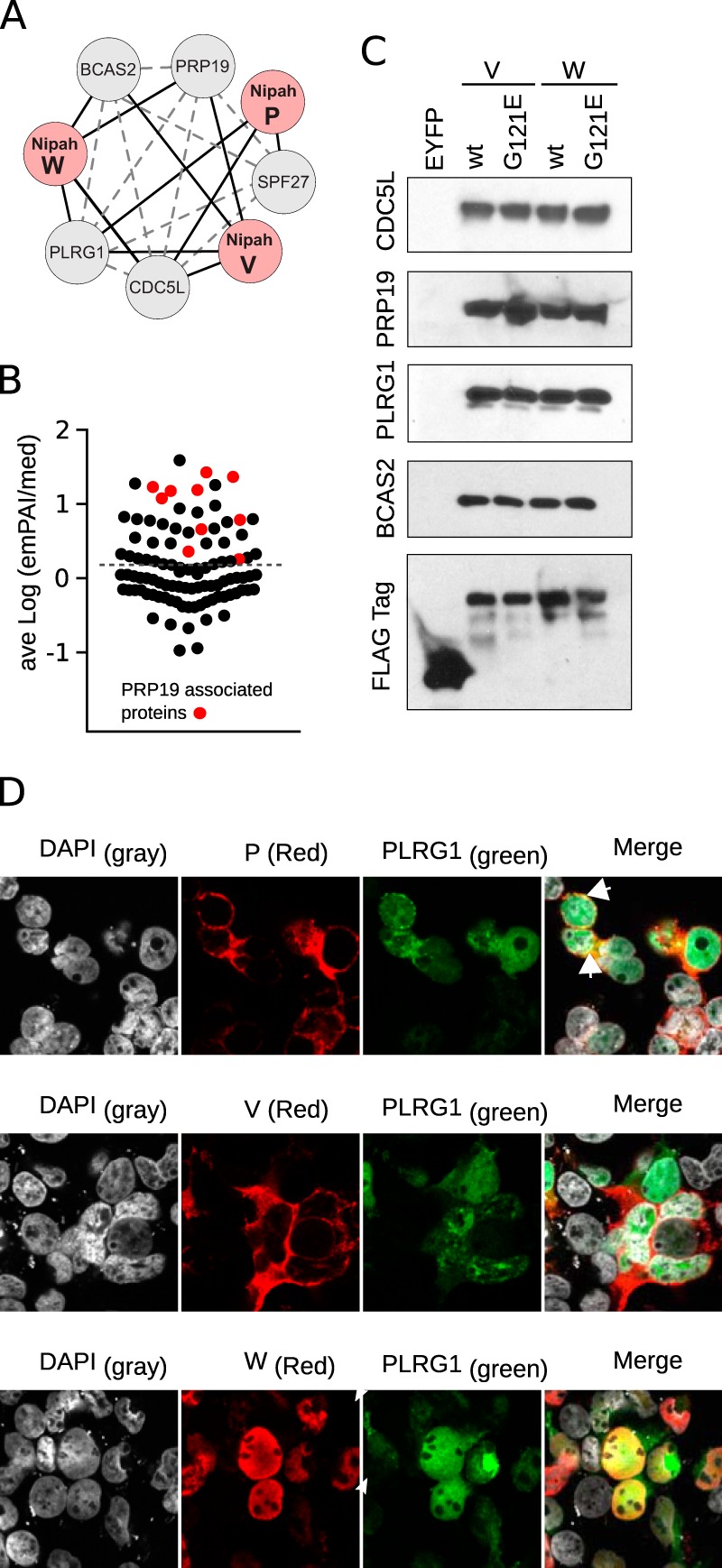
Interactions between the Nipah virus and the PRP19 complex. (A) Network representation of TAP-MS-identified interactions between PRP19 complex members (gray nodes) and Nipah virus proteins (red nodes). Solid black lines indicate interactions found in the TAP-MS experiments. Gray dotted lines represent PPIs identified in the String and intACT databases. (B) Average log(emPAI/median) value of the experimentally found interactions. Red dots highlight the PRP19 complex-associated proteins. The dotted line indicates the average value for all samples. (C) Western blot identification of PRP19 complex members. Wild-type and mutant (G121E) V and W lysates, together with EYFP, were purified by TAP and the final eluates immunoblotted for CDC5L, PRP19, PLRG1, BCAS2, and the Flag tag. (D) Cellular localization of the V, W, and P proteins (red; immune labeled) with YFP-PLRG1 (green). DAPI staining is shown as light gray. Colocalization of red and green channels is highlighted in yellow. Arrowheads indicate perinuclear regions where green and red colocalization was found.
The PRP19 complex manifests multiple activities in the nucleus. On the basis of nuclear localization signal (NLS) expression, only the Nipah virus W protein (of all the proteins encoded by the P gene) is predicted to be able to access this compartment. A robust nuclear colocalization between PLRG1 tagged with yellow fluorescent protein (YFP) and the W protein was detected (Fig. 4D). When the viral V or P proteins were coexpressed with YFP-PLRG1, only marginal colocalization at the perinuclear region could be detected (Fig. 4D, arrowheads). These results suggest that the strong interactions seen in the TAP-MS experiments between PRP19 and the V and P proteins occurred postlysis following disruption of the cellular nuclear envelope.
We next sought to discover whether the Nipah virus could interfere with the biologic functions of the PRP19 complex. First, we analyzed the activity of p53 in the presence of viral proteins. For this purpose we used a luciferase (Luc)-based p53-activity reporter assay (50). Briefly, a reporter plasmid containing a p53 DNA-binding site upstream of a luciferase reporter was cotransfected with the protein of interest into HCT116 cells (a human-derived cell line that retains p53 activity); EYFP was used to normalize the data (white bar in Fig. 5A). Interestingly, transfection and expression of the Nipah virus W (black bar, panels A, B and C) protein rendered higher luciferase values than EYFP (Fig. 5A). Conversely, transfections with P or V failed to alter p53 activity, as may be predicted from their cytosolic expression patterns, and showed luciferase values significantly lower than those obtained with W. However, based on our TAP-MS data, both the Nipah virus V and P proteins are capable of interacting with the PRP19 complex if present in the same compartment/space (as occurred postlysis). When we altered the expression pattern of the V protein by supplementing its sequence with an NLS (VNLS) (22), we observed an increase in p53-dependent luciferase expression. Conversely, elimination of the NLS in the W protein (WBR34 [22]) reduced p53 activity. In accordance with our pulldown assays, the elimination of STAT1 binding failed to influence the effects of the Nipah virus V or W protein on luciferase expression (Fig. 5A).
FIG 5.

Nipah virus modulation of p53 activity. To investigate p53 activity in the presence of Nipah virus protein, a luciferase reporter plasmid was transfected into HCT116 cells (A to C) and A549 (D to F). Posttransfection cells were left untreated (A and D), were infected with Newcastle disease virus (NDV) (B and E), or were exposed to doxorubicin (C and F). Luciferase values for control (untreated) cells transfected with the p53 reporter and EYFP were used to normalize the data (100%; indicated by dotted lines in each panel and by the white bar panel in A). Error bars indicate the standard deviations from at least three replicates. Significance (t test) in each panel was calculated by comparison with values obtained with the Nipah virus W samples (black bar). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
To analyze the role of Nipah virus W protein in p53 activity in the presence of stress/apoptotic stimuli, HCT116 cells either were treated with doxorubicin (a known inducer of p53 activity [51]) or were infected with Newcastle disease virus (NDV). NDV is a negative-sense ssRNA paramyxovirus with a genome structure similar to that of Nipah virus (52) that has been used for functional studies of Nipah virus proteins (13). Irrespective of the cellular stress (infection or apoptosis; Fig. 5B and C), the EYFP-associated values were higher than those for the mock-treated samples, confirming that p53 activity had been stimulated by the selected stimuli. In the cell stress scenarios (including viral infection), the Nipah virus W protein further enhanced p53 activity, with nuclear localization of the W or V proteins being key to achieving this activation, as confirmed in A549 cells (Fig. 5D to F).
We next decided to investigate the potential influence of the Nipah virus W protein on PRP19 complex-mediated gene expression and splicing. Briefly, W protein-Firefly luciferase (negative control) dual-expression plasmid EYFP constructs were transfected into HEK293T cells, and 48 h later, EYFP-positive cells were sorted and incubated (48 h) prior to extracting and sequencing mRNAs (Fig. 6A). This process was repeated in triplicate for each sample (Sequence Read Archive [SRA] accession number SRP116105). The consistency of replicate data is highlighted by the Pearson correlation analysis in which each set of three replicates can be seen clustered together (Fig. 6B). First, we assessed the differentially expressed genes, identifying 216, 708, and 308 differentially expressed genes using the Cuffdiff, EdgeR, and DESeq2 algorithms, respectively (53–55) (Fig. 6C). For subsequent analyses we considered only the 114 genes identified by all three algorithms (Fig. 6C and D; see also Table S1 in the supplemental material). The differential gene expression for W versus Luc samples identified by transcriptome sequencing (RNA-seq) then was validated by quantitative reverse transcription-PCR (qRT-PCR) (Fig. 6E). To accomplish this, we selected representative genes that were either downregulated (INHBA and HSP6) or upregulated (WFIKKN2, MUC19, and OR2B67). Interestingly, a GO analysis of the 114 differentially expressed genes (W versus Luc) revealed strong enrichment for terms associated with DNA packing, stability, and innate immune defense (Table 3). These data suggest that the W protein interferes with the PRP19 complex in terms of genomic maintenance or disrupts the innate antiviral response by altering gene regulation. We next sought to study the influence of nuclear localization on gene expression. Briefly, 48 h after transfecting HEK293T cells with W, V, or luciferase, mRNAs were extracted and the expression levels of INHBA and MUC19 analyzed by qRT-PCR. Despite an experimental design that excluded the selection of EYFP-positive cells and was restricted to a 48-h incubation posttransfection, the Nipah virus W protein induced a downregulation of INHBA and upregulation of MUC19 (versus the luciferase samples). In accordance with the p53 activity assay results, the V protein was incapable of altering the expression of any of the selected genes (Fig. 6F), indicating that nuclear localization is essential if gene expression is to be altered. Analyses of the differentially expressed transcripts (Table S2), together with the variation in transcript abundance (Fig. 6G) for W versus Luc, did not suggest a modification of cellular splicing in the presence of the Nipah virus W protein.
FIG 6.

Nipah virus modulation of gene expression. (A) Flowchart representation of the mRNA extraction and purification protocol. (B) Pearson correlation analyses of the sequenced samples. This analysis included the expression profiles of all of the genes. (C) Venn diagram indicating the number of differentially expressed genes comparing Nipah virus W and luciferase samples, identified by CuffDiff (pink), DESeq2 (light blue), and EdgeR (yellow). (D) Heat map of the values of log2 reads per million (RPM) for the 114 differentially expressed genes detected by all three algorithms. (E) qRT-PCR validation of RNA-seq data. The bar graph shows the fold induction provoked by the expression of W protein over the luciferase values. Error bars represent the standard deviations from three replicates. (F) qRT-PCR measurements of INHBA (red) and MUC19 (green) mRNAs, selected as examples of genes down- and upregulated, respectively, by the Nipah virus W protein. In these experiments, cells were transfected with luciferase (reference sample) and expression plasmids for either the W or V protein. After 48 h, mRNAs were extracted and measured by qRT-PCR. The bar graph shows the fold induction of mRNA achieved in the presence of either the W or V protein over values recorded for luciferase. Error bars indicate the standard deviations from three replicates. Significance (t test) was calculated by comparing the V and W samples. *, P < 0.05. (G) Average log2 RPM values for the Luc and W samples.
TABLE 3.
GO (biological process) of differentially expressed genes
| Gene set name | No. of genes | No. of genes in overlap | q value |
|---|---|---|---|
| DNA packing | 194 | 17 | 9.58 e−21 |
| Chromatin organization | 663 | 21 | 3.42 e−17 |
| Chromatin silencing | 95 | 9 | 9.29 e−11 |
| Innate immune response in mucosa | 23 | 6 | 1.31 e−9 |
| Defense response | 1,231 | 18 | 3.87 e−9 |
DICER1-TARBP2 interactions with Nipah virus matrix protein.
We were also extremely interested in the putative interactions identified in the TAP-MS experiments between the Nipah virus M protein and the DICER1-TARBP2 complex (Fig. 2 and 7A). DICER1 participates primarily in the biogenesis of (multiple classes of) small RNAs, including miRNAs (56). However, other functions have been attributed to this complex that include the processing of tRNAs and exogenous RNAs (57). To corroborate our TAP-MS data, we transfected HEK293T cells with the TAP-tagged versions of the M (Nt and Ct tagged) and V proteins. After 24 h, endogenous DICER1 was immunoprecipitated and probed with the Flag tag (inversely to the MS-TAP approach, where the bait was the Nipah M protein) (Fig. 7B, left), an experiment that was repeated with hemagglutinin (HA)-tagged M or V plasmid and including RFP as an additional negative control (Fig. 7, middle and right). As in the TAP-MS experiments, N-terminally tagged M was found to interact with DICER1 (Fig. 7B). At this point, we asked whether the Nipah virus M protein could influence DICER1 function. However, a luciferase-based DICER1 activity assay, with maturation analyses of exogenous miRNAs (Fig. 7C and D, respectively), showed that the M protein had no influence on the canonical functions of DICER1. Nonetheless, a role for M protein in other DICER1-TARBP2 activities cannot yet be ruled out.
FIG 7.

Interaction of Nipah virus M with the DICER1-TARBP2 complex. (A) Network representation of the Nipah virus M protein interaction with cellular proteins. (B, left) Immunoprecipitation (IP) of endogenous DICER1 in HEK293T cells transfected with Nipah virus M and V proteins (M-TAP and TAP-M and V-TAP) (TAP tagged either at the amino terminus, TAP-M, or the carboxyl end, M-TAP and V-TAP). (Middle and right) Immunoprecipitation of endogenous DICER1 in HEK293T cells transfected with Nipah virus M and V proteins and RFP tagged with an HA flag at the indicated terminus. IB, immunoblot. (C) DICER1 activity assay. HEK293T cells were transfected with luciferase plasmids containing mir124 target sites plus a plasmid expressing mir124. In the absence of mature miR124, expression of Luc is unhampered. Conversely, DICER1-dependent production of mature miR124 blocks Luc expression. The black bar indicates a negative control comprised of cells expressing Luc but no miR124 plasmid. White bars include EYFP (positive) and W samples. A Western blot was included to confirm the expression of EYFP and the Nipah virus M protein. Mw, molecular weight (in thousands). (D) Northern blot of pre-miR124 (pre-miR) and mature miR124 in cells expressing EYFP or the Nipah virus M protein.
DISCUSSION
Currently, only a few sporadic cases of Nipah virus human infection are reported each year. However, the potential for human-to-human transmission, and an elevated pathogenicity, makes this virus a major potential global health threat. Accordingly, Nipah virus has been classified as a biosafety level 4 (BSL4) pathogen. Surprisingly, few efforts have been made toward the development of a therapeutic or prophylactic treatment. In terms of antivirals, there are two main approaches. Historically, virologists have focused on the design of compounds that target viral proteins, and this approach has seen many successes. However, this strategy can present some disadvantages. For example, rapidly evolving viruses, such as RNA viruses, may develop drug resistance over a relatively short time period. In some cases (e.g., the neuraminidase inhibitors of the influenza A virus) the antiviral compound interferes with a viral protein/process at a late stage of the viral life cycle. This type of late blockade can severely reduce drug efficacy and narrow the treatment window. These and other hurdles have prompted a strategic shift toward examining, and potentially targeting, host proteins that are manipulated by the virus in order to fulfill a productive viral life cycle. By interfering with these interactions, either functionally (i.e., disrupting the host protein function) or upstream, by blocking the host-pathogen PPI, viral propagation might be reduced or even stopped. In the current work, we focused on identifying PPIs between human proteins and the Nipah virus. Our detailed TAP-MS analyses revealed 101 PPIs, most of them previously unreported. These interactions represent a valuable set of potential drug targets for the treatment of Nipah virus infection. Further, by utilizing the DrugBank database, we could explore the drugability of our interactor data set and provide a list of potential strategies to counter viral infection. It is important to mention that the experiments presented in this work were not conducted in a viral infection context. Transient expression from a plasmid-driven system yields large quantities of proteins which might alter the cellular interactome. Therefore, any result should be validated in a viral context before the functional relevance of the described PPIs is concluded.
The Nipah virus W protein participates in evading the innate immune response by binding STAT1 and preventing its phosphorylation in response to IFN. Additionally, protein W has been reported to block the TLR and RLR pathways. Here, we report a previously undetected interaction between the Nipah virus W protein and the PRP19 complex. Most likely, this interaction occurs through the N-terminal region of the protein that is shared with the V and P proteins. All three proteins (W, V, and P) have the potential to interact with the PRP19 complex, as shown by our TAP-MS experiments. However, in vivo, only the W protein was able to colocalize with PLRG1 (a member of the PRP19 complex). Furthermore, only the Nipah virus W protein was capable of modulating cellular processes that are controlled (partially) by the PRP19 complex. These results indicate that the interactions between P, V, and the PRP19 complex that were captured in our MS-TAP and pulldown assays have occurred after cell lysis.
Our results showed a change in p53 activity and gene expression in the presence of the Nipah virus W protein. These changes correlate with the nuclear localization of the protein, as analyses of mutated V and W proteins revealed. The Nipah virus V protein, despite its capacity to interact with the PRP19 complex, could alter neither p53 activity nor gene expression compared to controls. However, when localized to the nucleus, we detected a restoration of the ability of the V protein to activate p53.
We should mention that our RNA sequencing data (differential expression of transcripts and transcript abundance for W versus Luc samples) suggested no major modification of cellular splicing in the presence of W. Nonetheless, collectively, our data indicate that the Nipah virus W protein is capable of altering the function of the PRP19 complex, and that its nuclear localization is required for this activity. Furthermore, the binding of STAT1 does not seem to be related to these effects, as indicated by our analyses of the STAT1 binding-deficient V and W proteins. These data suggest that further analyses of the persistence of the protein W-PRP19 complex interaction, in the context of a viral infection, are now warranted.
Due to the multifunctionality of the PRP19 complex, it is currently impossible to pinpoint (with the present data) which of its multiple functions the virus is manipulating. Many viruses modulate the cellular apoptotic response (58), and it is possible that Nipah virus could, at some point during its life cycle, induce apoptosis by interfering with a negative regulator of p53, such as the PRP19 complex. There is also an extensive literature on the viral modulation of cellular gene expression. The Nipah virus interaction with PRP19 might represent an additional level of control over the cellular antiviral response. Whether this gene regulation is specific for a particular set of genes or is dependent on the genes expressed in the infected cell cannot yet be deduced (with the current data). It would be useful to investigate the influence of the W protein on gene expression in cells treated with IFN or stimulated with viral RNA. GO enrichment analyses revealed that the differentially expressed genes are linked with the innate immune response, which leads us to think that the Nipah virus is selectively hampering the cellular response to viral infection. However, besides the defense response, the gene set analyzed seems to participate in DNA packing, chromatin organization, and silencing, which indicate an indirect mechanism of altering the cellular response to viral infection. Since the PRP19 complex is heavily involved in genome maintenance and DNA stability (47), an enrichment of GO terms related to those functions could reflect the cellular response to the Nipah virus protein W-PRP19 complex interaction.
Finally, we focused on the DICER1-Nipah M PPI. This interaction was confirmed by the pulldown of endogenous DICER1 in the presence of TAP-tagged Nipah virus M protein and V protein (as a negative control). As expected, only Nt-tagged Nipah virus M protein could be seen interacting with DICER1. To corroborate this result the DICER1 pulldown was repeated in the presence of the HA-tagged versions of the Nipah virus M protein and using the RFP and Nipah virus V protein as a negative control. Once again, only the Nt-tagged forms of the Nipah virus M protein were pulled down together with DICER1. However, when DICER1 activity, in terms of miRNA production, was assessed in the presence of protein M, we detected no noticeable effects. Nonetheless, DICER1 has been described to participate in other functions. In a Nipah virus infection, DICER1 could conceivably act as a pattern recognition receptor, as described in reference 59. In that context, the interaction of Nipah virus M with DICER1 could prevent the cellular recognition of the virus without altering DICER1's canonical function.
Our work suggests multiple new strands of research. We not only describe new PPIs between the Nipah virus and host but also identify new functions for the viral W and M proteins. These data constitute a significant advance in our understanding of Nipah virus biology, and we anticipate that these results will contribute to the development of a much-needed antiviral treatment.
MATERIALS AND METHODS
Protein expression and purification.
For the identification of Nipah-human PPIs, DNAs encoding each viral protein fused to a tandem affinity purification (TAP) tag sequence at either the amino or carboxy terminus were transfected into HEK293T cells. Briefly, 30 μg of DNA was added to 1.5 ml of Opti-MEM medium (Gibco) (solution A). Separately, 1.5 ml Opti-MEM was supplemented with Lipofectamine 2000 (2 μl/μg DNA; Life Technologies) (solution B). Solutions A and B were then mixed and incubated at room temperature for at least 10 min. The mixture was then overlaid onto a 15-cm tissue culture dish. Approximately 24 h posttransfection, cells were harvested and washed with phosphate-buffered saline (PBS) (3×). Cells were then lysed with 300 μl of lysis buffer (30 mM Tris-HCl, 150 mM NaCl, 0.5% NP-40, protease inhibitor [cOmplete protease inhibitor cocktail; Roche], and phosphatase inhibitor [PhosSTOP; Roche]), and the lysate was passed through a 30-gauge syringe (3×). The cell extract was clarified by centrifugation (10 min, 10,000 × g) and the supernatant transferred to a new tube. TAP was performed as described in reference 28. Briefly, Nipah virus proteins were transiently expressed (24 to 30 h) in HEK293T cells. Cells next were lysed and viral proteins purified using the Strep and Flag tags in tandem. Protein digestion and identification (liquid chromatography-tandem mass spectrometry [LC-MS/MS] using an LTQ Orbitrap) was completed by the W. M. Keck Biotechnology Resource Laboratory (http://medicine.yale.edu/keck/index.aspx). Constructs analyzed by MS (number of replicates) included the following: empty (2), Nt-EYFP (2), EYFP-Ct (2), Nt-RFP (1), RFP-Ct (1), Nt-N (3), N-Ct (3), Nt-P (2), P-Ct (2), Nt-V (2), V-Ct (2), Nt-W (2), W-Ct (2), Nt-EYFP-C (2), C-EYFP-Ct (2), Nt-M (2), M-Ct (2), Nt-F (2), F-Ct (2), Nt-F plus F-Ct (1), Nt-G (2), G-Nt (2), and Nt-G plus G-Ct (1). For Nt-F plus F-Ct and Nt-G plus G-Ct, Nt and Ct plasmids were mixed 1:1. These sample were not included in the analysis of the tag position.
Selection criteria and the abundance of interacting proteins.
The MS data were filtered according to the following selection criteria. All pseudogenes, predicted proteins, and immunoglobulin fragments were discarded. Common contaminants in MS experiments, such as keratins and trypsins, were also eliminated. Those proteins purified with any of the negative controls (EYFP, RFP, or empty plasmid) were deleted next. To further eliminate common artifacts, we utilized the CRAPome database (29) of common protein contaminants (i.e., identified from negative-control experiments) identified in affinity purification experiments followed by MS. We selected only those proteins with experimentally obtained found/total ratios that exceeded those recorded in the CRAPome database by (at least) 5-fold. Finally, proteins that interacted with 3 or more Nipah virus proteins were discarded as contaminants from the experimental conditions used in the affinity purification (in this analysis, the sequence similarities for the viral P, V, and W proteins led these to be assessed collectively [i.e., as one]). The abundance of each interaction was estimated using the exponentially modified protein abundance index (emPAI). The emPAI value offers an approximate, label-free quantification of the proteins in a mixture based on protein coverage obtained by peptide matches in a database search (60). To compare the intensities across experiments, the emPAIs for each protein were divided by the median emPAI value of the sample (emPAI/med). As some hits were obtained 2, 3, 4, or 5 times, comparisons were made using the average log(emPAI/med).
Cell culture.
Colon epithelial carcinoma cells (HCT116), human alveolar epithelial cells (A549), and human embryonic kidney 293T (HEK293T) cells were obtained from the American Type Culture Collection and authenticated by Bioidentity (http://bioidentity.es). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) (HyClone, Thermo Scientific), and 1% penicillin-streptomycin (P/S) (Gibco). All cells were grown at 37°C in 5% CO2. Mycoplasma contamination analysis of the cells was performed by the Tissue Culture Core Facility at the Universitat de València.
p53 reporter assay.
HCT116 and A549 cells were transfected (Lipofectamine 2000) with a p53 reporter plasmid (plasmid 16442; Addgene) (50), a pRL-SV40 vector (included to normalize the measurements; Promega), and a plasmid carrying the protein of interest. To induce p53 activation, 24 h after transfection, cells were either infected with Newcastle disease virus (NDV) or were exposed to doxorubicin (0.2 to 0.4 μM; Calbiochem). Twenty-four hours later, cells were lysed and the luciferase signal from firefly-renilla luciferase was measured using a Dual-Luciferase reporter assay system (Promega). NDV infections were as follows. Twenty-four hours after transfection, medium was removed and the cells washed with PBS. Infection was with 200 μl of PBS–0.5% bovine serum albumin (BSA) with NDV at a multiplicity of infection of 10. After a 1-h incubation at room temperature, the virus was aspirated and the cells washed with PBS. Fresh medium then was added to the cells.
Subcellular protein localization.
For the colocalization study of PLRG1 and the Nipah virus W, V, and P proteins, HEK293T cells were seeded onto poly-Lys-treated coverslips and, approximately 20 to 24 h later, transfected with YFP-PLRG1 and either the P, V, or W (flag-tagged) construct. After 24 h, cells were fixed (4% paraformaldehyde, 20 min), permeabilized (0.1% Triton X-100), and washed (3× in PBS). Immunofluorescence was with a mouse anti-flag antibody (Sigma-Aldrich); the secondary antibody was an anti-mouse Alexa 647 conjugate (Sigma-Aldrich), with 4′,6-diamidino-2-phenylindole (DAPI) used to stain nuclei (Fluoroshield; Sigma-Aldrich). Images were captured using an FV1000 confocal microscope (Olympus) at the microscopy core facility of the University of Valencia.
Immunoprecipitation and immunodetection.
Cells were transfected with the appropriate protein (M, V, or RFP) bearing the indicated antibody tag (HA or Flag). Approximately 48 h later, cells were lysed (20 mM Tris [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM Na3VO4, 1× protease cocktail [Pierce]) on ice (5 min). Samples were centrifuged (10 min at 14,000 × g) and the supernatant precleared by a 30- to 60-min incubation (at 4°C) with protein A. Protein A then was pelleted by centrifugation (2,500 × g, 2 to 3 min), and 1 to 10 μg of anti-DICER1 antibody was added to the clarified supernatant (Ab14601; incubated at 4°C overnight; Abcam). Twenty microliters of protein A then was added, and the samples were incubated for 1 h at 4°C and then passed through a microspin column. Columns were washed (3×) with lysis buffer. Finally, samples were incubated with elution buffer (0.2 M glycine-HCl at pH 2.5) for 10 min at room temperature and then centrifuged (2 min at 2,500 × g). Flowthroughs were subjected to Western blotting using either an anti-HA or an anti-Flag antibody (Sigma-Aldrich). For the identification of BCAS2, PRP19, and CDC5L, we used the WH0010286M1, SAB4501215, and HPA011361 antibodies, respectively (Sigma-Aldrich).
DICER1 luciferase activity assay.
To explore the activity of DICER1 in the presence of the Nipah virus W protein, we used a luciferase assay based on miR124 processing. Briefly, a pBT plasmid expressing Gaussia luciferase containing four miR124 target sites was transfected into HEK293T cells together with a pEM plasmid containing miR124. An empty pEM plasmid was used as a negative control. Successful production of mature miR124 via DICER1 should block luciferase expression. Conversely, if DICER1 activity is diminished, the level of the luciferase signal should increase.
RNA-seq and analyses.
Sequencing and sample preparation were completed at the Genomics Core Facility at the University of Valencia. Prior to sequencing, the appropriate RNA quality was verified using a Bioanalyzer (Agilent) with the Agilent RNA 6000 Nano kit (used according to the manufacturer's instructions). RNA libraries were prepared using the TruSeq stranded mRNA sample preparation kit (Illumina) by following the manufacturer's specifications. Sequencing was with an Illumina NextSeq5000 sequencer using the high-output kit (Illumina) (SRA accession number SRP116105). Data analyses were performed with CuffDiff (53), EdgeR (54), and DESeq2 (55) software.
Accession number(s).
Sequences were deposited in SRA under accession number SRP116105.
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Shaw, C. F. Basler, B. R. Tenoever, and B. H. Lee for plasmids and reagents. We thank A. Lamond for the YFP-PLRG1 construct. We also thank M. Sanchez del Pino for his advice on the analysis of the proteomic data. The RNA sequencing was performed by the Genomics Core Facility at the Universitat de València.
This work was supported by grants from the Spanish Ministry of Economy and Competitiveness (MINECO) (grant no. BFU2016-79487) and from the Generalitat Valenciana (GV/2016/139, Program Grupos Emergentes, and PROMETEOII/2014/061, Program Grupos de Excelencia). L.M.-G. is funded by the Spanish MiNECO (Program Juan de la Cierva).
L.M.-G. designed the research, L.M.-G. and N.M.V.-V. performed the research, and L.M.-G. and I.M. analyzed data and wrote the paper.
We have no competing financial interests to declare.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01461-17.
REFERENCES
- 1.Aguilar HC, Lee B. 2011. Emerging paramyxoviruses: molecular mechanisms and antiviral strategies. Expert Rev Mol Med 13:e6. doi: 10.1017/S1462399410001754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wong KT, Shieh WJ, Zaki SR, Tan CT. 2002. Nipah virus infection, an emerging paramyxoviral zoonosis. Springer Semin Immunopathol 24:215–228. doi: 10.1007/s00281-002-0106-y. [DOI] [PubMed] [Google Scholar]
- 3.Lee KE, Umapathi T, Tan CB, Tjia HT, Chua TS, Oh HM, Fock KM, Kurup A, Das A, Tan AK, Lee WL. 1999. The neurological manifestations of Nipah virus encephalitis, a novel paramyxovirus. Ann Neurol 46:428–432. [PubMed] [Google Scholar]
- 4.Rockx B, Winegar R, Freiberg AN. 2012. Recent progress in henipavirus research: molecular biology, genetic diversity, animal models. Antiviral Res 95:135–149. doi: 10.1016/j.antiviral.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 5.Escaffre O, Borisevich V, Rockx B. 2013. Pathogenesis of Hendra and Nipah virus infection in humans. J Infect Dev Ctries 7:308–311. doi: 10.3855/jidc.3648. [DOI] [PubMed] [Google Scholar]
- 6.Clayton BA. 2017. Nipah virus: transmission of a zoonotic paramyxovirus. Curr Opin Virol 22:97–104. doi: 10.1016/j.coviro.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 7.Luby SP, Hossain MJ, Gurley ES, Ahmed BN, Banu S, Khan SU, Homaira N, Rota PA, Rollin PE, Comer JA, Kenah E, Ksiazek TG, Rahman M. 2009. Recurrent zoonotic transmission of Nipah virus into humans, Bangladesh, 2001-2007. Emerg Infect Dis 15:1229–1235. doi: 10.3201/eid1508.081237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Negrete OA, Levroney EL, Aguilar HC, Bertolotti-Ciarlet A, Nazarian R, Tajyar S, Lee B. 2005. EphrinB2 is the entry receptor for Nipah virus, an emergent deadly paramyxovirus. Nature 436:401–405. [DOI] [PubMed] [Google Scholar]
- 9.Negrete OA, Wolf MC, Aguilar HC, Enterlein S, Wang W, Mühlberger E, Su SV, Bertolotti-Ciarlet A, Flick R, Lee B. 2006. Two key residues in ephrinB3 are critical for its use as an alternative receptor for Nipah virus. PLoS Pathog 2:e7. doi: 10.1371/journal.ppat.0020007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhondt KP, Mathieu C, Chalons M, Reynaud JM, Vallve A, Raoul H, Horvat B. 2013. Type I interferon signaling protects mice from lethal henipavirus infection. J Infect Dis 207:142–151. doi: 10.1093/infdis/jis653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Satterfield BA, Cross RW, Fenton KA, Borisevich V, Agans KN, Deer DJ, Graber J, Basler CF, Geisbert TW, Mire CE. 2016. Nipah Virus C and W proteins contribute to respiratory disease in ferrets. J Virol 90:6326–6343. doi: 10.1128/JVI.00215-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Basler CF. 2012. Nipah and Hendra virus interactions with the innate immune system. Curr Top Microbiol Immunol 359:123–152. [DOI] [PubMed] [Google Scholar]
- 13.Park M-S, Shaw ML, Muñoz-Jordan J, Cros JF, Nakaya T, Bouvier N, Palese P, García-Sastre A, Basler CF. 2003. Newcastle disease virus (NDV)-based assay demonstrates interferon-antagonist activity for the NDV V protein and the Nipah virus V, W, and C proteins. J Virol 77:1501–1511. doi: 10.1128/JVI.77.2.1501-1511.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shaw ML. 2009. Henipaviruses employ a multifaceted approach to evade the antiviral interferon response. Viruses 1:1190–1203. doi: 10.3390/v1031190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ludlow LE, Lo MK, Rodriguez JJ, Rota PA, Horvath CM. 2008. Henipavirus V protein association with Polo-like kinase reveals functional overlap with STAT1 binding and interferon evasion. J Virol 82:6259–6271. doi: 10.1128/JVI.00409-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodriguez JJ, Parisien J-P, Horvath CM. 2002. Nipah virus V protein evades alpha and gamma interferons by preventing STAT1 and STAT2 activation and nuclear accumulation. J Virol 76:11476–11483. doi: 10.1128/JVI.76.22.11476-11483.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodriguez JJ, Cruz CD, Horvath CM. 2004. Identification of the nuclear export signal and STAT-binding domains of the Nipah virus V protein reveals mechanisms underlying interferon evasion. J Virol 78:5358–5367. doi: 10.1128/JVI.78.10.5358-5367.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaw ML, García-Sastre A, Palese P, Basler CF. 2004. Nipah virus V and W proteins have a common STAT1-binding domain yet inhibit STAT1 activation from the cytoplasmic and nuclear compartments, respectively. J Virol 78:5633–5641. doi: 10.1128/JVI.78.11.5633-5641.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, Randall RE. 2004. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci U S A 101:17264–17269. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Childs K, Stock N, Ross C, Andrejeva J, Hilton L, Skinner M, Randall R, Goodbourn S. 2007. mda-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology 359:190–200. doi: 10.1016/j.virol.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 21.Parisien J-P, Bamming D, Komuro A, Ramachandran A, Rodriguez JJ, Barber G, Wojahn RD, Horvath CM. 2009. A shared interface mediates paramyxovirus interference with antiviral RNA helicases MDA5 and LGP2. J Virol 83:7252–7260. doi: 10.1128/JVI.00153-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaw ML, Cardenas WB, Zamarin D, Palese P, Basler CF. 2005. Nuclear localization of the Nipah virus W protein allows for inhibition of both virus- and toll-like receptor 3-triggered signaling pathways. J Virol 79:6078–6088. doi: 10.1128/JVI.79.10.6078-6088.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bharaj P, Wang YE, Dawes BE, Yun TE, Park A, Yen B, Basler CF, Freiberg AN, Lee B, Rajsbaum R. 2016. The matrix protein of Nipah virus targets the E3-ubiquitin ligase TRIM6 to inhibit the IKKε kinase-mediated type-I IFN antiviral response. PLoS Pathog 12:e1005880. doi: 10.1371/journal.ppat.1005880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ding S, Mooney N, Li B, Kelly MR, Feng N, Loktev AV, Sen A, Patton JT, Jackson PK, Greenberg HB. 2016. Comparative proteomics reveals strain-specific β-TrCP degradation via rotavirus NSP1 hijacking a host Cullin-3-Rbx1 complex. PLoS Pathog 12:e1005929. doi: 10.1371/journal.ppat.1005929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirohata Y, Kato A, Oyama M, Kozuka-Hata H, Koyanagi N, Arii J, Kawaguchi Y. 2015. Interactome analysis of herpes simplex virus 1 envelope glycoprotein H. Microbiol Immunol 59:331–337. doi: 10.1111/1348-0421.12255. [DOI] [PubMed] [Google Scholar]
- 26.Sangsuriya P, Huang J-Y, Chu Y-F, Phiwsaiya K, Leekitcharoenphon P, Meemetta W, Senapin S, Huang W-P, Withyachumnarnkul B, Flegel TW, Lo C-F. 2014. Construction and application of a protein interaction map for white spot syndrome virus (WSSV). Mol Cell Proteomics 13:269–282. doi: 10.1074/mcp.M113.029199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tarassov K, Messier V, Landry CR, Radinovic S, Serna Molina MM, Shames I, Malitskaya Y, Vogel J, Bussey H, Michnick SW. 2008. An in vivo map of the yeast protein interactome. Science 320:1465–1470. doi: 10.1126/science.1153878. [DOI] [PubMed] [Google Scholar]
- 28.Gloeckner CJ, Boldt K, Schumacher A, Roepman R, Ueffing M. 2007. A novel tandem affinity purification strategy for the efficient isolation and characterisation of native protein complexes. Proteomics 7:4228–4234. doi: 10.1002/pmic.200700038. [DOI] [PubMed] [Google Scholar]
- 29.Mellacheruvu D, Wright Z, Couzens AL, Lambert J-P, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY, Halim VA, Bagshaw RD, Hubner NC, Al-Hakim A, Bouchard A, Faubert D, Fermin D, Dunham WH, Goudreault M, Lin Z-Y, Badillo BG, Pawson T, Durocher D, Coulombe B, Aebersold R, Superti-Furga G, Colinge J, Heck AJR, Choi H, Gstaiger M, Mohammed S, Cristea IM, Bennett KL, Washburn MP, Raught B, Ewing RM, Gingras A-C, Nesvizhskii AI. 2013. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods 10:730–736. doi: 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harcourt BH, Tamin A, Ksiazek TG, Rollin PE, Anderson LJ, Bellini WJ, Rota PA. 2000. Molecular characterization of Nipah virus, a newly emergent paramyxovirus. Virology 271:334–349. doi: 10.1006/viro.2000.0340. [DOI] [PubMed] [Google Scholar]
- 31.Horie R, Yoneda M, Uchida S, Sato H, Kai C. 2016. Region of Nipah virus C protein responsible for shuttling between the cytoplasm and nucleus. Virology 497:294–304. doi: 10.1016/j.virol.2016.07.013. [DOI] [PubMed] [Google Scholar]
- 32.Pentecost M, Vashisht AA, Lester T, Voros T, Beaty SM, Park A, Wang YE, Yun TE, Freiberg AN, Wohlschlegel JA, Lee B. 2015. Evidence for ubiquitin-regulated nuclear and subnuclear trafficking among Paramyxovirinae matrix proteins. PLoS Pathog 11:e1004739. doi: 10.1371/journal.ppat.1004739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Law V, Knox C, Djoumbou Y, Jewison T, Guo AC, Liu Y, Maciejewski A, Arndt D, Wilson M, Neveu V, Tang A, Gabriel G, Ly C, Adamjee S, Dame ZT, Han B, Zhou Y, Wishart DS. 2014. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res 42:D1091–D1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J. 2006. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res 34:D668–D672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Orchard S, Ammari M, Aranda B, Breuza L, Briganti L, Broackes-Carter F, Campbell NH, Chavali G, Chen C, del-Toro N, Duesbury M, Dumousseau M, Galeota E, Hinz U, Iannuccelli M, Jagannathan S, Jimenez R, Khadake J, Lagreid A, Licata L, Lovering RC, Meldal B, Melidoni AN, Milagros M, Peluso D, Perfetto L, Porras P, Raghunath A, Ricard-Blum S, Roechert B, Stutz A, Tognolli M, van Roey K, Cesareni G, Hermjakob H. 2014. The MIntAct project–IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res 42:D358–D363. doi: 10.1093/nar/gkt1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C. 2015. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43:D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pichlmair A, Kandasamy K, Alvisi G, Mulhern O, Sacco R, Habjan M, Binder M, Stefanovic A, Eberle C-A, Goncalves A, Bürckstümmer T, Müller AC, Fauster A, Holze C, Lindsten K, Goodbourn S, Kochs G, Weber F, Bartenschlager R, Bowie AG, Bennett KL, Colinge J, Superti-Furga G. 2012. Viral immune modulators perturb the human molecular network by common and unique strategies. Nature 487:486–490. doi: 10.1038/nature11289. [DOI] [PubMed] [Google Scholar]
- 38.Bauer A, Neumann S, Karger A, Henning A-K, Maisner A, Lamp B, Dietzel E, Kwasnitschka L, Balkema-Buschmann A, Keil GM, Finke S. 2014. ANP32B is a nuclear target of henipavirus M proteins. PLoS One 9:e97233. doi: 10.1371/journal.pone.0097233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parvege MM, Rahman M, Nibir YM, Hossain MS. 2016. Two highly similar LAEDDTNAQKT and LTDKIGTEI epitopes in G glycoprotein may be useful for effective epitope based vaccine design against pathogenic Henipavirus. Comput Biol Chem 61:270–280. doi: 10.1016/j.compbiolchem.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Popa A, Carter JR, Smith SE, Hellman L, Fried MG, Dutch RE. 2012. Residues in the Hendra virus fusion protein transmembrane domain are critical for endocytic recycling. J Virol 86:3014–3026. doi: 10.1128/JVI.05826-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bonaparte MI, Dimitrov AS, Bossart KN, Crameri G, Mungall BA, Bishop KA, Choudhry V, Dimitrov DS, Wang L-F, Eaton BT, Broder CC. 2005. Ephrin-B2 ligand is a functional receptor for Hendra virus and Nipah virus. Proc Natl Acad Sci U S A 102:10652–10657. doi: 10.1073/pnas.0504887102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun W, McCrory TS, Khaw WY, Petzing S, Myers T, Schmitt AP. 2014. Matrix proteins of Nipah and Hendra viruses interact with beta subunits of AP-3 complexes. J Virol 88:13099–13110. doi: 10.1128/JVI.02103-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ciancanelli MJ, Volchkova VA, Shaw ML, Volchkov VE, Basler CF. 2009. Nipah virus sequesters inactive STAT1 in the nucleus via a P gene-encoded mechanism. J Virol 83:7828–7841. doi: 10.1128/JVI.02610-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mahajan K. 2016. hPso4/hPrp19: a critical component of DNA repair and DNA damage checkpoint complexes. Oncogene 35:2279–2286. doi: 10.1038/onc.2015.321. [DOI] [PubMed] [Google Scholar]
- 45.Cheng SC, Tarn WY, Tsao TY, Abelson J. 1993. PRP19: a novel spliceosomal component. Mol Cell Biol 13:1876–1882. doi: 10.1128/MCB.13.3.1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chanarat S, Seizl M, Sträßer K. 2011. The Prp19 complex is a novel transcription elongation factor required for TREX occupancy at transcribed genes. Genes Dev 25:1147–1158. doi: 10.1101/gad.623411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu Q, Wang F, Xiang Y, Zhang X, Zhao Z-A, Gao Z, Liu W, Lu X, Liu Y, Yu X-J, Wang H, Huang J, Yi Z, Gao S, Li L. 2015. Maternal BCAS2 protects genomic integrity in mouse early embryonic development. Development 142:3943–3953. [DOI] [PubMed] [Google Scholar]
- 48.Chen P-H, Lee C-I, Weng Y-T, Tarn W-Y, Tsao Y-P, Kuo P-C, Hsu P-H, Huang C-W, Huang C-S, Lee H-H, Wu J-T, Chen S-L. 2013. BCAS2 is essential for Drosophila viability and functions in pre-mRNA splicing. RNA 19:208–218. doi: 10.1261/rna.034835.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kleinridders A, Pogoda H-M, Irlenbusch S, Smyth N, Koncz C, Hammerschmidt M, Brüning JC. 2009. PLRG1 is an essential regulator of cell proliferation and apoptosis during vertebrate development and tissue homeostasis. Mol Cell Biol 29:3173–3185. doi: 10.1128/MCB.01807-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. 1993. WAF1, a potential mediator of p53 tumor suppression. Cell 75:817–825. doi: 10.1016/0092-8674(93)90500-P. [DOI] [PubMed] [Google Scholar]
- 51.Hientz K, Mohr A, Bhakta-Guha D, Efferth T. 2017. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 8:8921–8946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Palese P, Shaw ML. 2007. Orthomyxoviridae viruses their replication, p 1647–1689. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 53.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Svobodova E, Kubikova J, Svoboda P. 2016. Production of small RNAs by mammalian Dicer. Pflugers Arch 468:1089–1102. doi: 10.1007/s00424-016-1817-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Song M-S, Rossi JJ. 2017. Molecular mechanisms of Dicer: endonuclease and enzymatic activity. Biochem J 474:1603–1618. doi: 10.1042/BCJ20160759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G. 2008. Viral control of mitochondrial apoptosis. PLoS Pathog 4:e1000018. doi: 10.1371/journal.ppat.1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rawling DC, Pyle AM. 2014. Parts, assembly and operation of the RIG-I family of motors. Curr Opin Struct Biol 25:25–33. doi: 10.1016/j.sbi.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ishihama Y, Oda Y, Tabata T, Sato T, Nagasu T, Rappsilber J, Mann M. 2005. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteomics 4:1265–1272. doi: 10.1074/mcp.M500061-MCP200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.