

ABSTRACT
Dengue virus (DENV) is a major global pathogen that causes significant morbidity and mortality in tropical and subtropical areas worldwide. An improved understanding of the regions within the DENV genome and its encoded proteins that are required for the virus replication cycle will expedite the development of urgently required therapeutics and vaccines. We subjected an infectious DENV genome to unbiased insertional mutagenesis and used next-generation sequencing to identify sites that tolerate 15-nucleotide insertions during the virus replication cycle in hepatic cell culture. This revealed that the regions within capsid, NS1, and the 3′ untranslated region were the most tolerant of insertions. In contrast, prM- and NS2A-encoding regions were largely intolerant of insertions. Notably, the multifunctional NS1 protein readily tolerated insertions in regions within the Wing, connector, and β-ladder domains with minimal effects on viral RNA replication and infectious virus production. Using this information, we generated infectious reporter viruses, including a variant encoding the APEX2 electron microscopy tag in NS1 that uniquely enabled high-resolution imaging of its localization to the surface and interior of viral replication vesicles. In addition, we generated a tagged virus bearing an mScarlet fluorescent protein insertion in NS1 that, despite an impact on fitness, enabled live cell imaging of NS1 localization and traffic in infected cells. Overall, this genome-wide profile of DENV genome flexibility may be further dissected and exploited in reporter virus generation and antiviral strategies.
IMPORTANCE Regions of genetic flexibility in viral genomes can be exploited in the generation of reporter virus tools and should arguably be avoided in antiviral drug and vaccine design. Here, we subjected the DENV genome to high-throughput insertional mutagenesis to identify regions of genetic flexibility and enable tagged reporter virus generation. In particular, the viral NS1 protein displayed remarkable tolerance of small insertions. This genetic flexibility enabled generation of several novel NS1-tagged reporter viruses, including an APEX2-tagged virus that we used in high-resolution imaging of NS1 localization in infected cells by electron microscopy. For the first time, this analysis revealed the localization of NS1 within viral replication factories known as “vesicle packets” (VPs), in addition to its acknowledged localization to the luminal surface of these VPs. Together, this genetic profile of DENV may be further refined and exploited in the identification of antiviral targets and the generation of reporter virus tools.
KEYWORDS: dengue virus, NS1, mutagenesis, virus replication, virus assembly, electron microscopy
INTRODUCTION
Dengue virus (DENV) is a mosquito-borne flavivirus that causes approximately 100 million symptomatic infections and 25,000 deaths each year (1). No antiviral drugs are available, and the only approved vaccine is partially limited in efficacy, only available in certain countries, and not recommended for young children or the elderly (2). Accordingly, there remains an urgent need for the development of safe and effective antiviral therapies and vaccines—challenges that are made more difficult by significant gaps in our understanding of the precise functions of the individual viral proteins and their domains.
After entry into susceptible cells via clathrin-dependent endocytosis, the plus-strand RNA viral genome of DENV is released into the cytosol and translated by the host ribosome machinery at the rough endoplasmic reticulum (ER). The encoded polyprotein is then proteolytically cleaved co- and posttranslationally by host and viral proteases to liberate the individual structural proteins capsid, prM, and E and the nonstructural (NS) proteins NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5. The NS proteins are essential for replication of the viral genome via a negative-strand RNA intermediate in virus-induced membranous organelles know as replication factories (3). Specifically, viral RNA replication mediated by the RNA-dependent RNA polymerase NS5 is thought to take place within invaginations of the ER membrane known as vesicle packets (VPs), while pores in these VPs may enable exchange of metabolites and export of newly synthesized genomes for encapsidation (4). Accordingly, virus particle assembly is thought to take place in close proximity to VP pores and virus particles can be observed by electron microscopy (EM) in ordered arrays that are encased by “virion bags” at these sites (4). The other major morphotype of DENV-induced membrane rearrangements are convoluted membranes (CMs), which are drastic rearrangements of ER membranes that are thought to serve polyprotein expression and maturation and may also serve as membrane reservoirs for further VP biogenesis (4, 5). Although NS4A may be principally required for these rearrangements (6), other NS proteins are also likely to be involved. NS proteins, such as NS1, NS2A, and NS3, also play essential roles in virus particle production, possibly by coordinating budding of newly formed nucleocapsids into the ER lumen and their envelopment with membranes that are enriched with the structural proteins prM and E (7–10). The conventional secretory pathway is then exploited in the release of virions, which involves a number of posttranslational modifications of structural proteins that confer infectivity to virus particles, including cleavage of prM by the host protease furin in the trans-Golgi network (TGN) (11).
Reverse-genetics analyses have been essential for our current understanding of the DENV replication cycle and the functions of the individual viral proteins and RNA elements. However, these studies are laborious and time-consuming and are therefore typically limited to the analysis of discrete regions of a viral genome and/or encoded protein in a single study. One approach to overcome this bottleneck involves the combination of high-throughput random mutagenesis of a cloned viral genome with next-generation sequencing (NGS) to quantify the degree to which regions within viral genomes tolerate mutations in cell culture models of viral replication. This approach has recently been used to provide global maps of genetic flexibility for viruses such as hepatitis C virus, influenza A virus, and measles virus and show how this genetic flexibility or inflexibility relates to functions of viral proteins and their targeting by host immune responses (12–16). Here, we combined transposon-mediated random insertional mutagenesis and NGS to generate a global map of genetic flexibility for a cloned DENV serotype 2 genome (DENV-2; strain 16881). We reveal for the first time that capsid- and NS1-encoding regions and the 3′ untranslated region (UTR) display the greatest overall genetic flexibility. In particular, NS1 was highly tolerant of insertions in regions surrounding N-glycosylation sites in the Wing and β-ladder domains and in a region surrounding a site in the second connector domain that is N-glycosylated for several other flaviviruses. In contrast, other regions, such as those encoding prM and NS2A, were highly intolerant of insertions. Building on these insights, we then generated a panel of infectious epitope- and reporter-tagged DENV-2 isolates, including a variant encoding the APEX2 EM reporter in NS1 that enabled high-resolution imaging of NS1 at the membrane and interior of VPs and, less strongly, at the Golgi and modified ER membranes. We also generated a novel DENV2 derivative encoding an mScarlet fluorescent protein insertion in NS1 that enabled visualization of NS1-mScarlet localization and traffic in infected cells and revealed that intense juxtanuclear NS1 foci are relatively static, whereas small and weakly fluorescent structures frequently display rapid, long-range, bidirectional traffic. Together, our data provide new insights into the localization and traffic of NS1 and provide a resource that may be exploited in future generation of tagged reporter viruses and in development of antiviral strategies directed toward genetically inflexible regions of the DENV genome and encoded proteins.
RESULTS
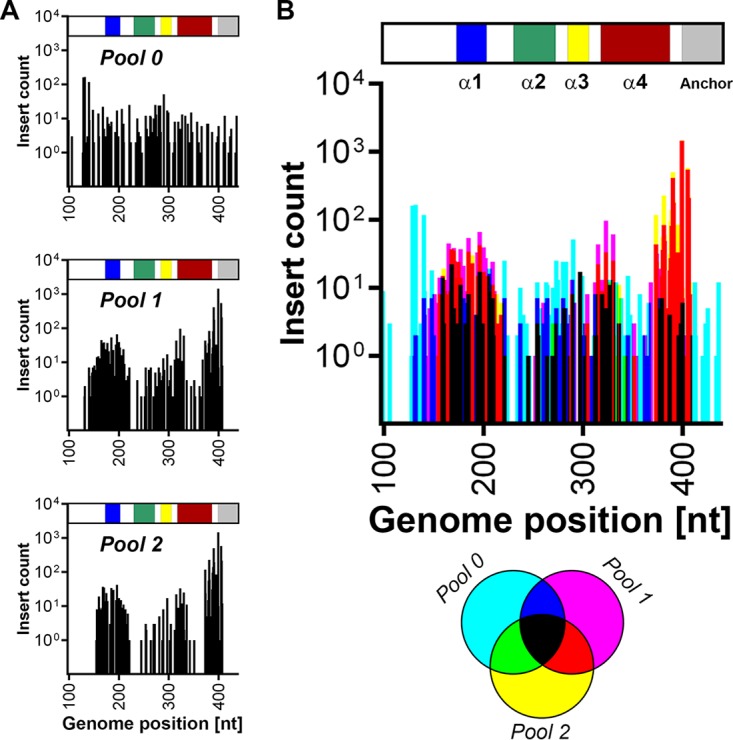
High-throughput transposon mutagenesis coupled to NGS reveals regions of genetic flexibility within the DENV-2 genome.
We subjected a cloned DENV-2 genome (strain 16681) to random transposon mutagenesis using Mu transposase to generate a mutant pool comprised of approximately 250,000 plasmid clones. The transposon body encoding a kanamycin resistance cassette was then excised, and the plasmid pool was religated to generate the final mutant pool comprised of a similar number of plasmid clones, each bearing a single 15-nucleotide (nt) insertion of which 10 nt are transposon derived (5′-TGCGGCCGCA-3′) and 5 nt are duplicated from the target site. When inserted in a coding region of the genome, this results in a 5-amino-acid (aa) insertion whose sequence is dependent on the frame and insertion site (C-G-R-I/M/T/N/K/S/R, L/M/V-R-P-H/Q, or X-A-A-A). From this mutant plasmid pool, we generated in vitro-transcribed viral RNA, referred to as pool 0 (“input”) for large-scale electroporation of Huh-7.5 hepatoma cells. After culture of electroporated cells for 6 days, total cell RNA was extracted from remaining cell monolayers (pool 1 [“replication competent”]), and cell culture supernatants were collected, clarified, and applied to naive cells. Infected target cells were then cultured for 2 days before extraction of total cell RNA to generate pool 2 (“infectious”) (Fig. 1A). Immunofluorescence microscopy analysis of parallel cell cultures confirmed robust DENV-2 replication in these cultures and an expected delay in replication and spread of the mutant pool compared to wild-type DENV-2 (not shown). Total RNA from each pool was then subjected to reverse transcription-PCR (RT-PCR) using DENV-specific primers that covered the genome in six overlapping fragments. In parallel, this PCR was also performed to determine insertion distribution in the initial mutant plasmid pool. NGS libraries for each pool were prepared using Nextera XT (Illumina) and sequenced on the NextSeq500 platform. Sequence reads were mapped to the reference DENV-2 genome, and the frequency of transposon insertions at each position was measured for each pool (Fig. 1 and see Data Set S1 in the supplemental material). This revealed that insertions were generally evenly distributed across the DENV-2 genome in both the mutant plasmid pool (not shown) and the mutant DENV-2 RNA library, pool 0, used for transfection (Fig. 1B), with the exception of “hot spots” for insertions in the NS2A-encoding region that may relate to disruption of cryptic bacterial promoter-driven expression or activity of toxic genes that otherwise limit propagation of flavivirus clones in bacteria (17). In total, 74.2, 71.4, 59.2, and 63.4 million reads were generated for the plasmid pool, pool 0, pool 1, and pool 2, respectively, whereas digital counting of transposon insertions revealed 7,867, 4,700, 1,122, and 1,003 unique insertions in the DENV2 genome for these respective pools.
FIG 1.

Insertional mutagenesis of the DENV-2 genome and identification of tolerated insertions. (A) Schematic overview of the transposon mutagenesis-coupled next-generation sequencing (NGS) approach to identify sites within the DENV-2 genome that tolerate small (15-nt) insertions. Briefly, Huh-7.5 cells were transfected by electroporation with in vitro-transcribed RNA for the DENV-2 insertional mutant pool (pool 0). At 6 days posttransfection (pool 1), virus-containing cell culture supernatants were collected and applied to naive target cells for an additional 2 days (pool 2). Total RNA was extracted from transfected and infected cell monolayers (pools 1 and 2, respectively) and, together with the input mutant RNA library (pool 0), was used for RT-PCR and NGS-based quantitation of each insertion mutant. Bar graphs displaying the number of reads containing transposon insertions at each nucleotide position are depicted for the input mutant library (pool 0) (B), replication-competent viruses (pool 1) (C), and infectious viruses (D). (E) Graphical overlay of insert counts for each pool. The colors assigned to each pool are depicted in the legend shown in the lower panel of panel A.
We compared genome insertion counts in the initial input mutant pool (pool 0; Fig. 1B) with those capable of viral RNA replication (pool 1; Fig. 1C) and those capable of viral RNA replication and infectious virus production (pool 2; Fig. 1D). This revealed that capsid- and NS1-encoding regions and the 3′ UTR were most tolerant of 15-nt insertions, whereas other regions such as prM- and NS2A-encoding regions were largely intolerant of these insertions. Figure 1E shows insertions that were most strongly selected against (cyan peaks) and insertion mutants capable of being sustained during viral RNA replication but not infectious virus production (blue and magenta peaks). All other peaks, and particularly yellow and red peaks, represent insertion mutants that are viable throughout the viral life cycle. We also assessed the frequency of insertions in replication-competent and infectious pools (pools 1 and 2, respectively) as a percentage of those counted in the initial input pool (pool 0), and this normalized view also highlighted sites in capsid- and NS1-encoding regions that were most tolerant of insertions and, in particular, the high sensitivity of prM- and NS2A-encoding regions to insertions (Fig. 2). In this context, the encoded amino acid sequences of the 25 most-tolerated insertions are displayed in Data Set S1 in the supplemental material. More detailed analysis is clearly required before firm conclusions can be drawn as to which frame-dependent transposon-encoded peptide sequences are better tolerated at different sites within the viral proteins. Nevertheless, of the 25 most-tolerated insertions, 60% (15/25) encoded C-G-R-I/M/T/N/K/S/R insertions, 28% (7/25) encoded X-A-A-A insertions and only 12% (3/25) encoded L/M/V-R-P-H/Q insertions (Data Set S1, column “N”), highlighting that the impact of a 5-aa insertion at a given site can be strongly influenced by factors such as the charge, hydrophobicity, and bulkiness of the inserted peptide. Given the high degree of overlap between pool 1 and pool 2, it is likely that spread of infectious virus during the 6-day culture period following electroporation of the mutant library strongly contributed to representation of infectious mutant viruses in pool 1. This possibility is also consistent with the unexpected selection against regions encoding structural proteins that are not required for viral RNA replication in pool 1 (Fig. 1C and 2B).
FIG 2.

Tolerance of transposon insertions in DENV-2 expressed as a percentage of the initial input mutant RNA pool. (A) Graph depicting the frequency of insertions in the replication-competent pool (pool 1; P1), expressed as a percentage of the frequency of the corresponding mutants in the initial mutant RNA pool (pool 0; P0). (B) Similarly, the frequency of insertions in the infectious pool (pool 2; P2) was expressed as a percentage of the frequency of the corresponding mutants in the initial mutant RNA pool (pool 0; P0). Nucleotide positions along the x axis indicate the junctions of the coding sequences of the individual DENV-2 proteins. For clarity, the regions encoding the respective DENV-2 proteins are indicated as colored backgrounds. Note that one insertion count was added to the raw counts for pool 0 to enable representation of insertions that only appeared in pools 1 and/or 2, but not in pool 0. See also Data Set S1 in the supplemental material.
It was also apparent that peaks representing tolerated mutations were often observed near the termini of viral proteins, for example, at the C termini of the capsid, E, NS1, and NS2B proteins, at the N terminus of NS3, and at both the N and the C termini of NS4B (Fig. 1E and 2B). Provided that the sites of DENV polyprotein cleavage are not disrupted by these insertions, the propensity of viral proteins to better tolerate insertions at their termini is consistent with a reduced likelihood of such insertions to disrupt the overall structure and in-turn interactions and functions of these viral proteins. Furthermore, for the coding portion of the genome, regions that were most tolerant of insertions were generally less strongly conserved between different DENV serotypes and other flaviviruses, although these regions were not characterized by unusually long stretches of sequence variation when the amino acid sequences of different flaviviruses were aligned and compared (see Fig. S1 in the supplemental material). Given that compact viral genomes rarely maintain nonfunctional or nonessential sequences, regions that are highly tolerant of insertions may play roles in immune evasion, pathogenesis, or virus-host interactions that are not recapitulated in Huh-7.5 cell culture.
DENV NS1 protein is highly tolerant of small insertions in regions surrounding N-glycosylation sites.
Of the DENV proteins, the multifunctional NS1 protein showed the greatest tolerance to transposon insertions in the context of the complete infectious virus replication cycle. Closer examination of the sites that tolerated insertions, with respect to the recently solved crystal structure of NS1 (18), revealed that sites within the second half of the Wing domain, in the second connector, and in the N-terminal region of the β-ladder domain were most tolerant of transposon insertions, whereas sites near the C terminus of the β-ladder were moderately tolerant of insertions (Fig. 3A). In contrast, the N-terminal β-roll domain, the first half of the Wing domain, and the first connector that separates these domains were highly sensitive to transposon insertions, an observation consistent with a recent alanine scanning mutagenesis study that identified several replication-lethal mutations in these domains, including C4A, W8A, Y32A, C55A, and R62A (8). Notably, the regions that were most tolerant of insertions were clustered in close proximity to the N-glycosylation sites in the Wing and β-ladder domains, Asn-130 and Asn-207, respectively, and surrounding a site in the second connector domain (Gln-175) that is a glycosylated Asn residue for several other flaviviruses, including West Nile virus (WNV), Saint Louis encephalitis virus, and Murray Valley encephalitis virus (19). Since N-glycosylation of NS1 is likely to be important to its functions as a secreted hexameric lipoparticle that modulates immune responses and causes vascular leakage, insertions in the regions surrounding these N-glycosylation sites may impact upon functions of secreted NS1 in immunomodulation and vascular leakage that are not recapitulated in hepatoma cell culture.
FIG 3.

The DENV-2 NS1 protein is highly tolerant of insertions. (A) The insertional mutagenesis profile within the NS1-encoding region of the DENV-2 genome was examined with respect to NS1 protein domains. In the NS1 schematic the β-roll, Wing, and β-ladder domains are colored blue, yellow, and red, respectively, while connector subdomains are depicted in orange and the location of N-glycosylation sites (N-glyc) are indicated in green. The graph shows an overlay of the number of raw reads for each transposon insertion site in pool 0 (cyan), pool 1 (magenta), and pool 2 (yellow) and where they overlap, according to the legend to the right of the graph. The x axis shows the DENV-2 nucleotide positions of the NS1-encoding region (nt 2422 to 3477). (B) The location of the 10 most tolerated transposon insertion sites in the NS1 protein, provided that at least two nucleotides within the given codon preceded tolerated insertions, are shown as green van der Waals spheres in a ribbon representation of the DENV NS1 dimer structure (PDB accession no. 4O6B) (also see Data Set S1 in the supplemental material). One monomer is shown in gray, and the other monomer is colored according to domains as in panel A. Insertions at Lys-174, Thr-126, and Ser-204 were highly tolerated. As shown in the perpendicular view (right panel), the most tolerated mutations are relatively distant from the inner hydrophobic face that is pointed downward.
To better understand the location of tolerated transposon insertions as they relate to the three-dimensional structure of NS1 dimers, we visualized the locations of the 10 most tolerated insertion sites (pool 2/pool 0) in the crystal structure of DENV-2 NS1 dimer (PDB 4O6B), provided that at least two positions in the given codon preceded tolerated insertions (Fig. 3B). This approach, focusing on sites that tolerated insertions in at least two alternative codon positions and hence resulting in alternative encoded peptide insertions (C-G-R-I/M/T/N/K/S/R, L/M/V-R-P-H/Q, or X-A-A-A), was used to highlight sites that were broadly tolerant of small insertions. This revealed that the sites most tolerant of insertions were predicted to be solvent-exposed and distant from the lipid bilayer-interacting inner hydrophobic face of the β-roll domain and adjacent “greasy finger” loop (18). The two residues that preceded the most tolerated insertions were Lys-174 (∼550-fold enrichment from input) and Thr-126 (∼93-fold enrichment from input), and these sites were flanked by several other top-ranking tolerated insertion sites, including Glu-174 (∼81-fold), Asn-130 (∼75-fold), Ser-125 (∼64-fold), Ser-128 (∼58-fold), and Glu-127 (∼44-fold).
Another region in the DENV-2 genome that was broadly tolerant of insertions was the capsid-encoding region, although the degree to which insertions were tolerated was considerably lower than that of NS1 (see Fig. 2). Capsid is the least conserved of the flavivirus proteins, although its general structural properties and charge distribution are well conserved. Structural studies of this highly basic protein have shown that the capsid monomer contains four α-helices (α1 to α4), while the dimer has an asymmetric charge distribution such that the highly basic α4-α4′ region may interact with viral RNA while the hydrophobic cleft, comprised of α1-α1′ and α2-α2′ regions, on the opposite side of the molecule forms an apolar surface that is predicted to interact with membranes (20, 21). Examination of the location of tolerated insertions revealed that the C terminus of capsid toward the end of the α4 region and, in particular, the region immediately downstream of the NS2B/3 cleavage site and adjacent to the transmembrane anchor were most tolerant of insertions (Fig. 4). In addition the region surrounding the α1 region was broadly and moderately tolerant of insertions, consistent with the flexibility of this region implied by the differing orientations of DENV and WNV α1 and mutational studies demonstrating that substitutions of uncharged amino acids in the α1 helix and α1-α2 connecting loop do not impair DENV propagation (22, 23). In contrast, the N terminus of capsid and the α2 and α3 regions were more sensitive to insertions. Taken together, the high tolerance of capsid for insertions is consistent with it being the least conserved of the flavivirus proteins.
FIG 4.
Tolerance of transposon insertions within the capsid-encoding sequence. (A) The insertional mutagenesis profile within the capsid-encoding region of the DENV-2 genome was examined with respect to capsid protein domains. In the capsid schematic the α1, α2, α3, and α4 alpha helices are colored blue, green, yellow, and red, respectively, while the transmembrane anchor is colored gray. The graph in the upper panel shows the distribution of insertions in the input mutant RNA pool (pool 0). The graph in the middle panel shows the number of raw sequencing reads for each insertion in the replication competent pool (pool 1), while the graph in the lower panel shows the number of reads for each insertion in the infectious virus pool (pool 2). (B) Graphical overlay of the number of raw reads for each transposon insertion site in pool 0 (cyan), pool 1 (magenta), and pool 2 (yellow) and where they overlap, according to the legend below the graph. The x axis shows the DENV-2 nucleotide positions of the capsid-encoding region (nt 97 to 342).
The influence of host cell type and species on the sensitivity of DENV-2 to 15-nt transposon insertions.
To investigate how host cell type and species impacts upon the tolerance of DENV-2 to transposon-derived 15-nt insertions, the infectious virus-containing cell culture supernatant from Huh-7.5 cells that were transfected with RNA transcripts for the mutant DENV-2 RNA library was applied in parallel to naive mammalian cell lines Huh-7.5 and Vero cells and the insect C6/36 cell line. At 48 h postinfection, RNA was then extracted from these cells and subjected to RT-PCR for Illumina sequencing (Fig. 5A). The frequency of 15-nt transposon insertions was measured and compared for DENV-2 genomes recovered from infected Huh-7.5, Vero, and C6/36 cells (Fig. 5B to D), using an overlay of the respective maps to highlight which transposon insertions are differentially tolerated in the alternative host cell types (Fig. 5E). Despite the appreciable differences in infection rates for revealed by parallel immunofluorescent staining of infected cells (Fig. 5F), our analysis revealed very close overlap between regions that displayed tolerance of insertions in the DENV-2 genome in these highly divergent host cells (Fig. 5E). However, subtle but clear differences were apparent (see Data Set S1 in the supplemental material), including modest increases in the frequency of tolerated transposon insertions in the 3′ UTR and sites within E for C6/36 host cells compared to Huh-7.5 and Vero host cells (see yellow peaks for Fig. 5E). In contrast, C6/36 host cells were associated with modest decreases in the frequency of tolerated insertions in regions surrounding the Asn-207 glycosylation site in NS1 and in the C terminus of NS2B (see blue peaks for Fig. 5E). Taken together, these experiments indicate that, following initial selection in Huh-7.5 human hepatoma cells, host cell selective pressures do not dramatically alter regions of genetic flexibility within the DENV-2 genome.
FIG 5.

The influence of host cell type on the tolerance of DENV-2 to transposon mutagenesis. (A) Infectious virus-containing cell culture supernatants collected from Huh-7.5 cells that were electroporated with the DENV-2 mutant library RNA (see Fig. 1A) were applied to naive Huh-7.5, Vero and C6/36 cells in parallel for 48 h prior to RNA extraction, RT-PCR, and Illumina (NextSeq500) sequencing, according to the depicted schematic diagram. The frequencies of transposon-derived insertions in the DENV-2 genome in infected Huh-7.5 cells (B), Vero cells (C), and C6/36 cells (D) were determined by sequencing. (E) Graphical overlay of normalized insert counts for each pool. The colors assigned to each pool are depicted in the legend shown in the lower panel of panel A. (F) Representative immunofluorescent micrographs of parallel cultures of mutant DENV-2-infected cells at 48 h postinfection. Anti-E staining and DAPI (4′,6′-diamidino-2-phenylindole)-counterstained nuclei are shown in red and blue, respectively. Scale bars, 100 μm.
Generation of tagged reporter viruses to study the DENV replication cycle and viral protein localization and interactions.
Epitope- and reporter-tagged viruses are valuable tools for studying viral protein localization and interactions in the context of productive viral infections. However, their development is challenging and requires prediction of sites that may tolerate insertions and empirical testing of candidate tagged viruses. Guided by our transposon mutational profile of sites that tolerate 15-nt (5-aa) insertions in the DENV2 genome, we generated and tested a panel of epitope- and reporter-tagged DENV-2 constructs, specifically focusing on the NS1 and capsid proteins given their high degree of tolerance of insertions and their essential but incompletely understood functions in the viral replication cycle (Fig. 5A). We found that the region at the C terminus of the capsid precursor and adjacent to the transmembrane anchor region (“CAPmem”), following Gly-103, could readily tolerate small epitope tags. This included the recently developed NanoBiT luciferase complementation component SmBiT (11 aa) for use in studying protein-protein interactions in living cells (24) and the split fluorescent protein component GFP11 (16 aa) for use in live cell imaging in cells expressing the complementary GFP 1-10 fragment (25). Tolerance of this site for insertions was influenced by the overall charge of the inserted peptide since incorporation of the smaller but highly charged FLAG tag at this site severely attenuated viral replication and spread (results not shown). We also generated tagged viruses encoding FLAG or GFP11 epitope tag insertions within NS1 between Lys-174 and Gln-175 (Fig. 6A). Similarly, we generated virus constructs bearing insertions of the larger APEX2 EM tag (26, 27) or the extremely bright NanoLuc (NLuc) luciferase reporter (28) at this same site in NS1.
FIG 6.

Characterization of epitope- and reporter-tagged DENV-2 constructs. (A) Guided by the insertional map, a panel of tagged viruses were generated that featured the indicated epitope or reporter protein insertions in capsid, adjacent to the membrane anchor (CAPmem), or NS1, immediately downstream of Lys-174. (B) Huh7.5 cells were electroporated with in vitro-transcribed RNA for the indicated DENV-2 constructs and cultured for 4 days prior to Western blot analysis of NS1 protein. Detection of β-actin served as a loading control. Similarly, supernatants from these cells were cleared by centrifugation and subjected to SDS-PAGE under nonreducing, nondenaturing conditions and Western blotting with anti-NS1 antibody. The numbers below the NS1 Western blots indicate the levels of NS1 protein in whole-cell lysates (normalized to β-actin and expressed as a percentage of wild-type levels [%-WT]; upper panel) and extracellular NS1 protein (expressed as a ratio to intracellular NS1 bands, with the wild-type ratio set to 1 [Ext:Int]; lower panel). (C) Automated immunofluorescence analysis of the proportion of capsid protein-positive Huh-7.5 cells at 4 days postelectroporation with the indicated DENV-2 RNA transcripts. Data are means + the standard deviations (SD; n = 3 for >1,500 cells/electroporation). (D) Infectivity titers were determined by focus forming unit (FFU) assays at 24 to 120 h postelectroporation of Huh-7.5 cells with the indicated DENV-2 RNA transcripts. Data are means + the SD (n = 3). The dashed line indicates the limit of detection of the assay.
Western blot analysis of Huh-7.5 cells transfected with the respective tagged DENV-2 or wild-type DENV-2 RNA transcripts revealed ready detection of NS1 for all virus constructs and expected increased molecular weights of tagged NS1 proteins (Fig. 6B). We also noted a moderate increase in the levels of tagged NS1 protein that may reflect minor effects of tag insertions at this site on NS1 stability. Importantly, however, analysis of NS1 protein levels in cell culture supernatants revealed no major tag-associated defects in NS1 secretion (Fig. 6B), although further analysis is required to definitively clarify whether the efficiency and kinetics of NS1 secretion are altered. Analysis of the replicative fitness of these tagged viruses compared to wild-type DENV-2 revealed that DENV2-NS1-FLAG displayed wild-type levels of viral replication and spread, as determined by automated immunofluorescence analysis (Fig. 6C), and wild-type levels of infectious virus production (Fig. 6D). Similarly, viruses bearing GFP11 or SmBiT insertions adjacent to the capsid membrane anchor or a GFP11 insertion in NS1 displayed robust replication and spread and infectious virus production that was only moderately impaired compared to wild-type DENV2 (Fig. 6C and D). Although viruses encoding larger APEX2 (783 nt, ∼28 kDa) or NLuc (516 nt, ∼19 kDa) insertions in NS1 were appreciably attenuated compared to wild-type DENV-2, they nonetheless displayed robust levels of viral replication and infectious virus production (Fig. 6C and D), supporting their utility in detailed analysis of NS1 localization, using DENV2-NS1-APEX2, and highly sensitive and simple monitoring of protein levels, using DENV2-NS1-NLuc (see below). Interestingly, the tolerance of insertions at this site in NS1 appears to be highly dependent on the sequence and/or structure of the inserted protein as several other reporter protein insertions at this site were not tolerated, including those of green fluorescent protein (GFP) and the self-labeling SNAP-tag (results not shown).
Given the potential for tag insertions to alter NS1 conformation, glycosylation, secretion, and localization, we further investigated these properties for our panel of epitope- and reporter tagged DENV-2 derivatives. First, in our hands nonreducing, nondenaturing conditions were required for reactivity of the anti-NS1 MAb 4G4 toward secreted DENV-2 NS1 (Fig. 7A), with results suggesting that the native epitope recognized by this antibody is similarly retained for all NS1-tagged viruses. Furthermore, we assessed whether N-glycosylation of NS1 is affected by the presence of tag insertions in NS1 by digesting cell culture supernatants with PNGase F prior to Western blotting (Fig. 7B). This revealed a similar shift in the apparent molecular weight of wild-type and tagged NS1 proteins, indicating no major defects in the N-glycosylation of NS1-tagged variants. Given the potential for cell type- and/or species-specific effects on the replicative fitness of these tagged viruses, we also compared infectious virus production for wild-type and tagged viruses following viral RNA transfection of Aedes albopictus-derived C6/36 cells and African green monkey-derived Vero cells (Fig. 7C and D). Consistent with observations in Huh-7.5 cells (Fig. 6D), in both C6/36 and Vero cells DENV2-NS1-FLAG produced wild-type levels of infectious virus production, other small insertions in capsid (SmBiT and GFP11) and NS1 (GFP11) were associated with moderate impairment of infectious virus levels, and larger insertions in NS1 (APEX2 and NLuc) were associated with more marked reductions in infectious virus levels (Fig. 7C and D). Importantly, all tagged viruses outlined in Fig. 6 displayed unaltered localization of NS1 and capsid proteins in Huh-7.5 cells and minimal changes to the degree of their colocalization with double-stranded RNA (dsRNA), a marker of viral replication factories (Fig. 8). Taken together, this panel of infectious tagged viruses, and future tagged viruses that are rationally generated using the insertional mutation map of DENV-2 as a guide will serve as important tools for studying viral protein localization, traffic, and interactions in the context of a productive viral infection.
FIG 7.

Impact of epitope and reporter insertions on NS1 secretion and glycosylation in Huh-7.5 cells and infectious virus production in C6/36 and Vero cell lines. (A) Supernatants were collected at 96 h posttransfection from Huh-7.5 cells transfected with the indicated DENV2 RNA transcripts and cleared of cells and debris by low-speed centrifugation (5,000 × g, 5 min). Samples were subjected to SDS-PAGE under reducing and denaturing conditions (βME/heat, +) or nonreducing, nondenaturing conditions (βME/heat, −), as indicated, and Western blotting was performed using anti-NS1 MAb 4G4. Secreted NS1 was strongly detected under nondenaturing, nonreducing conditions for wild-type and tagged viruses alike. (B) Supernatant samples were also subjected to deglycosylation using PNGase F under nondenaturing conditions (4 h at 37°C) or mock treated, before SDS-PAGE under nonreducing, nondenaturing conditions and Western blotting with anti-NS1 MAb 4G4. A similar shift in electrophoretic mobility was observed for wild-type and tagged NS1 proteins after PNGase F treatment. (C and D) Infectious virus production by C6/36 cells and Vero cells transfected with epitope- and reporter-tagged DENV2 RNA transcripts. C6/36 cells (C) and Vero cells (D) were transfected with the indicated DENV2 RNA transcripts and returned to culture before collection of supernatant samples at the indicated time points (24 to 120 h). Infectivity of supernatants were determined by focus-forming assay. Data are means + the SD (n = 3). The dashed line indicates the limit of detection.
FIG 8.

Localization of NS1 with respect to dsRNA and capsid is unaltered for epitope- and reporter-tagged DENV-2 viruses. Huh-7.5 cells were transfected with in vitro-transcribed RNA for DENV2 and NS1- and capsid-tagged DENV2 derivatives and cultured for 96 h prior to fixation and sequential indirect immunofluorescent labeling using anti-dsRNA (MAb 3G1) and Alexa Fluor 555-conjugated anti-mouse IgM (red), followed by either anti-NS1 (MAb 4G4) and Alexa Fluor 488-conjugated anti-mouse IgG (green) (A) or anti-capsid (MAb 6F3.1) and Alexa Fluor 488-conjugated anti-mouse IgG (green) (B). Samples were counterstained with DAPI (blue) and analyzed by confocal fluorescence microscopy. Yellow in the merged images indicates colocalization. Pearson's colocalization coefficients are indicated in white in the merged images (means ± the SD for n = 3 to 5 [stitched] fields of 203 × 203 μm, each containing >10 cells). The insets represent zoomed images of the boxed areas in the “merge” panels. Scale bars, 10 μm.
Sensitive luminescence-based monitoring of intracellular and secreted NS1 protein levels.
We next explored the utility of the DENV2-NS1-NLuc virus in monitoring intracellular and secreted NS1 protein levels during viral infection in cell culture. For this, Huh-7.5 cells in 96-well plates were infected with serial dilutions of DENV2-NS1-NLuc, washed, and returned to culture for 48 h prior to determination of extracellular and intracellular NS1-associated NLuc activity (Fig. 9A). This revealed simple and extremely sensitive detection of NS1-NLuc levels over a large dynamic range (∼4 orders of magnitude). As expected, extracellular NS1-NLuc was also strongly detected with levels closely reflecting those of intracellular NS1-NLuc. Furthermore, we confirmed the utility of this luminescent virus in antiviral drug testing by measuring the impact of the recently identified pan-flaviviral inhibitor nanchangmycin on viral infection and replication (29). Consistent with that study, nanchangmycin pretreatment for 1 h was associated with dose-dependent inhibition of virus encoded NS1-NLuc, measured in both intracellular and extracellular samples at 48 h postinfection (Fig. 9B). Taken together, DENV2-NS1-NLuc enables sensitive monitoring of NS1 protein levels during viral infection and may be well suited to high-throughput screening.
FIG 9.
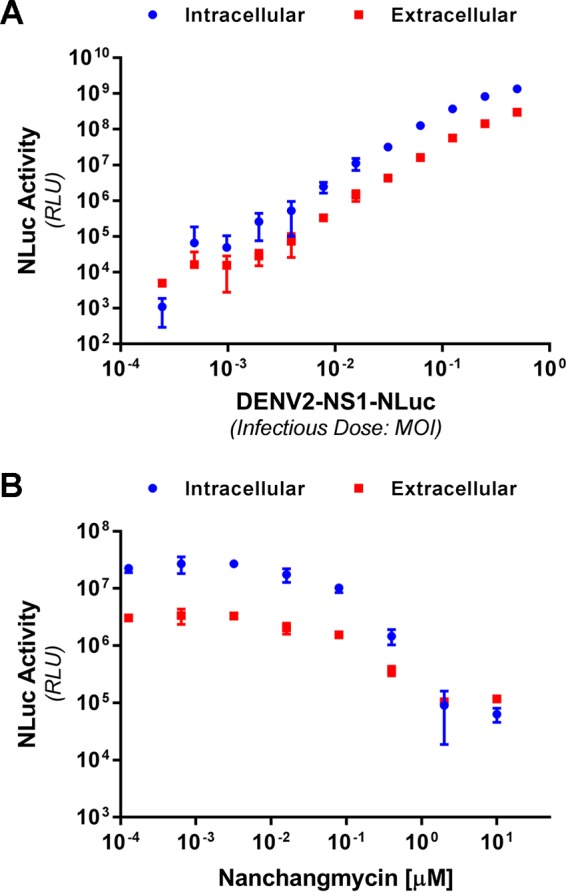
Sensitive monitoring of intracellular and secreted levels of NanoLuc-tagged NS1 following infection with DENV2-NS1-NLuc. (A) Huh-7.5 cells in 96-well plates were infected with DENV2-NS1-NLuc at the indicated MOI for 4 h, washed, and returned to culture for 48 h prior to the determination of intracellular and extracellular NanoLuc activity. (B) Semiconfluent Huh-7.5 cells in 96-well trays were treated with the indicated concentration of nanchangmycin for 1 h prior to infection with DENV2-NS1-NLuc (MOI of ∼0.05) for 4 h. Cells were then washed and returned to culture for 48 h prior to measurement of intracellular and extracellular NLuc activity. Data are means ± the (n = 4). The results are representative of similar repeat experiments.
High-resolution imaging of NS1 localization using APEX EM and STimulated Emission Depletion (STED) super-resolution microscopy.
Early studies using immuno-EM revealed the localization of NS1 to vesicle packets in DENV-infected cells and its colocalization with dsRNA at these sites (30); this is consistent with the essential role of NS1 in viral genome replication. Furthermore, recent studies demonstrated colocalization and interaction of NS1 with capsid and E proteins at putative viral assembly sites and/or assembled virions (8). However, traditional antibody-based approaches are limited in their ability to examine protein localization at high resolution in the context of clear visualization of cellular membranes and, accordingly, many details about the exact nature of NS1 localization remain unclear. We therefore examined NS1 localization by APEX EM, which allows high-resolution imaging of tagged protein localization by EM while maintaining excellent ultrastructural preservation (26, 27). For this, Huh-7.5 cells were infected with untagged DENV-2 or DENV2-NS1-APEX2 (multiplicity of infection [MOI], ∼0.01) or mock infected and returned to culture for 4 days prior to fixation, labeling with DAB/H2O2, and processing for EM. Bright-field microscopy following DAB/H2O2 labeling confirmed strong and specific staining of NS1 that was unique to DENV2-NS1-APEX2-infected cells (Fig. 10A). Samples were subsequently labeled with electron-dense OsO4, which stains the osmiophilic DAB polymer, and further processed for EM. As observed in standard EM analysis of DENV-infected cells, untagged virus infection was associated with typical induction of CMs and VPs that were clearly but not intensely stained (Fig. 10Bi and ii, respectively) and were absent in mock-infected cells (not shown). In contrast, in cells that were infected with DENV2-NS1-APEX2, intense staining of NS1-APEX2 could be observed in clusters of VPs, with somewhat diffuse staining localized to the outer membrane of VPs and intense, tightly localized staining of punctae that were asymmetrically distributed within VPs (Fig. 10Biv). We also observed clear but less intense staining of ER membranes that in many instances were proximal to lipid droplets (Fig. 10Biv). This approach also revealed clear staining of NS1-APEX2 in the Golgi (Fig. 10Biii). Although we could not clearly discern virus particles in these cells, we observed occasional examples of intensely stained clusters adjacent to VPs (Fig. 10Bv) and NS1-APEX2-positive highly ordered arrays (Fig. 10Bvi) that may be relevant to the function(s) of NS1 in virus particle assembly and secretion (8). Although we observed no unanticipated effects of the APEX2 insertion on NS1 protein processing (Fig. 6B), secretion (Fig. 6B and 7A), glycosylation (Fig. 7B), or localization (as determined by confocal microscopy; Fig. 8A), given the reduced fitness of the DENV2-NS1-APEX2 virus further studies using complementary imaging techniques are required to support our findings regarding NS1 localization using APEX EM.
FIG 10.

APEX electron microscopy analysis of NS1 localization. Huh-7.5 cells were mock infected or infected with untagged DENV2 or DENV2-NS1-APEX2 (MOI of ∼0.01) and cultured for 96 h prior to fixation and labeling with DAB-H2O2. (A) Light microscopy revealed the presence of DAB polymerization only in DENV2-NS1-APEX2-infected cells (right panel). Scale bars, 100 μm. (B) Samples were then processed for EM. Compared to mock-infected cells (not shown), DENV2-infected cells (upper panels) displayed characteristic virus-induced ER rearrangements including CMs (see subpanel i) and VPs (see subpanel ii). Scale bars are 500, 200, 500, and 100 nm, left to right, as indicated. In contrast, DENV2-NS1-APEX2-infected cells revealed discernible DAB deposition in the Golgi apparatus (subpanel iii), ER membranes (see “ER” arrowheads in subpanel iv), and intense diffuse staining of VP membranes and punctate staining of the VP interior (see “zoom inset”). Scale bars are 1,000, 100, 500, and 100 nm, left to right, as indicated. Enlarged views are shown to the right of each boxed area. In addition to localization to the surface and interior of VPs (see yellow arrows in subpanel v), NS1-APEX2 was also detected in irregular punctae that were adjacent to VPs (see yellow arrowheads in subpanel v). Furthermore, NS1-APEX2 was less frequently identified in regular arrays (see cyan arrowheads in subpanel vi) that featured more diffuse membrane staining and punctate luminal staining. The scale bars for subpanels v and vi are 100 and 200 nm, respectively. CM, convoluted membranes; mit, mitochondrium; VP, vesicle packet; ER, endoplasmic reticulum; LD, lipid droplet.
In this context, to complement the APEX EM imaging analysis of NS1 localization we also applied the DENV2-NS1-FLAG recombinant virus, which encodes a single FLAG tag within NS1, to STED super-resolution imaging of the localization of FLAG-tagged NS1 and dsRNA, given that this virus displayed wild-type levels of replication and infectious virus production and enables particularly strong and specific labeling of NS1-FLAG, with anti-FLAG antibodies, in combination with anti-dsRNA labeling. Although NS1-FLAG and dsRNA displayed near-complete colocalization when imaged by standard laser scanning confocal microscopy (Fig. 8A), even when image deconvolution was applied (Fig. 11A), STED imaging in combination with deconvolution revealed that dsRNA foci were frequently immediately adjacent to NS1 foci, which displayed a more reticular localization pattern compared to the intense and largely discrete dsRNA foci (Fig. 11B). Taken together, these experiments clarify the localization of NS1 with respect to VPs and provide powerful new tools to further examine NS1 localization as it relates to its roles viral RNA replication and virus particle production and as a secreted mediator of immune evasion and pathogenesis.
FIG 11.

STED super-resolution imaging of NS1 and dsRNA localization in infected cells. Huh-7.5 cells were infected with DENV2-NS1-FLAG (MOI of ∼0.1) and returned to culture for 48 h prior to fixation and indirect immunofluorescent labeling using anti-FLAG (Alexa Fluor 488, green) and anti-dsRNA (Alexa Fluor 647, red) as described in Materials and Methods. Samples were then mounted and imaged using a Leica TCS SP8 STED 3× microscope system (Leica Microsystems) equipped with 592-, 660-, and 775-nm STED lasers using a 100×, NA 1.4 oil objective lens at a 4× zoom. Confocal and STED images (in 2D STED mode) were acquired sequentially, as described in Materials and Methods. Imaging data for both confocal imaging and STED imaging were deconvolved using Huygens professional deconvolution software (v14.10; Scientific Volume Imaging), applying default settings. A single optical section for confocal (A) and STED (B) channels is depicted. Scale bars are 5 μm for main images and 500 nm for the inset images.
Live cell imaging of mScarlet-tagged NS1.
Although the GFP11-tagged viruses, DENV2-CAPmem-GFP11 and DENV2-NS1-GFP11, displayed robust viral replication and infectious virus production, we could not reliably detect strong GFP11-associated fluorescence upon complementation with heterologously expressed GFP(1–10) (N. S. Eyre and M. R. Beard, unpublished results). This may be attributable to inefficient fluorescence complementation and/or inaccessibility of the incorporated GFP11 peptides to GFP(1–10), obstacles that may be overcome by the development of analogous viruses bearing multiple tandem repeats of GFP11, targeting of GFP(1–10) to the ER lumen, and/or the application of newly developed split fluorescent protein systems based on GFP11/GFP(1–10) with improved complementation efficiencies (25, 31). Therefore, although our GFP11-tagged viruses provide important proof-of-concept that capsid and NS1 proteins can tolerate small functional insertions designed for fluorescent imaging, further optimization is required to improve their utility in demanding applications such as live cell imaging. We therefore developed a novel DENV-2 construct featuring an extremely bright and monomeric red fluorescent protein, mScarlet (32), inserted within NS1 (Fig. 12A). This recombinant virus supported infectious virus production and dictated strong NS1-associated fluorescence (Fig. 12B), despite appreciable attenuation compared to wild-type DENV-2 (Fig. 12C). We next investigated NS1 localization and traffic by live cell imaging analysis of Huh-7.5 cells transfected with DENV2-NS1-mScarlet RNA transcripts. At 6 days posttransfection, NS1-mScarlet fluorescence was observed throughout the culture (Fig. 12D), while imaging at high magnification revealed characteristic localization of NS1-mScarlet to large, intensely labeled juxtanuclear foci and smaller, weakly fluorescent foci throughout the cytoplasm (Fig. 12E). Live cell imaging over 5-min periods revealed that the intensely labeled NS1-mScarlet foci were largely static (Fig. 12E, cyan arrows and see Movie S1 in the supplemental material), while a minority of small and weakly fluorescent foci displayed rapid, long-range bidirectional traffic that is characteristic of microtubule-dependent transport (Fig. 12E, cyan arrowheads and see Movie S1 in the supplemental material). Importantly, similar NS1-mScarlet localization and trafficking patterns were observed for cells infected with DENV2-NS1-mScarlet, including in longer-term (∼45 min) live cell imaging acquisitions (see Movie S2 in the supplemental material). Taken together these results and the DENV2-NS1-mScarlet virus provide the basis for more detailed analysis of NS1 localization and traffic in live infected cells and indicate that intense NS1 foci that likely represent replication complexes are relatively static, while weakly fluorescent NS1 foci that may be involved in other aspects of the viral replication cycle can display rapid long-range traffic. Further characterization of the DENV2-NS1-mScarlet recombinant virus and development of adapted variants that closely mirror the replication, infectious virus production, and NS1 secretion kinetics of wild-type DENV2 will enable detailed analysis of NS1 localization, traffic, and interactions with relevant viral and host cell factors during a productive infection.
FIG 12.

Live cell imaging of mScarlet-tagged NS1 in infected cells. (A) Schematic diagram of the DENV2-NS1-mScarlet construct. (B and C) Infectivity of DENV2-NS1-mScarlet compared to wild-type DENV-2. Huh-7.5 cells were transfected with wild-type DENV-2 or DENV2-NS1-mScarlet RNA transcripts, and cell culture supernatants were collected at 24 to 120 h posttransfection for determination of virus infectivity by focus-forming assay (FFA). (B) The immunofluorescent micrographs show representative images from a FFA of cells infected with undiluted DENV-2 or DENV2-NS1-mScarlet supernatants collected at 120 h posttransfection. Cells were labeled with anti-E antibody (green) and mScarlet-associated epifluorescence was also visualized (red). Scale bars, 100 μm. (C) Quantitation of infectivity by FFA. Data are means + the SD (n = 3). The dashed line indicates the limit of detection. n.d., not detectable. (D and E) Live cell imaging of Huh-7.5 cells transfected with DENV2-NS1-mScarlet transcripts (6 days posttransfection). (D) Detection of mScarlet autofluorescence in live Huh-7.5 cells after mock transfection (“Mock”) or transfection DENV2-NS1-mScarlet RNA transcripts. Scale bars, 100 μm. (E) Live cell imaging of NS1-mScarlet localization and traffic. Insets depict examples of intensely fluorescent, relatively static NS1-mScarlet foci (cyan arrows) and weakly fluorescent, highly motile NS1-mScarlet foci (cyan arrowheads). See also Movie S1 in the supplemental material. Scale bars are 10 μm for the main image and 5 μm for the insets.
DISCUSSION
Reverse-genetics studies have contributed greatly to our understanding of the DENV replication cycle and the functions of the individual viral proteins and genetic elements. However, these studies are highly laborious and are typically limited to analysis of discrete regions of individual viral proteins. Our study used random transposon mutagenesis and high-throughput sequencing to provide a global overview of regions of genetic flexibility within the DENV-2 genome and its encoded proteins. These data provide a resource identifying genetically flexible regions that should arguably be avoided in antiviral and vaccine development strategies, since these sites may be prone to escape mutations with minimal impact on viral replicative fitness. Conversely, sites that are broadly tolerant of insertions can be exploited in the generation of infectious epitope- and reporter-tagged viruses. These viruses can be used in advanced applications such as high-resolution imaging and the interrogation of protein-protein interactions.
Our transposon mutagenic profile of DENV2 genetic flexibility indicates that the regions encoding prM, NS2A, and NS4A are highly intolerant of 15-nt insertions. For NS2A, an eight-transmembrane protein with essential roles in viral RNA replication and infectious virus production, our observations of intolerance to insertions are consistent with recent alanine scanning mutagenesis studies that have revealed numerous mutations, particularly in transmembrane regions, that are lethal to viral RNA replication or infectious virus production (9, 10). Similarly, recent site-directed mutagenesis studies of NS4A, which is essential for DENV-induced membrane rearrangements, have identified numerous conserved residues that are required for its oligomerization and viral RNA replication (33, 34). However, the lack of detection of replication-competent (pool 1) viruses bearing insertions in the prM-encoding region was more surprising since this region has no known involvement in viral RNA replication. Although it is possible that the spread of infectious virus in pool 1 has masked the presence of replication-competent prM mutants, it is also possible that insertion mutations within this region have unexpected dominant-negative effects on viral replication. Further studies are required to explore the apparent impact of prM mutations on DENV replication.
Overall, the regions encoding capsid, NS1 and the 3′ UTR were most tolerant of 15-nt insertions. As discussed above, capsid is the least conserved of all flavivirus proteins, although its charge distribution is strongly conserved and structural features are highly similar for capsid proteins of different flaviviruses (22). Accordingly, its relatively high tolerance for 15-nt insertions is not unexpected, especially for insertions that minimally alter its overall charge. In this context, none of the possible insertions encode negatively charged peptides (C-G-R-I/M/T/N/K/S/R, L/M/V-R-P-H/Q, or X-A-A-A). Similarly, region I of the 3′ UTR, where transposon insertions are most tolerated, is the most variable of all DENV nucleotide sequences with various deletions and point mutations identified in mosquito cell-adapted populations (35, 36). Although moderately conserved across flavivirus species, the NS1 protein was remarkably tolerant of 15-nt insertions. Largely consistent with a recent mutagenesis study (8), we found that the N-terminal β-roll, which contains a di-amino acid motif that may mediate interaction with NS4B and ER membrane association (37), the N-terminal half of the Wing domain and the C-terminal half of the β-ladder domain were highly sensitive to insertions. In contrast, relatively broad regions surrounding the N-glycosylation sites in the Wing (N130) and β-ladder (N207) domains and sites in close proximity to an N-glycosylation site in the connector domain of other flaviviruses (N175) were highly tolerant of insertions. Examination of the location of these sites in the crystal structure of the NS1 dimer demonstrated that they were all solvent exposed and relatively distant from the putative ER membrane-associating β-roll and greasy finger loop of the β-ladder (18). Likewise, in the context of the secreted NS1 hexamer these regions of genetic flexibility are distant from the lipoparticle interior and central lipid core that are contained by six copies of the β-roll (18). Accordingly, we hypothesize that the regions of high tolerance of insertions identified in our study are not required for functions of NS1 in viral RNA replication or infectious virus production but, rather, may be required for extracellular functions of NS1 in immune evasion and pathogenesis that are not recapitulated in hepatoma cell culture. If so, such replication-competent and infectious mutants that lack the immune evasion and vascular permeability functions of wild-type NS1 may potentially be combined and exploited in future attenuated vaccine strategies. In this context, a live-attenuated Zika virus (ZIKV) strain with engineered mutations to abolish NS1 glycosylation has very recently been shown to protect mice against virus-induced placental damage and fetal demise (38). Alternatively or additionally, regions of genetic flexibility in NS1 may be inherently flexible to skew humoral immune responses toward these epitopes and away from regions that are susceptible to function-neutralizing antibody responses and/or facilitate rapid viral adaptation in alternative host species. Such roles have been suggested for genetically flexible regions of influenza A virus NS1 protein in similar high-throughput insertional mutagenesis studies (13).
Although our mutant library did not achieve saturation, it nonetheless enabled generation of a comprehensive profile of regions in the DENV-2 genome that are broadly tolerant of small (15-nt) insertions and nonessential for viral RNA replication and infectious virus production in hepatoma cells. In this context, sequencing analysis of the initial mutant DENV-2 RNA pool indicated that ∼44% of all possible insertions were present. However, additional transposon insertions frequently emerged in sequencing analysis of the replication-competent and infectious virus pools (pools 1 and 2, respectively), such that the percentage of all possible unique transposon insertions in the DENV-2 genome in our study rose to ∼49%. Future studies of this nature involving near-saturation mutagenesis will help to resolve gaps in our mutational profile of DENV-2 but will not likely alter the overall appearance of this profile. Furthermore, additional approaches are required to better delineate regions that are essential to viral RNA replication and those that are required uniquely for infectious virus production. For example, blockade of infectious virus spread or parallel comparison of genetic flexibility in an analogous subgenomic replicon could resolve regions in the NS proteins that are required for replication versus those required for infectious virus production.
As detailed above, another application of our mutational profile of DENV2 is in the rational generation of epitope- and reporter-tagged viruses. Although the viability of such viruses is predictably dependent on the size and structure of the insertion, our mutational profile nonetheless provides a valuable resource for prediction of sites that may tolerate insertions for advanced imaging, proteomics, and molecular applications. For example, our APEX EM analysis indicates that NS1 is localized to both the luminal membrane of VPs and in discrete clusters within VPs, in contrast to longstanding models of replication complexes with respect to VPs, which depict NS1 as an exclusively luminal viral protein. Furthermore, live cell imaging studies using mScarlet-tagged NS1 revealed that intensely labeled NS1 foci, which may reflect clusters of vesicle packets, are relatively static. In contrast, small and less intensely labeled NS1 foci infrequently display rapid bidirectional traffic. Although further investigations are required, including confirmation that NS1 secretion is not perturbed by the mScarlet insertion, these motile foci may reflect pools of NS1 that are involved in other functions of NS1 such as virus assembly and as a secreted mediator of vascular damage and immune evasion. Further studies using the NS1-tagged viruses developed in this study and additional variations of these tagged viruses may help to further resolve the localization, traffic, interactions and functions of this enigmatic multifunctional viral protein.
The transposon mutagenesis-coupled high-throughput sequencing approach applied to DENV-2 here also provides a basis for numerous additional extensions that could rapidly improve our understanding of flavivirus- and host cell-specific functions of viral proteins and genetic elements. For example, this approach could be readily applied to other DENV serotypes, related flaviviruses or alternative cell types to unveil flavivirus- and host cell type- and species-specific differences in the functions and interactions of viral proteins. In this context, our analysis of cell type- and/or species-specific determinants of genetic flexibility indicate that the diverse host cells that we examined do not impart markedly different selective pressures that alter the relative fitness of viable transposon mutants. Nevertheless, it is possible that continued passage of the transposon mutant pool in these different cell types may further unveil regions of the DENV-2 genome that are differentially susceptible to host cell-specific selective pressures. In regard to flavivirus-specific determinants of viral replicative fitness, Fulton et al. very recently reported the impact of high-throughput transposon mutagenesis of a cloned ZIKV genome on viral replication and infectious virus production (39). Largely consistent with our findings for DENV-2, Fulton et al. demonstrated that NS1 and the structural genes of ZIKV displayed the greatest overall flexibility, although key differences in the mutational maps of DENV-2 and ZIKV were apparent. For example, the flexibility of prM appeared much higher for ZIKV compared to DENV-2 prM, while regions surrounding N-glycosylation sites of DENV-2 NS1 displayed greater apparent flexibility compared to the corresponding regions of ZIKV NS1. Further studies are required to define the commonalities and differences in sites of genetic flexibility between different flaviviruses and DENV serotypes and the impact of different host cell types and species on this flexibility. In a further refinement of the transposon mutagenesis approach, several recent studies have combined random point mutagenesis with NGS-based analysis of viral fitness to provide comprehensive profiles of the impact of all possible amino acid substitutions in a viral protein with respect to analysis of corresponding high-resolution protein structures (reviewed in reference 15). This approach could similarly be applied to great effect for DENV and related flaviviruses. In summary, we propose that variations of the high-throughput mutagenesis-coupled genetic profiling approach applied here, in conjunction with increased availability of high-resolution viral protein structural information, may rapidly increase our understanding of the DENV replication cycle and expedite the development of urgently required antiviral therapies and vaccines.
MATERIALS AND METHODS
Cell culture.
Huh-7.5 cells (40) were generously provided by Charles M. Rice (Rockefeller University, New York, NY) and were maintained as described previously (41). C6/36 cells, derived from Aedes albopictus mosquitoes, were generously provided by Jillian M. Carr (Flinders University, Adelaide, Australia) and were cultured at 28°C in a 5% CO2 atmosphere in basal medium Eagle supplemented with minimal essential medium nonessential amino acids, sodium pyruvate, GlutaMAX, penicillin-streptomycin, and 10% fetal bovine serum (FBS). Vero cells were generously provided by Jillian M. Carr (Flinders University, Adelaide, Australia) and were cultured as described previously (42). All cell culture media and additives were purchased from Thermo Fisher Scientific.
Antibodies and chemicals.
Mouse anti-NS1 monoclonal antibody (MAb) 4G4 and mouse anti-dsRNA MAb 3G1 (IgM) were generously provided by Roy Hall (University of Queensland, Brisbane, Australia) (43, 44). Mouse anti-capsid MAb 6F3.1 was kindly provided by John Aaskov (Queensland University of Technology, Brisbane, Australia) (45). Mouse anti-β-actin MAb (AC-74) was purchased from Sigma-Aldrich. Rabbit anti-FLAG MAb (D6W5B) was purchased from Cell Signaling Technologies. Alexa Fluor 488-, 555-, and 647-conjugated secondary antibodies and horseradish peroxidase-conjugated secondary antibodies were purchased from Thermo Fisher Scientific. Nanchangmycin was purchased from Selleck Chemicals and dissolved in dimethyl sulfoxide (DMSO) to 10 mM, divided into aliquots, and stored at −80°C.
Viruses and plasmids.
Plasmid pFK-DVs containing a full-length DENV-2 genome (strain 16681) was generously provide by Ralf Bartenschlager (University of Heidelberg, Heidelberg, Germany) (46). Exact details about reporter- and epitope-tagged virus generation are available upon request. To initiate viral RNA replication, DENV plasmids were linearized with XbaI before use as the templates in in vitro transcription reactions using an mMessage mMachine SP6 transcription kit (Thermo Fisher Scientific) and transfection of viral RNA into Huh-7.5 cells by electroporation or transfection with DMRIE-C reagent (Thermo Fisher Scientific), as described previously (41). Virus infectivity was measured by a focus-forming assay. Briefly, Huh-7.5 cells were seeded at 2 × 104 cells/well into 96-well plates and returned to culture overnight prior to inoculation with 40 μl/well of 10-fold serial dilutions of virus-containing cell culture supernatants. After infection for 3 h at 37°C and 5% CO2, the cells were washed once with phosphate-buffered saline (PBS) and returned to culture in fresh media for 72 h prior to fixation and immunofluorescent labeling with anti-capsid antibody. Clusters (foci) of infected cells were then enumerated, and virus infectivity was expressed as focus-forming units (FFU) per ml.
Generation of the DENV insertional mutant library.
Plasmid pFK-DVs was mutagenized using the mutation generation system (Thermo Fisher Scientific) according to the manufacturer's recommendations. Three independent in vitro transposon insertion reactions were performed using 500 ng of plasmid per reaction, pooled, and transformed into XL-10 Gold Ultracompetent cells (Agilent Technologies). Transformants were plated onto 15-cm plates with Luria-Bertani (LB) agar containing ampicillin and kanamycin and grown for 18 h at 37°C. Bacterial colonies (∼2.5 × 105) were then scraped and pooled before extraction of plasmid DNA using a NucleoBond Xtra Midi kit (Macherey-Nagel). Plasmid DNA was then digested with NotI-HF (New England BioLabs) to remove the transposon body, gel extracted, and religated using T4 DNA ligase (Promega). Approximately 150 ng of ligated plasmid was then transformed into XL-10 Gold cells, as described above, and the cells were plated onto LB agar plates containing ampicillin and cultured for 18 h at 37°C. Plasmid DNA was extracted from pooled colonies (∼2.5 × 105 colonies), as described above, and verified by diagnostic restriction digest (data not shown).
Mutant virus library passage and analysis of transposon insertion frequency by RT-PCR and Illumina sequencing.
Huh-7.5 cells were electroporated with in vitro-transcribed transposon mutant DENV library RNA. For this, 13 electroporations were performed with 4 × 106 cells and 10 μg of RNA per electroporation using 0.4-cm cuvettes and a Gene Pulser Xcell electroporation system (Bio-Rad) to deliver a single pulse (270 V, 100 Ω, 970 μF). Cells were then resuspended in complete media, pooled, and plated into 13 75-cm2 flasks and returned to culture. Virus replication and spread was monitored in parallel cultures by immunofluorescence and at 6 days postelectroporation, when ∼50% of cells were infected, cell culture supernatants were collected, cleared by centrifugation, and diluted in an equal volume of fresh medium before they were applied to naive target cells, seeded the previous day into 13 75-cm2 flasks at 1.6 × 106 cells per flask. Total RNA was isolated from electroporated cell monolayers (6 days postelectroporation) and infected cell monolayers (2 days postinfection) using TRIzol (Thermo Fisher Scientific) according to the manufacturer's instructions. This cellular RNA (pool 1 and pool 2) and the “input” RNA used for electroporation (pool 0) were reverse transcribed into cDNA using Superscript III (Thermo Fisher Scientific) and the oligonucleotide DV2NGS6R (see below) according to the manufacturer's instructions. Alternatively, the infectious virus-containing supernatant from mutant library-electroporated Huh-7.5 cells (see above) was diluted (1:3) in appropriate media and applied to naive Huh-7.5, Vero, or C6/36 cells that had been seeded the previous day into 75-cm2 flasks (three flasks per cell type, with 1.2 × 106 cells/flask). As above, at 2 days postinfection the total RNA was extracted from these cells and used to prepare cDNA. In both instances, this cDNA then served as the template for PCR to amplify the entire genome in six overlapping fragments using Q5 high-fidelity DNA polymerase (New England BioLabs) and the six following oligonucleotide pairs: DV2NGS1F (5′-AGTTGTTAGTCTACGTGGACCG-3′) and DV2NGS1R (5′-CGAATGGAGGTTCTGCTTCTATGT-3′), DV2NGS2F (5′-GCAGAAACACAACATGGAACAATAG-3′) and DV2NGS2R (5′-CCTAAGGCTAACGCATCAGTC-3′), DV2NGS3F (5′-TGTCCTTTAGAGACCTGGGAAG-3′) and DV2NGS3R (5′-ATGTCAGTTGTAACCACGAAGTCC-3′), DV2NGS4F (5′-CAGCAAGTATAGCAGCTAGAGGA-3′) and NGS4R (5′-TTTCCCTTCTGGTGTGACCATG-3′), DV2NGS5F (5′-CTCAAGTATTGATGATGAGGACTACATG-3′) and DV2NGS5R (5′-ACTTGTGTCCAATCATTCCATCC-3′), and DV2NGS6F (5′-CCGCAGGATGGGATACAAGA-3′) and DV2NGS6R (5′-AGAACCTGTTGATTCAACAGCAC-3′). PCR products were then gel-extracted, quantified using a Qubit dsDNA HS assay kit (Thermo Fisher Scientific) and combined in equimolar amounts for each pool. Samples were then processed using a Nextera XT library preparation kit (Illumina) and sequenced using a NextSeq500 (Illumina) and 150-nt paired-end reads. For analysis, trimmed sequencing reads were filtered for sequences containing the transposon-derived insertion sequence (TGCGGCCGCA), and the transposon sequence was annotated and mapped against the DENV-2 reference sequence (GenBank accession number NC_001474 with minor modifications, as described previously [46]) using Bowtie 2 (47). The locations of the annotated transposon insertions were then identified from the alignments and counted using Geneious version 8 software (48). Original sequencing files are accessible from the NCBI Sequence Reads Archive under the series record PRJNA400339.
Immunofluorescence microscopy, immunoblotting, and luciferase assays.
Immunofluorescent labeling was performed as described previously (41). Widefield fluorescence microscopy for infectivity assays was performed using a Nikon TiE inverted fluorescence microscope system. Confocal fluorescence microscopy was performed using a Zeiss LSM 700 confocal microscope system equipped with a 60× NA 1.4 water-immersion objective lens. Images were processed using NIS Elements AR v.3.22 (Nikon) and Photoshop 6.0 (Adobe) software. For STED super-resolution imaging, Huh-7.5 cells were cultured overnight on coverslips (18-mm round no. 1.5 glass) that were precoated with 0.2% gelatin. Cells were then infected with DENV2-NS1-FLAG (MOI of ∼0.1) and returned to culture for 48 h prior to fixation (ice-cold methanol-acetone [1:1], 5 min), washing, blocking (5% BSA in PBS for 30 min at room temperature), and labeling for 1 h at room temperature with rabbit anti-FLAG MAb (D6W5B [Cell Signaling Technologies], diluted 1:200) and anti-dsRNA (MAb 3G1.1 hybridoma supernatant, diluted 1:5) diluted in PBS–1% BSA. Samples were then washed twice with PBS before incubation for 1 h at 4°C with Alexa Fluor 488-conjugated anti-rabbit IgG and Alexa Fluor 647-conjugated anti-mouse IgG (cross-reactive to IgM) antibodies (Thermo Fisher Scientific) diluted 1:200 in 1% BSA-PBS. Samples were then washed three times and mounted with ProLong Gold (Thermo Fisher Scientific). Samples were then imaged using a Leica TCS SP8 STED 3× microscope system (Leica Microsystems) equipped with 592-, 660-, and 775-nm STED lasers, using a 100×, NA 1.4 oil objective lens at 4× zoom. For STED, Alexa Fluor 488 labels were excited with a 488-nm wavelength of a pulsed white light (WL) laser (80 MHz) and depleted with a CW 592 STED laser with a maximum power of 1,500 mW (typically operating at ∼30%). Similarly, for STED Alexa Fluor 647 labels were excited with a 647-nm wavelength of a WL laser and depleted with a CW 775 nm STED laser with a maximum power of 1,500 mW (typically operating at ∼50%). Images were acquired in two-dimensional (2D) STED mode with settings optimized for maximum gains in lateral resolution. Time gates were 0.5 to 6 ns. A total of four z-sections were acquired (0.142-μm z-steps) for both confocal and STED channels and a line averaging of 7 was applied. Deconvolution of confocal and STED data (see Fig. 9) was performed using Huygens Professional Deconvolution software (v14.10; Scientific Volume Imaging) and default settings. Immunoblotting was performed as described previously (49). Where indicated, precleared supernatants were treated with PNGase F (New England BioLabs) for 4 h at 37°C under nonreducing conditions in accordance with the manufacturer's instructions. Western blots were imaged using a ChemiDoc MP Imaging System (Bio-Rad) and, where applicable, the band intensities were quantified using Image Lab software (v5.2.1; Bio-Rad). Assays of NanoLuc (NLuc) activity were performed as described previously (50). In brief, Huh-7.5 cells were seeded into 96-well plates at 2 × 104 cells per well and cultured overnight prior to infection with DENV2-NS1-NLuc at the indicated MOI, with or without pretreatment with nanchangmycin for 1 h at the indicated concentration (or vehicle control: DMSO at 0.1%). Four hours later, the cells were washed and returned to culture for 48 h prior to the collection of supernatants, the washing of monolayers with PBS, and lysis using passive lysis buffer (Promega). Supernatant samples were cleared by centrifugation and mixed 1:1 with 2× passive lysis buffer before measurement of the NLuc activity in cell lysates and supernatants using a Nano-Glo luciferase assay system (Promega) and a GloMax 20/20 luminometer (Promega).
Live cell imaging.
Live cell imaging was performed as described previously (51). Briefly, DENV2-NS1-mScarlet RNA-transfected Huh-7.5 cells at 4 days posttransfection were seeded onto 0.2% gelatin-coated cover glass-bottom dishes (MatTek) and cultured for 2 days in phenol red-free Dulbecco modified Eagle medium containing 10% FBS. Alternatively, Huh-7.5 cells were seeded into these dishes at 1.5 × 105 cells per dish, infected the following day at an MOI of ∼0.01, and returned to culture for 3 days. Imaging was performed at 37°C using a Nikon TiE inverted fluorescence microscope system equipped with a heated stage (Okolab), a Plan Apochromat 60× NA 1.4 oil immersion objective lens (Nikon), BrightLine single-band filter sets (DAPI-5060C-NTE-ZERO, FITC-3540C-NTE-ZERO, and TxRed-4040C-NTE-ZERO; Semrock), a Perfect Focus System (Nikon), and a monochrome 12-bit cooled charge-coupled device camera with a maximum resolution of 1,280 × 1,024 (DS-Qi1; Nikon). Illumination was provided by an Intensilight C-HGFIE precentered fiber illuminator mercury light source (Nikon). Images were acquired every 1.5 s for 5 min (see Movie S1 in the supplemental material) or every 10 s for 50 min (see Movie S2 in the supplemental material). Image processing was performed using NIS Elements v 3.22 software (Nikon), as described in the figure legends and supplementary movie legends.
APEX electron microscopy.
Huh-7.5 cells were seeded into 150-mm cell culture dishes at 1.5 × 106 cells per dish and cultured overnight before mock infection or infection with DENV2 or DENV2-NS1-APEX2 viruses (MOI of ∼0.01), prepared as cell culture supernatants collected from electroporated Huh-7.5 cells. At 4 days postinfection, the cells were fixed in EM fixative (1.25% glutaraldehyde, 4% paraformaldehyde, and 4% sucrose in PBS [pH 7.2]) for 30 min at 4°C, washed, stained with DAB/H2O2, and processed for electron microscopy as described previously (41). Samples were imaged using a Tecnai G2 Spirit transmission electron microscope (FEI) operating at 100 kV.
Molecular graphics and sequence alignments.
Molecular graphics were performed on the DENV-2 NS1 crystal structure (Protein Data Bank [PDB] accession number 4O6B) using PyMOL version 1.8 molecular visualization system (Schrodinger). Sequence alignments of DENV polyprotein sequences were performed using JalView Desktop software with the ClustalW scoring algorithm. Flavivirus isolates with the following UniProt KB/Swiss-Prot accession numbers were compared: DENV-1, P27909 (Brazil/97-11/1997); DENV-2, P29991 (Thailand/16681-PDK53); DENV-3, Q6YMS3 (Martinique/1243/1999); DENV-4 Q2YHF0 (Thailand/0348/1991); West Nile virus P06935; Yellow Fever virus, Q6J3P1 (Ivory Coast/1999); and Japanese encephalitis virus P27395 (SA-14).
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Ralf Bartenschlager (University of Heidelberg) for the pFK-DVs construct, Charles Rice (The Rockefeller University) for Huh-7.5 cells, Jillian Carr (Flinders University) for Vero cells and C6/36 cells, Roy Hall (University of Queensland) for anti-NS1 and anti-dsRNA MAbs, and John Aaskov (Queensland University of Technology) for anti-capsid MAb. We also thank Kate Pilkington (The Detmold Family Trust Cell Imaging Centre, SA Pathology) for assistance with confocal microscopy, Lyn Waterhouse and Ruth Williams (Adelaide Microscopy, University of Adelaide) for assistance with electron microscopy, and Stephen Thompson (Leica Microsystems) for assistance with STED image acquisition and processing. DNA sequencing was performed at the Ramaciotti Centre for Genomics (University of New South Wales).
This study was funded by grants from the National Health and Medical Research Council (NHMRC) (1027641 and 1053206 to M.R.B.). A.A.E. and R.A.B. are supported by fellowships from the NHMRC (grants 1130128 and 1084706, respectively). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01455-17.
REFERENCES
- 1.Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GR, Simmons CP, Scott TW, Farrar JJ, Hay SI. 2013. The global distribution and burden of dengue. Nature 496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deen J. 2016. The dengue vaccine dilemma: balancing the individual and population risks and benefits. PLoS Med 13:e1002182. doi: 10.1371/journal.pmed.1002182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chatel-Chaix L, Bartenschlager R. 2014. Dengue virus- and hepatitis C virus-induced replication and assembly compartments: the enemy inside–caught in the web. J Virol 88:5907–5911. doi: 10.1128/JVI.03404-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P, Fuller SD, Antony C, Krijnse-Locker J, Bartenschlager R. 2009. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 5:365–375. doi: 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Westaway EG, Mackenzie JM, Kenney MT, Jones MK, Khromykh AA. 1997. Ultrastructure of Kunjin virus-infected cells: colocalization of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J Virol 71:6650–6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller S, Kastner S, Krijnse-Locker J, Buhler S, Bartenschlager R. 2007. The nonstructural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J Biol Chem 282:8873–8882. doi: 10.1074/jbc.M609919200. [DOI] [PubMed] [Google Scholar]
- 7.Gebhard LG, Iglesias NG, Byk LA, Filomatori CV, De Maio FA, Gamarnik AV. 2016. A proline-rich N-terminal region of the dengue virus NS3 is crucial for infectious particle production. J Virol 90:5451–5461. doi: 10.1128/JVI.00206-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scaturro P, Cortese M, Chatel-Chaix L, Fischl W, Bartenschlager R. 2015. Dengue virus nonstructural protein 1 modulates infectious particle production via interaction with the structural proteins. PLoS Pathog 11:e1005277. doi: 10.1371/journal.ppat.1005277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu RH, Tsai MH, Chao DY, Yueh A. 2015. Scanning mutagenesis studies reveal a potential intramolecular interaction within the C-terminal half of dengue virus NS2A involved in viral RNA replication and virus assembly and secretion. J Virol 89:4281–4295. doi: 10.1128/JVI.03011-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie X, Zou J, Puttikhunt C, Yuan Z, Shi PY. 2015. Two distinct sets of NS2A molecules are responsible for dengue virus RNA synthesis and virion assembly. J Virol 89:1298–1313. doi: 10.1128/JVI.02882-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roby JA, Setoh YX, Hall RA, Khromykh AA. 2015. Posttranslational regulation and modifications of flavivirus structural proteins. J Gen Virol 96:1551–1569. doi: 10.1099/vir.0.000097. [DOI] [PubMed] [Google Scholar]
- 12.Fulton BO, Sachs D, Beaty SM, Won ST, Lee B, Palese P, Heaton NS. 2015. Mutational analysis of measles virus suggests constraints on antigenic variation of the glycoproteins. Cell Rep 11:1331–1338. doi: 10.1016/j.celrep.2015.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heaton NS, Sachs D, Chen CJ, Hai R, Palese P. 2013. Genome-wide mutagenesis of influenza virus reveals unique plasticity of the hemagglutinin and NS1 proteins. Proc Natl Acad Sci U S A 110:20248–20253. doi: 10.1073/pnas.1320524110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qi H, Olson CA, Wu NC, Ke R, Loverdo C, Chu V, Truong S, Remenyi R, Chen Z, Du Y, Su SY, Al-Mawsawi LQ, Wu TT, Chen SH, Lin CY, Zhong W, Lloyd-Smith JO, Sun R. 2014. A quantitative high-resolution genetic profile rapidly identifies sequence determinants of hepatitis C viral fitness and drug sensitivity. PLoS Pathog 10:e1004064. doi: 10.1371/journal.ppat.1004064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qi H, Wu NC, Du Y, Wu TT, Sun R. 2015. High-resolution genetic profile of viral genomes: why it matters. Curr Opin Virol 14:62–70. doi: 10.1016/j.coviro.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 16.Remenyi R, Qi H, Su SY, Chen Z, Wu NC, Arumugaswami V, Truong S, Chu V, Stokelman T, Lo HH, Olson CA, Wu TT, Chen SH, Lin CY, Sun R. 2014. A comprehensive functional map of the hepatitis C virus genome provides a resource for probing viral proteins. mBio 5:e01469-14. doi: 10.1128/mBio.01469-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aubry F, Nougairede A, Gould EA, de Lamballerie X. 2015. Flavivirus reverse genetic systems, construction techniques and applications: a historical perspective. Antiviral Res 114:67–85. doi: 10.1016/j.antiviral.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akey DL, Brown WC, Dutta S, Konwerski J, Jose J, Jurkiw TJ, DelProposto J, Ogata CM, Skiniotis G, Kuhn RJ, Smith JL. 2014. Flavivirus NS1 structures reveal surfaces for associations with membranes and the immune system. Science 343:881–885. doi: 10.1126/science.1247749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Watterson D, Modhiran N, Young PR. 2016. The many faces of the flavivirus NS1 protein offer a multitude of options for inhibitor design. Antiviral Res 130:7–18. doi: 10.1016/j.antiviral.2016.02.014. [DOI] [PubMed] [Google Scholar]
- 20.Jones CT, Ma L, Burgner JW, Groesch TD, Post CB, Kuhn RJ. 2003. Flavivirus capsid is a dimeric alpha-helical protein. J Virol 77:7143–7149. doi: 10.1128/JVI.77.12.7143-7149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma L, Jones CT, Groesch TD, Kuhn RJ, Post CB. 2004. Solution structure of dengue virus capsid protein reveals another fold. Proc Natl Acad Sci U S A 101:3414–3419. doi: 10.1073/pnas.0305892101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Byk LA, Gamarnik AV. 2016. Properties and functions of the dengue virus capsid protein. Annu Rev Virol 3:263–281. doi: 10.1146/annurev-virology-110615-042334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samsa MM, Mondotte JA, Iglesias NG, Assuncao-Miranda I, Barbosa-Lima G, Da Poian AT, Bozza PT, Gamarnik AV. 2009. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog 5:e1000632. doi: 10.1371/journal.ppat.1000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dixon AS, Schwinn MK, Hall MP, Zimmerman K, Otto P, Lubben TH, Butler BL, Binkowski BF, Machleidt T, Kirkland TA, Wood MG, Eggers CT, Encell LP, Wood KV. 2016. NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem Biol 11:400–408. doi: 10.1021/acschembio.5b00753. [DOI] [PubMed] [Google Scholar]
- 25.Kamiyama D, Sekine S, Barsi-Rhyne B, Hu J, Chen B, Gilbert LA, Ishikawa H, Leonetti MD, Marshall WF, Weissman JS, Huang B. 2016. Versatile protein tagging in cells with split fluorescent protein. Nat Commun 7:11046. doi: 10.1038/ncomms11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lam SS, Martell JD, Kamer KJ, Deerinck TJ, Ellisman MH, Mootha VK, Ting AY. 2015. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods 12:51–54. doi: 10.1038/nmeth.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martell JD, Deerinck TJ, Sancak Y, Poulos TL, Mootha VK, Sosinsky GE, Ellisman MH, Ting AY. 2012. Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat Biotechnol 30:1143–1148. doi: 10.1038/nbt.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hall MP, Unch J, Binkowski BF, Valley MP, Butler BL, Wood MG, Otto P, Zimmerman K, Vidugiris G, Machleidt T, Robers MB, Benink HA, Eggers CT, Slater MR, Meisenheimer PL, Klaubert DH, Fan F, Encell LP, Wood KV. 2012. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem Biol 7:1848–1857. doi: 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rausch K, Hackett BA, Weinbren NL, Reeder SM, Sadovsky Y, Hunter CA, Schultz DC, Coyne CB, Cherry S. 2017. Screening bioactives reveals nanchangmycin as a broad spectrum antiviral active against Zika virus. Cell Rep 18:804–815. doi: 10.1016/j.celrep.2016.12.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mackenzie JM, Jones MK, Young PR. 1996. Immunolocalization of the dengue virus nonstructural glycoprotein NS1 suggests a role in viral RNA replication. Virology 220:232–240. doi: 10.1006/viro.1996.0307. [DOI] [PubMed] [Google Scholar]
- 31.Feng S, Sekine S, Pessino V, Li H, Leonetti MD, Huang B. 2017. Improved split fluorescent proteins for endogenous protein labeling. Nat Commun 8:370. doi: 10.1038/s41467-017-00494-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bindels DS, Haarbosch L, van Weeren L, Postma M, Wiese KE, Mastop M, Aumonier S, Gotthard G, Royant A, Hink MA, Gadella TW Jr. 2017. mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nat Methods 14:53–56. doi: 10.1038/nmeth.4074. [DOI] [PubMed] [Google Scholar]
- 33.Lee CM, Xie X, Zou J, Li SH, Lee MY, Dong H, Qin CF, Kang C, Shi PY. 2015. Determinants of dengue virus NS4A protein oligomerization. J Virol 89:6171–6183. doi: 10.1128/JVI.00546-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stern O, Hung YF, Valdau O, Yaffe Y, Harris E, Hoffmann S, Willbold D, Sklan EH. 2013. An N-terminal amphipathic helix in dengue virus nonstructural protein 4A mediates oligomerization and is essential for replication. J Virol 87:4080–4085. doi: 10.1128/JVI.01900-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Villordo SM, Carballeda JM, Filomatori CV, Gamarnik AV. 2016. RNA structure duplications and flavivirus host adaptation. Trends Microbiol 24:270–283. doi: 10.1016/j.tim.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Villordo SM, Filomatori CV, Sanchez-Vargas I, Blair CD, Gamarnik AV. 2015. Dengue virus RNA structure specialization facilitates host adaptation. PLoS Pathog 11:e1004604. doi: 10.1371/journal.ppat.1004604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Youn S, Li T, McCune BT, Edeling MA, Fremont DH, Cristea IM, Diamond MS. 2012. Evidence for a genetic and physical interaction between nonstructural proteins NS1 and NS4B that modulates replication of West Nile virus. J Virol 86:7360–7371. doi: 10.1128/JVI.00157-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Richner JM, Jagger BW, Shan C, Fontes CR, Dowd KA, Cao B, Himansu S, Caine EA, Nunes BTD, Medeiros DBA, Muruato AE, Foreman BM, Luo H, Wang T, Barrett AD, Weaver SC, Vasconcelos PFC, Rossi SL, Ciaramella G, Mysorekar IU, Pierson TC, Shi PY, Diamond MS. 2017. Vaccine-mediated protection against Zika virus-induced congenital disease. Cell 170:273–283. doi: 10.1016/j.cell.2017.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fulton BO, Sachs D, Schwarz MC, Palese P, Evans MJ. 2017. Transposon mutagenesis of the Zika virus genome highlights regions essential for RNA replication and restricted for immune evasion. J Virol 91:e00698-17. doi: 10.1128/JVI.00698-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eyre NS, Hampton-Smith RJ, Aloia AL, Eddes JS, Simpson KJ, Hoffmann P, Beard MR. 2016. Phosphorylation of NS5A serine-235 is essential to hepatitis C virus RNA replication and normal replication compartment formation. Virology 491:27–44. doi: 10.1016/j.virol.2016.01.018. [DOI] [PubMed] [Google Scholar]
- 42.Van der Hoek KH, Eyre NS, Shue B, Khantisitthiporn O, Glab-Ampi K, Carr JM, Gartner MJ, Jolly LA, Thomas PQ, Adikusuma F, Jankovic-Karasoulos T, Roberts CT, Helbig KJ, Beard MR. 2017. Viperin is an important host restriction factor in control of Zika virus infection. Sci Rep 7:4475. doi: 10.1038/s41598-017-04138-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clark DC, Lobigs M, Lee E, Howard MJ, Clark K, Blitvich BJ, Hall RA. 2007. In situ reactions of monoclonal antibodies with a viable mutant of Murray Valley encephalitis virus reveal an absence of dimeric NS1 protein. J Gen Virol 88:1175–1183. doi: 10.1099/vir.0.82609-0. [DOI] [PubMed] [Google Scholar]
- 44.O'Brien CA, Hobson-Peters J, Yam AW, Colmant AM, McLean BJ, Prow NA, Watterson D, Hall-Mendelin S, Warrilow D, Ng ML, Khromykh AA, Hall RA. 2015. Viral RNA intermediates as targets for detection and discovery of novel and emerging mosquito-borne viruses. PLoS Negl Trop Dis 9:e0003629. doi: 10.1371/journal.pntd.0003629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bulich R, Aaskov JG. 1992. Nuclear localization of dengue 2 virus core protein detected with monoclonal antibodies. J Gen Virol 73(Pt 11):2999–3003. [DOI] [PubMed] [Google Scholar]
- 46.Fischl W, Bartenschlager R. 2013. High-throughput screening using dengue virus reporter genomes. Methods Mol Biol 1030:205–219. doi: 10.1007/978-1-62703-484-5_17. [DOI] [PubMed] [Google Scholar]
- 47.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eyre NS, Drummer HE, Beard MR. 2010. The SR-BI partner PDZK1 facilitates hepatitis C virus entry. PLoS Pathog 6:e1001130. doi: 10.1371/journal.ppat.1001130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eyre NS, Aloia AL, Joyce MA, Chulanetra M, Tyrrell DL, Beard MR. 2017. Sensitive luminescent reporter viruses reveal appreciable release of hepatitis C virus NS5A protein into the extracellular environment. Virology 507:20–31. doi: 10.1016/j.virol.2017.04.003. [DOI] [PubMed] [Google Scholar]
- 51.Eyre NS, Fiches GN, Aloia AL, Helbig KJ, McCartney EM, McErlean CS, Li K, Aggarwal A, Turville SG, Beard MR. 2014. Dynamic imaging of the hepatitis C virus NS5A protein during a productive infection. J Virol 88:3636–3652. doi: 10.1128/JVI.02490-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.