

ABSTRACT
Epstein-Barr virus (EBV), a member of human gammaherpesvirus, infects mainly B cells. EBV has two alternative life cycles, latent and lytic, and is reactivated occasionally from the latent stage to the lytic cycle. To combat EBV-associated disorders, understanding the molecular mechanisms of the EBV lytic replication cycle is also important. Here, we focused on an EBV lytic gene, BKRF4. Using our anti-BKRF4 antibody, we revealed that the BKRF4 gene product is expressed during the lytic cycle with late kinetics. To characterize the role of BKRF4, we constructed BKRF4-knockout mutants using the bacterial artificial chromosome (BAC) and CRISPR/Cas9 systems. Although disruption of the BKRF4 gene had almost no effect on viral protein expression and DNA synthesis, it significantly decreased progeny virion levels in HEK293 and Akata cells. Furthermore, we show that BKRF4 is involved not only in production of progeny virions but also in increasing the infectivity of the virus particles. Immunoprecipitation assays revealed that BKRF4 interacted with a virion protein, BGLF2. We showed that the C-terminal region of BKRF4 was critical for this interaction and for efficient progeny production. Immunofluorescence analysis revealed that BKRF4 partially colocalized with BGLF2 in the nucleus and perinuclear region. Finally, we showed that BKRF4 is a phosphorylated, possible tegument protein and that the EBV protein kinase BGLF4 may be important for this phosphorylation. Taken together, our data suggest that BKRF4 is involved in the production of infectious virions.
IMPORTANCE Although the latent genes of EBV have been studied extensively, the lytic genes are less well characterized. This study focused on one such lytic gene, BKRF4, which is conserved only among gammaherpesviruses (ORF45 of Kaposi's sarcoma-associated herpesvirus or murine herpesvirus 68). After preparing the BKRF4 knockout virus using B95-8 EBV-BAC, we demonstrated that the BKRF4 gene was involved in infectious progeny particle production. Importantly, we successfully generated a BKRF4 knockout virus of Akata using CRISPR/Cas9 technology, confirming the phenotype in this separate strain. We further showed that BKRF4 interacted with another virion protein, BGLF2, and demonstrated the importance of this interaction in infectious virion production. These results shed light on the elusive process of EBV progeny maturation in the lytic cycle. Notably, this study describes a successful example of the generation and characterization of an EBV construct with a disrupted lytic gene using CRISPR/Cas9 technology.
KEYWORDS: BAC, BKRF4, CRISPR/Cas9, Epstein-Barr virus, lytic replication
INTRODUCTION
Epstein-Barr virus (EBV), a member of herpesviruses, infects mainly B cells. About 90% of the world's population is infected with EBV asymptomatically, but it can cause infectious mononucleosis and some malignancies such as Burkitt lymphoma, T/NK cell lymphoma, nasopharyngeal carcinoma, and gastric carcinoma (1, 2). Like other herpesviruses, EBV has two alternative life cycles, latent and lytic (3). Generally, infected B cells undergo a latent state under which they express several viral genes without production of progeny virions (4). Reactivation occasionally occurs, switching from latency to lytic states. In cultured cells, reactivation can be induced by chemical/biological agents or exogenous expression of one of the viral immediate-early (IE) genes, BZLF1 (Zta, Z, ZEBRA, and EB1) and BRLF1 (Rta, R). Expression of the IE genes induces viral early (E) genes, which include genes involved in viral DNA replication, such as BALF5 (a DNA polymerase catalytic subunit), BALF2 (a single-stranded DNA binding protein), and BMRF1 (an accessory factor of viral DNA polymerase). After viral genome DNA replication, viral late (L) genes are expressed. The L genes include capsid, tegument, and glycoproteins, such as major capsid protein (MCP) or glycoprotein B (gB), and these proteins contribute to the morphogenesis of progeny virus particles that are capable of infecting new cells (5, 6).
The tegument, or the space between the nucleocapsid and the envelope of the virion, is composed of viral proteins. Compared to glycoproteins or capsid proteins, tegument proteins are less well studied, particularly in the case of EBV. However, analyzing EBV tegument proteins is important because these proteins play critical roles in virion morphogenesis and enforcement of de novo infection in all members of the herpesvirus subfamily (7–9).
EBV BKRF4 is a possible tegument protein conserved among gammaherpesvirus but not among alpha- or betaherpesviruses (10). The strict homology between EBV BKRF4 and its homologs in other gammaherpesviruses lies only in the 15 amino acids of carboxyl-terminal ends, and the sequence similarities in the other portions of the proteins are limited. The EBV BKRF4 is only 217 amino acids long, whereas its homologs in Kaposi sarcoma-associated herpesvirus (KSHV) and murine gammaherpesvirus 68 (MHV-68) are 407 and 206 amino acids, respectively.
KSHV open reading frame 45 (ORF45), a homolog of EBV BKRF4, is a virion-associated, multifunctional tegument protein that has been studied extensively (11). ORF45 knockout in KSHV markedly reduces the yield of the progeny virus, while viral DNA replication is unaffected (12). KSHV ORF45 binds to interferon regulatory factor 7 (IRF-7) and kinesin-2, leading to viral immune evasion and transport of viral particles, respectively (13, 14). It can associate with viral tegument proteins, such as ORF33 and ORF36, and contribute to the efficient production of viral particles (15, 16). A recent report showed that ORF45 mediates the association between KSHV particles and internal lipid rafts for viral assembly (17).
MHV-68 ORF45 has also been identified as a tegument protein that plays important roles in viral gene expression during de novo infection (18, 19). An ORF45 knockout MHV-68 mutant is incapable of virion production, and MHV-68 ORF45 is essential for virion morphogenesis, particularly in the cytoplasm (20).
The role of EBV BKRF4 remains completely unreported. Because of the low similarities in amino acid sequences, it is assumed that the roles of EBV BKRF4 and KSHV/MHV-68 ORF45s may not be the same.
To characterize EBV BKRF4, we generated knockout viruses using the bacterial artificial chromosome (BAC) and CRISPR/Cas9 systems. Compared to the wild type, BKRF4 deficiency in both systems had almost no effect on viral gene expression and DNA synthesis, but it significantly decreased production of progeny virions. In addition, our data showed that BKRF4 is a phosphorylated virion protein that interacts with another EBV protein, BGLF2, and that this interaction played a critical role in progeny production.
RESULTS
Expression of the BKRF4 gene with late kinetics.
We prepared the rabbit antiserum against the BKRF4 polypeptide and subjected it to affinity purification. To induce the lytic cycle, B95-8 cells were treated with TPA (12-O-tetradecanoylphorbol-13-acetate), A23187 (calcium ionophore), and sodium butyrate (TAB) with or without phosphonoacetic acid (PAA) for 2 days. The cell lysates were analyzed by immunoblotting (IB) with the indicated antibodies (Fig. 1A). As shown in the right panel in Fig. 1A, BKRF4 protein was expressed in TAB-induced B95-8 cells, as detected by our anti-BKRF4 antibody. BKRF4 protein expression was strongly inhibited by the addition of a DNA polymerase inhibitor, PAA, in a manner similar to the inhibition shown by the L gene product, BALF4(gB) (Fig. 1A, left panel). PAA had less restriction on the expression of the IE and E genes, BRLF1 and BALF5, than it did on BKRF4 and gB genes (Fig. 1A).
FIG 1.

Identification of EBV BKRF4 protein. (A) B95-8 cells were treated with TPA (20 ng/ml), A23187 (1 μM), and TAB (5 mM) to induce reactivation. The DNA polymerase inhibitor PAA (400 μg/ml) was added (TAB+PAA) to inhibit late viral gene expression. After 2 days, cell lysates were subjected to IB using anti-BKRF4, anti-BRLF1, anti-BALF5, anti-BALF4(gB), and tubulin antibodies. (B) Total RNAs from cells treated as in panel A were subjected to qRT-PCR. The means ± the standard deviations (SD) of three independent biological replicates are shown.
The viral gene expression was also analyzed by qRT-PCR. As shown in Fig. 1B, the BKRF4 mRNA was induced by TAB treatment and then inhibited by PAA to the background level in a manner similar to another L gene, BALF4(gB); in contrast, the levels of BALF2 and BMRF1 (E genes) were inhibited very little, if at all (Fig. 1B). These results indicate that the BKRF4 gene is expressed with L kinetics during lytic replication of EBV.
Construction of the BKRF4 knockout mutants using the EBV-BAC system.
To study the role of BKRF4 in EBV lytic replication, we constructed BKRF4-deficient EBV-BAC mutants, as shown in Fig. 2A. We generated an insertion mutant (dBKRF4-ins) by inserting a Neo/St cassette at nucleotides 184 and 187 of the BKRF4 ORF. A revertant virus (dBKRF4-ins/R) was generated by replacing the cassette with the wild-type BKRF4 sequence. In addition, we prepared a nonsense mutant of the BKRF4 gene (dBKRF4-stop) by introducing two consecutive stop codons (G70AA to TAA and G73AG to TAG). The intended mutations in the recombinant BAC DNAs were confirmed by sequence analysis (not shown). To confirm that the recombinant viruses did not carry obvious deletions or insertions, the integrity of the viral genomes was checked by BamHI and EcoRI restriction digestion, followed by agarose gel electrophoresis (Fig. 2B). The BamHI fragments in the wild type, revertant, and nonsense-mutant constructs were identical. However, the BamHI-K fragment found in the wild-type, revertant, and nonsense-mutant constructs (4.7 kbp, lower arrow) migrated slower in the dBKRF4-ins mutant due to the size of the inserted cassette (higher arrow). The EcoRI digestion patterns of the recombinant viruses were almost identical even in the case of the insertion mutant, since the cassette was inserted into the second largest fragment, and thus shifting was not distinguishable in the gel.
FIG 2.
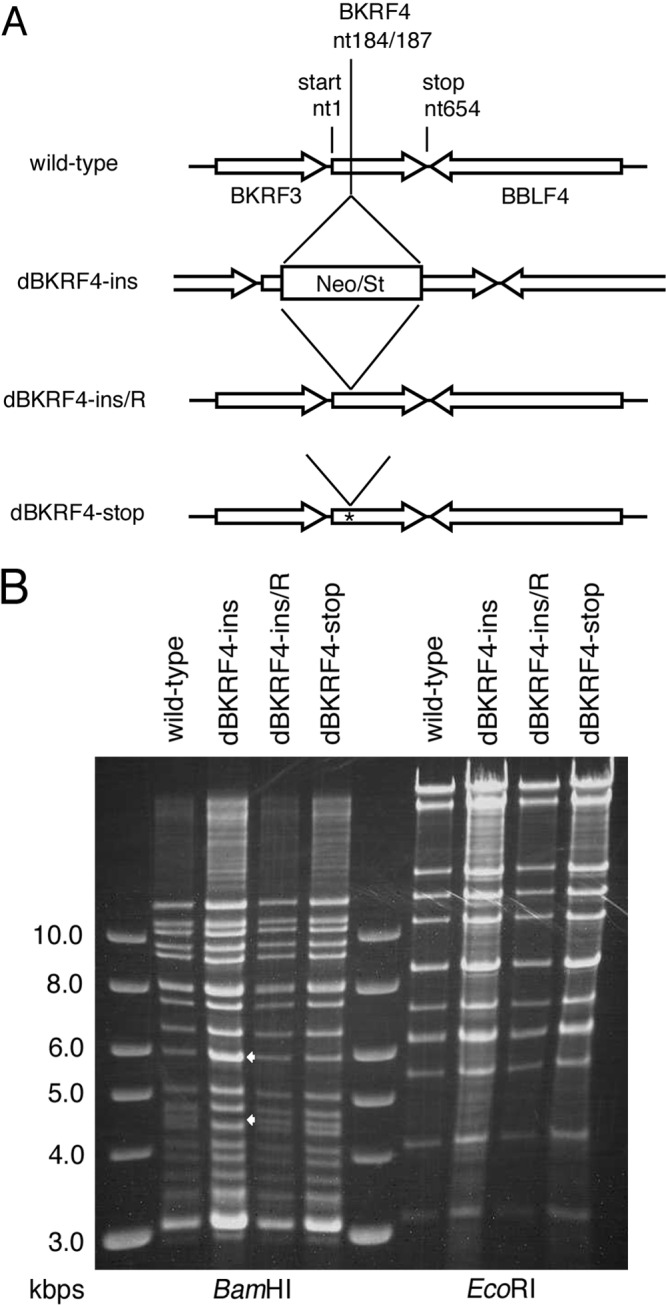
Construction of the EBV BKRF4 mutants. (A) Schematic representation of the recombination of the EBV-BAC genome using the tandemly arranged neomycin resistance gene and streptomycin sensitivity genes (Neo/St) to obtain BKRF4 mutants. The dBKRF4-ins insertion mutant was produced by inserting a Neo/St cassette at nucleotides 184 and 187 of the BKRF4 gene. To construct the revertant virus (dBKRF4-ins/R) and stop mutant (dBKRF4-stop), the cassette was replaced by the wild-type BKRF4 sequence and the BKRF4 sequence with two stop codons, respectively. (B) Electrophoresis of the recombinant EBV-BAC. Recombinant BAC DNAs were treated with BamHI and EcoRI and separated in an agarose gel. The lower arrow indicates the BamHI-K fragment, which contains the BKRF4 gene, and the upper arrow indicates the size of the BamHI-K fragment plus the Neo/St cassette.
The recombinant EBV-BAC DNAs were introduced into HEK293 cells. Then, green fluorescent protein (GFP)-positive, hygromycin-resistant cells were cloned, in which recombinant viruses were latently maintained as episomes.
Requirement of BKRF4 for efficient infectious progeny production but not for viral gene expression and DNA replication in HEK293 cells.
We investigated whether BKRF4 is required for lytic replication of EBV (Fig. 3). To induce the lytic replication cycle, the expression vector containing BZLF1 was transfected into HEK293 cells latently carrying EBV-BAC clones (wild type, dBKRF4-ins, dBKRF4-ins/R, and dBKRF4-stop).
FIG 3.

Viral protein expression, DNA replication, and progeny production of BKRF4-knockout EBV in HEK293 cells. (A) HEK293 cell clones that latently maintained the recombinant B95-8 EBV-BAC genome constructed above were transfected with the BZLF1 expression vector by electroporation. Cells were harvested after 0 and 2 days and subjected to IB with anti-BZLF1, anti-BRLF1, anti-BALF2, anti-BMRF1, anti-BGLF4, anti-BRRF2, anti-BALF4(gB), anti-LMP1, anti-EBNA1, anti-tubulin, and anti-BKRF4 antibodies. (B) Cells transfected as in panel A were harvested after 0 and 2 days and subjected to qPCR to detect levels of EBV DNA and genomic DNA of host cells. The means ± the SD of three independent biological replicates are shown after normalization to the value of host control. (C) Cells transfected as in panel A were cultured for 3 days, and the virus particles were collected from both the cells and media. After freezing/thawing and centrifugation, supernatants containing virus particles were used to infect Akata(−) cells. The percentage of GFP-positive cells was determined by FACS and is shown in log scale. The means ± the SD of three independent biological replicates are shown. A Student t test was performed. ***, P < 0.005. (D) Cells lytically induced as in panel A were cultured for 3 days, and virion particles were collected from culture media and treated with Turbo DNase I. After DNA extraction, EBV genomic DNA was quantified by qPCR, and the results are shown in log scale. (E) Using the same sample, Akata(−) cells were infected, and the percentage of GFP-positive cells was determined by FACS. (F) Virus stock solutions prepared as in panel D were attached to Akata(−) cells at room temperature with rotation for 3 h. EBV virion DNA levels bound to the cells were determined by qPCR. (G) Akata(−) cells were infected with the same virus stock solution after normalization to the data in panel F, and the percentage of GFP-positive cells was determined by FACS. The means ± the SD of three independent biological replicates are shown. Student t test was performed. **, P < 0.01; ***, P < 0.005; ****, P < 0.0001.
Protein samples were collected on days 0 and 2 and subjected to IB. As shown in Fig. 3A (right panel), BKRF4 was expressed in the day 2 samples of wild-type and revertant clones but not in the dBKRF4-ins and dBKRF4-stop clones. We detected a blurred band of BKRF4 protein in the day 0 wild-type and revertant samples, indicating that this protein can be produced to limited levels even without induction by BZLF1 in HEK293. Expression of the EBV lytic and latent genes—BZLF1 and BRLF1 (IE); BALF2, BMRF1, and BGLF4 (E); BRRF2 and BALF4(gB) (L); and LMP1 and EBNA1 (latent)—were almost equal in the wild-type, revertant, and BKRF4-deficient clones (Fig. 3A). Therefore, the expression of viral proteins, at least for the viral proteins tested here, was not affected markedly by the knockout of BKRF4.
Next, we examined the level of lytic viral DNA synthesis in the cell clones (Fig. 3B). Quantitative PCR (qPCR) analysis revealed that the viral DNA levels in all BZLF1-transfected clones increased about 1 order of magnitude and that the levels of viral DNA synthesis in the dBKRF4-ins and dBKRF4-stop mutants were comparable to those in the wild-type and revertant clones (Fig. 3B).
To determine whether infectious progeny virions were produced, virus particles in the cells and the media were harvested together at day 3 posttransfection with the BZLF1 expression vector. The virus solutions were used to inoculate the EBV-negative B cell line, Akata(−), and GFP-positive cells were counted by fluorescence-activated cell sorting (FACS). As recombinant EBV-BAC expressed GFP protein, Akata(−) cells infected with EBV became GFP positive. The level of infectious progeny virions was significantly lower in cells containing the dBKRF4-ins and dBKRF4-stop clones than in cells containing wild-type and revertant viruses (Fig. 3C).
To extend the progeny data, we then examined whether virus production from HEK293 EBV-BAC cells was inhibited by the BKRF4 knockout or whether progeny virus could be produced efficiently even in the knockout but successful infection and expression of GFP in Akata(−) cells were inhibited. EBV-BAC clones were transfected with BZLF1 expression vectors by electroporation and cultured for 3 days. Progeny virion DNA released into the culture medium was quantified by qPCR (Fig. 3D). Naked DNA that was not packaged into virion was eliminated by DNase treatment before DNA extraction. As a result, the dBKRF4-stop released significantly less progeny virion DNA than did the wild type (5.7-fold) (Fig. 3D), indicating that BKRF4 is involved in progeny secretion into the medium. Because the actual viral titer of the knockout measured by infection to Akata(−) cells was 10.5-fold lower (Fig. 3E), BKRF4 protein may be able to support efficient infection as well.
To exclude the possibility that EBV DNA might be included in the extracellular vesicles, we measured the EBV DNA that can attach to Akata(−) cells (Fig. 3F). Viruses (wild type and dBKRF4-stop mutant) in the stock solutions were inoculated with Akata(−) cells for 3 h with rotation, and the amount of viral DNA attached to the cells was determined. The level of virion DNA of the stop mutant attached to cells was 8.6% of that of the wild type (Fig. 3F). We then adjusted the virus solution to the attached virion DNA level so that the same numbers of virions could bind to the cells. Akata(−) cells were infected with wild-type or dBKRF4-stop mutant virus after normalization, and GFP expression was monitored after 2 days (Fig. 3G). Even though equivalent numbers of virions were infected, successful infection of dBKRF4-stop virus into Akata(−) cells was 0.45%, while that of the wild-type virus was 1.25% (Fig. 3G).
Taken together, our data suggest that BKRF4 deficiency does not affect EBV lytic gene expression and viral DNA synthesis; however, it markedly influenced the production of progeny by about 5- or 10-fold, and it also decreased infection efficiency by 2- or 3-fold upon de novo infection.
Interaction between BKRF4 and BGLF2, and requirement of the C-terminal region of BKRF4 for this interaction.
Assembly of the herpesvirus virion is regulated by multiple protein-protein interactions (7–9). Interestingly, KSHV ORF45 interacts with another viral tegument protein, ORF33, through its C terminus, and this interaction appears to play a fundamental role in the function of ORF45 (16). Because the 15-amino-acid sequence of the C terminus is highly conserved between BKRF4 and ORF45 of gammaherpesvirus, while other parts are not well conserved, we hypothesized that the EBV BKRF4 protein might interact with BGLF2 protein, the homolog of KSHV ORF33. To test the interaction, HEK293T cells were cotransfected with pcDNA (empty vector), Flag-BKRF4, Flag-BKRF4dc, and HA-BGLF2, as indicated in Fig. 4, and the transfected cells were subjected to immunoprecipitation (IP) assays with anti-FLAG antibody. A deletion mutant lacking the conserved C-terminal 14 amino acids of BKRF4 (Flag-BKRF4dc) lacked the following amino acids: PPIKGNNNYNWPWL. Our IP-IB data clearly indicate that HA-BGLF2 coprecipitated with wild-type Flag-BKRF4 but not with the C-terminal mutant of BKRF4 (Fig. 4).
FIG 4.

BKRF4 interacts with BGLF2. HEK293T cells were cotransfected with the indicated expression vectors by lipofection. After 24 h, whole-cell lysates were prepared, and a portion of the cell lysates was subjected to IB with anti-Flag and anti-HA antibodies. The remaining portions of the lysates were subjected to IP with anti-FLAG mouse antibody, followed by IB with the above-mentioned antibodies.
Successful restoration of infectious virion production of knockout virus by an exogenous supply of BKRF4.
We investigated whether an exogenous supply of BKRF4 in trans could rescue the reduced production of infectious progeny in the knockout virus. The expression vectors indicated in Fig. 5A were cotransfected into HEK293 cells latently infected with wild-type or BKRF4-knockout mutant (dBKRF4-stop). An exogenous supply of BKRF4 had no obvious effect in the wild type (Fig. 5A). However, an exogenous supply of wild-type BKRF4 fully complemented the previously reduced progeny levels in the knockout (Fig. 5A dBKRF4-stop, white bar) compared to the control (Fig. 5A, dBKRF4-stop, black bar). On the other hand, BKRF4 lacking the C terminus increased the progeny levels only partially (Fig. 5A, dBKRF4-stop, gray bar). Protein expression assays (Fig. 5B) revealed that although BKRF4 was undetectable in the stop mutant, as expected, it was detectable when wild-type BKRF4 was expressed in trans (Fig. 5B, third panel from the top). Our anti-BKRF4 antibody failed to detect the C-terminal deletion mutant of BKRF4 as the deleted region overlaps the immunized polypeptide sequence. Therefore, we used the anti-FLAG antibody to detect expression of the C-terminal deletion mutant of BKRF4 (Fig. 5B, panel 4). Other viral lytic proteins (BZLF1, BALF2, BMRF1, BALF4, and BRRF2) were expressed at almost equal levels in the knockout virus compared to wild type (Fig. 5B). Our data further confirm that BKRF4 is important for efficient production of progeny virions and that the interaction between BKRF4 and BGLF2 through its conserved C-terminal motif plays an important role in efficient progeny production, at least in part.
FIG 5.

Complementation of reduced progeny virions by exogenous supply of BKRF4. (A) HEK293 cells that carry recombinant EBV-BAC in the latent state were transfected with indicated expression vectors. Three days after transfection, cells and medium were collected together and processed as in Fig. 3C to determine the virus titer. Three independent samples were assayed, and the means ± the SD are shown. A Student t test was performed. *, P < 0.05; ***, P < 0.005. (B) Aliquots of the cells transfected for panel A were collected on the second day and subjected to IB with anti-BALF2, anti-BMRF1, anti-BKRF4, anti-Flag, anti-BZLF1, anti-BRRF2, anti-BALF4(gB), and anti-tubulin antibodies.
Localization of BKRF4 in the nucleus and perinuclear region.
We investigated the intracellular localization of BKRF4 by IF assay in AGS (EBV negative) (Fig. 6A), AGS/EGFP-EBV (Fig. 6B), and B95-8 cells (Fig. 6C). The lytic cycle was induced by transfection of BZLF1 and monitored by the staining of BMRF1 (green). We found neither BKRF4 nor BMRF1 signal in EBV-negative AGS cells, but in AGS/EGFP-EBV and B95-8 cells we found that the BKRF4 protein (red) localized in the nucleus and perinuclear region upon induction with BZLF1 (Fig. 6B and C). Although the BMRF1 protein formed typical replication compartments in the nucleus (30), the BKRF4 pattern did not match with that of BMRF1 (Fig. 6B and C).
FIG 6.

Subcellular localization of BKRF4 in infected cells. (A) AGS cells were transfected with the expression vectors (indicated on the left) by lipofection. After 2 days, cells were fixed and stained with anti-BKRF4 (red) and anti-BMRF1 (green) antibodies and DAPI (blue) and then analyzed by using a confocal laser microscope. (B) AGS/EGFP-EBV cells were transfected, fixed, stained, and analyzed as in panel A. (C) B95-8 cells were transfected, fixed, stained, and analyzed as in panel A.
Next, we examined the subcellular localization of exogenously expressed Flag-BKRF4 (red) and/or HA-BGLF2 (green) in HeLa cells. As shown in Fig. 7, BKRF4 predominantly localized in the nucleus in the punctate pattern and perinuclear region, whereas BGLF2 was located mostly in the cytoplasm when singly produced (Fig. 7). When BGLF2 was cotransfected with BKRF4, the BGLF2 protein was transported into the nucleus and BGLF2 merged with BKRF4 in the nucleus and peripheral region (Fig. 7). The nuclear targeting of BGLF2 protein by BKRF4, and colocalization of the two proteins was very efficient. BGLF2 protein redistributed predominantly from the cytoplasm to the nuclei and nuclear rim in 43 of 43 cotransfected cells (100%), and BGLF2 colocalized with BKRF4 in 43 (100%) of the cotransfected nuclei and the nuclear rim. These results indicate that BKRF4 protein localizes in the nucleus and perinuclear region and can colocalize with BGLF2 protein.
FIG 7.

Subcellular localization of BKRF4 in transfected cells. HeLa cells were transfected with the expression vectors (indicated on left) by lipofection. After 1 day, the cells were fixed and stained with anti-Flag (red) and anti-HA (green) antibodies and DAPI (blue) and then analyzed by confocal laser microscopy.
CRISPR-Cas9-mediated knockout of BKRF4 in Akata strain EBV.
As mentioned above, we used the B95-8 EBV-BAC system to demonstrate that the BKRF4 gene is involved in progeny production in HEK293 cells. To investigate whether this phenotype could be observed in a more natural host cell type for EBV, such as B cells or gastric epithelial cells, we tried to infect other cells with recombinant EBV produced from the HEK293 EBV-BAC. We successfully infected Akata(−) or AGS cells with B95-8 EBV-BAC and stably maintained the virus in the cells; however, induction of the lytic cycle in such cells could not be achieved efficiently, thereby hampering our research on the role of EBV lytic genes in a natural host cell type.
To circumvent this problem, we adopted the CRISPR/Cas9-mediated gene editing method. AGS/EGFP-EBV cells that contained recombinant Akata virus with the EGFP and G418 resistance genes were transfected with pX459-BKRF4, the vector designated for CRISPR/cas9-mediated BKRF4 knockout. After puromycin selection, the AGS cells were transfected with BZLF1 to induce progeny production, and the resultant viruses were used to infect Akata(−) cells. These cells were then cloned in the presence of G418. In this manner, we successfully obtained Akata cells with recombinant Akata virus with a mutation in the BKRF4 gene. We confirmed by sequencing that the Akata cell line with the BKRF4 knockout virus (BKRF4-KO) contained a frameshift mutation in the BKRF4 gene.
The Akata cell lines with BKRF4-KO or with wild-type BKRF4 virus were treated with anti-IgG, and protein samples were collected after 0 or 2 days for IB (Fig. 8A). As expected, BKRF4 protein was absent in the KO (Fig. 8A, right panel). Viral protein levels were comparable between the wild-type and BKRF4-KO samples (Fig. 8A, left panel), with the exception that expression of some viral proteins, such as BRLF1, BGLF4, BMRF1, BALF4(gB), and LMP1 may be marginally lower.
FIG 8.

Viral protein expression, viral DNA replication, and virion production of BKRF4-knockout EBV in Akata. (A to G) The BKRF4 gene in the Akata virus was mutated using the CRISPR/Cas9 system in AGS/EGFP-EBV cells, followed by infection of Akata(−) cells and cell cloning. A cell line latently infected with wild-type or BKRF4-knockout (BKRF4-KO) virus was used. (A) The Akata cell clones (wild-type and BKRF4-KO) were either treated with anti-IgG for lytic induction for 0 or 2 days. Cell proteins were subjected to IB with anti-BZLF1, anti-BRLF1, anti-BALF2, anti-BMRF1, anti-BGLF4, anti-BALF4(gB), anti-LMP1, anti-EBNA1, anti-tubulin, and anti-BKRF4 antibodies. (B) The Akata cell clones treated as in panel A were harvested after 0 or 2 days and subjected to qPCR to detect levels of EBV DNA and genomic DNA of host cells. The means ± the SD of three independent biological replicates are shown after normalization to the value of host control. (C) The Akata cell clones were treated with or without anti-IgG for 2 days. Progeny virions in the supernatant were determined as in Fig. 3C by FACS. Three independent samples were assayed and the means ± the SD are shown. A Student t test was performed. *, P < 0.05; ****, P < 0.0001. (D) Akata cell clones were lytically induced as in panel A and cultured for 2 days. Progeny virion DNA in the culture medium was determined as in Fig. 3D. (E) Using the same sample, Akata(−) cells were infected, and the percentage of GFP-positive cells was determined by FACS. The results are shown in log scale. (F) Virus stock solutions prepared as for panel D were attached to Akata(−) cells at room temperature with rotation for 3 h. EBV virion DNA levels bound to the cells were determined by qPCR. (G) Akata(−) cells were infected with the same virus stock solution after normalization to the data in panel F, and the percentage of GFP-positive cells was determined by FACS. The means ± the SD of three independent biological replicates are shown. A Student t test was performed. ****, P < 0.0001.
Viral DNA quantitation by qPCR revealed that the BKRF4-KO virus could successfully synthesize viral DNA almost as efficiently as wild-type virus in Akata cells (Fig. 8B), although the background viral DNA level was somewhat higher in the KO, due to unknown reasons.
Next, we examined the levels of progeny virions produced from the Akata cells. Cells were treated with or without anti-IgG for 2 days. Viruses in the media were used to infect Akata(−) cells, followed by quantification of GFP-positive cells by FACS analysis. The level of infectious progeny virions produced from BKRF4-KO was significantly lower than that from the wild type (Fig. 8C).
Then we analyzed the progeny virion release into the culture medium by means of qPCR. After anti-IgG treatment for 2 days, the culture supernatants from Akata wild-type and BKRF4-KO clones were subjected to DNase I treatment and DNA extraction, followed by qPCR. The progeny virion DNA levels produced in the media were significantly reduced in BKRF4-KO by 5.5-fold compared to the wild type (Fig. 8D). However, infectious progeny levels decreased even more to 26.2-fold compared to the wild type (Fig. 8E). Therefore, BKRF4 may increase the infectivity upon de novo infection to Akata(−) cells, too. Interestingly, the effect of the BKRF4 knockout on the efficiency of de novo infection in Akata cells (Fig. 8D and E, 26.2/5.5 = 4.8-fold) seems severer than that in HEK293 cells (Fig. 3D and E, 10.5/5.7 = 1.8-fold).
FIG 10.
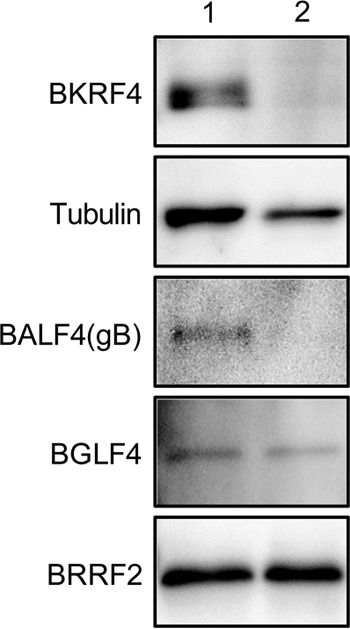
Identification of BKRF4 in the virion. (A) EBV from B95-8 cells was precleared by low-speed centrifugation and filtration. Virus particles were collected by ultracentrifugation in the presence (lane 2) or absence (lane 1) of NP-40, followed by IB with anti-BKRF4, anti-tubulin, anti-BALF4(gB), anti-BGLF4, and anti-BRRF2 antibodies.
To further confirm the result, we measured viral DNA that could attach to the Akata(−) cells (Fig. 8F). Viral DNA of the knockout virus was 6.4% of that of the wild type (Fig. 8F). Based on these DNA data, equivalents number of viruses were used to infect Akata(−) cells, followed by FACS analysis to check the GFP positivity ratio. Efficiency of de novo infection was significantly decreased in the knockout sample by 6.9-fold (Fig. 8G). Again, the effect of BKRF4 knockout was more prominent in Akata (Fig. 8G, 6.9-fold) than in HEK293 (Fig. 3G, 2.8-fold) cells. Our data confirmed in the context of Akata, too, that while BKRF4 did not affect viral gene expression and DNA synthesis, it played an important role in the production of progeny virus and in efficient de novo infection.
Phosphorylation of BKRF4 protein.
Since BKRF4 may be phosphorylated (10), we investigated its phosphorylation in both infected (Fig. 9A) and transfected cells (Fig. 9B). Here, we used Phos-tag gel electrophoresis (Wako Chemicals) and conventional SDS-PAGE, followed by IB. In the Phos-tag gel, phosphoproteins are trapped due to the bindings of the Phos-tag molecule to the phosphate group, and their migration during electrophoresis is reduced (31). HEK293 cell clones that latently maintain the recombinant wild-type or BGLF4 knockout (BGLF4-KO) EBV-BAC genome were transfected with the BZLF1 expression vector, and cells were harvested on day 0 or 2 posttransfection, treated with or without lambda phosphatase, and electrophoresed in a Phos-tag gel or by conventional SDS-PAGE (Fig. 9A). In infected cells, a part of BKRF4 protein migrated slowly in the Phos-tag gel (Fig. 9A, arrowhead), which was eliminated by lambda phosphatase treatment, indicating phosphorylation of the protein. Interestingly, this slowly migrating BKRF4 band in the Phos-tag gel was not detected in the BGLF4-knockout cells. Therefore, BGLF4, the only protein kinase encoded by EBV, may somehow be involved in the phosphorylation of BKRF4.
FIG 9.
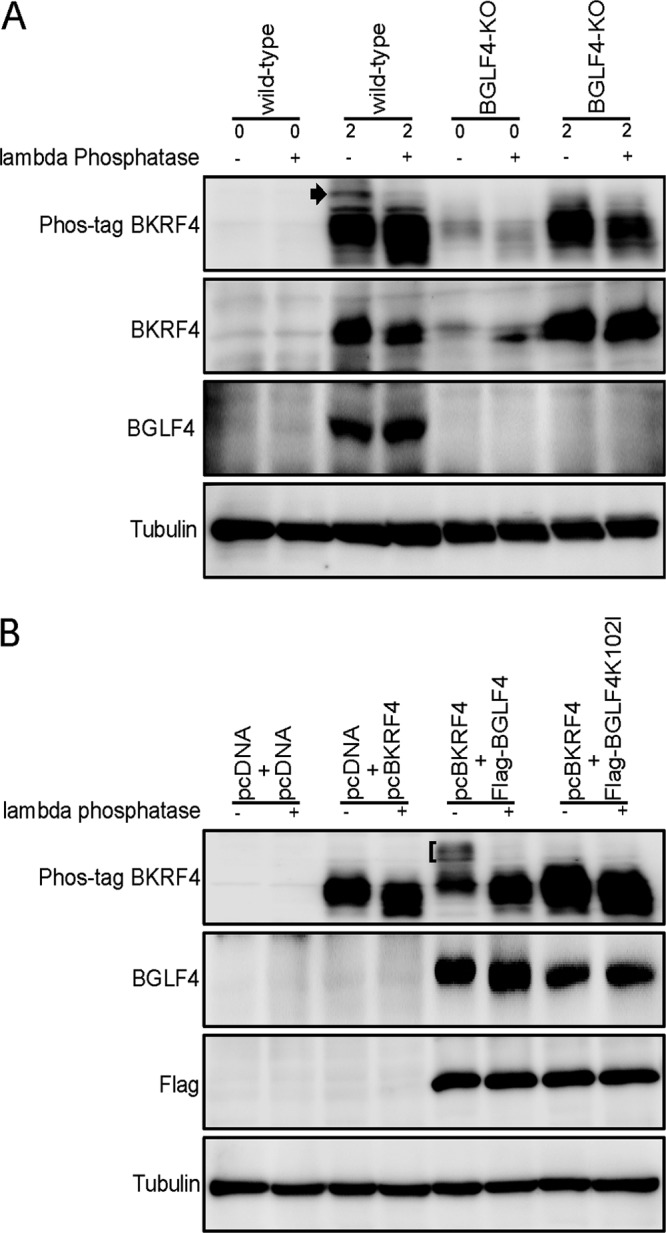
Phosphorylation of BKRF4. (A) HEK293 cell clones that latently maintain the wild type or BGLF4 knockout (BGLF4-KO) recombinant EBV-BAC genome were transfected with the BZLF1 expression vector by electroporation and harvested on days 0 and 2. Cell lysates were treated with or without lambda phosphatase and separated by Phos-tag gel electrophoresis (Phos-tag) or conventional SDS-PAGE, followed by IB with anti-BKRF4, anti-BGLF4, and anti-tubulin antibodies. (B) HEK293T cells were transfected with the expression vectors by lipofection. After 1 day, the cells were harvested and treated as in panel A and subjected to IB with anti-BKRF4, anti-BGLF4, anti-Flag, and anti-tubulin antibodies.
To extend this, HEK293T cells were transfected with the indicated expression vectors (Fig. 9B), harvested on the following day, and treated as described above. As expected, a part of the BKRF4 protein migrated slowly in the Phos-tag gel when BKRF4 was coexpressed with BGLF4 (Fig. 9B, bracket), which could be collapsed by phosphatase treatment. Importantly, the slowly migrating bands could not be observed when the kinase-dead form of BGLF4 (K102I) was cotransfected (Fig. 9B). Our data indicate that BKRF4 is phosphorylated in both infected and transfected cells and that the EBV protein kinase, BGLF4, may be involved in the phosphorylation.
BKRF4 was present as a virion component.
Since it has been suggested that BKRF4 is in the tegument (10), we examined its presence in purified virions using our anti-BKRF4 antibody. Virions collected in the presence (Fig. 10, lane 2) or absence (lane 1) of 1% NP-40 were subjected to SDS-PAGE, followed by IB. NP-40 completely removes envelope proteins and causes partial dissociation of the tegument proteins. As shown in Fig. 10, lane 1, BKRF4 could clearly be detected in the virion fraction like other virion proteins, such as tubulin, BALF4(gB), BGLF4, and BRRF2 (10). As expected, being an envelope protein, gB was washed out by NP-40 treatment (Fig. 10, lane 2). The BGLF4 and BRRF2 are tegument proteins and were clearly resistant to NP-40 treatment (Fig. 10, lane 2). Interestingly, BKRF4 could also be eliminated by NP-40, indicating that BKRF4 is likely associated with the envelope or, at least, is not very tightly attached to the nucleocapsid. This is not surprising because BKRF4 interacts with a tegument protein BGLF2, which is a myristoylated protein (BBLF1)-binding protein (32).
DISCUSSION
BKRF4 is one of the virion proteins of EBV conserved among gammaherpesviruses, but no homolog exists in alpha- or beta-herpesviruses. In this study, we analyzed the previously uncharacterized BKRF4 protein by generating BKRF4 knockouts using BAC (Fig. 2 and 3) and CRISPR/Cas9 (Fig. 8) systems. We found that deletion of BKRF4 in the B95-8 EBV-BAC or CRISPR/Cas9 systems had no marked effect on the expression of viral genes and DNA synthesis; however, it significantly reduced the production of progeny virions and infectivity of the progeny. Our results coincide with those from a previous study involving KSHV ORF45 (12), which showed that its null recombinant virus released fewer virions by 10-fold, and its infectivity was also lower but had no significant effects on viral gene expression and DNA replication. Although the role of EBV BKRF4 has not previously been analyzed, the ORF45 gene of KSHV has been extensively studied. KSHV ORF45 inhibits type I interferon by blocking phosphorylation and nuclear accumulation of IRF-7 (14, 33–35). It can efficiently activate RSK, eIF4B, and ERK (36–41). The interaction between KSHV ORF45 and SIAH-1 promotes ubiquitination and proteasomal degradation of the protein (42). It can associate with the ubiquitin-specific protease USP7 and stabilize ORF33 and ORF36 (15, 16). Monoubiquitinated ORF45 mediates the association between KSHV virus particles and lipid rafts for virus assembly and egress (17); it also interacts with kinesin-2 (13). However, with low sequence homology between EBV BKRF4 and KSHV ORF45, the amino acid sequences in the interacting domains with those partners are not well conserved in EBV BKRF4. In fact, we and others have found that EBV BKRF4 has no effect on IRF and ERK/RSK signaling (not shown). The only conserved residues are in the C-terminal region, a region where ORF45 and BKRF4 interact with ORF33 and BGLF2, respectively. Notably, EBV BGLF2 was recently found to activate AP-1 through activation of mitogen-activated proteins (MAPKs) (43). Although EBV BKRF4 cannot activate ERK/RSK by itself, it can associate with the activator of MAPK. Therefore, EBV BKRF4 may play a crucial role in activating MAPK signaling through the interaction with BGLF2. Moreover, EBV BGLF2, KSHV ORF33, and its HSV homolog UL16 have been reported to associate with other myristoylated and palmitoylated tegument proteins (BBLF1, ORF38, and UL11, respectively) (32, 44), and these interactions are crucial for efficient virus assembly, particularly via the secondary envelopment process involving certain cytoplasmic membrane structures, presumably the trans-Golgi network or endosomes. Since EBV BKRF4 was localized predominantly in the nucleus and nuclear rim in infected or transfected cells (Fig. 6 and 7), we speculate that the protein may attach to the nucleocapsid in the nucleus and may play a role in the initial envelopment and deenvelopment at the nuclear membrane. In addition, there may be an interaction between BKRF4 and the membrane-anchored BGLF2/BBLF1 complex in the cytoplasm, which may contribute to efficient secondary envelopment.
We also found that EBV BKRF4 protein can be phosphorylated and that the only EBV protein kinase, BGLF4, increased the phosphorylation either directly or indirectly (Fig. 9). It has been reported that the homolog of BKRF4 in KSHV, ORF45, can also be phosphorylated (11), and KSHV ORF36, the homolog of BGLF4, contributes to the phosphorylation (15). Thus, EBV BKRF4 and KSHV ORF45 proteins likely share certain functions or regulatory mechanisms.
In this study, we successfully generated BKRF4 knockout virus using the CRISPR/Cas9 system from EBV of the Akata strain, cloned the cells that contained the mutant virus, and analyzed the phenotype of the knockout virus in B cells, the natural host of EBV. Although this process may sound simple, cloning cells latently infected with the edited virus and triggering lytic induction from the latent cells is complex. A previous study edited the promoter region of BART miRNA and successfully collected cells with edited virus by FACS (45, 46). The present study is the second example of editing EBV by CRISPR/Cas9 for viral mutagenesis.
Our results indicate the importance of the BKRF4 gene in the production of infectious progeny virions. This function may occur, at least in part, via the interaction with BGLF2 through the C-terminal motif. The BKRF4/BGLF2 complex may somehow induce maturation or egress of EBV particles in virus-producing cells. Otherwise, the complex may somehow support fusion or transportation to the nucleus upon de novo infection. In addition, it may activate viral gene expression in newly infected cells as tegument components because BGLF2 can strongly activate the MAPK signal (43). Further studies are required to determine the underlying molecular mechanism of how BKRF4 is required for efficient infectivity of EBV.
MATERIALS AND METHODS
Cell culture and reagents.
HEK293, HEK293 EBV-BAC, HEK293T, and HeLa cells were maintained in Dulbecco modified Eagle medium (Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS). HEK293 cells with BGLF4-knockout EBV-BAC were prepared previously (21). B95-8, AGS, AGS/EGFP-EBV (contains recombinant Akata virus with EGFP and G418 resistance) (22), and Akata(−) cells were cultured in RPMI 1640 (Sigma-Aldrich) medium containing 10% FBS. TPA, A23187 (calcium ionophore), and PAA were purchased from Sigma-Aldrich, and sodium butyrate was from Wako Chemicals.
Antiserum against EBV BKRF4 was prepared by immunizing a rabbit with the synthetic peptide CETTPPIKGNNNYNWPWL, and the serum was purified by affinity purification using the same peptide. Antibodies against BZLF1, BRLF1, BMRF1, BALF2, BALF4(gB), BALF5, BRRF2, LMP1, and EBNA1 have been reported previously (23–25). Mouse anti-Flag, mouse anti-BMRF1, anti-α/β-tubulin, and anti-HA antibodies were purchased from Sigma-Aldrich, Novocastra, and Cell Signaling, respectively. Horseradish peroxidase (HRP)-linked goat antibodies to mouse or rabbit IgG were purchased from Amersham Biosciences. Secondary goat anti-rabbit and anti-mouse IgG antibodies conjugated with Alexa 488 and Alexa 546 and a Zenon mouse IgG labeling kit were obtained from Molecular Probes.
Construction of expression plasmids.
The expression vector pcDNABZLF1, pcDNAFlagBGLF4, and pcDNAFlagBGLF4K102I have previously been reported (21). The expression vector for BKRF4 (pcDNABKRF4) was constructed by inserting the BKRF4 ORF into pcDNA3 (Invitrogen) using the following amplification primers: forward (TAGAGAATTCATGGCCATGTTTCTGAAGTC) and reverse (CTATCTCGAGTTACAGCCATGGCCAATTGT). pcDNAFlagBKRF4 was constructed by inserting the BKRF4 ORF into pcDNAFlag. The BKRF4 C-terminal deletion mutant vector (pcDNABKRF4dc) was generated using a PCR-based method with forward (TAACTCGAGCATGCATCTAG) and reverse (GGTGGTTTCCATCATGTGTT) primers. Expression plasmid for HA-tagged BGLF2 (HA-BGLF2) was constructed using the following primers: (AAACGGGCCCTCTAGAATGGCATCCGCCGCGAACAG) and (CGTATGGGTAGAATTCATAAGAATGTAAGACCTGAC). Two oligonucleotide sequences, forward (CACCTGACTACTCAGATGAAGACG) and reverse (AAACCGTCTTCATCTGAGTAGTCA), were annealed and inserted into the BbsI site of the pX459 (Addgene) to create pX459-BKRF4, the vector for CRISPR/cas9-mediated BKRF4 knockout.
Genetic manipulation of EBV-BAC DNA and cloning of HEK293 cells with EBV-BAC.
B95-8 EBV-BAC DNA was provided by W. Hammerschmidt (26). Homologous recombination was carried out in E. coli as described previously (21).
To prepare recombinant viruses, a transfer DNA fragment for the first recombination was generated by PCR using rpsL-neo vector (Gene Bridges, Germany) as the template, with the following primers: Neo/St forward (TTTCTGAAGTCGCGTGGGGTCCGGTCTTGCAGGGACCGGCGCCTCTTGTCGGACGAGGAGGGCCTGGTGATGATGGCGGGATC) and Neo/St reverse (CCTCCTGGATAGACTGGGAGGCCTGAGACCCCAGAGTGTAGCTGCTGCTCTGTGAAGTCTTCAGAAGAACTCGTCAAGAAGG). To obtain the insertion mutant (dBKRF4-ins), kanamycin-resistant colonies were selected after recombination and checked by colony PCR using forward (GAGATAGATTGGAGGCTGTAG) and reverse (GACTCATCAGTGTCACTCAC) primers. The revertant mutants (dBKRF4-ins/R) were constructed by replacing the neomycin resistance and streptomycin sensitivity gene (Neo/St) cassette with a wild-type BKRF4 sequence. The wild-type PCR fragment was amplified using the above-mentioned primers. To generate the dBKRF4-stop mutant, the Neo/St cassette was replaced with the BKRF4 sequence containing a premature stop codon. The following oligonucleotides were annealed and used as a template to create the stop mutant: forward (GAGATAGATTGGAGGCTGTAGAGGGGTCATCACTATGGCCATGTTTCTGAAGTCGCGTGGGGTCCGGTCTTGCAGGGACCGGCGCCTCTTGTCGGACGAGGAGTAAT) and reverse (GACTCATCAGTGTCACTCACGTCCTCCTCCTGGATAGACTGGGAGGCCTGAGACCCCAGAGTGTAGCTGCTGCTCTGTGAAGTCTATTACTCCTCGTCCGACAAG). Electroporation of E. coli was performed using a Gene Pulser III (Bio-Rad), and EBV-BAC DNA was purified using NucleoBond Bac100 (Macherey-Nagel). Recombination was confirmed by PCR, sequence analysis, and electrophoresis of the BamHI- or EcoRI-digested viral genome.
HEK293 cells were transfected with the recombinant EBV-BAC DNA using Lipofectamine 2000 reagent, followed by culture on 10-cm dishes maintained with 150 μg/ml hygromycin B. After 2 weeks, GFP-positive, hygromycin-resistant cell colonies were cloned.
Disruption of the BKRF4 gene using the CRISPR/cas9 system.
AGS/EGFP-EBV cells were transfected with the CRISPR/cas9 vector pX459-BKRF4 using Lipofectamine 2000 reagent (Invitrogen). After puromycin selection, pcDNABZLF1 was transfected to induce progeny production by electroporation using the Neon transfection system (Thermo Fisher Scientific). Then the supernatant containing virus particles was harvested and used to infect Akata(−) cells. To obtain GFP-positive cell clones, infected cells were seeded after limiting dilution in 96-well plates and cultured with 750 μg/ml G418 for 3 to 4 weeks.
Transfection, immunoblotting, and immunoprecipitation.
Cells were transfected with the indicated plasmid DNAs using Lipofectamine 2000 reagent or by electroporation using the Neon transfection system. The total amount of plasmid DNA was standardized by the addition of an empty vector. At 2 days posttransfection, cells were washed with phosphate-buffered saline (PBS), harvested, and solubilized in sample buffer for IB, as described previously (21). Akata cells were induced with anti-IgG for 2 days. For immunoprecipitation (IP), HEK293T cells were transfected with the indicated plasmid DNA using Lipofectamine 2000 reagent; at 24 h posttransfection, the cells were solubilized in IP lysis buffer (20 mM Tris-HCl [pH 7.8], 150 mM NaCl, 0.5% NP-40, 1 mM EDTA, and protease inhibitor cocktail tablets [Complete mini, Roche]), followed by sonication and centrifugation at 15,000 rpm for 5 min. The supernatants were then mixed with anti-FLAG mouse antibody and protein G-Sepharose 4 Fast Flow (GE Healthcare), followed by incubation at 4°C for >2 h with rotation. Immunocomplexes were washed four times with the IP lysis buffer. Samples were subjected to SDS-PAGE, followed by IB with the antibodies indicated in the figures. TrueBlot goat anti-rabbit/mouse IgG HRP-conjugated antibodies (eBioscience) were used as the secondary antibodies. Phos-tag gel electrophoresis was carried out as described previously (23). Briefly, cells were lysed in phosphatase buffer with sonication and treated with or without lambda phosphatase (Santa Cruz) and separated on a SuperSep Phos-tag 7.5% gel (Wako Chemicals), before being subjected to IB.
Immunofluorescence analysis.
AGS/EGFP-EBV, EBV-negative AGS, and B95-8 cells were transfected with pcDNABZLF1 using Lipofectamine 2000 reagent or by electroporation using the Neon transfection system, and cells were fixed with 70% ethanol at 2 days posttransfection. Cells were permeabilized with 0.1% Triton X-100, blocked with 10% normal goat serum (Funakoshi) and incubated with anti-BKRF4 rabbit antibody and anti-BMRF1 mouse antibody. Next, the cells were washed with PBS and stained with secondary antibody (Alexa 546 anti-rabbit IgG and Alexa 488 anti-mouse IgG; Invitrogen).
HeLa cells were transfected with pcDNA (empty vector) or FlagBKRF4 with or without HA-BGLF2 using Lipofectamine 2000 reagent. Cells were fixed with 70% ethanol, permeabilized with 0.1% Triton X-100, and blocked as described above. The cells were incubated with anti-Flag mouse antibody and anti-HA rabbit antibody, washed with PBS, and then stained with secondary antibody (Alexa 546 anti-mouse IgG and Alexa 488 anti-rabbit IgG). After treatment with secondary antibody, the cells were washed with PBS and mounted in ProLong Gold antifade reagent with 4′, 6′-diamidino-2-phenylindole (DAPI; Molecular Probes). LSM880 confocal microscopy (Zeiss) was used to analyze the sample.
Quantification of viral DNA synthesis.
Levels of viral DNA synthesis were determined using qPCR, as described previously (28). Briefly, cells were washed with PBS, lysed in lysis buffer with sonication, and treated with proteinase K. After deactivation of the proteinase, qPCRs were performed using Fast Start Universal Probe Master (Rox; Roche Applied Science). A standard curve drawn using serial dilutions of DNA was used to quantify the amount of DNA. The probe and primers for detection of the viral genome were designed in the BALF2-coding region.
Virus titration by FACS analysis.
To induce lytic replication in HEK293 cells carrying EBV-BAC, cells were transfected with pcDNABZLF1 using the Neon transfection system. After 3 days, cells and culture media were collected and subjected to high-speed centrifugation. For the Akata CRISPR/cas9 mutant, cells were lytically induced using anti-IgG; after 2 days postinduction, the supernatants were collected, followed by centrifugation and filtration. Then, Akata(−) cells were infected with the virus solution for >2 h at room temperature with rotation. After 2 days, the cells were fixed with 1% formaldehyde, washed with PBS, and resuspended in PBS. GFP-positive cells were counted using the FACSCalibur G5 system (Becton Dickinson), according to the manufacturer's instructions.
Quantification of extracellular virion DNA.
Extracellular virions were quantified using qPCR as described previously with some modifications (17). Briefly, 200 μl of virus stock was pretreated with 2 μl of Turbo DNase I (Invitrogen) for 1 h at 37°C. The reaction was stopped by addition of EDTA (15 mM) and incubated at 75°C for 10 min to heat inactivate the DNase I. Then, 20 μl of proteinase K solution and 200 μl of buffer AL from the DNeasy blood and tissue kit (Qiagen) were added. The mixture was kept at 70°C for 15 min, and then the DNA was extracted using the kit according to the manufacturer's instructions. Finally, the relative levels of viral DNA were quantified as described above.
Quantification of virion DNA bound to cells.
Virion DNA levels bound to Akata(−) cells were quantified as described previously (29). Briefly, Akata(−) cells were inoculated with virus stock solutions for 3 h at room temperature with rotation and washed extensively with PBS. After Turbo DNase I treatment, cell-associated DNA was extracted and quantified as described above.
Purification of extracellular EBV virions.
Culture supernatants from EBV B95-8 cells were collected after 5 days of incubation. After low-speed centrifugation and filtration, the culture supernatants were treated with or without 1% Nonidet P-40 (NP-40) and then subjected to ultracentrifugation (Himac CP 60E; Hitachi) at 15,000 rpm for 1 h. Finally, pellets were solubilized in sample buffer and subjected to SDS-PAGE, followed by IB.
ACKNOWLEDGMENTS
We thank W. Hammerschmidt, H. J. Delecluse, E. Gershburg, Y. Narita, T. Tsurumi, T. Kanda, and H. Yoshiyama for materials and discussions.
This study was supported by grants-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (to T.M. [15K08494], H.K. [17H04081], and Y.S. [16H06231]), the Japan Agency for Medical Research and Development (to T.M. [Japanese Initiative for Progress of Research on Infectious Disease for Global Epidemic, 17fm0208016] and H.K. [Practical Research Project for Rare/Intractable Diseases, 16ek0109098]), and the Takeda Science Foundation and Toyoaki Scholarship Foundation (to T.M.).
REFERENCES
- 1.Murata T, Sato Y, Kimura H. 2014. Modes of infection and oncogenesis by the Epstein-Barr virus. Rev Med Virol 24:242–253. doi: 10.1002/rmv.1786. [DOI] [PubMed] [Google Scholar]
- 2.Young LS, Rickinson AB. 2004. Epstein-Barr virus: 40 years on. Nat Rev Cancer 4:757–768. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- 3.Murata T, Tsurumi T. 2014. Switching of EBV cycles between latent and lytic states. Rev Med Virol 24:142–153. doi: 10.1002/rmv.1780. [DOI] [PubMed] [Google Scholar]
- 4.Lieberman PM. 2015. Chromatin structure of Epstein-Barr virus latent episomes. Curr Top Microbiol Immunol 390:71–102. doi: 10.1007/978-3-319-22822-8_5. [DOI] [PubMed] [Google Scholar]
- 5.Hammerschmidt W, Sugden B. 2013. Replication of Epstein-Barr viral DNA. Cold Spring Harb Perspect Biol 5:a013029. doi: 10.1101/cshperspect.a013029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murata T. 2014. Regulation of Epstein-Barr virus reactivation from latency. Microbiol Immunol 58:307–317. doi: 10.1111/1348-0421.12155. [DOI] [PubMed] [Google Scholar]
- 7.Diefenbach RJ. 2015. Conserved tegument protein complexes: essential components in the assembly of herpesviruses. Virus Res 210:308–317. doi: 10.1016/j.virusres.2015.09.007. [DOI] [PubMed] [Google Scholar]
- 8.Owen DJ, Crump CM, Graham SC. 2015. Tegument assembly and secondary envelopment of alphaherpesviruses. Viruses 7:5084–5114. doi: 10.3390/v7092861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sathish N, Wang X, Yuan Y. 2012. Tegument proteins of Kaposi's sarcoma-associated herpesvirus and related gammaherpesviruses. Front Microbiol 3:98. doi: 10.3389/fmicb.2012.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johannsen E, Luftig M, Chase MR, Weicksel S, Cahir-McFarland E, Illanes D, Sarracino D, Kieff E. 2004. Proteins of purified Epstein-Barr virus. Proc Natl Acad Sci U S A 101:16286–16291. doi: 10.1073/pnas.0407320101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu FX, Yuan Y. 2003. The ORF45 protein of Kaposi's sarcoma-associated herpesvirus is associated with purified virions. J Virol 77:4221–4230. doi: 10.1128/JVI.77.7.4221-4230.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu FX, Li X, Zhou F, Gao SJ, Yuan Y. 2006. Functional characterization of Kaposi's sarcoma-associated herpesvirus ORF45 by bacterial artificial chromosome-based mutagenesis. J Virol 80:12187–12196. doi: 10.1128/JVI.01275-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sathish N, Zhu FX, Yuan Y. 2009. Kaposi's sarcoma-associated herpesvirus ORF45 interacts with kinesin-2 transporting viral capsid-tegument complexes along microtubules. PLoS Pathog 5:e1000332. doi: 10.1371/journal.ppat.1000332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu FX, King SM, Smith EJ, Levy DE, Yuan Y. 2002. A Kaposi's sarcoma-associated herpesviral protein inhibits virus-mediated induction of type I interferon by blocking IRF-7 phosphorylation and nuclear accumulation. Proc Natl Acad Sci U S A 99:5573–5578. doi: 10.1073/pnas.082420599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Avey D, Tepper S, Pifer B, Bahga A, Williams H, Gillen J, Li W, Ogden S, Zhu F. 2016. Discovery of a coregulatory interaction between Kaposi's sarcoma-associated herpesvirus ORF45 and the viral protein kinase ORF36. J Virol 90:5953–5964. doi: 10.1128/JVI.00516-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gillen J, Li W, Liang Q, Avey D, Wu J, Wu F, Myoung J, Zhu F. 2015. A survey of the interactome of Kaposi's sarcoma-associated herpesvirus ORF45 revealed its binding to viral ORF33 and cellular USP7, resulting in stabilization of ORF33 that is required for production of progeny viruses. J Virol 89:4918–4931. doi: 10.1128/JVI.02925-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Zhu N, Li W, Zhu F, Wang Y, Yuan Y. 2015. Mono-ubiquitylated ORF45 mediates association of KSHV particles with internal lipid rafts for viral assembly and egress. PLoS Pathog 11:e1005332. doi: 10.1371/journal.ppat.1005332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bortz E, Whitelegge JP, Jia Q, Zhou ZH, Stewart JP, Wu TT, Sun R. 2003. Identification of proteins associated with murine gammaherpesvirus 68 virions. J Virol 77:13425–13432. doi: 10.1128/JVI.77.24.13425-13432.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jia Q, Chernishof V, Bortz E, McHardy I, Wu TT, Liao HI, Sun R. 2005. Murine gammaherpesvirus 68 open reading frame 45 plays an essential role during the immediate-early phase of viral replication. J Virol 79:5129–5141. doi: 10.1128/JVI.79.8.5129-5141.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jia X, Shen S, Lv Y, Zhang Z, Guo H, Deng H. 2016. Tegument protein ORF45 plays an essential role in virion morphogenesis of murine gammaherpesvirus 68. J Virol 90:7587–7592. doi: 10.1128/JVI.03231-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murata T, Isomura H, Yamashita Y, Toyama S, Sato Y, Nakayama S, Kudoh A, Iwahori S, Kanda T, Tsurumi T. 2009. Efficient production of infectious viruses requires enzymatic activity of Epstein-Barr virus protein kinase. Virology 389:75–81. doi: 10.1016/j.virol.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 22.Katsumura KR, Maruo S, Wu Y, Kanda T, Takada K. 2009. Quantitative evaluation of the role of Epstein-Barr virus immediate-early protein BZLF1 in B-cell transformation. J Gen Virol 90:2331–2341. doi: 10.1099/vir.0.012831-0. [DOI] [PubMed] [Google Scholar]
- 23.Watanabe T, Tsuruoka M, Narita Y, Katsuya R, Goshima F, Kimura H, Murata T. 2015. The Epstein-Barr virus BRRF2 gene product is involved in viral progeny production. Virology 484:33–40. doi: 10.1016/j.virol.2015.05.010. [DOI] [PubMed] [Google Scholar]
- 24.Asai R, Kato A, Kato K, Kanamori-Koyama M, Sugimoto K, Sairenji T, Nishiyama Y, Kawaguchi Y. 2006. Epstein-Barr virus protein kinase BGLF4 is a virion tegument protein that dissociates from virions in a phosphorylation-dependent process and phosphorylates the viral immediate-early protein BZLF1. J Virol 80:5125–5134. doi: 10.1128/JVI.02674-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki M, Takeda T, Nakagawa H, Iwata S, Watanabe T, Siddiquey MN, Goshima F, Murata T, Kawada J, Ito Y, Kojima S, Kimura H. 2015. The heat shock protein 90 inhibitor BIIB021 suppresses the growth of T and natural killer cell lymphomas. Front Microbiol 6:280. doi: 10.3389/fmicb.2015.00470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Delecluse HJ, Hilsendegen T, Pich D, Zeidler R, Hammerschmidt W. 1998. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc Natl Acad Sci U S A 95:8245–8250. doi: 10.1073/pnas.95.14.8245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reference deleted.
- 28.Narita Y, Murata T, Ryo A, Kawashima D, Sugimoto A, Kanda T, Kimura H, Tsurumi T. 2013. Pin1 interacts with the Epstein-Barr virus DNA polymerase catalytic subunit and regulates viral DNA replication. J Virol 87:2120–2127. doi: 10.1128/JVI.02634-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neuhierl B, Feederle R, Hammerschmidt W, Delecluse HJ. 2002. Glycoprotein gp110 of Epstein-Barr virus determines viral tropism and efficiency of infection. Proc Natl Acad Sci U S A 99:15036–15041. doi: 10.1073/pnas.232381299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sugimoto A, Kanda T, Yamashita Y, Murata T, Saito S, Kawashima D, Isomura H, Nishiyama Y, Tsurumi T. 2011. Spatiotemporally different DNA repair systems participate in Epstein-Barr virus genome maturation. J Virol 85:6127–6135. doi: 10.1128/JVI.00258-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kinoshita-Kikuta E, Aoki Y, Kinoshita E, Koike T. 2007. Label-free kinase profiling using phosphate affinity polyacrylamide gel electrophoresis. Mol Cell Proteomics 6:356–366. doi: 10.1074/mcp.T600044-MCP200. [DOI] [PubMed] [Google Scholar]
- 32.Chiu YF, Sugden B, Chang PJ, Chen LW, Lin YJ, Lan YC, Lai CH, Liou JY, Liu ST, Hung CH. 2012. Characterization and intracellular trafficking of Epstein-Barr virus BBLF1, a protein involved in virion maturation. J Virol 86:9647–9655. doi: 10.1128/JVI.01126-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang Q, Fu B, Wu F, Li X, Yuan Y, Zhu F. 2012. ORF45 of Kaposi's sarcoma-associated herpesvirus inhibits phosphorylation of interferon regulatory factor 7 by IKKε and TBK1 as an alternative substrate. J Virol 86:10162–10172. doi: 10.1128/JVI.05224-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sathish N, Zhu FX, Golub EE, Liang Q, Yuan Y. 2011. Mechanisms of autoinhibition of IRF-7 and a probable model for inactivation of IRF-7 by Kaposi's sarcoma-associated herpesvirus protein ORF45. J Biol Chem 286:746–756. doi: 10.1074/jbc.M110.150920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu FX, Sathish N, Yuan Y. 2010. Antagonism of host antiviral responses by Kaposi's sarcoma-associated herpesvirus tegument protein ORF45. PLoS One 5:e10573. doi: 10.1371/journal.pone.0010573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Avey D, Tepper S, Li W, Turpin Z, Zhu F. 2015. Phosphoproteomic analysis of KSHV-infected cells reveals roles of ORF45-activated RSK during lytic replication. PLoS Pathog 11:e1004993. doi: 10.1371/journal.ppat.1004993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fu B, Kuang E, Li W, Avey D, Li X, Turpin Z, Valdes A, Brulois K, Myoung J, Zhu F. 2015. Activation of p90 ribosomal S6 kinases by ORF45 of Kaposi's sarcoma-associated herpesvirus is critical for optimal production of infectious viruses. J Virol 89:195–207. doi: 10.1128/JVI.01937-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuang E, Fu B, Liang Q, Myoung J, Zhu F. 2011. Phosphorylation of eukaryotic translation initiation factor 4B (EIF4B) by open reading frame 45/p90 ribosomal S6 kinase (ORF45/RSK) signaling axis facilitates protein translation during Kaposi sarcoma-associated herpesvirus (KSHV) lytic replication. J Biol Chem 286:41171–41182. doi: 10.1074/jbc.M111.280982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuang E, Tang Q, Maul GG, Zhu F. 2008. Activation of p90 ribosomal S6 kinase by ORF45 of Kaposi's sarcoma-associated herpesvirus and its role in viral lytic replication. J Virol 82:1838–1850. doi: 10.1128/JVI.02119-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuang E, Wu F, Zhu F. 2009. Mechanism of sustained activation of ribosomal S6 kinase (RSK) and ERK by Kaposi's sarcoma-associated herpesvirus ORF45: multiprotein complexes retain active phosphorylated ERK AND RSK and protect them from dephosphorylation. J Biol Chem 284:13958–13968. doi: 10.1074/jbc.M900025200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li X, Du S, Avey D, Li Y, Zhu F, Kuang E. 2015. ORF45-mediated prolonged c-Fos accumulation accelerates viral transcription during the late stage of lytic replication of Kaposi's sarcoma-associated herpesvirus. J Virol 89:6895–6906. doi: 10.1128/JVI.00274-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abada R, Dreyfuss-Grossman T, Herman-Bachinsky Y, Geva H, Masa SR, Sarid R. 2008. SIAH-1 interacts with the Kaposi's sarcoma-associated herpesvirus-encoded ORF45 protein and promotes its ubiquitylation and proteasomal degradation. J Virol 82:2230–2240. doi: 10.1128/JVI.02285-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu X, Cohen JI. 2015. Epstein-Barr virus (EBV) tegument protein BGLF2 promotes EBV reactivation through activation of the p38 mitogen-activated protein kinase. J Virol 90:1129–1138. doi: 10.1128/JVI.01410-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu JJ, Avey D, Li W, Gillen J, Fu B, Miley W, Whitby D, Zhu F. 2015. ORF33 and ORF38 of Kaposi's sarcoma-associated herpesvirus interact and are required for optimal production of infectious progeny viruses. J Virol 90:1741–1756. doi: 10.1128/JVI.02738-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yuen KS, Chan CP, Kok KH, Jin DY. 2017. Mutagenesis and genome engineering of Epstein-Barr virus in cultured human cells by CRISPR/Cas9. Methods Mol Biol 1498:23–31. doi: 10.1007/978-1-4939-6472-7_2. [DOI] [PubMed] [Google Scholar]
- 46.Yuen KS, Chan CP, Wong NH, Ho CH, Ho TH, Lei T, Deng W, Tsao SW, Chen H, Kok KH, Jin DY. 2015. CRISPR/Cas9-mediated genome editing of Epstein-Barr virus in human cells. J Gen Virol 96:626–636. doi: 10.1099/jgv.0.000012. [DOI] [PubMed] [Google Scholar]