

ABSTRACT
Apoptosis is an important antiviral host defense mechanism. Here we report the identification of a novel apoptosis inhibitor encoded by the vaccinia virus (VACV) M1L gene. M1L is absent in the attenuated modified vaccinia virus Ankara (MVA) strain of VACV, a strain that stimulates apoptosis in several types of immune cells. M1 expression increased the viability of MVA-infected THP-1 and Jurkat cells and reduced several biochemical hallmarks of apoptosis, such as PARP-1 and procaspase-3 cleavage. Furthermore, ectopic M1L expression decreased staurosporine-induced (intrinsic) apoptosis in HeLa cells. We then identified the molecular basis for M1 inhibitory function. M1 allowed mitochondrial depolarization but blocked procaspase-9 processing, suggesting that M1 targeted the apoptosome. In support of this model, we found that M1 promoted survival in Saccharomyces cerevisiae overexpressing human Apaf-1 and procaspase-9, critical components of the apoptosome, or overexpressing only conformationally active caspase-9. In mammalian cells, M1 coimmunoprecipitated with Apaf-1–procaspase-9 complexes. The current model is that M1 associates with and allows the formation of the apoptosome but prevents apoptotic functions of the apoptosome. The M1 protein features 14 predicted ankyrin (ANK) repeat domains, and M1 is the first ANK-containing protein reported to use this inhibitory strategy. Since ANK-containing proteins are encoded by many large DNA viruses and found in all domains of life, studies of M1 may lead to a better understanding of the roles of ANK proteins in virus-host interactions.
IMPORTANCE Apoptosis selectively eliminates dangerous cells such as virus-infected cells. Poxviruses express apoptosis antagonists to neutralize this antiviral host defense. The vaccinia virus (VACV) M1 ankyrin (ANK) protein, a protein with no previously ascribed function, inhibits apoptosis. M1 interacts with the apoptosome and prevents procaspase-9 processing as well as downstream procaspase-3 cleavage in several cell types and under multiple conditions. M1 is the first poxviral protein reported to associate with and prevent the function of the apoptosome, giving a more detailed picture of the threats VACV encounters during infection. Dysregulation of apoptosis is associated with several human diseases. One potential treatment of apoptosis-related diseases is through the use of designed ANK repeat proteins (DARPins), similar to M1, as caspase inhibitors. Thus, the study of the novel antiapoptosis effects of M1 via apoptosome association will be helpful for understanding how to control apoptosis using either natural or synthetic molecules.
KEYWORDS: Apaf-1, M1L, ankyrin repeat, apoptosis, apoptosome, caspase-9, poxvirus, vaccinia virus
INTRODUCTION
Apoptosis is a powerful antiviral mechanism (1, 2). There are two classical forms of apoptosis: extrinsic (mediated by caspase-8) and intrinsic (mediated by caspase-9) (3, 4). Intrinsic apoptosis often is triggered during virus infection of the host cell (2). In this case, there is depolarization and permeabilization of the outer mitochondrial membrane. Released cytochrome c (cyt c) and dATP then stimulate Apaf-1 oligomerization (3, 5–7). The apoptosome is next formed when monomeric, inactive procaspase-9 proteins are recruited to Apaf-1 oligomers via caspase recruitment domain (CARD)-CARD interactions (8, 9). In the apoptosome, procaspase-9 can exist as either homodimers or Apaf-1–procaspase-9 heterodimers. In both cases, procaspase-9 conformationally changes to an active state and cleaves procaspase-3 to trigger apoptosis. Autocleavage of procaspase-9 also occurs after activation, resulting in processed caspase-9 complexes that retain the ability to cleave procaspase-3 while associated with Apaf-1 (10, 11). Thus, both unprocessed and processed forms of active caspase-9 can cleave procaspase-3. Activated caspase-3, in turn, cleaves cellular PARP-1 and other protein substrates, culminating in cell death (4).
Poxviruses are master manipulators of the host, using multiple strategies to evade apoptosis and other antiviral immune responses (12–14). Wild-type vaccinia virus (VACV) strain WR is one of the best-studied poxviruses, and it expresses at least five intracellular antiapoptosis proteins, B13 (SPI-2), F1, N1, B22 (SPI-1), and E3, suggesting that apoptosis is an important host response to defend against during virus infection (12). A few other VACV strains (Lister, USSR, and Evans, but not WR) and camelpox virus encode vGAAP, a protein that inhibits endoplasmic reticulum (ER)-induced apoptosis (15–17). The current hypothesis is that VACV expresses multiple apoptosis antagonists to protect against a variety of proapoptotic pathways triggered in different host cells during an infection in vivo.
Modified vaccinia virus Ankara (MVA) is an attenuated VACV strain that was created by serially passaging wild-type VACV more than 500 times (18). As a result, approximately 15% of the VACV genome is deleted or truncated in MVA (19). With respect to antiapoptosis genes, MVA retains only E3L, F1L, and B22R (19). Despite the presence of these three genes, MVA infection nevertheless induces apoptosis in several immune cell types (20–23). Thus, MVA infection of immune cells provides an excellent platform to identify novel WR-encoded antiapoptosis proteins not encoded by MVA, which have mechanisms distinct from those of E3, F1, and B22 (24–27).
Ankyrin (ANK) repeats are one of the most abundant motifs in nature (28, 29). These are 33-residue motifs that form alpha-helical structures and provide platforms for protein-protein interactions (28). This property has led to the use of designed ANK repeat proteins (DARPins) as a drug development platform (30, 31). VACV strain WR encodes at least eight known or predicted ANK proteins, including 005-008 and 211-214 (Copenhagen B25 homologs), 014-017 (variola virus strain Bangladesh D8 homologs), 019 (Copenhagen C9 homolog), 030 (M1), 031 (K1), 186 (B4), 188 (B6), and 199 and 202 (B18) (32, 33). However, only three of the WR ANK proteins (K1, B4, and B18) have reported functions (34–42). Thus, the study of the remaining ANK proteins is likely to uncover novel aspects of poxvirus biology.
The goal of this study was to identify a function for the VACV ANK-encoding M1L gene, a gene with no previously ascribed function that is located within a region of the WR genome that was deleted during the derivation of MVA. Because multiple natural and synthetic ANK proteins (e.g., DARPins) inhibit apoptosis (31, 43–46), M1 may possess this same function. Interestingly, M1 inhibited intrinsic apoptosis under several conditions and in multiple cell lines. The biochemical hallmarks of the intrinsic apoptosis signal transduction pathway were next examined to define the step of the apoptosis pathway that M1 targets.
RESULTS
Creation and characterization of a recombinant MVA virus containing the M1L gene.
There is no reported function for the WR M1 protein, which is predicted to harbor 14 ANK repeat sequences (32, 33). ANK-containing proteins expressed by polydnavirus (44) and intracellular bacteria (43, 45) possess antiapoptotic properties, suggesting that M1 may have a similar function. An initial approach to answer this question was to capitalize on the fact that MVA infection induces apoptosis in immune cells (20–23). The M1L gene is present in wild-type VACV but absent in MVA (19). Thus, if M1L encodes an antiapoptosis protein, then a recombinant MVA virus engineered to express M1 (MVA/M1L) was expected to decrease MVA-induced apoptosis.
A two-gene cassette containing the GFP gene (under the control of the poxvirus p11 promoter) and M1L gene (under the control of its natural promoter) was inserted into the del III region of MVA, an area commonly used for placement of genetic material into the MVA genome (Fig. 1A). PCR analysis of viral genomes revealed that the M1L gene was indeed stably inserted into del III using DNA from MVA/M1L-infected cells but not MVA-infected cells (Fig. 1B). Additionally, a set of primers that are specific to the del III region (F1 and R1) PCR amplified a 3.3-kb product (Fig. 1B), which is the expected size of an amplicon if GFP and M1L are present.
FIG 1.

Creation and characterization of an M1L-expressing MVA virus. (A) Schematic showing insertion of tandem GFP and M1L genes into the del III region of the MVA genome, in which the GFP gene is under the control of the poxvirus p11 promoter (p11) and the M1L gene is under the control of its natural promoter (NP). Primers used for verifying insertion of this GFP-M1L cassette are shown. (B) CEF monolayers were mock infected or infected with MVA, MVA/M1L, or WR (MOI = 10). At 24 h p.i., infected cells were harvested and lysed. DNA was subjected to PCR amplification using either the M1F and M1R primer set to amplify the M1L gene or the F1 and R1 primers to PCR amplify the MVA del III region. A portion of each PCR amplification reaction mixture was separated by agarose gel electrophoresis, and DNA was visualized using ethidium bromide staining. Reactions were analyzed in the same gel, and gel images were spliced for labeling purposes. (C) Detection of M1L gene transcription using semiquantitative RT-PCR. PMA-stimulated THP-1 cells were mock infected or infected with the indicated viruses (MOI = 2). At 6 h p.i., cells were collected and total RNA was extracted from lysed cells. Total RNA was reverse transcribed into cDNA. A portion of cDNA was incubated with primers either nested inside the M1L gene or for the actin gene as a control. A portion of each PCR was analyzed by agarose gel electrophoresis, and PCR amplicons were detected by using ethidium bromide staining of the gel. Reactions were analyzed in the same gel, and gel images were spliced for labeling purposes. (D) CEF or RK13 cellular monolayers were infected with MVA, MVA/M1L, or WR (50 PFU/well of a six-well plate). At 24 h p.i., cells were fixed and incubated in a solution containing anti-vaccinia virus antiserum, followed by a solution containing HRP-conjugated goat anti-rabbit antiserum. The diameters of at least 10 foci per condition were measured, and results are presented as the mean focus size ± SEM for each sample.
Multiple attempts were made to raise polyclonal antiserum against M1 peptides in rabbits. Each attempt failed; raised antisera did not detect M1 from virus-infected cellular lysates. Thus, we analyzed virus-infected cells for M1L mRNA using semiquantitative reverse transcription-PCR (RT-PCR). M1L is an early gene (47, 48). M1L mRNA is detected as early as 30 min postinfection (47, 48) and remains detectable until 12 h postinfection (h p.i.) (47, 49). We chose to examine M1L transcription at 6 h p.i., a time at which M1L transcription was reported to occur (47, 48). As shown in Fig. 1C, an M1L-containing amplicon was detected when using RNA isolated from cells infected with MVA/M1L or the WR strain of wild-type vaccinia virus but not from MVA-infected or mock-infected cells. These data indicated that the M1L gene was expressed during infection.
MVA lacks about 15% of the parental (wild-type) vaccinia virus genome (19). MVA has a narrow host range due to this loss of genes and only replicates in chicken embryo fibroblasts (CEFs) and a few other avian cell lines (50). Another vaccinia virus ANK repeat protein, K1, increases the host range of MVA to include RK13 cells (50, 51). Consequently, we asked if M1L insertion also would increase the MVA host range in a fashion similar to that of K1L. The formation and size of foci were examined as an indirect readout for virus replication. This was performed in infected monolayers of CEFs (permissive for MVA infection) and rabbit RK13 cells (nonpermissive for MVA infection) (Fig. 1D). The addition of M1L neither increased the size of MVA-based foci in CEFs nor allowed focus formation in RK13 cells. Of course, this does not rule out the possibility that M1L may increase the host range in other cell types, and this possibility will be examined in the future. Nevertheless, the M1L gene product did not increase the host range of MVA in RK13 cells and therefore had properties distinct from the ANK repeat-containing K1 protein.
The M1L gene increases viability of MVA-infected cells.
MVA infection of primary antigen-presenting cells (APCs) induces apoptosis (20–23). We used the human monocytic THP-1 cell line to assess the effect of M1L on viability after virus infection. We used this cell line instead of primary human cells because it removes potential problems with donor-to-donor variation. For all experiments shown here, THP-1 cells were incubated with phorbol 12-myristate 13-acetate (PMA) for 48 h to differentiate cells into macrophage-like cells. As shown in Fig. 2A, 85% of the mock-infected cells were viable, as evaluated by trypan blue dye staining. In contrast, MVA infection reduced viability such that only 23% of cells remained viable. The viability of the cell population increased to 53% during MVA/M1L infection, implying that M1L encoded an inhibitor of cell death. As a control, a separate set of PMA-matured THP-1 cells was incubated in medium containing staurosporine (STS), a drug that triggers intrinsic apoptosis (52). As expected, nearly all THP-1 cells died following STS treatment.
FIG 2.

M1L increases viability during MVA infection. PMA-stimulated THP-1 cells were either mock infected or infected with MVA or MVA/M1L (MOI = 5) or incubated in medium containing 1 μM staurosporine (STS). (A) At 24 h p.i. or post-STS treatment, all cells were collected and cell death was quantified by using a trypan blue dye exclusion assay. Results are presented as the mean percentage of cells that excluded trypan blue (live) cells divided by the total number of live and dead cells. Cells from each sample were counted in triplicate, and data shown here are the mean ± SD from three independent experiments. (B) At 24 h p.i. or post-STS treatment, PrestoBlue reagent was added to the wells. The fluorescence of each well was quantified using a microplate reader. Results are presented as the mean fluorescence ± SD for each sample. PrestoBlue-based assays were performed in technical triplicate. The graphs shown here represent data obtained from at least three independent experiments. Asterisks indicate conditions in which the viability of MVA/M1L-infected cells was statistically different from that of MVA-infected cells (P < 0.05).
We repeated the above-described assay using a red fluorescent viability dye to quantify cell viability (Fig. 2B). Using this approach, we observed trends similar to those shown in Fig. 2A. Mock-infected cells had the highest fluorescence, indicating that a large percentage of the cell population was viable. This was dramatically decreased by incubation of cells with STS or infection of cells with MVA. In comparison, MVA/M1L infection provoked only a slight decrease in fluorescence values compared to those of mock-infected cells, demonstrating that M1L reduced MVA-induced cell death.
M1 inhibits MVA-induced apoptosis.
There are many types of cell death, including apoptosis, necrosis, and necroptosis (1). Apoptosis has unique biochemical hallmarks, including activation of procaspases, resulting in the cleavage of downstream substrates of caspases (4). For example, the 116-kDa PARP-1 protein is a preferred substrate for caspase-3, and PARP-1 cleavage into its 89-kDa and 24-kDa products is a downstream event in apoptosis that can be detected by immunoblotting (53).
To ask if M1L blocked apoptosis, we infected THP-1 cells with MVA or MVA/M1L and examined lysates of cells for full-length or cleaved PARP-1, using antiserum that recognizes both the full-length (116-kDa) and cleaved (24-kDa) PARP-1 (Fig. 3A). In mock-infected cells, the majority of the detected PARP-1 was the full-length form. In contrast, treatment of cells with STS, which induces intrinsic apoptosis, resulted in increased cleaved PARP-1 and decreased full-length PARP-1. Full-length and cleaved PARP-1 were detected in MVA-infected cells. However, cleaved PARP-1 (24-kDa) was dramatically decreased when lysates from MVA/M1L-infected cells were compared to those infected with MVA.
FIG 3.

M1L inhibits MVA-induced apoptosis. PMA-treated THP-1 cells (A and B), Jurkat cells (C and D), or caspase-8-deficient (C8−/−) Jurkat cells (E and F) were either mock infected or infected with MVA or MVA/M1L (MOI = 2). A separate set of uninfected cells was incubated in medium containing 1 μM staurosporine (STS). (A, C, and E) At 16 h p.i. or post-STS treatment, cells were collected and lysed in RIPA buffer. Thirty micrograms of each sample was separated by using SDS–12% PAGE, and proteins were transferred to a PVDF membrane. Membranes were probed with an anti-PARP-1 antibody that detects full-length (116-kDa) and cleaved (24-kDa) PARP-1. An asterisk denotes a nonspecific band. Blots subsequently were incubated with either anti-E3 or anti-actin antisera and redeveloped. Data are representative of at least three independent experiments. In panels A and C, reactions were analyzed in the same gel and gel images were spliced for labeling purposes. (B, D, and F) At 24 h p.i. or post-STS treatment, cells were lysed, and caspase-3 and -7 activities were quantified using the Caspase-Glo 3/7 assay kit. Results are presented as the mean fold induction of caspase activity for each sample ± SD above that for mock-infected cells, whose value was set to 1. Data were obtained from at least three independent experiments. Statistically significant changes in values between MVA and MVA/M1L are indicated by asterisks (P < 0.05).
Caspase-3 and -7 are executioner caspases, and their activation occurs upstream of PARP-1 cleavage. Activation of these two caspases can be quantified using a luciferase-based assay that detects their proteolytic activity. Figure 3B showed that caspase-3 and -7 activity was decreased when M1 was expressed during virus infection, indicating that M1 inhibited apoptosis. When PARP-1 cleavage and caspase-3/7 activity in infected Jurkat T cells were examined, similar results were observed (Fig. 3C and D). Namely, MVA/M1L infection decreased the levels of cleaved PARP-1 (Fig. 3C) and caspase-3 and caspase-7 (Fig. 3D) activities compared to those after MVA infection. Thus, this antiapoptotic function of M1L is not cell type specific. It should be noted that PARP-1 cleavage was more extensive in STS-treated cells than in MVA-infected cells. In contrast, caspase-3/7 activities were similar between STS-treated and MVA-infected cells. One expectation is that caspase-3/7 activity would also be increased in lysates from STS-treated cells compared to MVA-infected cells. One reason for this discrepancy may be as follows: PARP cleavage was examined at 16 h p.i., while caspase-3/7 activity was examined at 24 h p.i. For immunoblotting, infected cells were harvested at earlier time points because we observed reduced amounts of protein levels in samples (due to cell death) at later time points. Nevertheless, the presence of the M1L gene consistently decreased apoptotic events in both THP-1 and Jurkat cells.
Jurkat cells are type II cells, in which active caspase-8 feeds into the intrinsic pathway via its cleavage of Bid, leading to caspase-9-induced apoptosis (54). MVA infection is reported to activate both intrinsic and extrinsic apoptosis (20–23). We examined apoptosis in Jurkat cells that are deficient in caspase-8 (C8−/− Jurkat) to focus solely on M1 inhibition of intrinsic apoptosis (55). In these cells, MVA induced PARP-1 cleavage and executioner caspase activation (Fig. 3E and F), consistent with reports that MVA triggers intrinsic apoptosis (22). Because MVA induces both caspase-8 and caspase-9 activation, caspase-3/7 activity was lower in caspase-8-deficient Jurkat cells than in wild-type Jurkat cells since one pathway that contributes to the apoptotic phenotype was missing. While M1 inhibition of caspase-3/7 activity was stronger in wild-type Jurkat cells than in caspase-8−/− Jurkat cells, MVA/M1L inhibition was still significantly different from MVA inhibition, suggesting that M1 inhibits intrinsic apoptosis.
M1 inhibits procaspase-9 processing.
Procaspase-9 (47 kDa) is inactive until it interacts with Apaf-1 in the apoptosome (56). Apaf-1-associated procaspase-9 homodimers then autocleave at Asp315, resulting in processing to 35-kDa and 12-kDa forms (10, 57). In addition, once activated, caspase-3 can cleave procaspase-9 to produce a 37-kDa caspase-9 (57). To explore the impact of M1 on procaspase-9 cleavage, a step that follows procaspase-9 activation, lysates from virus-infected cells were probed by immunoblotting using an antibody that simultaneously detected the zymogen (47-kDa), autoprocessed (35-kDa), and caspase-3-cleaved (37-kDa) forms of caspase-9 in the same three cell lines used in Fig. 3. Note that procaspase-9 cleavage was examined at times postinfection different from those used in the studies illustrated in Fig. 3 and that procaspase-9 cleavage was examined at different times postinfection in each cell line. Time course assays were first performed to detect maximal procaspase-9 cleavage during MVA infection for each cell line (data not shown). Once this time point was determined for each cell line, we then compared procaspase-9 cleavage in MVA-infected cells with that in MVA/M1L-infected cells. As shown in Fig. 4, only the 47-kDa form of procaspase-9 was detected in mock-infected THP-1 or Jurkat cells. MVA infection triggered intrinsic apoptosis, as evidenced by the presence of the 35-kDa form of caspase-9 in THP-1 cells (Fig. 4A). Both the 35- and 37-kDa forms were detected in MVA-infected Jurkat cells (Fig. 4B and C), reflecting autoprocessing and caspase-3 cleavage of procaspase-9, respectively. The 37-kDa form was not detected in THP-1 cellular lysates; this may reflect more efficient procaspase-9 autoprocessing in these cells. Regardless, the intensities of the 37- and 35-kDa bands were greatly reduced in MVA/M1L-infected cells, implying that M1 blocked infection-induced capsase-9 activity.
FIG 4.

M1L inhibits MVA-induced procaspase-9 cleavage. PMA-treated THP-1 cells (A), Jurkat cells (B), or caspase-8-deficient (C8−/−) Jurkat cells (C) were either mock infected or infected with MVA or MVA/M1L (MOI = 2). A separate set of uninfected cells was incubated in medium containing 1 μM staurosporine (STS) for 12 h. At either 12 (A), 20 (B), or 16 (C) h p.i., cells were collected and lysed in RIPA buffer. Thirty micrograms of each sample was analyzed by SDS–10% PAGE, and proteins were transferred to a PVDF membrane. The membrane was probed with anti-caspase-9 antiserum, which detects the 47-, 37-, and 35-kDa forms of caspase-9. Blots were subsequently incubated with either anti-E3 or anti-actin antisera and developed. Data shown are representative of data obtained from at least three independent experiments.
M1 inhibits biochemical hallmarks of intrinsic apoptosis when it is expressed independently of infection.
We were interested in identifying the portion of the proapoptotic signal transduction pathway that M1 targets. For these experiments, we moved away from infections and expressed M1 ectopically. The reason for using this approach is that MVA encodes at least three other antiapoptotic proteins (F1, B22, and E3) (19). Thus, expression of these proteins during MVA/M1L infection may make it difficult to identify the molecular mechanism of M1. Additionally, MVA infection triggers apoptosis via both intrinsic and extrinsic apoptosis (20–23, 58, 59). Thus, moving away from an MVA infection system affords the opportunity to examine intrinsic apoptosis alone.
HeLa cells were used for transient expression of M1L. With this system, we observed a 90% transfection efficiency. Additionally, staurosporine (STS) treatment of HeLa cells stimulates intrinsic apoptosis (60). As observed in the infection system, the 116-kDa PARP-1 protein was cleaved to a 24-kDa form in STS-treated HeLa cells (Fig. 5). M1 expression greatly decreased the amount of detectable cleaved PARP-1. Similarly, M1-expressing HeLa cells also decreased detectable cleaved caspase-3 and caspase-9. Note that the detection of M1, the full-length PARP-1, and the zymogenic forms of procaspase-3 and -9 decreased slightly over time. Of these proteins, the decrease in procaspase-3 perhaps was the most severe. This decrease could not be attributed to overall protein degradation or uneven protein levels in gels, because actin levels remained similar between samples. At this time, it is not clear why procaspase-3 levels would decrease in M1-expressing cells. Nevertheless, in the apoptosis field, it is the cleaved forms of caspase-3 and -9 that are hallmarks of apoptosis. The presence of cleaved forms of cellular proteins responsible for apoptosis were lower in M1-transfected cells than in pCI-transfected cells. Thus, M1 blocked intrinsic apoptosis triggered by an infection-independent stimulus.
FIG 5.
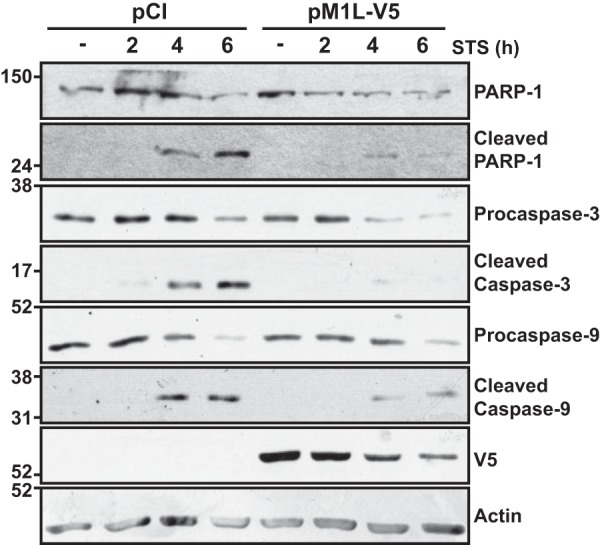
Biochemical hallmarks of staurosporine-induced apoptosis are reduced when M1 is expressed independently of infection. Subconfluent HeLa cellular monolayers were transfected with 1 μg of either empty vector (pCI) or pM1L-V5. At 24 h posttransfection, cells were treated with medium either lacking (−) or containing 0.5 μM staurosporine (STS). At 2, 4, or 6 h post-STS incubation, cells were collected and lysed in RIPA buffer. Lysates were separated by SDS–12% PAGE, and proteins were transferred to a PVDF membrane. Membranes were probed with either anti-PARP-1, anti-caspase-3, or anti-caspase-9 antisera. Membranes were developed and bands were detected using chemiluminescence. Blots were subsequently incubated with either anti-V5, anti-FLAG, or anti-actin antiserum and redeveloped. Data shown are representative of at least three independent experiments.
The M1 protein does not prevent mitochondrial depolarization.
Depolarization of the mitochondria often accompanies Bax/Bak-mediated permeabilization of the outer mitochondrial membrane and cyt c release to the cytoplasm in cells undergoing intrinsic apoptosis (61). M1-expressing HeLa cells were incubated with tetramethylrhodamine ethyl ester (TMRE) to gauge mitochondrial polarization during STS treatment. TMRE is a cell-permeable, positively charged dye that accumulates in active mitochondria, which have a relative negative charge (62). Thus, healthy cells have a higher fluorescence than cells with depolarized or inactive mitochondria. In untreated cells, no more than 20% of the population lost fluorescence, indicating that most of the cells were viable (Fig. 6). STS treatment of these cells increased membrane depolarization to 71% of the cellular population, concomitant with apoptosis. The VACV F1 protein binds to Bim to prevent mitochondrial outer membrane permeabilization and subsequent mitochondrial depolarization (25, 63, 64). Only 49% of the F1-transfected cells lost TMRE staining after exposure to STS, consistent with F1's previously reported antiapoptosis function. In contrast, M1 expression did not inhibit mitochondrial damage: 75% of M1-expressing cells lost TMRE fluorescence upon STS treatment, similar to empty vector (pCI)-expressing cells. Both F1 and M1 protein levels were detected by immunoblotting to show protein expression. Thus, the M1 protein, while inhibiting procaspase-9 cleavage, still allowed mitochondrial depolarization.
FIG 6.
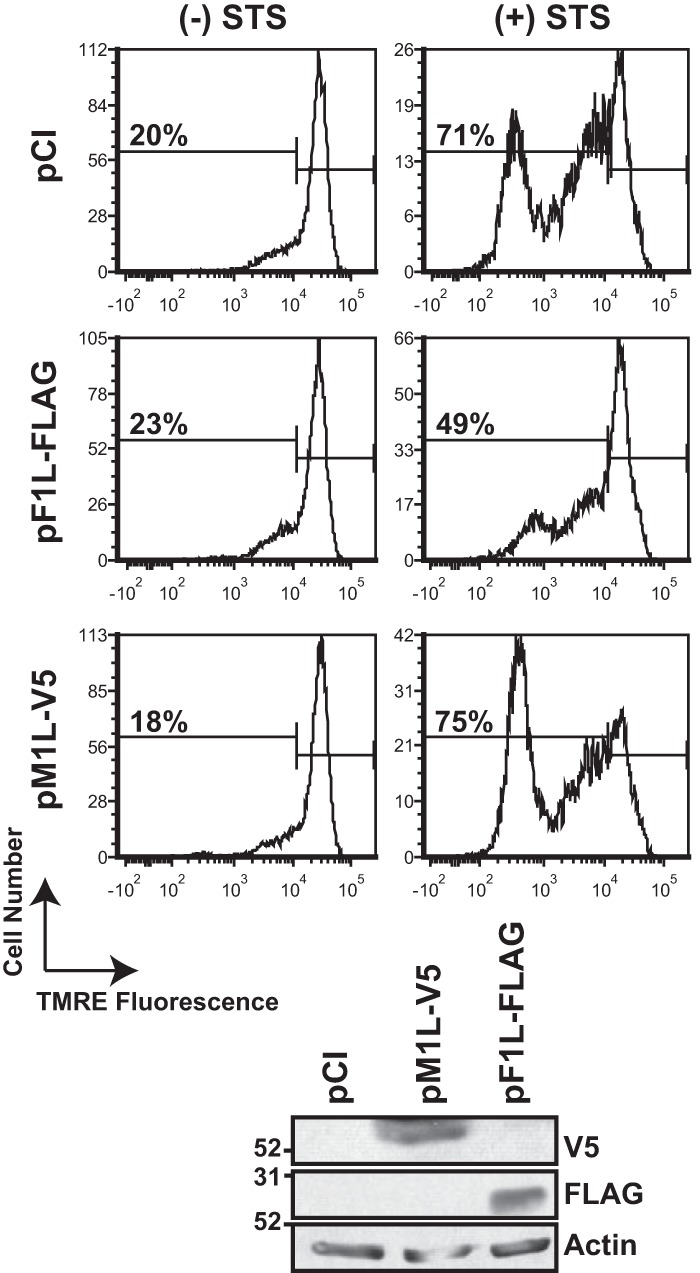
M1 does not block the loss of mitochondrial membrane potential during staurosporine-induced apoptosis. Subconfluent HeLa cellular monolayers were transfected with 1 μg of either empty vector (pCI), pM1L-V5, or pF1L-FLAG. At 24 h posttransfection, cells were treated with medium lacking (−) or containing (+) 5 μM staurosporine (STS) for 2 h. Cells were incubated in 0.2 μM TMRE for 30 min at 37°C. The percentage of cells with decreased TMRE fluorescence is shown in each histogram. Data shown are representative of at least three independent experiments. A portion of each cellular population was collected and lysed in RIPA buffer, and vaccinia virus protein levels were examined by immunoblotting. This same blot was incubated with anti-actin antiserum and subsequently developed.
M1 inhibits yeast cell death induced by expression of mammalian apoptosome components.
Data mentioned above suggested that M1 inhibited the intrinsic apoptosis signal transduction pathway at a step downstream of mitochondrial membrane permeabilization but upstream of procaspase-9 processing. The most likely possibility was that M1 interfered with apoptosome formation or function.
To further examine this, we used Saccharomyces cerevisiae as a system to reconstitute the apoptosome and study its regulation by M1. In this system, the overexpression of mammalian apoptosis effectors (e.g., caspases, Bax) are lethal to yeast (65). Thus, this system can identify direct inhibitors of these proteins by using suppression of lethality as a readout. For example, coexpression of mammalian Apaf-1, procaspase-9, and procaspase-3 proteins is sufficient to trigger cell death (Fig. 7A, lane 2) (66). This system identifies a candidate protein's ability to interact with mammalian apoptosis inducers because death is blocked only if an inhibitor acts directly on the proteins used to induce death (65). For example, the mammalian antiapoptotic Bcl-xL protein blocks mitochondrial outer membrane permeabilization by binding to Bax and Bak (3). Expression of Bcl-xL did not inhibit yeast death when Apaf-1, procaspase-9, and procaspase-3 were coexpressed, because Bcl-xL acts prior to apoptosome formation (Fig. 7A, lane 4). However, Bcl-xL inhibited Bax-induced death (Fig. 7A, lane 9), consistent with its mode of action in mammalian cells (3). The baculovirus P35 protein binds to caspase-9 and caspase-3 (67, 68) and therefore inhibited Apaf-1/procaspase-9/procaspase-3-induced death (Fig. 7A, lane 3) but not Bax-induced death (Fig. 7, lane 8). M1 inhibited lethality of Apaf-1/procaspase-9/procaspase-3 expression (Fig. 7A, lane 5) but not Bax-induced death (Fig. 7A, lane 10), suggesting that M1 inhibited death at the level of the apoptosome.
FIG 7.

M1 inhibits yeast lethality that is triggered by a reconstituted apoptosome or constitutively active caspase-9. Yeast cells were transformed with the indicated expression plasmids, and lethality was induced by overexpressing either Apaf-1, procaspase-9 (Casp-9), and procaspase-3 (Casp-3) or Bax (A) or reverse caspase-9 (rCasp-9) and procaspase-3 (B). Suspensions containing equivalent concentrations of each transformant were serially diluted, and 5 μl of each dilution was spotted onto plates containing galactose (to induce transgene expression) or glucose (to repress transgene expression). Growth on inducing plates indicates survival and proliferation of yeast cells expressing the transgenes.
A separate experiment was performed in which a constitutively active caspase-9 protein (rCasp-9) and procaspase-3 were coexpressed (Fig. 7B). This allows cell death in an Apaf-1-independent manner, because rCasp-9 has the small and large subunit orders swapped to mimic the structure of the active caspase (65, 66). Under these conditions, both M1 (Fig. 7B, lane 5) and P35 (Fig. 7B, lane 3) rescued yeast viability. These data suggest that M1 interacted with the active form of caspase-9 and that such interactions were sufficient to block yeast lethality.
The M1 protein coimmunoprecipitates with caspase-9–Apaf-1 complexes.
To examine the molecular targets of M1, we performed coimmunoprecipitations in 293T cells ectopically coexpressing epitope-tagged M1, a truncated Apaf-1 (N-Apaf-1; amino acids [aa] 1 to 559), and a dominant negative (DN) procaspase-9 (C9DN). N-Apaf-1 lacks the WD40 repressive domain; thus, this construct allows for the formation of an apoptosome complex in the absence of cyt c (69). C9DN was used instead of wild-type procaspase-9 to prevent apoptosis and maximize detection of protein-protein interactions. C9DN still binds to N-Apaf-1 and forms homodimers but is not catalytically active and is not processed (9).
Figure 8A showed that the myc-tagged N-Apaf-1 coimmunoprecipitated with FLAG-tagged C9DN regardless of M1 coexpression, demonstrating that M1 allowed apoptosome formation. Interestingly, the V5-tagged M1 protein also coimmunoprecipitated with FLAG-tagged C9DN, showing that M1 associated with the N-Apaf-1–C9DN complex. Another VACV ANK-containing protein (K1) did not coimmunoprecipitate with the N-Apaf-1–C9DN complex (Fig. 8A), suggesting the specificity of the M1 ANK repeats for the apoptosome. Reverse coimmunoprecipitations were performed in which M1 or K1 were immunoprecipitated (Fig. 8B and C). High backgrounds and nonspecific bands were observed in Fig. 8B because rabbit IgG was used as an isotype control and the anti-FLAG and anti-myc antibodies were also from rabbit. Nevertheless, a distinct C9DN-containing band was detected when M1 was immunoprecipitated, but this band was absent in empty vector (pCI)-transfected cells. This same blot was reprobed for the presence of myc-tagged N-Apaf-1, and a unique band was observed at the molecular weight expected for N-Apaf-1 (Fig. 8B). Figure 8C shows that the epitope-tagged version of C9DN and N-Apaf-1 did not coimmunoprecipitate with K1. Overall, these results demonstrate that M1 associates with the apoptosome.
FIG 8.

M1 interacts with and does not disrupt procaspase-9–Apaf-1 interactions. Subconfluent 293T cellular monolayers were cotransfected with 500 ng C9DN-FLAG, 500 ng N-Apaf-1-myc, and 1,000 ng of either pCI, pM1L-V5, or pHA-K1L. At 24 h posttransfection, cells were lysed in NP-40 lysis buffer. Some lysates were set aside to monitor protein expression. For the remaining clarified cellular lysates, immunoprecipitations (IP) were performed using murine IgG or anti-FLAG (A), rabbit IgG or anti-V5 (B), or murine IgG or anti-HA antibodies (C) conjugated to protein G-Sepharose beads. Immunoprecipitated samples or 20 μg of cellular lysates was analyzed by SDS–8% PAGE, and proteins were transferred to a PVDF membrane for immunoblotting. Membranes were probed with the indicated antibodies to detect the epitope-tagged versions of C9DN, N-Apaf-1, M1, K1 or cellular actin. For panel B, all immunoprecipitated and cellular lysates were analyzed in the same gel and gels were spliced for labeling purposes.
DISCUSSION
Apoptosis is an evolutionarily conserved mechanism to remove infected cells (1). Many intracellular pathogens possess strategies to prevent apoptosis of infected host cells. Here we show that M1 is a novel VACV apoptosis antagonist. Expression of M1 inhibited biochemical hallmarks of apoptosis, including PARP-1 and procaspase-3 cleavage induced by either infection with MVA or STS treatment.
Procaspase-9 is a unique initiator caspase. Other initiator caspases like procaspase-8 must be cleaved, separating active subunits in order to achieve optimal catalytic activity (70). However, a conformational change in procaspase-9 due to Apaf-1 interactions can activate apoptosis without caspase-9 cleavage. Cleavage will occur after activation but is not necessary for catalytic activity (10). We observed that M1 (i) coimmunoprecipitated with procaspase-9 and Apaf-1, (ii) prevented procaspase-9 processing, and (iii) blocked caspase-9-dependent yeast lethality in the absence of Apaf-1. Together, these data suggest that M1 interacts with the Apaf-1-associated form of procaspase-9. Our current model is that M1 allows formation of the apoptosome but inhibits apoptosome function (e.g., procaspase-9 processing and procaspase-3 cleavage). The most likely possibility is that M1 binds to a procaspase-9 region other than the CARD since M1 does not block procaspase-9–Apaf-1 interactions and M1 lacks an obvious CARD domain. These data do not preclude the possibility that M1 may also bind to Apaf-1. It is unlikely that M1 acts as a competitive substrate for caspase-9 because there are no obvious caspase-9 cleavage motifs in the M1 amino acid sequence.
To the best of our knowledge, M1 is the first poxviral protein that targets apoptosome function. VACV B13 was also reported to interact with caspase-9 in vitro (71, 72). However, unlike M1, B13 did not inhibit intrinsic apoptosis despite these interactions (72). Vaccinia virus F1 originally was reported to function upstream of M1, inhibiting mitochondrial membrane pore formation to prevent apoptosome formation (73). More recently, it was shown that the N terminus of F1 binds to procaspase-9 to inhibit recruitment of procaspase-9 to Apaf-1 (74, 75). The physiological relevance of this interaction was recently called into question when Caria et al. showed that this N terminus of F1 is dispensable for antiapoptosis function (76). Regardless, M1 appears to have an inhibitory strategy distinct from that of F1 because F1 prevents apoptosome formation and prevents mitochondrial depolarization (74, 75) while M1 allows Apaf-1–procapase-9 interactions and does not suppress depolarization of the mitochondrial membrane. In addition, the M1 and F1 mechanisms are likely biochemically distinct because MVA induces apoptosis despite the presence of F1 and the introduction of M1 into MVA results in a virus whose infection is much less apoptotic than MVA.
ANK repeats are one of the most abundant motifs found in nature (28, 29). Mammalian cells have ANK proteins that either induce or inhibit apoptosis. Cellular cardiac ankyrin repeat protein Ankrd1/CARP (ASPP1 and ASPP2) promotes apoptosis via interactions with cellular p53 (77, 78). Murine nucling, an ortholog of the human uveal autoantigen with coiled-coil domains and ankyrin repeats (UACA) protein, associates with and promotes apoptosome nuclear translocation to further promote stress-induced apoptosis (79). In contrast, proteins like human gankyrin, an ANK-containing oncoprotein often overexpressed in many hepatocellular carcinomas, inhibits apoptosis by binding to the Mdm2 E3 ubiquitin ligase, leading to ubiquitination and degradation of proapoptotic p53 (80, 81). Because these proteins are critical to the survival of healthy versus damaged cells, synthetic ANKs (DARPins) have been engineered as a means to control apoptosis (30, 31, 82). New information concerning ANK proteins that alter apoptosis, such as the novel way that M1 inhibits intrinsic apoptosis at the level of the apoptosome, could potentially benefit the development of novel DARPins.
There are reports of other ANK-containing proteins with antiapoptosis properties from viruses, bacteria, and eukaryotes. The Campoletis sonorensis ichnovirus (CsIV) P-vank-1 protein inhibits caspase-3 activity via an as-yet-characterized mechanism in the insect Sf9 cell line (44). Like M1, P-vank-1 retains this function independent of infection (44). Intracellular Coxiella burnetii encodes the AnkG protein, which binds to the proapoptotic cellular p32 protein to inhibit apoptosis through a currently uncharacterized nuclear mechanism (43). Ehrlichia chaffeensis and Ehrlichia canis each express p200, which is proposed to inhibit apoptosis by binding an adenine-rich motif found in Alu-Sx elements, a motif found in promoters and introns for various host genes, including those for apoptosis (83). Each of these ANK-containing proteins has strategies distinct from M1. This implies that intracellular pathogens have capitalized on manipulating ANK domains to inhibit the predominant proapoptosis signaling pathways triggered during the infection of different host cell types.
There are myriad non-ANK-containing proteins from viruses, bacteria, and eukaryotes that also inhibit intrinsic apoptosis. The cellular Bcl-2 family of proteins, along with their viral homologs, are well known for their inhibition of mitochondrial permeabilization (3). The mitochondria itself are also a popular target for inhibition of intrinsic apoptosis, with viruses and bacteria both encoding a plethora of proteins to block cytochrome c release (84, 85). Additionally, there are proteins that target caspases, which more closely relate to the function of M1. African swine fever virus A224 as well as effector proteins from Salmonella enterica serovar Typhimurium, Pseudomonas aeruginosa, Francisella tularensis, and Legionella pneumophila directly or indirectly inhibits caspase-3 (86–90); however, we demonstrated that M1 inhibited procaspase-9 and therefore functions in a distinct manner. Other proteins function, like M1, directly on the apoptosome. For example, cellular APIP, Aven, Hsp70, Hsp90, and TUCAN bind Apaf-1 or procaspase-9 to prevent either Apaf-1 oligomerization or caspase-9 binding to Apaf-1 oligomers (91–96). M1, on the other hand, allows apoptosome formation and therefore functions downstream of these inhibitors. Baculovirus P49 interacts with caspase-9 and works as a direct competitive substrate (97). M1 lacks an obvious caspase-9 cleavage site, suggesting that M1 functions in a manner distinct from P49. Mammalian X-linked inhibitor of apoptosis (XIAP) is likely the closest in function to M1, as it also inhibits apoptosis after formation of the apoptosome. However, XIAP can bind only to the processed/cleaved form of caspase-9 (98), while M1 inhibited caspase-9 cleavage. Therefore, while targeting the apoptosome is a tactic used by multiple organisms to inhibit apoptosis, the mechanism employed by M1 is a new poxviral strategy and gives important insight as to how viruses may control intrinsic apoptosis.
It is striking to observe that VACV WR has multiple weapons in its arsenal to neutralize myriad proapoptotic events, including mitochondrial membrane permeabilization (F1), caspase-8 activation (B13 and B22), Bid cleavage (N1), and PKR-induced apoptosis (E3) (99). M1 is now the sixth weapon of this intracellular arsenal, acting at the level of the apoptosome for its antagonistic function. One favored hypothesis is that each of these antiapoptosis proteins is necessary during infection of numerous different cell types in vivo to neutralize the multiple intracellular responses (e.g., virus infection itself) and extracellular responses (e.g., tumor necrosis factor [TNF], cytotoxic T cells) to infection. For example, while MVA encodes inhibitors of intrinsic apoptosis that work upstream of M1 function (F1 and N1), apoptosis can still be induced in certain immune cells by MVA (20–23). However, MVA does not induce apoptosis in other nonimmune cell types (59). This supports the hypothesis that each VACV inhibitor of apoptosis functions in a nonredundant fashion and highlights the key role M1 plays to dampen the host response during VACV infection.
MATERIALS AND METHODS
Cell lines.
Human cervical carcinoma (HeLa) cells, human embryonic kidney 293T (293T) cells, and human acute monocytic leukemia (THP-1) cells were obtained from the American Type Culture Collection (ATCC). Rabbit kidney (RK13) cells were a kind gift from Grant McFadden (Arizona State University). Chicken embryo fibroblasts (CEFs) were obtained from Charles River Laboratories. Wild-type and caspase-8 knockout Jurkat cells were obtained from Ralph Budd (University of Vermont). HeLa and 293T cells were cultured in Eagle's minimal essential medium (EMEM) supplemented with 10% fetal bovine serum (FBS) and 2 mM l-glutamine. HeLa and 293T cells were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% FBS and 2 mM l-glutamine. THP-1 and Jurkat lines were cultured in RPMI 1640 medium supplemented with 10% FBS and 2 mM l-glutamine. For all experiments using THP-1 cells, cells were differentiated with medium containing 100 ng/ml phorbol 12-myristate 13-acetate (PMA; Sigma) for 48 h prior to infection.
Plasmids and transfections.
Plasmid pM1L-V5 was created by PCR amplification of the VACV strain WR M1L gene in which a Kozak sequence was engineered onto the N terminus of M1L and a V5 epitope tag sequence was engineered onto the C terminus of the gene. The M1L open reading frame (ORF) was amplified using the primers 5′-TAAGAATTCGCCACCATGGCTATATTTGTTATAGAG-3′ and 5′-GGT ACC TTA tgt aga atc aag acc aag tag agg att agg gat agg ttt tcc AAA ATA ATC ATC-3′, containing the EcoRI and KpnI restriction enzyme sites (in italics), Kozak (in underlined italics) and stop codon (underlined) sequences, and the V5 sequence (lowercase). The amplicon was cloned into plasmid pCI (Promega). A synthetic M1L gene with codons optimized for E. coli expression was purchased from Bioneer Pacific (Kew East, Victoria, Australia), and the coding region was amplified using the following primers: 5′-GCGGATCCGCCATGATCTTCGTTATTGAAAG-3′ and 5′-GCCTCGAGCTAAAAATAGTCATCAAAAAC-3′. The product was cut with BamHI and XhoI and ligated into pGALL-(TRP1)-MCS for yeast expression (100). pF1L-FLAG encodes a FLAG-tagged F1 protein and has been described previously (73). Plasmid pC9DN-FLAG contains a C287S mutation of procaspase-9. Therefore, this plasmid expresses a dominant negative mutant procaspase-9 protein that interacts with Apaf-1 but lacks enzymatic activity. pN-Apaf-1-myc expresses the N-terminal region (aa 1 to 559) of Apaf-1 that interacts with caspase-9 (69). Both pC9DN-FLAG and pN-Apaf-1-myc were kind gifts from Gabriel Núñez (University of Michigan). Plasmid pHA-K1L expresses a hemagglutinin (HA)-tagged K1 protein and has been described previously (34, 37). All mammalian expression plasmids were transiently transfected into cells using the TransIT 2020 reagent (Mirus) in accordance with the manufacturer's instructions.
Creation and characterization of MVA/M1L viruses.
The modified vaccinia virus Ankara (MVA) strain of VACV was obtained from Bernard Moss (National Institutes of Health) (19). An MVA virus containing the M1L transcriptional unit (MVA/M1L) was created by using homologous recombination in CEFs. The M1L gene and its natural promoter were PCR amplified from purified vaccinia virus strain WR DNA using primers 5′-ACCGGATCCTTCTTGACATAAAAG-3′ and 5′-CTCTCTATGAATCATCATCTGTTCC-3′. The resultant PCR product was inserted into pLW44 (provided by Bernard Moss, National Institutes of Health) in a multiple-cloning site (MCS) directly downstream of the GFP gene to create pLW44/M1L. pLW44/M1L retained the GFP gene linked to the vaccinia virus p11 promoter but lacked the adjacent vaccinia virus modified H5 promoter that might have otherwise influenced the expression of the inserted M1L gene. Green fluorescent protein (GFP) expression was used as a stable screening marker during isolation. Sequence analysis of a representative plasmid indicated that no new mutations had been introduced into the amplicon. MVA/M1L viruses were generated when recombination occurred between the homologous del III regions in both the MVA genome and pLW44/M1L in cells that were infected and transfected. Cells were infected with MVA (multiplicity of infection [MOI] = 0.05 PFU/cell) and then transfected with pLW44/M1L. Using this approach, the M1L gene and a GFP gene would be inserted into the del III region of MVA. After a 48-h incubation period, cells were collected and subjected to three freeze-thaw cycles to release viruses. Serial dilutions of viruses were inoculated onto CEF monolayers, and then monolayers were incubated in medium containing 0.5% methylcellulose. Recombinant viruses were selected based on the development of GFP-expressing foci. Four rounds of plaque purification were performed. The presence of the M1L gene in the MVA del III region and the lack of contaminating parental viral DNA were verified by PCR analysis of the isolated viruses' genome, using primers that were specific for a site either flanking the del III region or present in and unique to the M1L gene. The primer sets used were the following: M1F (5′-CATCGAATACTTCGTAAGATACTCC-3′) and M1R (5′-GAGAGTAAATTGTTGCAAAATATACAGAAA-3′) to detect M1L, and F1 (5′-GTAACAAAAGTATTGGTAATCGTGTC-3′) and R1 (5′-GAATCTACTCATCTAAACGATTTAG-3′) to PCR amplify the del III region to ensure that M1L was inserted in the correct region. PCR products were analyzed by agarose gel electrophoresis, and amplicons were detected by staining the gel with an ethidium bromide solution (Pierce).
The expression of the M1L gene was detected by using semiquantitative RT-PCR. PMA-simulated THP-1 cells were either mock infected or infected with MVA, MVA/M1L, or WR at an MOI of 2. At 6 h p.i., RNA was collected using an RNeasy minikit (Qiagen), by following the manufacturer's instructions. From each sample, 500 ng of RNA was reverse transcribed into cDNA as follows. First, total RNA for each sample was incubated with 0.5 μg of oligo(dT) primer (IDT) at 70°C for 5 min, followed by a 5-min incubation at 4°C to anneal primers. Next, samples were reverse transcribed into cDNA using 10 U/μl of Moloney murine leukemia virus (M-MuLV) reverse transcriptase (New England BioLabs). Ten microliters of template cDNA from each sample was used for PCR amplification, using a Mastercycler (Eppendorf) and Go Taq Flexi DNA polymerase (Promega), per the manufacturer's instructions. Primers designed to amplify M1L were 5′-CGAGTATTATCTATCTCTTTATG-3′ and 5′-GTTTATATGAAGTAAAGTATC-3′. Primers to amplify β-actin were 5′-AGTTGCGTTACACCCTTTCT-3′ and 5′-ACCTTCACCGTTCCAGTTT-3′. PCR conditions for M1L were 1 cycle at 95°C for 2 min; 25 cycles at 95°C for 45 s, 41°C for 30 s, and 72°C for 90 s; and 1 cycle at 72°C for 5 min. PCR conditions for β-actin were 1 cycle at 95°C for 2 min; 25 cycles at 95°C for 45 s, 54°C for 45 s, and 72°C for 20 s; and 1 cycle at 72°C for 5 min. PCR products were analyzed by agarose gel electrophoresis, and amplicons were detected by staining the gel with an ethidium bromide solution (Pierce).
Immunostaining and quantification of focus formation in infected cellular monolayers.
CEF or RK13 cellular monolayers in six-well plates were infected (MOI = 50 PFU/well) with either MVA, MVA/M1L, or WR. After 24 h, infected monolayers were fixed and permeabilized with methanol-acetone (1:1) for 2 min at room temperature. Cells were then washed with phosphate-buffered saline (PBS) and incubated for 1 h at room temperature with anti-vaccinia virus antiserum (1:1,000; Accurate Chemical and Scientific Corp., catalog no. YVS8101) in PBS containing 3% FBS. After two washes with PBS, fixed monolayers were incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit antiserum (1:1,000; Calbiochem) in PBS containing 3% FBS for 1 h at room temperature. Cells were washed two times with PBS, and foci were visualized using PBS containing o-dianisidine (Sigma) and 0.2% H2O2. Images of foci were taken using a Powershot SD1300 IS camera (Cannon). The diameters of foci were measured by using Image J software. Pixels were scaled to millimeters by determining the number of pixels needed to draw 1 mm using a ruler as a reference, and foci were subsequently measured using this scale. Focus size is represented as the mean size ± standard error of the mean (SEM) of 35 (CEF) or 10 (RK13) foci per sample. The Student t test was used to determine statistically significant differences in size among experimental conditions.
Viability assays.
For trypan blue exclusion assays, PMA-stimulated THP-1 (1 × 106) cells in 12-well plates were mock infected, infected at an MOI of 5, or treated with 1 μM staurosporine (STS; Selleck Chemicals) for 24 h. Detached and adherent cells were collected, centrifuged (250 × g, 5 min), and resuspended in complete medium. An aliquot was mixed 1:1 with trypan blue dye (Thermo Scientific). Cells were counted by using a hemocytometer (Fisher), and viability was calculated as the percentage of cells that excluded trypan blue divided by the total number of cells. Results are represented as the mean ± standard deviation (SD). The Student t test was used to determine statistically significant differences in cellular viability among experimental conditions.
For PrestoBlue assays, PMA-stimulated THP-1 (5 × 104) cells in a 96-well plate were infected at an MOI of 5 or were treated with 1 μM STS in a total volume of 90 μl. At 24 h after infection or STS incubation, 10 μl of PrestoBlue dye (Invitrogen) was added to each well and incubated at 37°C for 5 h before fluorescence was recorded (560/590 excitation/emission) using a SpectraMax M2 microplate reader (Molecular Devices). Viability is represented as the mean fluorescence intensity ± SD. The Student t test was used to determine statistically significant differences in cellular viability among experimental conditions.
Detection of PARP-1 and procaspase cleavage by using immunoblotting.
For assays involving infections, cells were either mock infected or infected with MVA or MVA/M1L for the times indicated in each figure legend. Infections of adherent PMA-stimulated THP-1 cells were performed by removing PMA-containing medium and inoculating monolayers with medium containing viruses. For infection of the Jurkat suspension cell lines, cells were first concentrated by centrifugation (250 × g, 5 min), resuspended, and then counted prior to infection. The absorption phase of infection for Jurkat cells occurred in a small volume at 37°C. After the absorption phase, Jurkat cells were centrifuged (250 × g, 5 min), supernatants were removed, and infected cells were resuspended in complete medium and transferred to 12-well tissue culture plates. For transfection assays, subconfluent HeLa cellular monolayers in six-well plates were transfected with 1,000 ng of either pCI, pM1L-V5, or pF1L-FLAG. At 24 h posttransfection, cells were incubated in medium lacking or containing 0.5 μM STS (Selleck Chemicals) for 2, 4, or 6 h.
For both infected and transfected cells, cells were collected and cellular pellets were lysed in 100 μl RIPA buffer (150 mM NaCl, 1% sodium deoxycholate, 1% NP-40, 0.1% SDS, and 0.01 M Tris-HCl) containing Halt protease inhibitor (Thermo Scientific) for 20 min at 4°C. Cellular lysates were then centrifuged (18,000 × g, 10 min). Clarified supernatants were collected and transferred to new tubes. The protein concentration of lysates from each sample was determined using the BCA protein assay kit (Pierce). Thirty micrograms of protein from each lysate was incubated with 5× nonreducing lane marker (Thermo Scientific) and 2 μl of 2-mercaptoethanol (Fisher Scientific), boiled for 5 min, and then separated using SDS-PAGE. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore), and membranes were incubated in 5% (wt/vol) milk in Tris-buffered saline and Tween 20 (TBST; 150 mM NaCl, 50 mM Tris base, and 0.05% Tween 20) for at least 30 min at room temperature. Membranes were incubated with the indicated primary antibodies overnight at 4°C. Next, membranes were washed three times in large volumes of TBST and incubated with the appropriate HRP-conjugated secondary antibodies. Immunoblots were developed by using chemiluminescence reagents (Thermo Scientific and Amersham) according to the manufacturer's instructions, and images were detected by autoradiography.
Primary antibodies used in these experiments were the following: anti-PARP-1 (1:1,000; Santa Cruz, sc-25780), anti-caspase-3 (1:1,000; Cell Signaling, 9665), and anti-caspase-9 (1:1,000; Cell Signaling, 9502). These antibodies were chosen because they detect both nonprocessed and cleaved forms of PARP-1, caspase-3, and caspase-9, respectively. Other antibodies used included mouse monoclonal anti-E3L (1:5,000; a gift from Stuart Isaacs, University of Pennsylvania), anti-V5 (1:5,000; Millipore, AB3792), anti-FLAG (1:2,000; Sigma, F7425), and anti-β-actin (1:5,000; Sigma, A5060). Secondary HRP-conjugated antibodies were obtained from either Calbiochem (goat anti-rabbit IgG; 1:10,000) or Thermo Scientific (goat anti-mouse IgG; 1:5,000).
Detection of caspase-3/7 activity.
Wild-type or caspase-8-deficient Jurkat cells (1 × 103) or PMA-stimulated THP-1 cells (1 × 104) were infected at an MOI of 2 or treated with 1 μM STS (Selleck Chemicals) in a total volume of 50 μl in a 96-well plate. At 24 h p.i., 50 μl of Caspase-Glo 3/7 substrate (Promega) was added to each well. Plates were incubated in the dark at 37°C for 1 h before luciferase activity was recorded using a SpectraMax M2 microplate reader (Molecular Devices). All experiments were performed in triplicate. For each sample, background medium levels were subtracted and triplicate results were averaged. The resultant values were then normalized to that of mock-infected cells, whose value was set to 1. Values are shown as the mean normalized luminescence ± SD. The Student t test was used to determine the statistical significance of decreased luminescence of MVA/M1L-infected cells compared to the luminescence of MVA-infected cells.
TMRE staining to detect mitochondrial membrane potential.
Subconfluent HeLa cells in six-well plates were transfected with 1,000 ng of either pCI, pM1L-V5, or pF1L-FLAG. At 24 h posttransfection, cells were incubated in medium either lacking or containing 5 μM STS (Selleck Chemicals) at 37°C. After 2 h, medium was removed and cells were incubated with fresh medium containing 0.2 μM tetramethylrhodamine ethyl ester (TMRE; Thermo Fisher) for 30 min at 37°C. Cells were harvested by trypsinization, collected by centrifugation (500 × g, 5 min), resuspended in PBS containing 2% FBS, and incubated on ice. Next, samples were analyzed by using a BD FACSCanto II (BD Biosciences) equipped with 488-nm and 633-nm lasers. TMRE dye was excited by the 488-nm laser, and its fluorescence was captured using the 585/42 nm bandpass filter. At least 10,000 events (live and apoptotic cells) were collected for each sample. Cell doublets and debris, identified on the basis of their scatter characteristics, were excluded from analysis. Data analysis was performed using FCS Express 6 (De Novo Software). The percent apoptosis was calculated as the percentage of cells that had low fluorescence divided by the total number of cells. A separate portion of each sample was instead lysed in 100 μl RIPA buffer (150 mM NaCl, 1% sodium deoxycholate, 1% NP-40, 0.1% SDS, and 0.01 M Tris-HCl) containing Halt protease inhibitor (Thermo Scientific) for 20 min at 4°C. Cellular lysates were then centrifuged (18,000 × g, 10 min), clarified supernatants were collected, and 30 μg of protein was used to assess protein expression levels via immunoblotting.
Yeast death assays.
The Saccharomyces cerevisiae yeast strain W303α was transformed and analyzed in survival assays as previously described (65). Plasmids directing yeast expression of caspase-3 (101), caspase-9 (102), reverse caspase-9 (66), Bcl-xL (103), and Bax (100) have been previously described. Construction of the M1L yeast expression plasmid is outlined above. pGALL-(HIS3)-ApafFL was created by first inserting an internal HindIII fragment of the Apaf-1 coding region into pGALL-(URA3)-Apaf1–530 (102) and then adding a PCR-generated SpeI/NotI fragment encompassing the 3′ portion of the coding sequence. An EcoRI/NotI fragment containing the 3′ part of the compiled open reading frame was then ligated into pGALL-(HIS3), which followed a EcoRI/EcoRI fragment containing the remaining 5′ section.
Coimmunoprecipitations.
Subconfluent 293T cellular monolayers in six-well plates were transfected with 500 ng C9DN-FLAG, 500 ng N-Apaf-1-myc, and 1,000 ng of either pCI, pM1L-V5, or pHA-K1L. At 24 h posttransfection, cells were detached from plates by scraping and collected by centrifugation (18,000 × g, 1 min). Cellular pellets were resuspended in 150 μl NP-40 lysis buffer (142.5 mM NaCl, 5 mM MgCl2, 10 mM HEPES [pH 7.2], 1 mM EGTA, and 0.2% NP-40) containing Halt protease inhibitor (Thermo Scientific) for 30 min at 4°C. Cellular lysates were then centrifuged (18,000 × g, 10 min). Fifty-microliter volumes of the supernatants were set aside and used to detect protein expression levels via immunoblotting. The remaining 100 μl of the clarified lysates was used for immmunoprecipitations (IP) to detect protein-protein interactions. For IPs, clarified lysates were precleared for 1 h with 50 μl of protein G-Sepharose beads (Invitrogen) at 4°C with rotation. Beads were collected by centrifugation (18,000 × g, 1 min), and clarified lysates were incubated overnight with 1 μl of either anti-FLAG (Sigma, F3165), anti-V5 (Cell Signaling, 13202), or anti-HA (Sigma, H9658) antibody or the appropriate IgG isotype control (mouse [Sigma, I5381]; rabbit [Cell Signaling, 3900]) with constant rotation at 4°C. The next day, 50 μl of protein G-Sepharose beads (Invitrogen) was added to the samples and rotated at 4°C for another 4 h. Beads were collected by centrifugation (18,000 × g, for 1 min) and washed four times with 750 μl of NP-40 lysis buffer. Pelleted bead-protein complexes were resuspended in 33 μl of 2× Laemmli buffer containing 10% 2-mercaptoethanol and boiled for 5 min. Fifteen microliters of each sample was then analyzed for the presence of proteins using immunoblotting. Primary antibodies and concentrations used for these experiments were as follows: anti-FLAG, 1:2,000 (Sigma, F7425); anti-myc, 1:2,000 (Cell Signaling, 2272); anti-V5, 1:5,000 (Millipore, AB3792); anti-HA, 1:10,000 (Cell Signaling, 3724); and anti-β-actin, 1:5,000 (Sigma, A5060).
ACKNOWLEDGMENTS
This work was supported by the University of Illinois at Urbana—Champaign (M.R.R. and J.L.S.) and the Australian Research Council (fellowship no. FT130101349 to M.K.).
We thank Sunetra Biswas, Lauren Gates, Ariana Bravo Cruz, Chris Brooke, Ed Roy, and Daniel Ryerson for helpful discussions.
REFERENCES
- 1.Jorgensen I, Rayamajhi M, Miao EA. 2017. Programmed cell death as a defence against infection. Nat Rev Immunol 17:151–164. doi: 10.1038/nri.2016.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Danthi P. 2016. Viruses and the diversity of cell death. Annu Rev Virol 3:533–553. doi: 10.1146/annurev-virology-110615-042435. [DOI] [PubMed] [Google Scholar]
- 3.Birkinshaw RW, Czabotar PE. 8 April 2017. The BCL-2 family of proteins and mitochondrial outer membrane permeabilisation. Semin Cell Dev Biol doi: 10.1016/j.semcdb.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 4.Kiraz Y, Adan A, Kartal Yandim M, Baran Y. 2016. Major apoptotic mechanisms and genes involved in apoptosis. Tumour Biol 37:8471–8486. doi: 10.1007/s13277-016-5035-9. [DOI] [PubMed] [Google Scholar]
- 5.Genini D, Budihardjo I, Plunkett W, Wang X, Carrera CJ, Cottam HB, Carson DA, Leoni LM. 2000. Nucleotide requirements for the in vitro activation of the apoptosis protein-activating factor-1-mediated caspase pathway. J Biol Chem 275:29–34. doi: 10.1074/jbc.275.1.29. [DOI] [PubMed] [Google Scholar]
- 6.Kim J, Parrish AB, Kurokawa M, Matsuura K, Freel CD, Andersen JL, Johnson CE, Kornbluth S. 2012. Rsk-mediated phosphorylation and 14-3-3epsilon binding of Apaf-1 suppresses cytochrome c-induced apoptosis. EMBO J 31:1279–1292. doi: 10.1038/emboj.2011.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saleh A, Srinivasula SM, Acharya S, Fishel R, Alnemri ES. 1999. Cytochrome c and dATP-mediated oligomerization of Apaf-1 is a prerequisite for procaspase-9 activation. J Biol Chem 274:17941–17945. doi: 10.1074/jbc.274.25.17941. [DOI] [PubMed] [Google Scholar]
- 8.Li Y, Zhou M, Hu Q, Bai X-C, Huang W, Scheres SHW, Shi Y. 2017. Mechanistic insights into caspase-9 activation by the structure of the apoptosome holoenzyme. Proc Natl Acad Sci U S A 114:1542–1547. doi: 10.1073/pnas.1620626114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shiozaki EN, Chai J, Shi Y. 2002. Oligomerization and activation of caspase-9, induced by Apaf-1 CARD. Proc Natl Acad Sci U S A 99:4197–4202. doi: 10.1073/pnas.072544399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu C-C, Lee S, Malladi S, Chen M-D, Mastrandrea NJ, Zhang Z, Bratton SB. 2016. The Apaf-1 apoptosome induces formation of caspase-9 homo- and heterodimers with distinct activities. Nat Commun 7:13565. doi: 10.1038/ncomms13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu CC, Bratton SB. 2017. Caspase-9 swings both ways in the apoptosome. Mol Cell Oncol 4:e1281865. doi: 10.1080/23723556.2017.1281865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Veyer DL, Carrara G, Maluquer de Motes C, Smith GL. 2017. Vaccinia virus evasion of regulated cell death. Immunol Lett 186:68–80. doi: 10.1016/j.imlet.2017.03.015. [DOI] [PubMed] [Google Scholar]
- 13.Shisler JL. 2015. Immune evasion strategies of molluscum contagiosum virus. Adv Virus Res 92:201–252. doi: 10.1016/bs.aivir.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Nichols D, De Martini W, Cottrell J. 2017. Poxviruses utilize multiple strategies to inhibit apoptosis. Viruses 9:215. doi: 10.3390/v9080215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gubser C, Smith GL. 2002. The sequence of camelpox virus shows it is most closely related to variola virus, the cause of smallpox. J Gen Virol 83:855–872. doi: 10.1099/0022-1317-83-4-855. [DOI] [PubMed] [Google Scholar]
- 16.Gubser C, Bergamaschi D, Hollinshead M, Lu X, van Kuppeveld FJ, Smith GL. 2007. A new inhibitor of apoptosis from vaccinia virus and eukaryotes. PLoS Pathog 3:e17. doi: 10.1371/journal.ppat.0030017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saraiva N, Prole DL, Carrara G, Maluquer de Motes C, Johnson BF, Byrne B, Taylor CW, Smith GL. 2013. Human and viral Golgi anti-apoptotic proteins (GAAPs) oligomerize via different mechanisms and monomeric GAAP inhibits apoptosis and modulates calcium. J Biol Chem 288:13057–13067. doi: 10.1074/jbc.M112.414367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mayr A, Hochstein-Mintzel V, Stickl H. 1975. Abstammung, Eigenschaften und Verwendung des attenuierten Vaccinia-Stammes MVA. Infection 3:6–14. doi: 10.1007/BF01641272. [DOI] [Google Scholar]
- 19.Antoine G, Scheiflinger F, Dorner F, Falkner FG. 1998. The complete genomic sequence of the modified vaccinia Ankara strain: comparison with other orthopoxviruses. Virology 244:365–396. doi: 10.1006/viro.1998.9123. [DOI] [PubMed] [Google Scholar]
- 20.Chahroudi A, Garber DA, Reeves P, Liu L, Kalman D, Feinberg MB. 2006. Differences and similarities in viral life cycle progression and host cell physiology after infection of human dendritic cells with modified vaccinia virus Ankara and vaccinia virus. J Virol 80:8469–8481. doi: 10.1128/JVI.02749-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu L, Chavan R, Feinberg MB. 2008. Dendritic cells are preferentially targeted among hematolymphocytes by modified vaccinia virus Ankara and play a key role in the induction of virus-specific T cell responses in vivo. BMC Immunol 9:15. doi: 10.1186/1471-2172-9-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guzman E, Cubillos-Zapata C, Cottingham MG, Gilbert SC, Prentice H, Charleston B, Hope JC. 2012. Modified vaccinia virus Ankara-based vaccine vectors induce apoptosis in dendritic cells draining from the skin via both the extrinsic and intrinsic caspase pathways, preventing efficient antigen presentation. J Virol 86:5452–5466. doi: 10.1128/JVI.00264-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Royo S, Sainz B Jr, Hernandez-Jimenez E, Reyburn H, Lopez-Collazo E, Guerra S. 2014. Differential induction of apoptosis, interferon signaling, and phagocytosis in macrophages infected with a panel of attenuated and nonattenuated poxviruses. J Virol 88:5511–5523. doi: 10.1128/JVI.00468-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brooks MA, Ali AN, Turner PC, Moyer RW. 1995. A rabbitpox virus serpin gene controls host range by inhibiting apoptosis in restrictive cells. J Virol 69:7688–7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Campbell S, Thibault J, Mehta N, Colman PM, Barry M, Kvansakul M. 2014. Structural insight into BH3 domain binding of vaccinia virus antiapoptotic F1L. J Virol 88:8667–8677. doi: 10.1128/JVI.01092-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia MA, Guerra S, Gil J, Jimenez V, Esteban M. 2002. Anti-apoptotic and oncogenic properties of the dsRNA-binding protein of vaccinia virus, E3L. Oncogene 21:8379–8387. doi: 10.1038/sj.onc.1206036. [DOI] [PubMed] [Google Scholar]
- 27.Macen JL, Garner RS, Musy PY, Brooks MA, Turner PC, Moyer RW, McFadden G, Bleackley RC. 1996. Differential inhibition of the Fas- and granule-mediated cytolysis pathways by the orthopoxvirus cytokine response modifier A/SPI-2 and SPI-1 protein. Proc Natl Acad Sci U S A 93:9108–9113. doi: 10.1073/pnas.93.17.9108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J, Mahajan A, Tsai M-D. 2006. Ankyrin repeat: a unique motif mediating protein-protein interactions. Biochemistry 45:15168–15178. doi: 10.1021/bi062188q. [DOI] [PubMed] [Google Scholar]
- 29.Bork P. 1993. Hundreds of ankyrin-like repeats in functionally diverse proteins: mobile modules that cross phyla horizontally? Proteins 17:363–374. doi: 10.1002/prot.340170405. [DOI] [PubMed] [Google Scholar]
- 30.Pluckthun A. 2015. Designed ankyrin repeat proteins (DARPins): binding proteins for research, diagnostics, and therapy. Annu Rev Pharmacol Toxicol 55:489–511. doi: 10.1146/annurev-pharmtox-010611-134654. [DOI] [PubMed] [Google Scholar]
- 31.Flutsch A, Schroeder T, Barandun J, Ackermann R, Buhlmann M, Grutter MG. 2014. Specific targeting of human caspases using designed ankyrin repeat proteins. Biol Chem 395:1243–1252. doi: 10.1515/hsz-2014-0173. [DOI] [PubMed] [Google Scholar]
- 32.Herbert MH, Squire CJ, Mercer AA. 2015. Poxviral ankyrin proteins. Viruses 7:709–738. doi: 10.3390/v7020709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sonnberg S, Fleming SB, Mercer AA. 2011. Phylogenetic analysis of the large family of poxvirus ankyrin-repeat proteins reveals orthologue groups within and across chordopoxvirus genera. J Gen Virol 92:2596–2607. doi: 10.1099/vir.0.033654-0. [DOI] [PubMed] [Google Scholar]
- 34.Bravo Cruz AG, Shisler JL. 2016. Vaccinia virus K1 ankyrin repeat protein inhibits NF-kappaB activation by preventing RelA acetylation. J Gen Virol 97:2691–2702. doi: 10.1099/jgv.0.000576. [DOI] [PubMed] [Google Scholar]
- 35.Bravo Cruz AG, Han A, Roy EJ, Guzman AB, Miller RJ, Driskell EA, O'Brien WD Jr, Shisler JL. 12 July 2017. Deletion of the K1L gene results in a vaccinia virus that is less pathogenic due to muted innate immune responses, yet still elicits protective immunity. J Virol doi: 10.1128/JVI.00542-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Willis KL, Patel S, Xiang Y, Shisler JL. 2009. The effect of the vaccinia K1 protein on the PKR-eIF2alpha pathway in RK13 and HeLa cells. Virology 394:73–81. doi: 10.1016/j.virol.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Willis KL, Langland JO, Shisler JL. 2011. Viral double-stranded RNAs from vaccinia virus early or intermediate gene transcripts possess PKR activating function, resulting in NF-kappaB activation, when the K1 protein is absent or mutated. J Biol Chem 286:7765–7778. doi: 10.1074/jbc.M110.194704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meng X, Jiang C, Arsenio J, Dick K, Cao J, Xiang Y. 2009. Vaccinia virus K1L and C7L inhibit antiviral activities induced by type I interferons. J Virol 83:10627–10636. doi: 10.1128/JVI.01260-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Drillien R, Koehren F, Kirn A. 1981. Host range deletion mutant of vaccinia virus defective in human cells. Virology 111:488–499. doi: 10.1016/0042-6822(81)90351-2. [DOI] [PubMed] [Google Scholar]
- 40.Perkus ME, Goebel SJ, Davis SW, Johnson GP, Limbach K, Norton EK, Paoletti E. 1990. Vaccinia virus host range genes. Virology 179:276–286. doi: 10.1016/0042-6822(90)90296-4. [DOI] [PubMed] [Google Scholar]
- 41.Burles K, Irwin CR, Burton R-L, Schriewer J, Evans DH, Buller RM, Barry M. 2014. Initial characterization of vaccinia virus B4 suggests a role in virus spread. Virology 456-457: 108–120. doi: 10.1016/j.virol.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 42.Alcami A, Symons JA, Collins PD, Williams TJ, Smith GL. 1998. Blockade of chemokine activity by a soluble chemokine binding protein from vaccinia virus. J Immunol 160:624–633. [PubMed] [Google Scholar]
- 43.Eckart RA, Bisle S, Schulze-Luehrmann J, Wittmann I, Jantsch J, Schmid B, Berens C, Luhrmann A. 2014. Antiapoptotic activity of Coxiella burnetii effector protein AnkG is controlled by p32-dependent trafficking. Infect Immun 82:2763–2771. doi: 10.1128/IAI.01204-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fath-Goodin A, Kroemer JA, Webb BA. 2009. The Campoletis sonorensis ichnovirus vankyrin protein P-vank-1 inhibits apoptosis in insect Sf9 cells. Insect Mol Biol 18:497–506. doi: 10.1111/j.1365-2583.2009.00892.x. [DOI] [PubMed] [Google Scholar]
- 45.Luhrmann A, Nogueira CV, Carey KL, Roy CR. 2010. Inhibition of pathogen-induced apoptosis by a Coxiella burnetii type IV effector protein. Proc Natl Acad Sci U S A 107:18997–19001. doi: 10.1073/pnas.1004380107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schweizer A, Roschitzki-Voser H, Amstutz P, Briand C, Gulotti-Georgieva M, Prenosil E, Binz HK, Capitani G, Baici A, Pluckthun A, Grutter MG. 2007. Inhibition of caspase-2 by a designed ankyrin repeat protein: specificity, structure, and inhibition mechanism. Structure 15:625–636. doi: 10.1016/j.str.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 47.Tamin A, Villarreal EC, Weinrich SL, Hruby DE. 1988. Nucleotide sequence and molecular genetic analysis of the vaccinia virus HindIII N/M region encoding the genes responsible for resistance to α-amanitin. Virology 165:141–150. doi: 10.1016/0042-6822(88)90667-8. [DOI] [PubMed] [Google Scholar]
- 48.Yang Z, Bruno DP, Martens CA, Porcella SF, Moss B. 2010. Simultaneous high-resolution analysis of vaccinia virus and host cell transcriptomes by deep RNA sequencing. Proc Natl Acad Sci U S A 107:11513–11518. doi: 10.1073/pnas.1006594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiang Y, Simpson DA, Spiegel J, Zhou A, Silverman RH, Condit RC. 1998. The vaccinia virus A18R DNA helicase is a postreplicative negative transcription elongation factor. J Virol 72:7012–7023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carroll MW, Moss B. 1997. Host range and cytopathogenicity of the highly attenuated MVA strain of vaccinia virus: propagation and generation of recombinant viruses in a nonhuman mammalian cell line. Virology 238:198–211. doi: 10.1006/viro.1997.8845. [DOI] [PubMed] [Google Scholar]
- 51.Wyatt LS, Carroll MW, Czerny CP, Merchlinsky M, Sisler JR, Moss B. 1998. Marker rescue of the host range restriction defects of modified vaccinia virus Ankara. Virology 251:334–342. doi: 10.1006/viro.1998.9397. [DOI] [PubMed] [Google Scholar]
- 52.Voth DE, Howe D, Heinzen RA. 2007. Coxiella burnetii inhibits apoptosis in human THP-1 cells and monkey primary alveolar macrophages. Infect Immun 75:4263–4271. doi: 10.1128/IAI.00594-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE, Poirier GG. 1993. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res 53:3976–3985. [PubMed] [Google Scholar]
- 54.Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH, Peter ME. 1998. Two CD95 (APO-1/Fas) signaling pathways. EMBO J 17:1675–1687. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Juo P, Kuo CJ, Yuan J, Blenis J. 1998. Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade. Curr Biol 8:1001–1008. doi: 10.1016/S0960-9822(07)00420-4. [DOI] [PubMed] [Google Scholar]
- 56.Rodriguez J, Lazebnik Y. 1999. Caspase-9 and APAF-1 form an active holoenzyme. Genes Dev 13:3179–3184. doi: 10.1101/gad.13.24.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES. 1998. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol Cell 1:949–957. doi: 10.1016/S1097-2765(00)80095-7. [DOI] [PubMed] [Google Scholar]
- 58.Wenzel M, Wunderlich M, Besch R, Poeck H, Willms S, Schwantes A, Kremer M, Sutter G, Endres S, Schmidt A, Rothenfusser S. 2012. Cytosolic DNA triggers mitochondrial apoptosis via DNA damage signaling proteins independently of AIM2 and RNA polymerase III. J Immunol 188:394–403. doi: 10.4049/jimmunol.1100523. [DOI] [PubMed] [Google Scholar]
- 59.Fischer SF, Ludwig H, Holzapfel J, Kvansakul M, Chen L, Huang DC, Sutter G, Knese M, Hacker G. 2006. Modified vaccinia virus Ankara protein F1L is a novel BH3-domain-binding protein and acts together with the early viral protein E3L to block virus-associated apoptosis. Cell Death Differ 13:109–118. doi: 10.1038/sj.cdd.4401718. [DOI] [PubMed] [Google Scholar]
- 60.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. 1997. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 61.Tait SW, Green DR. 2010. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 62.Cottet-Rousselle C, Ronot X, Leverve X, Mayol J-F. 2011. Cytometric assessment of mitochondria using fluorescent probes. Cytometry A 79:405–425. doi: 10.1002/cyto.a.21061. [DOI] [PubMed] [Google Scholar]
- 63.Taylor JM, Quilty D, Banadyga L, Barry M. 2006. The vaccinia virus protein F1L interacts with Bim and inhibits activation of the pro-apoptotic protein Bax. J Biol Chem 281:39728–39739. doi: 10.1074/jbc.M607465200. [DOI] [PubMed] [Google Scholar]
- 64.Wasilenko ST, Meyers AF, Vander Helm K, Barry M. 2001. Vaccinia virus infection disarms the mitochondrion-mediated pathway of the apoptotic cascade by modulating the permeability transition pore. J Virol 75:11437–11448. doi: 10.1128/JVI.75.23.11437-11448.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bloomer DT, Kitevska T, Brand IL, Jabbour AM, Nguyen H, Hawkins CJ. 2016. Modeling metazoan apoptotic pathways in yeast. Methods Mol Biol 1419:161–183. doi: 10.1007/978-1-4939-3581-9_13. [DOI] [PubMed] [Google Scholar]
- 66.Hawkins CJ, Silke J, Verhagen AM, Foster R, Ekert PG, Ashley DM. 2001. Analysis of candidate antagonists of IAP-mediated caspase inhibition using yeast reconstituted with the mammalian Apaf-1-activated apoptosis mechanism. Apoptosis 6:331–338. doi: 10.1023/A:1011329917895. [DOI] [PubMed] [Google Scholar]
- 67.Clem RJ, Fechheimer M, Miller LK. 1991. Prevention of apoptosis by a baculovirus gene during infection of insect cells. Science 254:1388–1390. doi: 10.1126/science.1962198. [DOI] [PubMed] [Google Scholar]
- 68.Brand IL, Green MM, Civciristov S, Pantaki-Eimany D, George C, Gort TR, Huang N, Clem RJ, Hawkins CJ. 2011. Functional and biochemical characterization of the baculovirus caspase inhibitor MaviP35. Cell Death Dis 2:e242. doi: 10.1038/cddis.2011.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hu Y, Benedict MA, Wu D, Inohara N, Núñez G. 1998. Bcl-X(L) interacts with Apaf-1 and inhibits Apaf-1-dependent caspase-9 activation. Proc Natl Acad Sci U S A 95:4386–4391. doi: 10.1073/pnas.95.8.4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oberst A, Pop C, Tremblay AG, Blais V, Denault J-B, Salvesen GS, Green DR. 2010. Inducible dimerization and inducible cleavage reveal a requirement for both processes in caspase-8 activation. J Biol Chem 285:16632–16642. doi: 10.1074/jbc.M109.095083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garcia-Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW, Thornberry NA. 1998. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J Biol Chem 273:32608–32613. doi: 10.1074/jbc.273.49.32608. [DOI] [PubMed] [Google Scholar]
- 72.Ryan CA, Stennicke HR, Nava VE, Burch JB, Hardwick JM, Salvesen GS. 2002. Inhibitor specificity of recombinant and endogenous caspase-9. Biochem J 366:595–601. doi: 10.1042/bj20020863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wasilenko ST, Stewart TL, Meyers AF, Barry M. 2003. Vaccinia virus encodes a previously uncharacterized mitochondrial-associated inhibitor of apoptosis. Proc Natl Acad Sci U S A 100:14345–14350. doi: 10.1073/pnas.2235583100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yu E, Zhai D, Jin C, Gerlic M, Reed JC, Liddington R. 2011. Structural determinants of caspase-9 inhibition by the vaccinia virus protein, F1L. J Biol Chem 286:30748–30758. doi: 10.1074/jbc.M111.280149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhai D, Yu E, Jin C, Welsh K, Shiau CW, Chen L, Salvesen GS, Liddington R, Reed JC. 2010. Vaccinia virus protein F1L is a caspase-9 inhibitor. J Biol Chem 285:5569–5580. doi: 10.1074/jbc.M109.078113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Caria S, Marshall B, Burton RL, Campbell S, Pantaki-Eimany D, Hawkins CJ, Barry M, Kvansakul M. 2016. The N terminus of the vaccinia virus protein F1L is an intrinsically unstructured region that is not involved in apoptosis regulation. J Biol Chem 291:14600–14608. doi: 10.1074/jbc.M116.726851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shen L, Chen C, Wei X, Li X, Luo G, Zhang J, Bin J, Huang X, Cao S, Li G, Liao Y. 2015. Overexpression of ankyrin repeat domain 1 enhances cardiomyocyte apoptosis by promoting p53 activation and mitochondrial dysfunction in rodents. Clin Sci 128:665–678. doi: 10.1042/CS20140586. [DOI] [PubMed] [Google Scholar]
- 78.Kojic S, Nestorovic A, Rakicevic L, Belgrano A, Stankovic M, Divac A, Faulkner G. 2010. A novel role for cardiac ankyrin repeat protein Ankrd1/CARP as a co-activator of the p53 tumor suppressor protein. Arch Biochem Biophys 502:60–67. doi: 10.1016/j.abb.2010.06.029. [DOI] [PubMed] [Google Scholar]
- 79.Sakai T, Liu L, Teng X, Mukai-Sakai R, Shimada H, Kaji R, Mitani T, Matsumoto M, Toida K, Ishimura K, Shishido Y, Mak TW, Fukui K. 2004. Nucling recruits Apaf-1/Pro-caspase-9 complex for the induction of stress-induced apoptosis. J Biol Chem 279:41131–41140. doi: 10.1074/jbc.M402902200. [DOI] [PubMed] [Google Scholar]
- 80.Higashitsuji H, Higashitsuji H, Itoh K, Sakurai T, Nagao T, Sumitomo Y, Masuda T, Dawson S, Shimada Y, Mayer RJ, Fujita J. 2005. The oncoprotein gankyrin binds to MDM2/HDM2, enhancing ubiquitylation and degradation of p53. Cancer Cell 8:75–87. doi: 10.1016/j.ccr.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 81.Higashitsuji H, Liu Y, Mayer RJ, Fujita J. 2005. The oncoprotein gankyrin negatively regulates both p53 and RB by enhancing proteasomal degradation. Cell Cycle 4:1335–1337. doi: 10.4161/cc.4.10.2107. [DOI] [PubMed] [Google Scholar]
- 82.Flutsch A, Ackermann R, Schroeder T, Lukarska M, Hausammann GJ, Weinert C, Briand C, Grutter MG. 2014. Combined inhibition of caspase 3 and caspase 7 by two highly selective DARPins slows down cellular demise. Biochem J 461:279–290. doi: 10.1042/BJ20131456. [DOI] [PubMed] [Google Scholar]
- 83.Zhu B, Nethery KA, Kuriakose JA, Wakeel A, Zhang X, McBride JW. 2009. Nuclear translocated Ehrlichia chaffeensis ankyrin protein interacts with a specific adenine-rich motif of host promoter and intronic Alu elements. Infect Immun 77:4243–4255. doi: 10.1128/IAI.00376-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Faherty CS, Maurelli AT. 2008. Staying alive: bacterial inhibition of apoptosis during infection. Trends Microbiol 16:173–180. doi: 10.1016/j.tim.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G. 2008. Viral control of mitochondrial apoptosis. PLoS Pathog 4:e1000018. doi: 10.1371/journal.ppat.1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nogal ML, González de Buitrago G, Rodríguez C, Cubelos B, Carrascosa AL, Salas ML, Revilla Y. 2001. African swine fever virus IAP homologue inhibits caspase activation and promotes cell survival in mammalian cells. J Virol 75:2535–2543. doi: 10.1128/JVI.75.6.2535-2543.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Santic M, Pavokovic G, Jones S, Asare R, Kwaik YA. 2010. Regulation of apoptosis and anti-apoptosis signalling by Francisella tularensis. Microbes Infect 12:126–134. doi: 10.1016/j.micinf.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ashare A, Monick MM, Nymon AB, Morrison JM, Noble M, Powers LS, Yarovinsky TO, Yahr TL, Hunninghake GW. 2007. Pseudomonas aeruginosa delays Kupffer cell death via stabilization of the X-chromosome-linked inhibitor of apoptosis protein. J Immunol 179:505–513. doi: 10.4049/jimmunol.179.1.505. [DOI] [PubMed] [Google Scholar]
- 89.Knodler LA, Finlay BB, Steele-Mortimer O. 2005. The Salmonella effector protein SopB protects epithelial cells from apoptosis by sustained activation of Akt. J Biol Chem 280:9058–9064. doi: 10.1074/jbc.M412588200. [DOI] [PubMed] [Google Scholar]
- 90.Abu-Zant A, Jones S, Asare R, Suttles J, Price C, Graham J, Kwaik YA. 2007. Anti-apoptotic signalling by the Dot/Icm secretion system of L. pneumophila. Cell Microbiol 9:246–264. doi: 10.1111/j.1462-5822.2006.00785.x. [DOI] [PubMed] [Google Scholar]
- 91.Pathan N, Marusawa H, Krajewska M, Matsuzawa S-I, Kim H, Okada K, Torii S, Kitada S, Krajewski S, Welsh K, Pio F, Godzik A, Reed JC. 2001. TUCAN, an antiapoptotic caspase-associated recruitment domain family protein overexpressed in cancer. J Biol Chem 276:32220–32229. doi: 10.1074/jbc.M100433200. [DOI] [PubMed] [Google Scholar]
- 92.Pandey P, Saleh A, Nakazawa A, Kumar S, Srinivasula SM, Kumar V, Weichselbaum R, Nalin C, Alnemri ES, Kufe D, Kharbanda S. 2000. Negative regulation of cytochrome c-mediated oligomerization of Apaf-1 and activation of procaspase-9 by heat shock protein 90. EMBO J 19:4310–4322. doi: 10.1093/emboj/19.16.4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Saleh A, Srinivasula SM, Balkir L, Robbins PD, Alnemri ES. 2000. Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat Cell Biol 2:476–483. doi: 10.1038/35019510. [DOI] [PubMed] [Google Scholar]
- 94.Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR. 2000. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol 2:469–475. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- 95.Chau BN, Cheng EHY, Kerr DA, Hardwick JM. 2000. Aven, a novel inhibitor of caspase activation, binds Bcl-xL and Apaf-1. Mol Cell 6:31–40. doi: 10.1016/S1097-2765(05)00021-3. [DOI] [PubMed] [Google Scholar]
- 96.Cho D-H, Hong Y-M, Lee H-J, Woo H-N, Pyo J-O, Mak TW, Jung Y-K. 2004. Induced inhibition of ischemic/hypoxic injury by APIP, a novel Apaf-1-interacting protein. J Biol Chem 279:39942–39950. doi: 10.1074/jbc.M405747200. [DOI] [PubMed] [Google Scholar]
- 97.Zoog SJ, Schiller JJ, Wetter JA, Chejanovsky N, Friesen PD. 2002. Baculovirus apoptotic suppressor P49 is a substrate inhibitor of initiator caspases resistant to P35 in vivo. EMBO J 21:5130–5140. doi: 10.1038/sj.emboj.7594736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J, Lee R-A, Robbins PD, Fernandes-Alnemri T, Shi Y, Alnemri ES. 2001. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature 410:112–116. doi: 10.1038/35065125. [DOI] [PubMed] [Google Scholar]
- 99.Smith GL, Benfield CT, Maluquer de Motes C, Mazzon M, Ember SW, Ferguson BJ, Sumner RP. 2013. Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. J Gen Virol 94:2367–2392. doi: 10.1099/vir.0.055921-0. [DOI] [PubMed] [Google Scholar]
- 100.Jabbour AM, Puryer MA, Yu JY, Lithgow T, Riffkin CD, Ashley DM, Vaux DL, Ekert PG, Hawkins CJ. 2006. Human Bcl-2 cannot directly inhibit the Caenorhabditis elegans Apaf-1 homologue CED-4, but can interact with EGL-1. J Cell Sci 119:2572–2582. doi: 10.1242/jcs.02985. [DOI] [PubMed] [Google Scholar]
- 101.Ho PK, Jabbour AM, Ekert PG, Hawkins CJ. 2005. Caspase-2 is resistant to inhibition by inhibitor of apoptosis proteins (IAPs) and can activate caspase-7. FEBS J 272:1401–1414. doi: 10.1111/j.1742-4658.2005.04573.x. [DOI] [PubMed] [Google Scholar]
- 102.Hawkins CJ, Wang SL, Hay BA. 1999. A cloning method to identify caspases and their regulators in yeast: identification of Drosophila IAP1 as an inhibitor of the Drosophila caspase DCP-1. Proc Natl Acad Sci U S A 96:2885–2890. doi: 10.1073/pnas.96.6.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Beaumont TE, Shekhar TM, Kaur L, Pantaki-Eimany D, Kvansakul M, Hawkins CJ. 2013. Yeast techniques for modeling drugs targeting Bcl-2 and caspase family members. Cell Death Dis 4:e619. doi: 10.1038/cddis.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]