

Abstract
A suitable liquid chromatography quadrupole time-of-flight mass spectrometric (LC–Q-TOF–MS) method was developed for separation and characterization of related substances in bacitracin test drug. The separation was performed on LiChrospher RP-18 column using methanol as mobile phase A and 0.2% ammonium acetate buffer solution as mobile phase B in gradient elution. A total of 12 related substances were detected through high resolution mass spectrometric determination in a positive electrospray ionization mode. They were identified as co-existing active components and degradation products of bacitracin through the analysis and elucidation of both the protonated parents and the product ions of all the related substances and their fragmentation pathways were also proposed.
Keywords: Bacitracin, Degradation products, Fragmentation pathways, Related substances, LC–Q-TOF–MS
1. Introduction
Bacitracin (Fig. 1) is an antibiotic that is produced by certain strains of bacteria, Bacillus licheniformis and Bacillus subtilis [1], [2], [3], [4], [5], [6], [7]. It has a potent antimicrobial activity against gram-positive bacteria, more specifically against cocci and bacilli bacteria [1], [6], [7], [8]. Its antimicrobial activity is due to its interference with the bacterial cell wall synthesis as well as its inhibition of the regeneration of phospholipid receptors involved in peptidoglycan synthesis [1], [5], [6]. Bacitracin is used in human medicine as well as for veterinary purposes especially in animal feeds [1], [4].
Fig. 1.

Structure of bacitracin.
Bacitracin belongs to a class of antibiotics called polymyxins that have characteristic cyclic peptidic structures with hydrophobic side chains [5]. Bacitracin is available as a mixture of several closely related polypeptides components differing in some amino acids [2], [3], [4]. Bacitracin is a highly polar compound and is often available in more stable salt form as bacitracin zinc [1], [3], [4].
Bacitracin is composed of heptapeptide ring that is connected to a pentapeptide side chain and its related substances are governed by the type of N-terminal thiazoline ring and amino acids at positions X and Y in the polypeptide chain [6], [7]. The components designated as A, B1 and B2 are the most microbiologically active while the main degradation product is an inactive component F [2], [4], [6]. Bacitracin F is known to be nephrotoxic and is the oxidative deaminated derivative of bacitracin containing a ketothiazole instead of an amino-thiazoline moiety [2], [4], [9].
In the 1980s, mass spectrometry (MS) became important in peptide chemistry and was capable of determining the molecular masses of peptides. Techniques like tandem MS could even determine the amino acid sequence. The widespread use of MS led to further studies on bacitracin. The earliest of these studies used fast atom bombardment tandem MS (FAB–MS/MS) coupled with different separation techniques. They managed to obtain the molecular masses but little information concerning the structures and peptide sequences of bacitracin and its related substances was available from FAB–MS/MS analysis [10].
From 1992, electrospray ionization tandem MS (ESI–MS/MS) became a widely used technique. This is a powerful and efficient method for peptide sequencing, and can be used independently or in conjunction with wet chemical methods. The successive studies that used ESI–MS/MS succeeded in sequencing the linear side-chain portion of bacitracin A and three bacitracins. In some cases FAB–MS/MS and ESI–MS/MS were used together. They also managed to sequence the cyclic portion of the purified bacitracins A, A2a, B1, B2 and F, for the other minor components. The structures of the minor components were proposed based on the sequence information obtained for the linear side chain and the mass observed for the ring portion. In these studies, the order of the amino acid residues in the ring of the minor components was not sequenced [10], [11].
In 2003, Govaerts et al. [10] did an extensive study on the fragmentation of bacitracin components using ion trap mass spectrometer in positive and negative ESI modes. Ion trap provides MSn capabilities, which allows for more profound study of the fragmentation pattern, besides MS3 and MS4 experiments. In their work, they managed to confirm the structures of the main components and the related components from earlier literature.
However, there are still some components not identified. The aim of this work was to prepare a chromatographic method suitable for mass spectrometric analysis and to identify the structures of the related substances in bacitracin using liquid chromatography coupled with quadrupole time-of-flight MS (LC–Q-TOF–MS) in positive ESI mode.
2. Materials and methods
2.1. Chemicals and reagents
Bacitracin test drug (Batch No. 130902BA) was obtained from Jiangsu Jiuyang Biopharmaceutical Co., Ltd. (China). The standard sample was United States Pharmacopeia (USP) bacitracin reference standard (CAT. No. 1047503, Lot No. IOK222). High-performance liquid chromatography (HPLC) grade methanol was purchased from Tedia (Fairfield, OH, USA). All other chemicals were of analytical grades. Ammonium acetate was supplied by Sinopharm Chemical Reagent Co., Ltd. (China). Formic acid, ammonia solution, 30% hydrogen peroxide and acetic acid were purchased from Nanjing Chemical Reagents Co., Ltd. (Nanjing, China). All aqueous solutions were prepared with highly purified water.
2.2. Instrumentation and conditions
MS analysis was carried out on an Agilent 1260 LC system coupled with Agilent 6520 Q-TOF (Agilent technologies, Santa Clara, CA, USA) and Finnigan TSQ Quantum Ultra AM Triple-quadrupole tandem mass spectrometer (Thermo electron corporation, San Jose, CA, USA).
Q-TOF provided MS data and the initial MS/MS data. It was operated with dual ESI source in positive mode using protonated adduct ions of purine and fluorinated phosphazine HP-921 with m/z 121.050873 (C5H5N4+) and 922.009798 (C18H19O6N3P3F24+) as the internal calibration reference ions, respectively. The nitrogen drying gas temperature was set at 300 °C and the nebulizer pressure at 345 kPa with a flow rate of 10 L/min. The skimmer was set at 65 V, the fragmentor was 175 V and capillary voltage was set at 4 kV. The MS spectra were collected over a range of m/z 100−2000 with the acquisition rate for MS set at 4 spectra/s and MS/MS set at 2 spectra/s. The collision energy was fixed at 40 V.
More liquid chromatography tandem MS (LC–MS/MS) analyses were carried out on the triple-quadruple tandem mass spectrometer in positive ESI mode as well. The product mass spectra were collected over the m/z range from 50 to 1500 Da with the following conditions: spray voltage set at 4 kV, heated capillary temperature at 350 °C, and the nitrogen sheath gas and auxiliary gas set at 350 and 35 kPa, respectively. Argon gas collision induced dissociation was used with a pressure of 0.2 Pa and collision energy of 20–45 eV for optimum fragmentation for each impurity.
2.3. LC analysis
LC analysis was performed on an Agilent 1260 LC system equipped with PDA detector. The separation was carried out on a LiChrospher (250 mm×4.6 mm, 5 µm, 100 Å, Merck, Darmstadt, Germany) by linear gradient elution with mobile phases A and B. Mobile phase A was a mixture of 100 volumes of methanol and 500 volumes of 0.2% ammonium acetate aqueous solution with the pH adjusted to 6.20 by formic acid, which was pre-filtered through 0.45 µm membrane and sonicated before use. Mobile phase B was HPLC grade methanol. The gradient (A:B, v/v) elution program of 0 min (42:58)→35 min (42:58)→40 min (38:62)→55 min (25:75)→55.1 min (42:58)→60 min (42:58) was employed at a flow rate of 1.0 mL/min. The column temperature was maintained at 30 °C, the injection volume was 10 µL and the detector wavelength was set at 254 nm.
2.4. Sample preparation
To prepare the sample solution, about 20 mg of bacitracin test drug was weighed accurately and transferred into a clean 10 mL volumetric flask, dissolved with 6 mL methanol, and then diluted to volume with the buffer solution. The solution with the initial mobile phase ratio was sonicated for about 5 min and then filtered through a 0.45 µm membrane.
Similarly, the standard solution was prepared by weighing about 20 mg of USP bacitracin reference standard and the rest of the procedure was the same as that of the sample solution to produce a final concentration of about 2 mg/mL.
2.5. Stress conditions
The bacitracin test drug underwent the stress conditions of oxidation and epimerization. For oxidation, about 50 mg of bacitracin test drug was dissolved in a mixture of 5 mL formic acid and 0.25 mL 30% hydrogen peroxide. The solution was allowed to stay at room temperature for 30 min. Then, an aliquot of 1 mL of the solution was diluted to 50 mL with water. The pH was adjusted to 7.0 with 25% ammonia solution. The solution was analyzed using the above LC conditions.
For epimerization, about 50 mg of bacitracin test drug was dissolved in 10 mL 3% (m/v) acetic acid solution and it was allowed to stay heated at 37 °C. After 12, 24 and 36 h, 2 mL aliquots were taken and diluted to 50 mL with water (0.2 mg/mL), respectively. The solutions were then analyzed by LC.
3. Results and discussions
3.1. Development of the LC method
The USP [12] and British Pharmacopeia (BP) [13] use phosphate buffer solution as mobile phase for the separation of bacitracin. The phosphate salt used to make the buffer solution is non-volatile. Non–volatile salts are not compatible with ESI–MS because they tend to clog the sampling capillaries and also interfere with the electrospray process by changing the surface tension of the droplets, which in turn affects the evaporation process [10].
Therefore, a mobile phase compatible with MS had to be developed to replace the non-volatile salt. In the development of the MS compatible method, ammonium acetate was chosen as the mobile phase additive with the pH adjusted to 6.20 in a gradient mode on a LiChrospher RP-18 column. The baseline of the gradient method as seen in Fig. 2A was improved by adding about 100 mL of the organic mobile phase to the buffer solution in order to stabilize the buffer and to reduce the bubbles produced during the elution. The related substances numbered in Fig. 2B were all above 0.1% area relative to the main bacitracin component A.
Fig. 2.

(A) Chromatograms of bacitracin test drug (a) and the reference standard (b). (B) Chromatograms of main components of bacitracin test drug (A, B1, B2 and B3) and the related substances (1-12).
3.2. Structural elucidation of bacitracins
The structures of the cyclic polypeptides are determined by the accurate mass matching of the high resolution TOF mass data of their protonated parent molecules, fragments elucidation as well as the fragmentation pattern comparison of the unknowns with known components. The structure of each bacitracin component was obtained by applying this principle. This can work because most of the unknown components are derived from the known components through simple amino acid substitution. In this case the MS data of known components of bacitracin could be used as an interpretive guide to help in the elucidation of the fragmentation spectra for the unknown related substances of bacitracin [10].
Some of the peaks that eluted early in Fig. 2B despite having relative areas higher than 0.1% with UV detection were not recognized as the related substances of bacitracin according to the pharmacopeias. These were also confirmed by the ESI positive MS detection as shown in Fig. 3 because they did not have obvious responses like other detectable polypeptides. In some cases the doubly protonated adduct ions of some components were not detected while their singly protonated adduct ions are represented in Table 1.
Fig. 3.

Total ion chromatograms (ESI+) of bacitracin test drug by (A) LC–Q-TOF–MS and (B) LC–MS/MS.
Table 1.
Structures of the main components and related substances of bacitracin identified by LC–MS and LC–MS/MS mass spectrometric techniques.
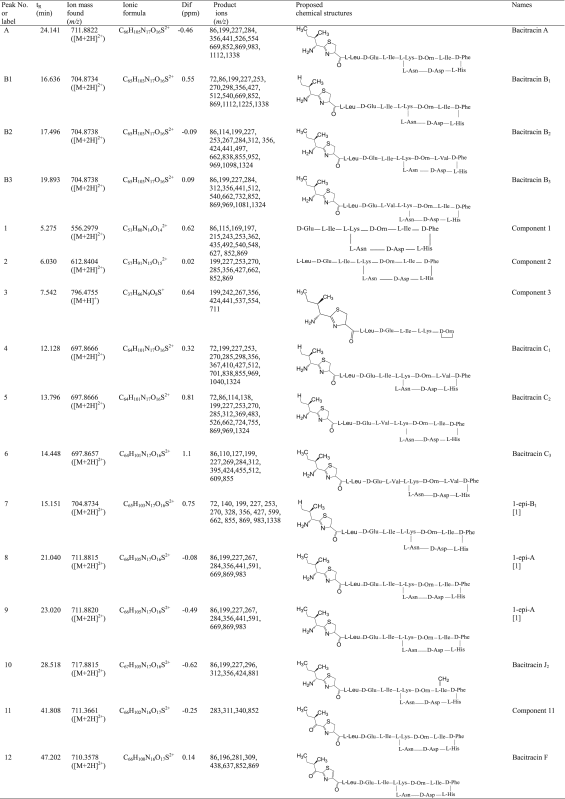 |
3.2.1. Fragmentation patterns of bacitracin's main components (A, B1, B2 and B3)
Bacitracins A, B1, B2 and B3 are regarded as the main active components in bacitracin, so these components are well researched and their tandem mass fragmentation patterns are well documented in literature. The fragments obtained from MS/MS determination shown in Table 1 were elucidated to be consistent with both the structures of the corresponding components and that reported in the literature. Characteristic fragments indicating intact cyclic polypeptide ring were used to identify the amino acid residue type that is found in position X (Fig. 1) of the ring. Then, the characteristic fragments of the side chain provided additional information of position R and Y to complete the structural elucidation processes. Following these processes we were able to observe that bacitracins A and B1 (X and Y=Ile) (Table 2) produced the characteristic fragment ions of m/z 669 (z=2) and 1338 (z=1) when the isoleucine amino acid residue at the N-terminus was cleaved. Similarly, when isoleucine at the N-terminus of bacitracins B2 (X=Val, Y=Ile) and B3 (X=Ile, Y=Val) was cleaved, they produced the characteristic fragment ions of m/z 662 (z=2) and 1324 (z=1). A series of fragments with m/z as 1225, 1112, 983, and 869 which tended to be found in the spectra of these components indicated the cleavage of the peptide bonds between individual amino acid residues on the side chain. However, in the case of bacitracin B3 which has valine at Y, the corresponding fragments would be 14 Da less which was equivalent to the mass difference of Val and Ile. When the cyclic ring was left intact, a series of fragments produced by the side chain for bacitracins A and B2 (Y=Ile, R=CH3) were m/z 554, 441, 199, and 86. Similarly, m/z 540, 427, and 298 were produced by bacitracin B1 (Y=Ile, R=H) and m/z 540, 441, 312, 199, and 86 were produced by bacitracin B3 (Y=Val, R= CH3). Fragments with m/z 356 and 227 found in bacitracins A, B1, B2 and B3 were produced by the neutral loss of the thiazoline ring on the side chain.
Table 2.
The amino acids at positions X and Y and R at the N-terminus for some of the identified bacitracin epimers.
| Name | X | Y | R |
| Bacitracin A | L-Ile | L-Ile | CH3 |
| Bacitracin B1 | L-Ile | L-Ile | H |
| Bacitracin B2 | L-Val | L-Ile | CH3 |
| Bacitracin B3 | L-Ile | L-Val | CH3 |
| Bacitracin C1 | L-Val | L-Ile | H |
| Bacitracin C2 | L-Ile | L-Val | H |
| Bacitracin C3 | L-Val | L-Val | CH3 |
3.2.2. Identification of related substances of bacitracin
3.2.2.1. Bacitracin F (component 12)
Bacitracin F (component 12) is the main related impurity. It was formed when isoleucine and cysteine were condensed to form the thiazoline ring found on the N-terminus with further oxidation to form 2-thiazole [10]. This is the main structural difference between bacitracin F and the main components A, B1, B2 and B3. Bacitracin F has the same cyclic polypeptide ring structure as bacitracins A, B1 and B3, which is proved by the presence of fragment ion m/z 852. A distinct series of fragments of m/z 281 and 309 indicating the N-terminus containing thiazole ring were observed, leading us to the conclusion that component 12 is bacitracin F.
3.2.2.2. Bacitracin C1, C2 and C3 (components 4, 5 and 6)
These components are part of the known related substances of bacitracin documented in literature [10]. Peaks 4, 5 and 6 produced the signature fragments of bacitracins C1, C2 and C3. Since bacitracin C1 has X=Val, Y=Ile, and R=H, its fragmentation was observed to have common fragments of m/z 1324, 969, and 855 with bacitracin B2 and m/z 1324, 969 with bacitracin B3. Fragments characteristic to R=H like m/z 512, 427, 270, 253 and 72 were also the same as bacitracin B1. Similarly, bacitracin C2 has X=Ile, Y=Val and R=H, so a fragmentation pattern corresponding to these was observed such as m/z 1324, 969, 869, and 662. These fragments were the same as bacitracin B2, while fragment ions with m/z 1324, 969, and 662 were similar to bacitracin B3. Other characteristic fragment ions similar to B1 were characteristic to R=H of m/z 270, 253 and 72. Bacitracin C3 has X=Y=Val and R=CH3, so a fragmentation pattern corresponding to this was observed. The fragment ion m/z 855 was the same as bacitracin B2 indicative of the presence of valine in the cyclic ring. Fragment ions same as bacitracin B3 were m/z 1324, 969, and 662. Fragment ions characteristic to R=CH3 like m/z 312, 284, 199 and 86 were similarly observed in bacitracin B3. Bacitracin C3 was also observed to have a molecular weight of 28 Da less than bacitracin B1.
3.2.2.3. Bacitracin epimers (components 7, 8 and 9)
Components of bacitracin that are regarded as epimers tend to produce identical list of product ions as their corresponding counterparts because they have similar structures. For example, an epimer of bacitracin A is called 1-epibacitracin A and its trivial name is allo-bacitracin A. It has d-Ile instead of l-Ile in the N-terminus. Epimers were produced when bacitracin was stressed in acidic media by shifting its double bond in the thiazoline ring to the exocyclic position. This process gave rise to transient intermediate that opened a pathway for the epimerization at α-C of N-terminal Ile. So all related components of bacitracin A should have their own epimers because of the formation of d-Ile or d-Val in the N-terminus [10].
Component 7 had the same mass and fragmentation pattern as bacitracin B1. Similarly, components 8 and 9 had the same mass and fragmentation pattern as bacitracin A. These were all explained to be the isomers of bacitracins B1 and A, respectively. The results of the epimerization experiment in Fig. 4 suggested that these components were all epimers. An increase in peak areas was observed for components 7 and 9. On the other hand, component 8 was found to be made of two overlapping components with one component increasing at a higher rate than the other. Therefore, this experiment suggests that components 7, 8 and 9 are epimers.
Fig. 4.

The chromatogram of bacitracin test drug under the conditions of epimerization after 12 (a), 24 (b) and 36 h (c).
3.2.2.4. Bacitracin J2 (component 10)
It was observed that the molecular weight of component 10 was 12 Da higher than that of bacitracin A. The characteristic fragment m/z 869 of bacitracin A corresponding to the cyclic polypeptide ring was also bigger in component 10, i.e., m/z 881. This means that the amino acid, isoleucine at X position, was replaced by an unknown amino acid residue with 12 Da higher than isoleucine. Component 10 was observed to have the same fragment ions of m/z 312, 199, and 86 as bacitracin B2 and bacitracin B3, which suggests that R, at the N terminal, is a methyl group. The unknown amino acid residue and the new bacitracin component were identified by Mansson et al. [14]. In their work they called this unknown amino acid 5-methylene-isoleucine residue. They found this bacitracin to be an active antimicrobial like the other main components of bacitracin (A, B1, B2 and B3) and named this specific bacitracin bacitracin J2 [14].
3.2.2.5. Component 11
Component 11 was observed to be similar to bacitracins A, B1 and B3, in the cyclic amino acid ring and at the fragment ion m/z 852. However, it was observed that fragments m/z 283 and 311 of component 11 were greater than their counterpart fragments m/z 281 and 309 of bacitracin F by 2 Da. This can be explained that the component 11 was partially oxidized, i.e., the thiazoline ring was not oxidized to form thiazole ring like bacitracin F. The results of the oxidation experiment in Fig. 5 showed that component 11 and bacitracin F were unaffected by the oxidation test.
Fig. 5.

The chromatogram of bacitracin test drug under the conditions of oxidation.
3.2.2.6. Components 1, 2 and 3
The peaks labeled 1, 2 and 3 had low relative abundances and their responses were very low on the MS data. These components had lower molecular masses than other components in the bacitracin test sample. From the MS/MS analysis, it was observed that these peaks were actually fragments of the main components of bacitracin. The MS data of component 1 displayed a doubly protonated ion of m/z 556.2979 as the accurate mass. After studying the fragmentation of component 1, it was observed that it had the same fragment ions of m/z 852, and 869 as bacitracin B3 representing the cyclic polypeptide ring structure. Also the presence of fragment ion m/z 1112 found in bacitracins A and B1, which was equivalent to the singly protonated adduct ion of component 1, could only mean that component 1 was a product of a peptide bond fracture between glutamic acid and leucine amino acid residues of bacitracins A and B1. Component 2, on the other hand, was observed to have the same fragment ion of m/z 869 as bacitracin B3 corresponding to the cyclic polypeptide ring structure found in bacitracins A and B1. Also the presence of fragment ion m/z 1225 found in bacitracin A and B1, which is similar to singly protonated adduct ion of component 2, means that component 2 was a product of a peptide bond fracture between the carbonyl attached to the thiazoline ring and leucine of bacitracins A and B1. Component 3 was observed to have the same fragment ions m/z 554, 356, and 199 as bacitracins A and m/z 441, and 356 as bacitracins A, B1, B2 and B3, indicating that the structure of component 3 is the same as that of bacitracin A except the cyclic polypeptide ring. Fig. 6, Fig. 7, Fig. 8 show proposed fragmentation pathways of components 1, 2 and 3.
Fig. 6.

Proposed fragmentation pathway of component 1.
Fig. 7.

Proposed fragmentation pathway of component 2.
Fig. 8.

Proposed fragmentation pathway of component 3.
4. Conclusion
The HPLC method developed for mass spectrometry was effective and all components were determined for the bacitracin test drug. The known bacitracin components were determined as bacitracins A, B1, B2, B3, C1, C2, C3, F and less known component of bacitracin J2. The exact structures of the epimers (7, 8 and 9) of bacitracins B1 and A could not be determined. The structure of component 11 was identified. Components 1, 2 and 3 were identified as the degradant fragment impurities and their structures and fragmentation pathways were all proposed.
Acknowledgments
This work was financially supported by both the National Natural Science Foundation (NO. 81402900) and the Fundamental Research Funds for the Central Universities of the Ministry of Education (NO. 2015PT043) of China.
Footnotes
Peer review under responsibility of Xi'an Jiaotong University.
References
- 1.Sarri A.K., Megoulas N.C., Koupparis M.A. Development of a novel liquid chromatography–evaporative light scattering detection method for bacitracins and applications to quality control of pharmaceuticals. Anal. Chim. Acta. 2006;573–574:250–257. doi: 10.1016/j.aca.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 2.Pavli V., Kmetec V. Fast separation of bacitracin on monolithic silica columns. J. Pharm. Biomed. Anal. 2004;36:257–264. doi: 10.1016/j.jpba.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 3.Capitan-Vallvey L.F., Titos A., Checa R. High-performance liquid chromatography determination of Zn-bacitracin in animal feed by post-column derivatization and fluorescence detection. J. Chromatogr. A. 2002;943:227–234. doi: 10.1016/s0021-9673(01)01442-x. [DOI] [PubMed] [Google Scholar]
- 4.Pavli V., Kmetec V. Optimization of HPLC method for stability testing of bacitracin. J. Pharm. Biomed. Anal. 2001;24:977–982. doi: 10.1016/s0731-7085(00)00569-0. [DOI] [PubMed] [Google Scholar]
- 5.Potts A.R., Psurek T., Jones C. Validation of a quantitative HPLC method for bacitracin and bacitracin zinc using EDTA as a mobile-phase modifier. J. Pharm. Biomed. Anal. 2012;70:619–623. doi: 10.1016/j.jpba.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 6.Sin D.W.M., Wong Y.C. Analytical methodologies for identifying a polypeptide antibiotic. Trends Anal. Chem. 2003;22:799–809. [Google Scholar]
- 7.Sin D.W., Ho C., Wong Y.C. Analysis of major components of residual bacitracin and colistin in food samples by liquid chromatography tandem mass spectrometry. Anal. Chim. Acta. 2005;535:23–31. [Google Scholar]
- 8.Ming L.J., Epperson J.D. Metal binding and structure–activity relationship of the metalloantibiotic peptide bacitracin. J. Inorg. Biochem. 2002;91:46–58. doi: 10.1016/s0162-0134(02)00464-6. [DOI] [PubMed] [Google Scholar]
- 9.Mascher D.G., Unger C.P., Mascher H.J. Determination of neomycin and bacitracin in human or rabbit serum by HPLC–MS/MS. J. Pharm. Biomed. Anal. 2007;43:691–700. doi: 10.1016/j.jpba.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 10.Govaerts C., Li C., Orwa J. Sequencing of bacitracin A and related minor components by liquid chromatography/electrospray ionization ion trap tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2003;17:1366–1379. doi: 10.1002/rcm.1058. [DOI] [PubMed] [Google Scholar]
- 11.Ikai Y., Oka H., Hayakawa J. Total structures and antimicrobial activity of bacitracin minor components. J. Antibiot. 1995;48:233–242. doi: 10.7164/antibiotics.48.233. [DOI] [PubMed] [Google Scholar]
- 12.The United States Pharmacopeia 38-National Formulary 33, the United States Pharmacopeial Convention, Inc., 12061, Twinbrook Parkway, Rockville, MD 20852, 2015.
- 13.British Pharmacopoeia, Specific monograph: British Pharmacopoeia Commission, London, 2015.
- 14.M. Mansson, C. Senstad, Bacitracin antibiotics, United States Patent, Patent No.:8410044 B2, 2013.