

Abstract
Background:
There are more than 500 different mutations on phenylalanine hydroxylase (PAH) gene that is responsible for phenylketonuria (PKU) diseases and the spectrum of these mutations is varied in different populations. The main clinical manifestation of untreated patients is severe mental retardation. The PAH gene, that is 90 kb long, is consisted of 13 exons and 12 introns. The aim of the present study was to identify the frequency of five common mutations on PAH gene among patients with PKU in Mazandaran and Golestan provinces including c.1066-11G>A, p. R261Q, p. R252W, p. R261X, and c.1200 + 1G>C.
Methods:
Forty unrelated PKU patients, that 22 of them, were from Mazandaran and 18 of them from Golestan provinces were enrolled in the study. Genomic DNA was extracted from leukocytes using Qiagen DNA extraction kit and polymerase chain reaction - restriction fragment length polymorphism method was applied to detect five common mutations.
Results:
Three out of the 5 investigate mutations were identified among the patients. The c.1066-11G>A mutation has the highest frequency (27.5%) among the patients and the frequency of p. R261Q and p. R261X mutations were 3.75 and 1.25%, respectively. In Golestan province, only c.1066-11G>A mutation was observed in investigated alleles.
Conclusions:
The high frequency of c.1066-11G>A mutation in Golestan province may be related to genetic drift, founder effect, and consanguinity.
Keywords: Mutation, phenylalanine hydroxylase, phenylketonuria
Introduction
Phenylketonuria (PKU) is an autosomal recessive genetic disorder which is caused by lack or reduced activity of phenylalanine hydroxylase (PAH) enzyme that converts phenylalanine to tyrosine. Any defects on PAH activity leads to toxic accumulation of phenylalanine on patient's body fluids, and the severity of the disease depends on the level of enzyme activity.[1] In the most severe form of the disorder which is named classic PKU, the serum level of phenylalanine reaches more than 20 mg/dl.[2] Severe mental retardation is the most noticeable clinical manifestation of the disease in untreated subjects.[3] Early diagnosis of the PKU and adopting low-phenylalanine dietary treatment to control the serum level of phenylalanine is the main approach in preventing mental retardation.[3]
The frequency of PKU is 1 in 10,000 and 1 in 16,500 in Caucasians and Oriental live births, respectively.[3,4] Based on screening results, the frequency of PKU is 1 in 10,000 live births in Iran.[5]
The gene responsible for the disease PAH is located on 12 q22–q24.1. The PAH gene, that is 90 kb long, is consisted of 13 exons and 12 introns. To date, more than 500 different mutations have been identified on PAH gene.[4,6] The spectrum of these mutations is varied in different populations, and it is necessary to identify the common mutations in each distinct area to facilitate prenatal diagnosis procedure and genetic counseling.
The aim of this study was to identify the frequency of c.1066-11G>A, p. R261Q, p. R252W, p. R261X, and c.1200 + 1G>C mutations on PAH gene among patients with PKU in Mazandaran and Golestan provinces.
Methods
Forty unrelated PKU patients that 22 of them were from Mazandaran and 18 of them from Golestan provinces were enrolled the study. The parents of all patients signed an informed consent and agreed that their children can share participation in the study. Genomic DNA was extracted from 200 μl of peripheral blood using Qiagen DNA extraction kit.
Polymerase chain reaction (PCR) - restriction fragment length polymorphism method was used to detect five mutations including c.1066-11G>A, p. R261Q, p. R252W, p. R261X, and c.1200 + 1G>C.[6] The regions on PAH gene containing sites of mutations were amplified using PCR with specific primers derived from a published article.[6] The PCR mixture included 200 ng of DNA, 12.5 μ of a 2X PCR master mix containing Taq enzyme, buffer and dNTPs (Takara, Japan), and 10 pmol of reverse and forward primers in total of 25 μ. The amplification program was as follows 5 min at 95°C, 35 cycles of 1 min at 95°C, 1 min at an appropriate annealing temperature, and 1 min at 72°C, ended by a final extension step for 7 min at 72°C.
For the detection of the mentioned mutations, the amplified fragments were treated with specific restriction enzymes as published before (DdeI for c.1066-11G>A, c.1200 + 1G>C and p. R261X, HifI for p. R261Q, and AvaI for p. R252W mutations) and subsequently were analyzed on 2% gel agarose [Figure 1].[6]
Figure 1.
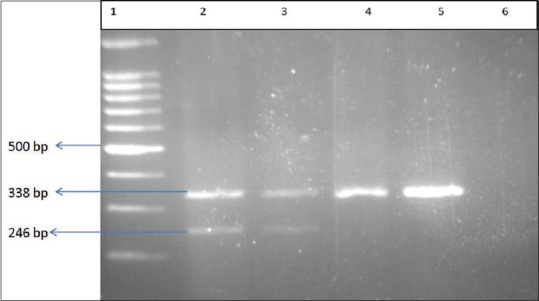
The results of polymerase chain reaction-restriction fragment length polymorphism test for the identification of IVS10-11G>A mutation. Lane 1: Ladder (100 bp), Lanes 2 and 3: heterozygote samples, Lanes 4 and 5: normal samples, Lane 6: Nontemplate control
Results
Of the investigated cases, 16 were male, and 24 were female. Three out of the five common investigated mutations were identified among the patients. In 26 alleles (39.9%), the mutation was detected, and 54 alleles did not carry the investigated mutations. The c.1066-11G>A mutation had the highest frequency in patients and p. R261X mutation was identified just in one allele [Table 1]. Twenty four patients did not have the five common mutations. In patients with Golestani origin only c.1066-11G>A mutation was found (homozygote state in six patients and heterozygote state in two cases) while in Mazandaran province three different mutations were identified. In Mazandaran province in 4 patients, the mutation was identified in both alleles (three cases homozygote for c.1066-11G>A and one subject homozygote for p. R261Q mutations) and in four cases, the mutation was detected just in one allele (two cases with c.1066-11G>A, one with p. R261Q, and one with p. R261X mutations).
Table 1.
The allelic frequency of investigated mutations in Mazandaran and Golestan provinces

Discussion
Mazandaran and Golestan are two neighboring provinces of Iran located on Southern coastlines of the Caspian Sea with a population of around 3 and 2 million, respectively. In Mazandaran, the majority of the population has Mazandarani origin while in Golestan province, Turkmens are one of the major ethnic groups. This is the first report investigating the mutations responsible for PKU disorder in Mazandaran and Golestan provinces.
Bonyadi et al. at 2010 showed that in Azeri Turkish patients of Iran and among 44 PKU patients, c.1066-11G>A and p. P261 L mutations had the highest incidence rate (19.3% and 19.3%) and the frequencies of p. R261Q, p. R261X, and c.1200 + 1G>C mutations were 5.7, 4.5, and 1.1%, respectively.[7] The results of our study showed that among the 80 investigated alleles, c.1066-11G>A was the most common mutation (27.5%) and the frequency of this mutation in Golestani patients was higher than patients with Mazandarani origin (43.75 vs. 18.3%). The p. R261Q and p. R261X mutations were only found in patients from Mazandaran with close frequencies to Azeri patients.
Zare-Karizi Sh et al. at 2011 investigated the mutation spectrum of PAH gene among 248 unrelated chromosomes of Iranian patients.[6] The results showed that c.1066-11G>A is the most frequent mutation in Iran with the frequency of 24.6% and p. R261Q and p. R261X were found in 12.1% and 6% of the alleles, respectively while in our study, the frequency of c.1066-11G>A in Golestani patients was much higher than other provinces and the frequency of p. R261Q mutation on Mazandarani patients were almost half of what was reported in the mentioned study.
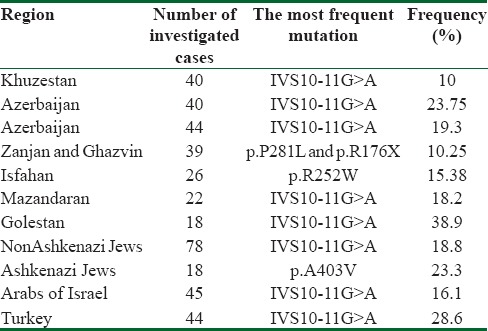
Fifteen different mutations were observed on molecular analysis of PAH gene in 51 of the 54 alleles in Kurdish PKU patients in West of Iran.[8] The c.168 + 5G>C mutation was the most prevalent mutation with the frequency of 26%. In Kermanshah province – located at West of Iran with mainly Kurd population-p. R261Q was observed just in one chromosome of 25 PKU patients.[9] In another study in Qazvin and Zanjan provinces at 2015, Biglari et al. identified 74% of the mutations in 70 investigated chromosomes. P. R176X (10.25%) and p. P281 L (10.25%) were the most common mutations and c.1066-11G>A was not found in any of the patients.[10] PAH mutation analysis of PKU patients in Isfahan Province – Central Iran showed that p. R252W (15.38%) was the most frequent mutation among 26 cases[11] while in our study, none of the patients from Mazandaran and Golestan provinces had that mutation. Besides, in Isfahan province c.1066-11G>A was observed just in 5.7% of the alleles. In Khuzestan province-southeast of Iran-13 different mutations with mutation detection rate of 53.75% was observed on 40 PKU patients and c.1066-11G>A splice site mutation had the highest relative frequency (10%).[12] These differences in the frequencies of mutations represent the variety of ethnic groups and high heterogeneity of the PAH locus in Iran.
The mutation analysis of 163 patients in Israel showed that like the results of our study c.1066-11G>A mutation is the most common mutations among non-Ashkenazi Jews (18.8%) and Arab (16.1%) patients with PKU while this mutation was not identified in Ashkenazi Jews and p. A403V mutation (23.3%) was the most frequent one.[13] In China, p. R243Q mutation is the most frequent mutation and the frequency of c.1066-11G>A mutation is 0.25%.[14]
The results of the present study showed that in comparison to other parts of Iran and the Middle East c.1066-11G>A has the highest frequency in Golestan province [Table 2] that may be related to genetic drift, founder effect, and consanguinity.
Table 2.
Frequency of the most common mutations in different provinces of Iran and Middle East
Although the present study have investigated the frequencies of 5 mutations on PAH gene, the mutations were found only on 32.5% of the chromosomes. Hence, different mutations are responsible for PKU disease in north of Iran, and further studies are recommended to identify all of the mutations on PAH gene in the region.
Financial support and sponsorship
Mazandaran University of Medical Sciences.
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
The author would like to thank the staffs of thalassemia research center for technical support.
References
- 1.Williams RA, Mamotte CD, Burnett JR. Phenylketonuria: An inborn error of phenylalanine metabolism. Clin Biochem Rev. 2008;29:31–41. [PMC free article] [PubMed] [Google Scholar]
- 2.Hanley WB. Adult phenylketonuria. Am J Med. 2004;117:590–5. doi: 10.1016/j.amjmed.2004.03.042. [DOI] [PubMed] [Google Scholar]
- 3.Williams RA, Mamotte CD, Burnett JR. Phenylketonuria: An inborn error of phenylalanine metabolism. Clin Biochem Rev. 2008;29:31–41. [PMC free article] [PubMed] [Google Scholar]
- 4.Scriver CR, Kaufman S, Eisensmith RC, Woo SLC, Vogelstein B, Childs B. The hyperphenylalaninemias. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 1995. pp. 1015–75. [Google Scholar]
- 5.Karamifar H, Ordoei M, Karamizadeh Z, Amirhakimi GH. Incidence of neonatal hyperphenylalaninemia in Fars Province, South Iran. Iran J Pediatr. 2010;20:216–20. [PMC free article] [PubMed] [Google Scholar]
- 6.Zare-Karizi Sh, Hosseini-Mazinani SM, Khazaei-Koohpar Z, Seifati SM, Shahsavan-Behboodi B, Akbari MT, et al. Mutation spectrum of phenylketonuria in Iranian population. Mol Genet Metab. 2011;102:29–32. doi: 10.1016/j.ymgme.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 7.Bonyadi M, Omrani O, Moghanjoghi SM, Shiva S. Mutations of the phenylalanine hydroxylase gene in Iranian Azeri Turkish patients with phenylketonuria. Genet Test Mol Biomarkers. 2010;14:233–5. doi: 10.1089/gtmb.2009.0153. [DOI] [PubMed] [Google Scholar]
- 8.Alibakhshi R, Moradi K, Mohebbi Z, Ghadiri K. Mutation analysis of PAH gene in patients with PKU in Western Iran and its association with polymorphisms: Identification of four novel mutations. Metab Brain Dis. 2014;29:131–8. doi: 10.1007/s11011-013-9432-0. [DOI] [PubMed] [Google Scholar]
- 9.Moradi K, Alibakhshi R, Ghadiri K, Khatami SR, Galehdari H. Molecular analysis of exons 6 and 7 of phenylalanine hydroxylase gene mutations in phenylketonuria patients in Western Iran. Indian J Hum Genet. 2012;18:290–3. doi: 10.4103/0971-6866.107978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Biglari A, Saffari F, Rashvand Z, Alizadeh S, Najafipour R, Sahmani M, et al. Mutations of the phenylalanine hydroxylase gene in Iranian patients with phenylketonuria. Springerplus. 2015;4:542. doi: 10.1186/s40064-015-1309-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vallian S, Barahimi E, Moeini H. Phenylketonuria in Iranian population: A study in institutions for mentally retarded in Isfahan. Mutat Res. 2003;526:45–52. doi: 10.1016/s0027-5107(03)00015-0. [DOI] [PubMed] [Google Scholar]
- 12.Ajami N, Kazeminezhad SR, Foroughmand AM, Hasanpour M, Aminzadeh M. A preliminary mutation analysis of phenylketonuria in Southwest Iran. Genet Mol Res. 2013;12:4958–66. doi: 10.4238/2013.October.24.7. [DOI] [PubMed] [Google Scholar]
- 13.Bercovich D, Elimelech A, Yardeni T, Korem S, Zlotogora J, Gal N, et al. A mutation analysis of the phenylalanine hydroxylase (PAH) gene in the Israeli population. Ann Hum Genet. 2008;72:305–9. doi: 10.1111/j.1469-1809.2007.00425.x. [DOI] [PubMed] [Google Scholar]
- 14.Li N, Jia H, Liu Z, Tao J, Chen S, Li X, et al. Molecular characterisation of phenylketonuria in a Chinese mainland population using next-generation sequencing. Sci Rep. 2015;5:15769. doi: 10.1038/srep15769. [DOI] [PMC free article] [PubMed] [Google Scholar]