

The abrupt and irreversible transitions that drive cells through the DNA replication-division cycle are governed by molecular mechanisms that function as bistable “toggle” switches. A common theme of these switches is a network motif consisting of a “beleaguered” enzyme and its “domineering” substrate, locked in a feedback amplification loop.
Abstract
The cell division cycle is the process by which eukaryotic cells replicate their chromosomes and partition them to two daughter cells. To maintain the integrity of the genome, proliferating cells must be able to block progression through the division cycle at key transition points (called “checkpoints”) if there have been problems in the replication of the chromosomes or their biorientation on the mitotic spindle. These checkpoints are governed by protein-interaction networks, composed of phase-specific cell-cycle activators and inhibitors. Examples include Cdk1:Clb5 and its inhibitor Sic1 at the G1/S checkpoint in budding yeast, APC:Cdc20 and its inhibitor MCC at the mitotic checkpoint, and PP2A:B55 and its inhibitor, alpha-endosulfine, at the mitotic-exit checkpoint. Each of these inhibitors is a substrate as well as a stoichiometric inhibitor of the cell-cycle activator. Because the production of each inhibitor is promoted by a regulatory protein that is itself inhibited by the cell-cycle activator, their interaction network presents a regulatory motif characteristic of a “feedback-amplified domineering substrate” (FADS). We describe how the FADS motif responds to signals in the manner of a bistable toggle switch, and then we discuss how this toggle switch accounts for the abrupt and irreversible nature of three specific cell-cycle checkpoints.
INTRODUCTION
The cell division cycle is the process by which a growing cell replicates all its components and divides them more or less evenly between two daughter cells, so the daughter cells receive all the information and machinery necessary to repeat the process (Mitchison, 1971; Morgan, 2007). The cell’s chromosomes (genome) are the most important components that need to be carefully replicated and accurately partitioned to the daughter cells. Eukaryotic cells replicate their chromosomes during S phase of the cell cycle and partition the sister chromatids to the two daughter cells during mitosis (M phase) and cell division. Proliferating cells undergo repeated cycles of cell division, for which S phase and M phase strictly alternate. Of crucial importance to the integrity of the genome, proliferating cells must be able to block progression through the division cycle at key transition points if there have been problems in the replication or partitioning of the chromosomes. These essential controls are achieved by switchlike cell-cycle transitions, where cells move abruptly and irreversibly from one phase of the division cycle to the next, in response to “wait” and “go” signals (Novak et al., 2007).
Abrupt cell-cycle transitions are governed by underlying biochemical regulatory networks consisting of phase-specific cell-cycle activators and inhibitors (Verdugo et al., 2013). (Here “activator” refers to a component, usually an enzyme, whose activity is required to promote the transition into a specific phase of the cell cycle, regardless of whether its effect on specific substrates is to activate or inhibit them.) Before the transition, an inhibitor (I) maintains the activator (A) in an inactive state and blocks the transition to the next phase of the cell cycle until the requirements for the transition are satisfied. Once the requirements are met, the inhibition is lifted, and the activator promotes the transition into the next phase of the cycle. In addition, the activator prevents the cell from slipping back into the previous phase of the cycle (i.e., the activator–inhibitor motif assures that the transition is abrupt and irreversible).
The “master regulators” of the eukaryotic cell cycle are cyclin-dependent protein kinases (Cdk), which, when active, phosphorylate multiple target proteins at specific sites. As their name implies, activity of a Cdk depends on binding to a cyclin subunit, which activates the kinase subunit and directs its activity toward specific substrates. In addition, Cdk:cyclin dimers can bind to cyclin-dependent kinase inhibitors (CKIs) to form inactive Cdk:cyclin:CKI complexes (Sherr and Roberts, 1999). In this manner, the temporal pattern of Cdk activity is regulated through synthesis and degradation of both cyclins and CKIs. Temporal regulation of Cdk activity, together with anti-correlated regulation of Cdk counteracting protein phosphatases (PP1, PP2, etc.), determines the phosphorylation states of Cdk-targeted substrates, which vary from one phase of the cell cycle to the next (Barr et al., 2011).
Cdk activity is low during G1 phase of the cycle, which precedes DNA replication (Morgan, 2007). Rising Cdk activity, in conjunction with “S-phase cyclins” (e.g., cyclin A), triggers the G1/S transition. Progression through S and G2 phases and into mitosis is characterized by gradual buildup of further Cdk activity, in conjunction with “M-phase cyclins” (cyclin B). As the cell exits mitosis (metaphase-anaphase-telophase), all existing cyclin subunits are destroyed by a two-step process: polyubiquitylation by an E3 ubiquitin ligase, the anaphase promoting complex (APC; also called the cyclosome) in association with a targeting subunit Cdc20, followed by degradation within proteasomes (Peters, 2006). APC-dependent degradation of mitotic cyclins and securin promotes the metaphase-anaphase transition. To successfully exit from mitosis and return to G1, it is not enough just to remove Cdk activity by cyclin degradation. Cdk counteracting phosphatases must also become active to remove phosphate groups from mitotic-Cdk substrates. In mammalian cells in particular, activation of PP2A:B55 is important for mitotic exit and full transition into G1 phase of the next cell cycle (Cundell et al., 2013).
Cdk:CycA, APC:Cdc20, and PP2A:B55 are all examples of phase-specific cell-cycle activators (for G1/S, M/A and mitotic exit, respectively). The phase-specific cell-cycle inhibitors that oppose these particular activators are CKIs (like p27Kip1) for Cdk:CycA, alpha-endosulfine (ENSA) for PP2A:B55, and the mitotic checkpoint complex (MCC) for APC:Cdc20. Of special interest to us in this paper is the fact that the production (or actuation) of each of these inhibitors is promoted by a regulatory protein that is itself inhibited by the cell-cycle activator, creating a characteristic “network motif” illustrated in Figure 1. In this paper, we first describe how this motif responds to signals in the manner of a bistable toggle switch. Then we discuss in detail how this toggle switch accounts for the abrupt and irreversible nature of the three specific cell-cycle transitions governed by Cdk:CycA, APC:Cdc20, and PP2A:B55.
FIGURE 1:
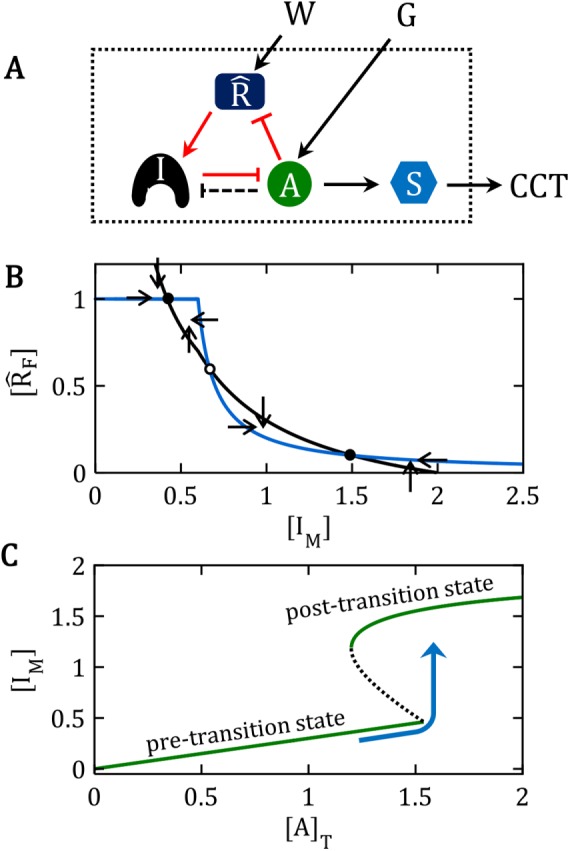
A generic network motif for the bistable switches discussed in this paper. (A) Influence diagram. In an “influence diagram,” barbed arrows (e.g., A → S) mean that “substance A activates substance S,” whereas blunt connectors (e.g., I ⊣ A) mean that “I inhibits A,” without any implications about the molecular mechanism of activation or inhibition. Such influence diagrams are often called “network motifs” because they indicate the “topology” of network interactions without specifying the biochemical mechanism. In this particular influence diagram, A activates S, which promotes a cell-cycle transition (CCT). The CCT is held off by I, which inhibits A. I, A, and 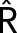 are locked in a positive-feedback amplification loop (the red interactions), which is responsible for the bistable switching properties of the motif. The double-negative feedback loop between A and I is responsible for the nonlinear activation of A by a mechanism of “stoichiometric inhibitor ultrasensitivity,” as will be described later. The double-negative feedback loop by itself is not capable of generating bistability, which we indicate by using a dashed connector from A to I (A – – –| I). By activating , the “wait” signal (W) holds off the CCT and by driving the production of A, the “go” signal (G) promotes the CCT. The motif as a whole is a “signal processing” system (enclosed in the dotted box), which receives “wait” and “go” signals and determines whether the cell will pass the next cell-cycle transition (a binary decision). (B) Phase plane for the network motif in A. In Supplemental Text S1, we propose a simple mathematical model for the influence diagram in A. In the model, the inhibitor I has three forms: Ifree, Imodified, and A:I complex, with [Ifree] + [Imodified] + [A:I] = [I]total = constant. Similarly, [AF] + [A:I] = [A]T = constant and [F] + [M] = []T = constant, where we have introduced the abbreviations F for “free,” M for “modified,” and T for “total.” The mathematical model is described by a pair of ordinary differential equations for d[IM]/dt and d[F]/dt. In the “phase plane” (the Cartesian coordinate system spanned by [IM] and [F]), we plot the curves (called “nullclines”), where d[IM]/dt = 0 (black curve) and where d[F]/dt = 0 (blue curve), for a particular choice of parameter values (see Supplemental Text S1). The small arrows indicate the directions of the vector field along the nullclines. The nullclines intersect in three places (steady states): two stable steady states (•) separated by an unstable steady state (⚬). (C) Signal–response curve. In the simple mathematical model underlying the phase plane in B, the go-signal is the total concentration of A, and the response variable is the steady-state concentration of IM (thinking of IM as representative of all the substrates modified by A). In this diagram, solid green lines represent stable steady states of the bistable switch, and the black dashed line is the locus of unstable steady states. When the signal is small, 0 < [A]T < 1.5, the concentration of free activator is very small, [AF] ≈ 0, and substrates of the activator are sparsely modified (e.g., [IM]/[I]T < 0.25 in this figure). Hence, the lower branch of green curves represents the pretransition state. When the signal is large, [A]T > 1.2, the concentration of free activator is large, [AF] ≈ [A]T, and substrates of the activator are heavily modified (e.g., [IM]/[I]T > 0.75), which represents the posttransition state. The system is “bistable” for 1.2 < [A]T < 1.5. As [A]T (the go-signal) increases from 0 toward a final value of 2 (blue curve), the system makes an abrupt transition from the pretransition state to the posttransition state at [A]T ≈ 1.5. See Supplemental Text S1 for details of this calculation.
are locked in a positive-feedback amplification loop (the red interactions), which is responsible for the bistable switching properties of the motif. The double-negative feedback loop between A and I is responsible for the nonlinear activation of A by a mechanism of “stoichiometric inhibitor ultrasensitivity,” as will be described later. The double-negative feedback loop by itself is not capable of generating bistability, which we indicate by using a dashed connector from A to I (A – – –| I). By activating , the “wait” signal (W) holds off the CCT and by driving the production of A, the “go” signal (G) promotes the CCT. The motif as a whole is a “signal processing” system (enclosed in the dotted box), which receives “wait” and “go” signals and determines whether the cell will pass the next cell-cycle transition (a binary decision). (B) Phase plane for the network motif in A. In Supplemental Text S1, we propose a simple mathematical model for the influence diagram in A. In the model, the inhibitor I has three forms: Ifree, Imodified, and A:I complex, with [Ifree] + [Imodified] + [A:I] = [I]total = constant. Similarly, [AF] + [A:I] = [A]T = constant and [F] + [M] = []T = constant, where we have introduced the abbreviations F for “free,” M for “modified,” and T for “total.” The mathematical model is described by a pair of ordinary differential equations for d[IM]/dt and d[F]/dt. In the “phase plane” (the Cartesian coordinate system spanned by [IM] and [F]), we plot the curves (called “nullclines”), where d[IM]/dt = 0 (black curve) and where d[F]/dt = 0 (blue curve), for a particular choice of parameter values (see Supplemental Text S1). The small arrows indicate the directions of the vector field along the nullclines. The nullclines intersect in three places (steady states): two stable steady states (•) separated by an unstable steady state (⚬). (C) Signal–response curve. In the simple mathematical model underlying the phase plane in B, the go-signal is the total concentration of A, and the response variable is the steady-state concentration of IM (thinking of IM as representative of all the substrates modified by A). In this diagram, solid green lines represent stable steady states of the bistable switch, and the black dashed line is the locus of unstable steady states. When the signal is small, 0 < [A]T < 1.5, the concentration of free activator is very small, [AF] ≈ 0, and substrates of the activator are sparsely modified (e.g., [IM]/[I]T < 0.25 in this figure). Hence, the lower branch of green curves represents the pretransition state. When the signal is large, [A]T > 1.2, the concentration of free activator is large, [AF] ≈ [A]T, and substrates of the activator are heavily modified (e.g., [IM]/[I]T > 0.75), which represents the posttransition state. The system is “bistable” for 1.2 < [A]T < 1.5. As [A]T (the go-signal) increases from 0 toward a final value of 2 (blue curve), the system makes an abrupt transition from the pretransition state to the posttransition state at [A]T ≈ 1.5. See Supplemental Text S1 for details of this calculation.
RESULTS
A design principle of cell-cycle transitions
Bistable toggle switches are common features of biochemical regulatory networks characterized by positive feedback in the network and sufficient nonlinearity in the kinetics of the biochemical reactions (Griffith, 1968; Tyson and Othmer, 1978; Thomas, 1998; Cherry and Adler, 2000; Tyson et al., 2003; Tyson and Novak, 2010). Our network motif (Figure 1) provides the requisite nonlinearity by a mechanism of stoichiometric inhibitor ultrasensitivity and the requisite positive feedback by the amplification loop evident in Figure 1A. In Supplemental Text S1, we analyze a simple mathematical model of the regulatory motif in Figure 1A. In this model, we think of the activator, A, as an enzyme that “modifies” its substrates, S, I, and . The model consists of a pair of nonlinear ordinary differential equations, (S1) and (S2), that describe the interactions of F and IM (the “unmodified” form of the regenerating enzyme and the “modified” form of inhibitor, respectively), assuming that “free” activator is the difference between “total” activator and activator bound in tight complexes with “unmodified” inhibitor ([AF] = [A]T – [A:I]). Figure 1B shows a phase plane for the simple model for a particular choice of [A]T. In this case, the nullclines (d[F]/dt = 0, blue curve, and d[IM]/dt = 0, black curve) intersect in three points, two stable steady states (•) separated by an unstable steady state (o), that is, the model exhibits bistability. If we plot the values of [IM] at the stable and unstable steady states as functions of [A]T, then we obtain an S-shaped “signal-response” curve (Figure 1C), which shows how the model executes an irreversible transition from a pretransition steady state ([IM] small, [AF] small) to a posttransition steady state ([IM] large, [AF] large) as [A]T increases. These generic properties of the network motif in Figure 1A will be characteristic features of the molecular mechanisms that underlie the specific cell-cycle transitions to be described in detail later in this paper.
In this section, by a sequence of reaction steps in Figure 2, A–C, we convert the influence diagram (Figure 1A) into a detailed reaction mechanism (Figure 3A) to show how ultrasensitivity and positive feedback arise from molecular interactions in the network.
FIGURE 2:
Properties of reaction mechanisms that lack the positive-feedback amplification loop. (A) The reaction mechanism for stoichiometric inhibition of A by strong binding to I. In a reaction mechanism, as opposed to an influence diagram, solid arrows indicate chemical reactions (reactants at one end and products at the other), and dashed arrows indicate catalytic influences (enzyme at one end and catalyzed reaction at the other). In some instances, a dashed arrow points to a catalyzed “process” (e.g., CCT) rather than a specific chemical reaction. (B) The reaction mechanism by which a domineering substrate I is shut off by enzyme A, because A catalyzes the modification of I to an ineffectual form, IM. (C) The reaction mechanism for regenerating the domineering substrate I by the action of a demodifying enzyme . (D) Ultrasensitive “signal–response” curve for SM as a function of AT, for the reaction mechanism in A. The dashed line is the limiting case of the signal–response curve derived in the main text, for JA = 0.5. The solid curve is the ultrasensitive response curve for realistic values of rate constants, as derived in Supplemental Text S2. (E) For the reaction mechanism in B, I is steadily converted to IM over the course of time, and eventually the beleaguered enzyme A is able to modify its intended substrate (S → SM) and induce the cell-cycle transition. See Supplemental Text S3 for details.
FIGURE 3:
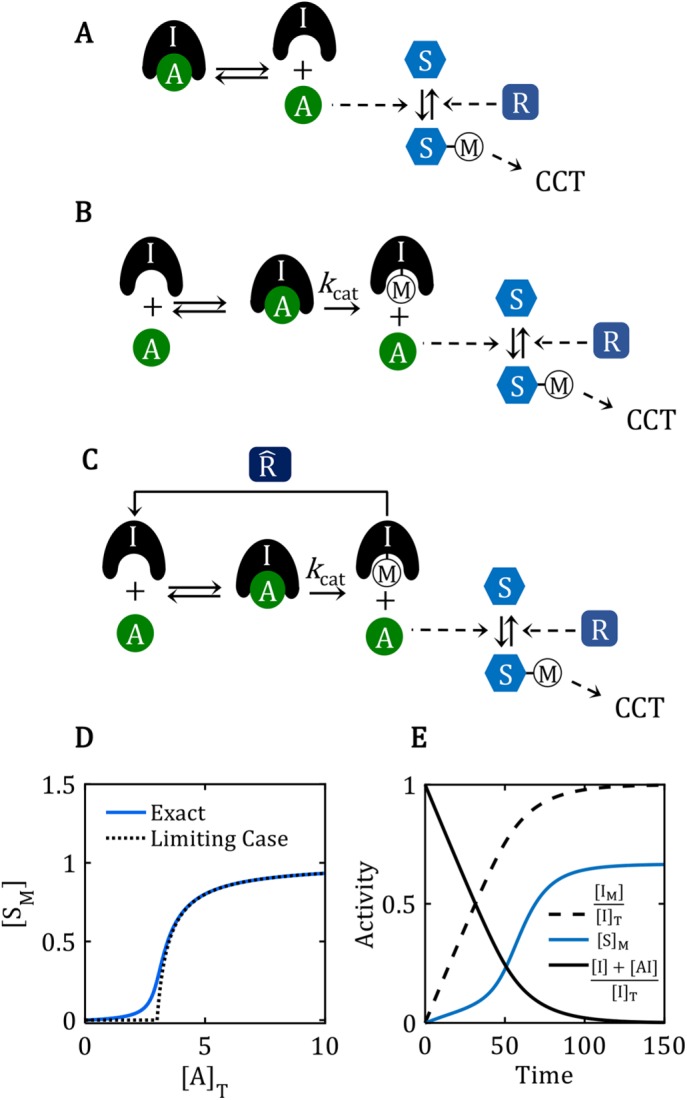
Bistability in the full network, with the positive-feedback amplification loop. (A) Reaction mechanism for a feedback-amplified domineering substrate. This reaction mechanism is a particular realization of the FADS motif illustrated in Figure 1A. The enzyme, W, that demodifies M is a “transition wait” signal. (B) Phase plane diagrams for the bistable switch. For AT = 0.5, there is a unique, stable steady state (•) with F ≈ T = 1, IM ≈ 0.3 << IT = 3, AF ≈ 0. For AT = 2, there is a unique, stable steady state (•) with F ≈ 0, IM ≈ IT = 3, AF ≈ 2. For AT = 1, the network is bistable: there are two stable steady states (•) separated by an unstable steady state (⚬). See Supplemental Text S4 for details of the calculations in B, C, and D. (C) Signal–response curve for a bistable switch. The go-signal is the total concentration of A; the response is the concentration of active (free) A. Solid lines plot the loci of stable steady states as functions of [A]T; dashed line plots the locus of unstable steady states. The network is bistable over the interval 0.70 < [A]T < 1.74. (D) The toggle-switch domain in parameter space. The FADS motif is bistable within the wedge-shaped region bounded by the green curves. [A]T is the go-signal for the cell-cycle transition; kmodR is the rate constant for the modification of by A. For kmodR = 0, the positive feedback amplification loop is broken, and the FADS motif is mono-stable.
Stoichiometric inhibitor ultrasensitivity
Consider first a generic cell-cycle activator (A, an enzyme) that initiates a cell-cycle transition by catalysing the posttranslational modification of a substrate S, as in Figure 2A. (The “modification” may be phosphorylation, dephosphorylation, or ubiquitylation in the case of a protein kinase, a protein phosphatase, or an E3 ubiquitin ligase, respectively. In general, S is one representative of a set of many substrates that must be modified by A to effect the transition.) Prior to the cell-cycle transition, the activator A is held in an inactive state by high-affinity binding to a competitive inhibitor, I; that is, I prevents A from modifying S by binding to A with much higher affinity than does S. In this case, the concentration of the A:I complex is approximately equal to the concentration of the subunit that is in short supply, CAI = [A:I] ≈ min(AT, IT), where AT and IT are the total concentrations of A and I, respectively. We refer to I as a “stoichiometric” inhibitor of A, because the activity of A toward S is nearly zero so long as I is in stoichiometric excess over A.
For simplicity, we assume that the total concentration of S is much less than the Km for binding of S to A, ST << Km. In this case, the concentration of the A:S complex is negligible, CAS = [A:S] << ST, and we can use a “mass action” rate law (rather than a “Michaelis-Menten” rate law) to describe the kinetics of substrate modification by A;
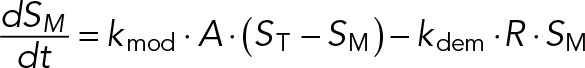 |
where SM is the concentration of the modified form of S, and ST − SM ≈ S = concentration of unmodified substrate. R is the concentration of a “demodifying” (or “regenerating”) enzyme, and kmod and kdem are second-order rate constants for the modification and demodification reactions, respectively.
A consequence of enzyme binding to a high-affinity stoichiometric inhibitor is an ultrasensitive dependence of substrate modification on total enzyme concentration (Figure 2D). By “ultrasensitive” we mean that, for low values of AT, there is little or no modification of the substrate, whereas, for values of AT greater than a certain threshold (AT > IT), the steady-state level of substrate modification rises abruptly to its maximum value, SM ≈ ST.
This ultrasensitive response of substrate modification to total activator concentration is easy to see. For AT < IT, most activator molecules are bound to inhibitor and unavailable to catalyze the conversion of S into SM; so SM ≈ 0 for AT < IT. As AT exceeds IT, increasing concentrations of “free” activator molecules (namely, AT − IT) are available to catalyze the reaction, and the steady-state level of substrate modification is given by
 |
These approximate equations are very close to the exact relation; see Supplemental Text S2 and Figure 2D. This mechanism for generating an ultrasensitive response was first described, to our knowledge, by Thron (1996). It was called “inhibitor ultrasensitivity” by Ferrell (Ferrell and Ha, 2014), but we suggest that “stoichiometric inhibitor ultrasensitivity” is a more precise description.
The inhibitor as a domineering substrate
So far we have considered I to be a competitive inhibitor of the “true” substrates of A. But, in fact, I is often a bona fide substrate of A, and the A:I complex catalyzes the conversion of I into IM, releasing A (Figure 2B). If the dissociation constant of the A:I complex is much less than the dissociation constants of all other A:S complexes, then A will be tied up, initially, converting an excess of I into IM and unavailable for modifying any other substrates (see Supplemental Text S3). Eventually A will rid itself of I, by converting it into IM, and then A will be available for modifying its other substrates (Figure 2E). In this case, the reaction mechanism in Figure 2B functions as a time-delayed, ultrasensitive response, where the duration of the time delay depends inversely on the value of kcat, the rate constant for the catalytic step A:I → A + IM.
Williams et al. (2014) were first to study the reaction mechanism in Figure 2B, which they called “unfair competition.” However, the essence of the mechanism, in our opinion, is not so much that I is an unfair competitor of S as that I is a “domineering substrate” of A. The “beleaguered enzyme” A is dominated by binding to I and unavailable to catalyze the modification of S. Only after A is able to shake off its tormentor (by converting I to IM) is it free to modify its other substrates, the ones involved in the cell-cycle transition (Figure 2E).
If I is a “poor” as well as a “domineering” substrate, that is, if kcat is small, then it will take a long time for A to free itself of I. Worse yet, if there is an enzyme, , that regenerates the domineering substrate, for example, by demodifying IM back to I (Figure 2C), then A may never be able to free itself from inhibition. By that statement, we mean that the mechanism in Figure 2C cannot exhibit bistability for any positive values of the rate constants in the elementary reaction mechanism, as confirmed by “chemical reaction network theory” (Craciun et al., 2006). Intuitively, it is impossible for reaction mechanism 2C to exhibit bistability, despite the presence of a double-negative feedback loop between A and I (I inhibits A by stoichiometric binding and A inhibits I by catalytic modification), because there is no capacity for activator amplification in mechanism 2C. The rate at which I is rendered ineffectual (I → IM) depends on the concentration of A:I complexes, not on the concentration of free A. Hence, the free activator does not feed back positively on its own production. To get positive feedback into the mechanism, we need an additional interaction in the mechanism.
The feedback-amplification loop
The last piece of the puzzle is the recognition that the regenerating enzyme is also a substrate of A and that M is an inactive form of the regenerating enzyme (Figure 3A). This interaction closes the positive feedback loop, I ⊣ A ⊣
→ I, which is a self-amplifying loop, as far as any one of the components is concerned. For example, A helps itself by inhibiting the “friend” () of its own “enemy” (I). This feedback amplification loop converts the ultrasensitive response mechanism (Figure 2D) into a robust, bistable, toggle switch (Figure 3, B and C). In Figure 3B, we plot phase plane portraits, [] versus [IM], for increasing values of [A]T, to show how the toggle switch passes from a pretransition state (left panel, [A]T = 0.5), through a region of bistability (central panel, [A]T = 1), to a posttransition state (right panel, [A]T = 2). In Figure 3C, we collect these steady-state calculations into a signal–response curve, which plots free activator, [A], as a function of total activator, [A]T. The control system undergoes an irreversible transition (red curve) from the pretransition state ([A] << [A]T) to the posttransition state ([A] ≈ [A]T) as [A]T increases past the saddle-node bifurcation point at [A]T = 1.7.
Bistability of the motif is dependent on the feedback amplification loop: if kmodR, the rate constant for modification of by A, is reduced to zero, then mechanism 3A reduces to mechanism 2E and bistability is lost (Figure 3D).
We may refer to the network motif (Figure 1A) (or the full reaction mechanism in Figure 3A) as a “feedback-amplified domineering substrate” (a FADS motif). A mathematical description of the full reaction mechanism (Figure 3A) and its function as a toggle switch is provided in the Supplemental Text.
Domineering substrates and beleaguered enzymes in cell-cycle regulation
We now present evidence that the FADS motif in Figure 1A is a design principle common to three cell-cycle transitions (Table 1) and is responsible for their abrupt and irreversible (switchlike) nature.
TABLE 1:
Activators and inhibitors of cell-cycle transitions.
| Cell-cycle transition | Inhibitor (I) | Activator (A) | Inhibitor-regenerating enzyme () |
Cell-cycle signals |
|---|---|---|---|---|
| G1/S transition in budding yeast | Cyclin-dependent kinase inhibitor (Sic1) | S-phase cyclin-dependent kinase (Cdk1:Clb5) | Transcription factor of Sic1 (Swi5) |
|
| Prometaphase-to-anaphase transition | Mitotic checkpoint complex (MCC) | Anaphase-promoting complex (APC:Cdc20) | M-phase cyclin-dependent kinase (Cdk1:CycB) |
|
| Mitotic exit in human cells | α-endosulfine (ENSA) | Protein phosphatase (PP2A:B55) | Greatwall-kinase (Gwl) |
|
Sic1 inhibition of Cdk1:Clb5 restrains the G1/S transition in budding yeast
While higher eukaryotes use multiple Cdks to control progression through their cell division cycles, yeasts rely on a single Cdk, called Cdk1 (Malumbres, 2014). Cdk1 is associated with specific B-type cyclins during DNA replication and mitosis. For budding yeast, these Cdk1:CycB complexes are Cdk1:Clb5 during S phase and Cdk1:Clb2 during M phase. (Due to a genome duplication event in the evolutionary history of budding yeast, these cyclins have “twins,” Clb5/Clb6 and Clb1/Clb2. We shall ignore this complication, using “Clb5” to refer to both Clb5 and Clb6 and “Clb2” to refer to both Clb1 and Clb2.) Hence, in our FADS motif, the activator (the beleaguered enzyme) of the G1/S transition in budding yeast is Cdk1:Clb5. The inhibitor of Cdk1:Clb5 in budding yeast is a protein called Sic1 (“substrate and inhibitor of Cdk1”) (Schwob et al., 1994). It is interesting to note that Sic1 was recognized, from its initial discovery, as a “domineering substrate” of Cdk1.
By binding to Cdk1:Clb5 complexes, Sic1 inhibits the phosphorylation of other S-phase Cdk1 substrates (Schwob et al., 1994). Since Sic1 is also phosphorylated by Cdk1:Clb5, targeting Sic1 for ubiquitin-mediated degradation (Koivomagi et al., 2011; Yang et al., 2013), the time-delay mechanism of Figure 2C is operating at the G1/S transition in yeasts. One of the substrates of active Cdk1:Clb5 is Swi5, the transcription factor for Sic1 in budding yeast. Because Cdk1-phosphorylated Swi5 is excluded from the nucleus (Moll et al., 1991), the triad Swi5–-Sic1–-Cdk1:Clb5 is locked in a feedback-amplification loop, as required for our generic FADS motif (compare Figure 4A to Figure 1A and Supplemental Figure S1a to Figure 3A).
FIGURE 4:
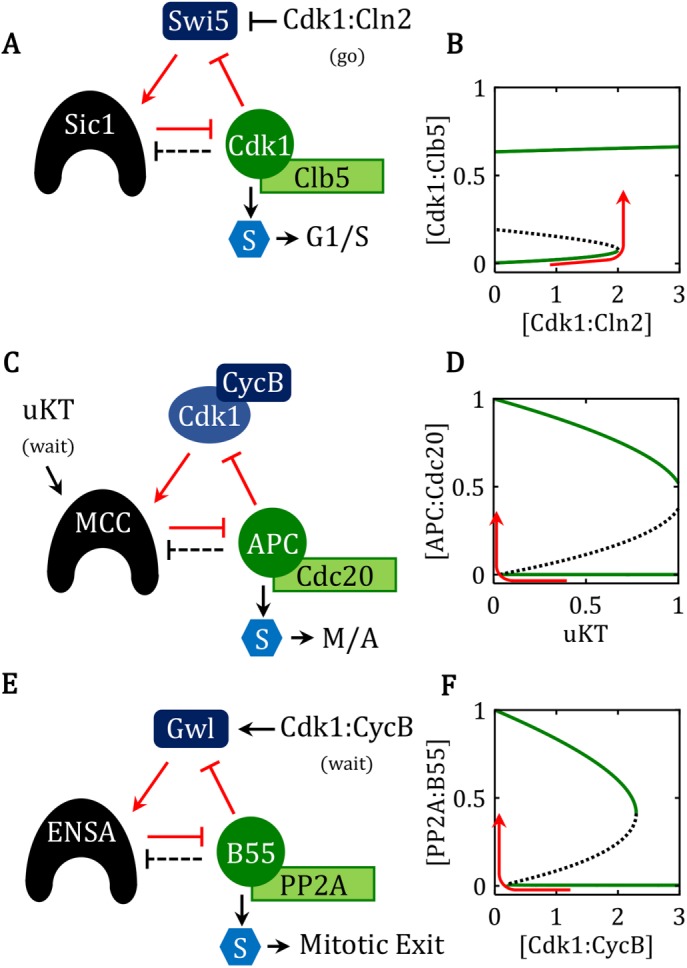
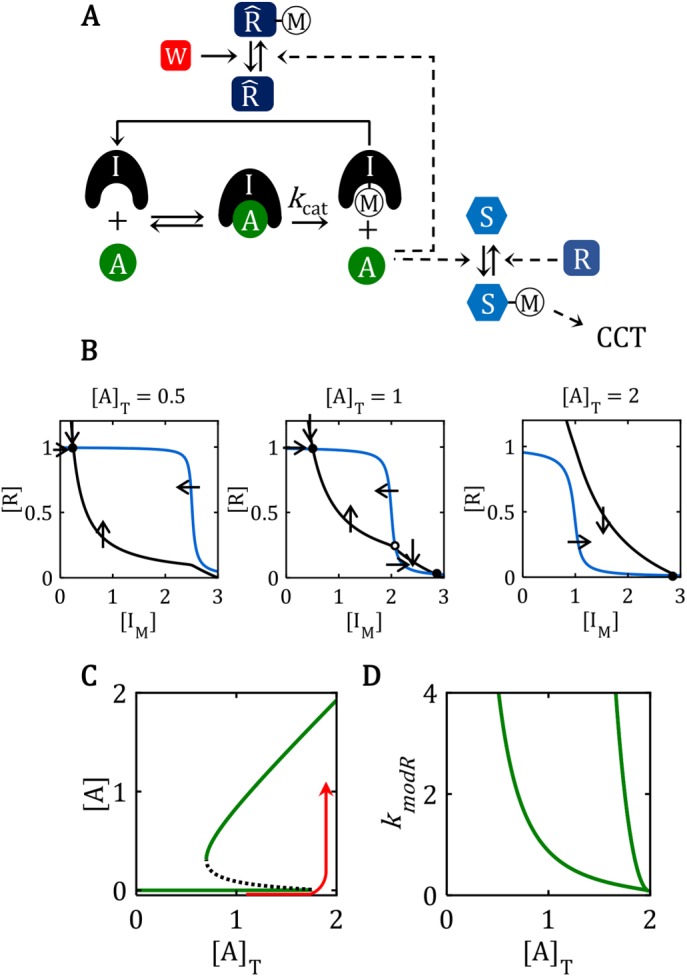
Cell-cycle transitions governed by FADS motifs. For each of the following three cases, the modeling details (reaction mechanisms, differential equations, parameter values, and phase plane diagrams) are given in Supplemental Texts S5–S7 and Supplemental Figures S1–S3. (A, B) The G1/S transition in budding yeast: motif and signal-response curve, respectively. The domineering substrate is Sic1, the beleaguered enzyme is Cdk1:Clb5, and the regenerating factor is Swi5. The steady-state activity of Cdk1:Clb5 is plotted as a function of the activity of Cdk1:Cln2, the go-signal for the G1/S transition in budding yeast. A newborn cell, with [Cdk1:Cln2] = 0, starts in the lower steady state (the G1 phase of the cell cycle; i.e., the pretransition state), with active Swi5, plenty of Sic1, and Cdk1:Clb5 silenced by binding to Sic1. The rise of Cln2-dependent kinase late in G1 phase triggers the irreversible transition into S phase (red curve), when Swi5 is silenced, Sic1 is degraded, and Clb5-dependent kinase activity is high. (C, D) The spindle assembly checkpoint (SAC): motif and signal–response curve, respectively. The domineering substrate is the MCC, the beleaguered enzyme is APC:Cdc20, and the regenerating factor is the mitotic Cdk activity (Cdk1:CycB). The steady-state activity of APC:Cdc20 is plotted as a function of the fraction of kinetochores that are unattached to the mitotic spindle (uKT, the wait-signal). As the cell enters prometaphase, it starts in the lower right corner of the diagram, with all kinetochores unattached, Cdk1:CycB activity high, MCC active, and APC:Cdc20 activity low (the SAC is on). As the cell proceeds through prometaphase, more and more kinetochores become correctly attached to the bipolar spindle, and uKT drops close to 0. For uKT small enough, the control system leaves the pretransition steady state and flips to the posttransition steady state (red curve), with APC:Cdc20 active, securin degraded, and cyclin B level dropping. The transition period comprises metaphase and early anaphase of the classical mitotic sequence; during this time it is possible to reverse the transition and force a cell back into a prometaphase-like state. However, at some point in late anaphase/telophase, the transition becomes irreversible, because Cdk1:CycB activity is so low that detached kinetochores can no longer activate the MCC. (E, F) Exit from mitosis: motif and signal–response curve, respectively. The domineering substrate is ENSA-P, the beleaguered enzyme is PP2A:B55, and the regenerating enzyme is Gwl-P. The steady-state activity of PP2A:B55 is plotted as a function of the activity of the mitotic cyclin-dependent kinase, Cdk1:CycB, the wait-signal for exit-from-mitosis. The late anaphase/telophase cell is waiting for [Cdk1:CycB] to be reduced sufficiently to allow PP2A:B55 to dephosphorylate Gwl-P, the regenerating enzyme. Then the control system fully activates PP2A:B55 (red curve), which dephosphorylates mitotic substrates and thereby returns the cell to G1 phase of the cell cycle.
The “go” signal for the G1/S transition in budding yeast is Cdk1:Cln2, a Cdk:cyclin complex that can phosphorylate Sic1 but is not inhibited by Sic1. Expression of the CLN2 gene is controlled by a different checkpoint (called Start in budding yeast), which is not the subject of this paper. Suffice it to say that as Cdk1:Cln2 activity begins to accumulate in the cell in late G1, some Sic1 gets phosphorylated and degraded, releasing Cdk1:Clb5 from inhibition and triggering the positive-feedback amplification loop.
This particular case of the FADS motif is described in detail in the Supplemental Material, and the signal–response curve for this case is presented in Figure 4B, where the go-signal is G = [Cdk1:Cln2] and the response variable is the activity of the free, S-phase cyclin-dependent kinase [Cdk1:Clb5]. For G = 0, the switch is in the bistable domain, with stable steady states at ≈ 1, IT >> AT, and AF = [Cdk1:Clb5] ≈ 0 (pretransition state, i.e., G1 phase of the cell cycle), and at ≈ 0, IT << AT, and AF = [Cdk1:Clb5] ≈ 0.65 (posttransition state, i.e., S phase of the cell cycle). As G = [Cdk1:Cln2] increases from 0 to just past the saddle-node bifurcation point at G ≈ 2 (the red line in Figure 4A), the control system executes an abrupt transition from G1 phase into S phase of the cell cycle. The transition is irreversible: even if the go-signal drops back to 0 after the transition, the control system stays in the posttransition state, with high activity of Cdk1:Clb5.
MCC inhibition of APC:Cdc20 restrains progression into metaphase
Activation of the Cdk1:CycB1 complex is the universal trigger of mitosis in eukaryotes, causing chromosome condensation and spindle formation. Further progression through mitosis (metaphase → anaphase → telophase) is triggered by the anaphase-promoting complex APC:Cdc20), a ubiquitin-ligase that promotes the degradation of securin and B-type cyclins (Peters, 2006). Securin degradation frees separase to cleave cohesin rings that hold sister chromatids together at centromeres, thereby allowing the mitotic spindle to segregate sister chromatids to opposite sides of the dividing cell. The degradation of all B-type cyclins allows the cell to reset into G1 phase of the next cell cycle.
Successful segregation of sister chromatids requires formation of proper microtubule attachments to the kinetochores of all sister chromatids, thereby achieving biorientation of all replicated chromosomes on the mitotic spindle. To prevent the cell from entering anaphase (the phase of sister chromatid separation) until biorientation of the chromosomes is complete, APC:Cdc20 is kept inactive during the early stages of mitosis (prometaphase) by the spindle assembly checkpoint (SAC) (Musacchio, 2015). We propose that the SAC is based on our generic FADS motif (Figure 1A), with APC:Cdc20 as the beleaguered enzyme, and the MCC as the domineering substrate; compare Figure 4C to Figure 1A and Supplemental Figure S2a to Figure 3A.
The MCC is assembled from four proteins (BubR1, Bub3, Mad2, and Cdc20) at kinetochores that are not attached to the mitotic spindle (Musacchio, 2015). Unattached kinetochores (uKT) are the upstream wait-signals that regulate the SAC. Because Cdk1:CycB1 activity is required for MCC production at unattached kinetochores (Clijsters et al., 2014; Rattani et al., 2014; Vazquez-Novelle et al., 2014), Cdk1:CycB1 is the APC:Cdc20 substrate () that is responsible for regenerating the domineering substrate (MCC). When B-type cyclin levels are high, MCC is produced at unattached kinetochores and outcompetes securin and CycB1 for binding to APC:Cdc20, thereby maintaining the cell in prometaphase. Consistent with the domineering substrate motif, MCC is also a substrate of APC:Cdc20, undergoing continuous disassembly after ubiquitylation of the Cdc20 subunit of the MCC (Reddy et al., 2007; Mansfeld et al., 2011; Varetti et al., 2011; Uzunova et al., 2012). But rapid reassembly of MCCs at unattached kinetochores continues to beleaguer the APC:Cdc20. Once MCC assembly is terminated, following microtubule attachments to every kinetochore, the APC:Cdc20 is able to rid itself of MCC involvement. MCC-free APC:Cdc20 then starts the ubiquitin-mediated degradation of securin and B-type cyclins, which is a hallmark of cell progression through metaphase.
Bistability of the SAC creates a “point-of-no-return” in APC:Cdc20 activation and mitotic progression (He et al., 2011; Verdugo et al., 2013). Once the fraction of unattached kinetochores (uKT) drops below a critical threshold (the red curve in Figure 4D), the domineering substrate (MCC) is no longer regenerated, the beleaguered enzyme (APC:Cdc20) is able to eliminate securin and mitotic cyclins, and the cell transits irreversibly from prometaphase into anaphase. (Notice that “metaphase” is simply the lag period during which this transition is carried out.)
As discussed in He et al. (2011), it is crucial that the saddle-node bifurcation point for activating APC:Cdc20 be close to uKT = 0 (“all centromeres under tension”), so the cell does not transition from prometaphase to anaphase prematurely, and that the saddle-node bifurcation point for inactivating APC:Cdc20 be greater than uKT = 1 (“no centromeres under tension”), so the cell does not revert to SAC-signaling when cohesins are cleaved and tension is lost at all centromeres. Constraints such as these help us to estimate parameter values in reaction mechanisms (He et al., 2011), once the basic switchlike behavior of the checkpoint mechanism is established.
ENSA inhibition of PP2A:B55 restrains exit from mitosis
Intense phosphorylation of hundreds of proteins by Cdk1:CycB1 is necessary to establish the mitotic state. Full mitotic phosphorylation of Cdk1-targeted proteins requires not only high kinase activity but also suppression of Cdk1:CycB1 counteracting phosphatases, like PP2A:B55 (Castilho et al., 2009; Mochida et al., 2009; Vigneron et al., 2009). Inhibition of this phosphatase is accomplished by the BEG (B55-ENSA-Greatwall) pathway (Cundell et al., 2013), which corresponds to our feedback-amplified I—A— loop in Figure 1A. The beleaguered enzyme is the phosphatase PP2A:B55, its domineering substrate is the phosphorylated form of ENSA (Endosulfine and Arpp19 [ Gharbi-Ayachi et al., 2010; Mochida et al., 2010]), and the demodifying/regenerating enzyme is Greatwall kinase (Gwl). PP2A:B55 continuously turns over phosphorylated ENSA into its ineffectual (unphosphorylated) form (Williams et al., 2014). Regeneration (phosphorylation) of ENSA requires Gwl, whose activity depends on Cdk1-dependent phosphorylation; hence, Cdk1:CycB1 is the upstream wait-signal (Blake-Hodek et al., 2012). Since phosphorylated Gwl is inactivated by PP2A:B55 (Mochida et al., 2016), the beleaguered enzyme (PP2A:B55) down-regulates the production of its domineering substrate (P-ENSA) by inactivating the regenerating enzyme (P-Gwl). Therefore, the mechanism of the BEG pathway is identical to the generic mechanism of a feedback-amplified domineering substrate (compare Supplemental Figure S3a and Figure 3A; also Figure 4E to Figure 1A) (Vinod and Novak, 2015).
In prometaphase, the upstream wait-signal (Cdk1:CycB1) is large, say, [Cdk1:CycB1] = 3 in Figure 4F. In this pretransition state, Gwl and ENSA are phosphorylated and PP2A:B55 is inactivated by binding to P-ENSA. Once all the chromosomes are bioriented along the mitotic spindle, the degradation of mitotic cyclins causes a steady drop of Cdk1:CycB1 activity (the red line in Figure 4F). Since the BEG network is bistable, PP2A:B55 is kept inactive until Cdk1:CycB1 drops below the lower threshold in Figure 4F, at the end of mitosis (Cundell et al., 2013), at which point the BEG switch flips to the posttransition (Gwl and ENSA unphosphorylated and PP2A:B55 active). As a consequence of low activity of Cdk1:CycB1 and high activity of PP2A:B55, substrates that were highly phosphorylated in M phase become dephosphorylated, and the cell returns to G1 phase of the cell cycle.
DISCUSSION
We have identified a regulatory motif (FADS) that is common to three eukaryotic cell-cycle transitions. The key features of this scheme are a tight-binding stoichiometric inhibitor of a cell-cycle activator, generating ultrasensitivity, coupled with a positive feedback loop whereby the activator inhibits regeneration of the inhibitor. This motif creates a bistable switch without requiring additional assumptions about ultrasensitivity arising from saturating enzyme kinetics (Goldbeter and Koshland, 1981) or multi-site phosphorylation reactions (Yang et al., 2004; Gunawardena, 2005; Kapuy et al., 2009; Salazar and Hofer, 2009; Ferrell and Ha, 2014). The assumptions made about the kinetic properties of the inhibitor (that it is a “domineering” substrate) are consistent with what might be expected of a stoichiometric inhibitor of an enzyme.
The pipette-rinser analogy
The positive feedback loop in this system (I —| A —|
→ I) provides a mechanism for rapid reversal of inhibition once an upstream cell-cycle signal promotes the transition to the next phase of the cell cycle. To see how this comes about, we compare the mechanisms in Figures 2A and 3A. For the case of stoichiometric-inhibitor ultrasensitivity (Figure 2A), as activator molecules accumulate in the cell, there is, at first, no conversion of S into SM because the activator molecules are sequestered inside a large “vessel” (the A:I complexes). The total amount of A must rise large enough to fill the vessel (AT = IT), and only then will excess A (AT − IT) pour over the top of the vessel and start modifying S.
The case of a feedback-amplified domineering substrate (Figure 3A) is quite different. Instead of a simple vessel into which we “pour” activator molecules until they overflow the top, think of the FADS motif as a pipette washer/rinser. (Readers unfamiliar with this simple piece of laboratory equipment might want to google the term.) Dirty pipettes are placed in a basket, which is placed into a plastic cylinder of soapy water. Then the pipettes are repeatedly rinsed by introducing clean (later, distilled) water into the cylinder. As the water level in the cylinder rises, rinsing the pipettes in the basket, it also rises in the siphon tube on the side of the vessel. When the water reaches the top of the siphon, it overflows, and the siphon tube rapidly drains all the water from the large vessel. The transition from filling to emptying is abrupt and irreversible. The positive feedback loop in the FADS motif functions like the siphon, rapidly freeing all the activator molecules that had been storing up in A:I complexes. The burst of free A rapidly modifies its “true substrates” S and triggers the cell-cycle transition. Down-regulation of by A makes the transition irreversible, because it is no longer possible to regenerate I and reimpose the checkpoint.
The presence of this positive feedback loop in three different inhibitor-activator pairings in vivo (Sic1—Cdk1:CycB, MCC—APC:Cdc20, ENSA—PP2A:B55) suggests that this type of feedback amplification is a well-established mechanism for rendering cell-cycle transitions abrupt and irreversible.
Other motifs
Nonetheless, we are not suggesting that all cell-cycle transitions are governed by FADS motifs. Other bistability-generating motifs play important roles at eukaryotic cell-cycle transitions. For example, the Start transition in budding yeast is governed by a positive-feedback amplification loop (Whi5 —| SBF → Cln2 —| Whi5), but the activator of the transition (SBF) is not an enzyme, and the inhibitor (Whi5) is not an SBF-substrate. Ultrasensitivity is provided, not by stoichiometric inhibition, as in Figure 2, but by multisite phosphorylation of Whi5 (Costanzo et al., 2004; de Bruin et al., 2004). Likewise, entry into mitosis in fission yeast, frog eggs, and mammalian cells is governed by a positive feedback loop (MPF → Cdc25 → MPF) paired with a double-negative feedback loop (Wee1 —| MPF —| Wee1). Although Wee1 is an inhibitor as well as a substrate of MPF, its inhibitory role is mediated by phosphorylation of MPF rather than by stoichiometric binding. Exit from mitosis in budding yeast is promoted by a phosphatase, Cdc14, that is kept inactive until metaphase by binding to a stoichiometric inhibitor, Net1, but this interaction is not a FADS motif because Net1 is not a substrate of Cdc14. Indeed, the phosphorylation of Net1 by Cdk1-, Cdc5-, and Cdc15-kinases during anaphase causes the release of Cdc14 from its complex with Net1 (Queralt and Uhlmann, 2008). If the phosphorylated form of Net1 is a substrate of Cdc14 (Bosl and Li, 2005), it must be a weak substrate; otherwise, their binding would counteract the release of Cdc14 from its complex with Net1 induced by Net1 phosphorylation during anaphase.
Even the G1/S transition in budding yeast is not controlled solely by the FADS motif involving Sic1 and Cdk1:Clb5. In addition, the Clb-type cyclins involved in the G1-to-S transition are destabilized by ubiquitylation by the APC in combination with Cdh1. Cdh1, in turn, is inactivated by phosphorylation on multiple sites by Cdk1:Clb(s) (Zachariae et al., 1998). Even in the absence of Sic1, the double-negative feedback loop between APC:Cdh1 and Cdk1:Clb(s) seems to be sufficient to generate an abrupt and irreversible transition into S phase, probably by a “SIMM” motif, as described by Verdugo et al. (2013).
The latter example (the interaction between Cdh1 and Cdk1:Clb) demonstrates that a double-negative feedback loop can generate bistability but not so in the case of the FADS motif. Although the FADS motif contains two mutually inhibitory species (I —| A and A —| I), the double-negative feedback loop is not the source of bistability in the motif. (The intervening is necessary for generating bistability.) This is because the rate at which the activator shuts down the inhibitor is proportional to the number of A:I complexes; so, with a fixed activator pool, as the concentration of free activator increases, the rate of inhibitor shutdown decreases. To constitute positive feedback, the rate of inhibitor shutdown would have to increase with increasing free-activator concentration. Hence, in the FADS motif, although activator and inhibitor are mutually antagonistic, bistability depends on the three-component, positive feedback loop between activator and inhibitor via the demodifying enzyme . The mutual antagonism of activator and inhibitor, however, is crucial to creating an ultrasensitive nonlinearity, which is also a crucial requirement for bistability.
Reversibility and irreversibility of cell-cycle transitions
In previous publications (Novak and Tyson, 1993; Novak et al., 2007; He et al., 2011; Verdugo et al., 2013), we have emphasized the importance of bistable switches at cell-cycle transitions, because the cell’s “decision” to move on to the next phase of the cell cycle must be resolute and irreversible. Bistability is also crucial to the checkpoint mechanisms that patrol each transition. Wait-signals stabilize the switch in the “off” state (the pretransition state), and go-signals flip the switch to the “on” state (posttransition). Once flipped on, the switch should not drift back into the off state. This sort of irreversibility is a characteristic feature of bistable “toggle” switches.
Nonetheless, there are some subtleties of “irreversibility” that must be acknowledged. The resistance of a molecular switching mechanism to moving backward to the pretransition state is based on biochemical changes that take some time to develop. For example, in the FADS motif, it takes some time for the activator to shake off the inhibitor and to inactivate . During this time, it is possible for a sufficiently strong perturbation to reverse the process and push the switch back to the off state. For example, in the SAC switch, all kinetochores are attached to microtubules at the beginning of metaphase, but it takes some time to activate the APC:Cdc20, to degrade securin, to cut cohesins (beginning of anaphase), and to degrade mitotic cyclins. Hence, there is a window of time (metaphase and early anaphase, at least) when it is still possible to reverse the switch and push the cell back into a prometaphase state, as revealed by a number of elegant experiments in a variety of eukaryotic cells (Clute and Pines, 1999; Hagting et al., 2002; Lopez-Aviles et al., 2009; Oliveira et al., 2010; Vazquez-Novelle et al., 2014). The apparent reversibility of the switch during this transitory period is not evidence against the bistability paradigm but rather a direct consequence of the reaction kinetics of the switch and further corroboration of the general principle.
Chaining the switches
Progression through the eukaryotic cell cycle is regulated at five checkpoints: 1) Start (in budding yeast; called the “restriction point” in mammalian cells), 2) the G1/S transition, 3) the G2/M transition, 4) the spindle assembly checkpoint, and 5) exit from mitosis. All five checkpoints exhibit the properties of bistable toggle switches, and they are all based on protein interaction networks characterized by “ultrasensitivity” and “positive feedback” (Tyson and Novak, 2013). We have shown that checkpoints 2, 4, and 5 are governed by a common motif based on a feedback-amplified domineering substrate. Start and G2/M are governed by other motifs with the common properties of ultrasensitivity and positive feedback. To get a complete picture of progression through the eukaryotic cell cycle, we must chain these toggle switches together in a big loop, so the transitions occur in numerical order, with the posttransition state of phase i being the pretransition state of phase i + 1. In particular, exit from mitosis must put newborn cells into the pretransition state for Start.
To construct a mathematical model of the full cell cycle, we would hook the toggle switches together by identifying the “activator” of transition i with the “inhibitor” (or the wait-signal) of transition i+1. For example, Cdk1:CycB, the activator of the G2/M checkpoint, is the “regenerating enzyme” for the SAC and the “wait-signal” for exit from mitosis. The cell enters mitosis when Cdk1:CycB activity rises at the end of G2 phase, and high activity of Cdk1:CycB promotes MCC assembly (in response to unattached kinetochores) and Greatwall activation (Gwl is the “regenerating enzyme” of the exit-from-mitosis FADS motif). When the mitotic checkpoint is satisfied (i.e., unattached kinetochores = 0), the beleaguered enzyme (APC:Cdc20) is activated, and active APC:Cdc20 promotes degradation of securin and of cyclin B. Falling activity of Cdk1:CycB is identified with loss of the wait-signal for exit-from-mitosis, allowing the Exit checkpoint to flip to the active state for PP2A:B55. Among the substrates dephosphorylated by PP2A:B55 is Cdh1, which, in its dephosphorylated form, targets the APC to ubiquitylation of B-type cyclins. In this role, APC:Cdh1 is a major stabilizer of G1 phase of the cell cycle, that is, a major wait-signal for the Start transition and the G1/S checkpoint.
By pursuing this line of reasoning, we could construct, in principle, a mathematical model of the full cell division cycle of a “generic” eukaryotic cell. For any particular type of cell (yeast, frog egg, fruit fly, mouse, or human cell), we would have to take into account the peculiar molecular features of the proteins making up the checkpoints in each of these cell types.
Conclusion
The conservation of the FADS motif across three cell-cycle transitions highlights both the similar requirements of the system at each transition and the simplicity and efficiency of the network motif, which is implemented using a different set of molecular components in each case. At each transition, the cell must be able to move from one cell-cycle stage to the next in a rapid and irreversible manner in response to specific upstream signals. The transition must otherwise be robust to perturbation. A bistable switch fulfills these requirements, and feedback-amplification of domineering substrates and their beleaguered enzymes creates the necessary conditions for switch-like behavior from a small set of components and interactions, while still maintaining the ability to promote a rapid transition in response to cell-cycle signals.
MATERIALS AND METHODS
All mathematical equations and computational methods are described and explained in detail in the Supplemental Text. We also provide the code for each model in the form of “.ode” files (see the Supplemental ODE Files) that allow the users to reproduce our figures with the free available software XPP/AUTO (www.math.pitt.edu/∼bard/xpp/xpp.html).
Supplementary Material
Acknowledgments
We acknowledge financial support from EPSRC grant EP/G03706X/1 (to M.H.), the National Institutes of Health (USA) grant GM078989-10 (to J.J.T.), and BBSRC Strategic LoLa grant BB/M00354X/1 (to B.N.).
Abbreviations used:
- APC
anaphase-promoting complex
- Cdk
cyclin-dependent kinase
- ENSA
alpha-endosulfine
- FADS
feedback-amplified domineering substrate
- MCC
mitotic checkpoint complex
- PP2A
protein phosphatase 2A.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-06-0349) on September 20, 2017.
REFERENCES
- Barr FA, Elliott PR, Gruneberg U. Protein phosphatases and the regulation of mitosis. J Cell Sci. 2011;124:2323–2334. doi: 10.1242/jcs.087106. [DOI] [PubMed] [Google Scholar]
- Blake-Hodek KA, Williams BC, Zhao Y, Castilho PV, Chen W, Mao Y, Yamamoto TM, Goldberg ML. Determinants for activation of the atypical AGC kinase Greatwall during M phase entry. Mol Cell Biol. 2012;32:1337–1353. doi: 10.1128/MCB.06525-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosl WJ, Li R. Mitotic-exit control as an evolved complex system. Cell. 2005;121:325–333. doi: 10.1016/j.cell.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Castilho PV, Williams BC, Mochida S, Zhao Y, Goldberg ML. The M phase kinase Greatwall (Gwl) promotes inactivation of PP2A/B55delta, a phosphatase directed against CDK phosphosites. Mol Biol Cell. 2009;20:4777–4789. doi: 10.1091/mbc.E09-07-0643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JL, Adler FR. How to make a biological switch. J Theor Biol. 2000;203:117–133. doi: 10.1006/jtbi.2000.1068. [DOI] [PubMed] [Google Scholar]
- Clijsters L, van Zon W, Riet BT, Voets E, Boekhout M, Ogink J, Rumpf-Kienzl C, Wolthuis RM. Inefficient degradation of cyclin B1 re-activates the spindle checkpoint right after sister chromatid disjunction. Cell Cycle. 2014;13:2370–2378. doi: 10.4161/cc.29336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clute P, Pines J. Temporal and spatial control of cyclin B1 destruction in metaphase. Nat Cell Biol. 1999;1:82–87. doi: 10.1038/10049. [DOI] [PubMed] [Google Scholar]
- Costanzo M, Nishikawa JL, Tang X, Millman JS, Schub O, Breitkreuz K, Dewar D, Rupes I, Andrews B, Tyers M. CDK activity antagonizes Whi5, an inhibitor of G1/S transcription in yeast. Cell. 2004;117:899–913. doi: 10.1016/j.cell.2004.05.024. [DOI] [PubMed] [Google Scholar]
- Craciun G, Tang Y, Feinberg M. Understanding bistability in complex enzyme-driven reaction networks. Proc Natl Acad Sci USA. 2006;103:8697–8702. doi: 10.1073/pnas.0602767103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cundell MJ, Bastos RN, Zhang T, Holder J, Gruneberg U, Novak B, Barr FA. The BEG (PP2A-B55/ENSA/Greatwall) pathway ensures cytokinesis follows chromosome separation. Mol Cell. 2013;52:393–405. doi: 10.1016/j.molcel.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin RA, McDonald WH, Kalashnikova TI, Yates J, 3rd, Wittenberg C. Cln3 activates G1-specific transcription via phosphorylation of the SBF bound repressor Whi5. Cell. 2004;117:887–898. doi: 10.1016/j.cell.2004.05.025. [DOI] [PubMed] [Google Scholar]
- Ferrell JE, Jr, Ha SH. Ultrasensitivity part II: multisite phosphorylation, stoichiometric inhibitors, and positive feedback. Trends Biochem Sci. 2014;39:556–569. doi: 10.1016/j.tibs.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharbi-Ayachi A, Labbe JC, Burgess A, Vigneron S, Strub JM, Brioudes E, Van-Dorsselaer A, Castro A, Lorca T. The substrate of Greatwall kinase, Arpp19, controls mitosis by inhibiting protein phosphatase 2A. Science. 2010;330:1673–1677. doi: 10.1126/science.1197048. [DOI] [PubMed] [Google Scholar]
- Goldbeter A, Koshland DE., Jr An amplified sensitivity arising from covalent modification in biological systems. Proc Natl Acad Sci USA. 1981;78:6840–6844. doi: 10.1073/pnas.78.11.6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith JS. Mathematics of cellular control processes. II. Positive feedback to one gene. J Theor Biol. 1968;20:209–216. doi: 10.1016/0022-5193(68)90190-2. [DOI] [PubMed] [Google Scholar]
- Gunawardena J. Multisite protein phosphorylation makes a good threshold but can be a poor switch. Proc Natl Acad Sci USA. 2005;102:14617–14622. doi: 10.1073/pnas.0507322102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagting A, Den Elzen N, Vodermaier HC, Waizenegger IC, Peters JM, Pines J. Human securin proteolysis is controlled by the spindle checkpoint and reveals when the APC/C switches from activation by Cdc20 to Cdh1. J Cell Biol. 2002;157:1125–1137. doi: 10.1083/jcb.200111001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He E, Kapuy O, Oliveira RA, Uhlmann F, Tyson JJ, Novak B. System-level feedbacks make the anaphase switch irreversible. Proc Natl Acad Sci USA. 2011;108:10016–10021. doi: 10.1073/pnas.1102106108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapuy O, Barik D, Sananes MR, Tyson JJ, Novak B. Bistability by multiple phosphorylation of regulatory proteins. Prog Biophys Mol Biol. 2009;100:47–56. doi: 10.1016/j.pbiomolbio.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivomagi M, Valk E, Venta R, Iofik A, Lepiku M, Balog ER, Rubin SM, Morgan DO, Loog M. Cascades of multisite phosphorylation control Sic1 destruction at the onset of S phase. Nature. 2011;480:128–131. doi: 10.1038/nature10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Aviles S, Kapuy O, Novak B, Uhlmann F. Irreversibility of mitotic exit is the consequence of systems-level feedback. Nature. 2009;459:592–595. doi: 10.1038/nature07984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M. Cyclin-dependent kinases. Genome Biol. 2014;15:122. doi: 10.1186/gb4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfeld J, Collin P, Collins MO, Choudhary JS, Pines J. APC15 drives the turnover of MCC-CDC20 to make the spindle assembly checkpoint responsive to kinetochore attachment. Nat Cell Biol. 2011;13:1234–1243. doi: 10.1038/ncb2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison JM. The Biology of the Cell Cycle. Cambridge, UK: Cambridge University Press; 1971. [Google Scholar]
- Mochida S, Ikeo S, Gannon J, Hunt T. Regulated activity of PP2A-B55 delta is crucial for controlling entry into and exit from mitosis in Xenopus egg extracts. EMBO J. 2009;28:2777–2785. doi: 10.1038/emboj.2009.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida S, Maslen SL, Skehel M, Hunt T. Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science. 2010;330:1670–1673. doi: 10.1126/science.1195689. [DOI] [PubMed] [Google Scholar]
- Mochida S, Rata S, Hino H, Nagai T, Novak B. Two bistable switches govern M phase entry. Curr Biol. 2016;26:3361–3367. doi: 10.1016/j.cub.2016.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll T, Tebb G, Surana U, Robitsch H, Nasmyth K. The role of phosphorylation and the CDC28 protein kinase in cell cycle-regulated nuclear import of the S. cerevisiae transcription factor SWI5. Cell. 1991;66:743–758. doi: 10.1016/0092-8674(91)90118-i. [DOI] [PubMed] [Google Scholar]
- Morgan DO. The Cell Cycle: Principles of Control. London: New Science Press; 2007. [Google Scholar]
- Musacchio A. The molecular biology of spindle assembly checkpoint signaling dynamics. Curr Biol. 2015;25:R1002–R1018. doi: 10.1016/j.cub.2015.08.051. [DOI] [PubMed] [Google Scholar]
- Novak B, Tyson JJ. Numerical analysis of a comprehensive model of M-phase control in Xenopus oocyte extracts and intact embryos. J Cell Sci. 1993;106:1153–1168. doi: 10.1242/jcs.106.4.1153. [DOI] [PubMed] [Google Scholar]
- Novak B, Tyson JJ, Gyorffy B, Csikasz-Nagy A. Irreversible cell-cycle transitions are due to systems-level feedback. Nat Cell Biol. 2007;9:724–728. doi: 10.1038/ncb0707-724. [DOI] [PubMed] [Google Scholar]
- Oliveira RA, Hamilton RS, Pauli A, Davis I, Nasmyth K. Cohesin cleavage and Cdk inhibition trigger formation of daughter nuclei. Nat Cell Biol. 2010;12:185–192. doi: 10.1038/ncb2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol. 2006;7:644–656. doi: 10.1038/nrm1988. [DOI] [PubMed] [Google Scholar]
- Queralt E, Uhlmann F. Cdk-counteracting phosphatases unlock mitotic exit. Curr Opin Cell Biol. 2008;20:661–668. doi: 10.1016/j.ceb.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattani A, Vinod PK, Godwin J, Tachibana-Konwalski K, Wolna M, Malumbres M, Novák B, Nasmyth K. Dependency of the spindle assembly checkpoint on Cdk1 renders the anaphase transition irreversible. Curr Biol. 2014;24:630–637. doi: 10.1016/j.cub.2014.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy SK, Rape M, Margansky WA, Kirschner MW. Ubiquitination by the anaphase-promoting complex drives spindle checkpoint inactivation. Nature. 2007;446:921–925. doi: 10.1038/nature05734. [DOI] [PubMed] [Google Scholar]
- Salazar C, Hofer T. Multisite protein phosphorylation–from molecular mechanisms to kinetic models. FEBS J. 2009;276:3177–3198. doi: 10.1111/j.1742-4658.2009.07027.x. [DOI] [PubMed] [Google Scholar]
- Schwob E, Bohm T, Mendenhall MD, Nasmyth K. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell. 1994;79:233–244. doi: 10.1016/0092-8674(94)90193-7. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Thomas R. Laws for the dynamics of regulatory networks. Int J Dev Biol. 1998;42:479–485. [PubMed] [Google Scholar]
- Thron CD. A model for a bistable biochemical trigger of mitosis. Biophys Chem. 1996;57:239–251. doi: 10.1016/0301-4622(95)00075-5. [DOI] [PubMed] [Google Scholar]
- Tyson JJ, Chen KC, Novak B. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr Opin Cell Biol. 2003;15:221–231. doi: 10.1016/s0955-0674(03)00017-6. [DOI] [PubMed] [Google Scholar]
- Tyson JJ, Novak B. Functional motifs in biochemical reaction networks. Annu Rev Phys Chem. 2010;61:219–240. doi: 10.1146/annurev.physchem.012809.103457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson JJ, Novak B. Irreversible transitions, bistability and checkpoint controls in the eukaryotic cell cycle: a systems-level understanding. In: Walhout M., Vidal M, Dekker J., editors. Handbook of Systems Biology. Concepts and Insights. London: Academic Press; 2013. [Google Scholar]
- Tyson JJ, Othmer HG. The dynamics of feedback control circuits in biochemical pathways. Prog Theor Biol. 1978;5:1–62. [Google Scholar]
- Uzunova K, Dye BT, Schutz H, Ladurner R, Petzold G, Toyoda Y, Jarvis MA, Brown NG, Poser I, Novatchkova M, et al. APC15 mediates CDC20 autoubiquitylation by APC/C(MCC) and disassembly of the mitotic checkpoint complex. Nat Struct Mol Biol. 2012;19:1116–1123. doi: 10.1038/nsmb.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varetti G, Guida C, Santaguida S, Chiroli E, Musacchio A. Homeostatic control of mitotic arrest. Mol Cell. 2011;44:710–720. doi: 10.1016/j.molcel.2011.11.014. [DOI] [PubMed] [Google Scholar]
- Vazquez-Novelle MD, Sansregret L, Dick AE, Smith CA, McAinsh AD, Gerlich DW, Petronczki M. Cdk1 inactivation terminates mitotic checkpoint surveillance and stabilizes kinetochore attachments in anaphase. Curr Biol. 2014;24:638–645. doi: 10.1016/j.cub.2014.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdugo A, Vinod PK, Tyson JJ, Novak B. Molecular mechanisms creating bistable switches at cell cycle transitions. Open Biol. 2013;3:120179. doi: 10.1098/rsob.120179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigneron S, Brioudes E, Burgess A, Labbe JC, Lorca T, Castro A. Greatwall maintains mitosis through regulation of PP2A. Embo J. 2009;28:2786–2793. doi: 10.1038/emboj.2009.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinod PK, Novak B. Model scenarios for switch-like mitotic transitions. FEBS Lett. 2015;589:667–671. doi: 10.1016/j.febslet.2015.02.007. [DOI] [PubMed] [Google Scholar]
- Williams BC, Filter JJ, Blake-Hodek KA, Wadzinski BE, Fuda NJ, Shalloway D, Goldberg ML. Greatwall-phosphorylated Endosulfine is both an inhibitor and a substrate of PP2A-B55 heterotrimers. eLife. 2014;3:e01695. doi: 10.7554/eLife.01695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, MacLellan WR, Han Z, Weiss JN, Qu Z. Multisite phosphorylation and network dynamics of cyclin-dependent kinase signaling in the eukaryotic cell cycle. Biophys J. 2004;86:3432–3443. doi: 10.1529/biophysj.103.036558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Lau KY, Sevim V, Tang C. Design principles of the yeast G1/S switch. PLoS Biol. 2013;11:e1001673. doi: 10.1371/journal.pbio.1001673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariae W, Schwab M, Nasmyth K, Seufert W. Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science. 1998;282:1721–1724. doi: 10.1126/science.282.5394.1721. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.