

Abstract
Lung cancer resists radiation therapy, making it one of the deadliest forms of cancer. Here we show that human lung cancer cell lines can be rendered sensitive to ionizing radiation (IR) by RNAi knockdown of C-terminus of Hsc70-interacting protein (CHIP/STUB1), a U-box-type E3 ubiquitin ligase that targets a number of stress-induced proteins. Mechanistically ubiquitin-dependent degradation of the cyclin-dependent kinase (CDK) inhibitor p21 protein is reduced by CHIP knockdown, leading to enhanced senescence of cells in response to exposure to IR. Cellular senescence and sensitivity to IR is prevented by CRISPR/Cas9-mediated deletion of the p21 gene (CDKN1A) in CHIP knockdown cells. Conversely, over-expression of CHIP potentiates p21 degradation and promotes greater radioresistance of lung cancer cells. In vitro and cell-based assays demonstrate that p21 is a novel and direct ubiquitylation substrate of CHIP that also requires the CHIP-associated chaperone heat shock protein 70 (HSP70). These data reveal that the inhibition of the E3 ubiquitin ligase CHIP promotes radiosensitivity; thus, suggesting a novel strategy for the treatment of lung cancer.
Implications
The CHIP-HSP70-p21 ubiquitylation/degradation axis identified here could be exploited to enhance the efficacy of radiotherapy in patients with non-small cell lung cancer.
Keywords: CHIP, ubiquitylation, p21, lung cancer, ionizing radiation
Introduction
Lung cancer is one of the most lethal forms of cancer, in part because of its resistance to therapy. Non-small cell lung cancer (NSCLC) is one of the leading causes of cancer death, and radiation therapy is an important component in the current treatment of NSCLC (1). Radiation combined with chemotherapy is the standard of care for patents with NSCLC with mediastinal involvement (Stage 3). Since the 5-year survival of Stage 3 lung cancer patients is under 25% (1) there is clearly an important need to understand the molecular basis of radiation resistance. Ionizing radiation (IR) is used therapeutically in about one half of cancer patients and induces extreme cellular stress, including oxidative stress plus mitochondrial and DNA damage. The molecular basis for resistance to IR is not well understood, posing a barrier to improved patient outcomes. At least one possibility for radioresistance is the action of proteins that alleviate cellular stresses.
Carboxyl terminus of Hsc70-interacting protein (CHIP; also known as STIP1 homology and U-box containing protein 1 or STUB1) is an E3 ubiquitin ligase that functions in association with HSP70 and HSP90 chaperones to target misfolded proteins for proteolytic degradation (2, 3). CHIP also is directly involved in the repair of single strand DNA breaks by regulating the base excision repair (BER) proteins XRCC1 and DNA Pol β (4). XRCC1 forms a complex with Pol β and DNA ligase III on damaged DNA to facilitate repair following radiation (5). CHIP may also regulate DNA damage repair through the stabilization of the histone deacetylase SirT6, which has been shown to interact with multiple DNA repair proteins including telomere-associated protein WRN and blocks KDAC-dependent transcription regulation (6).
Accumulating evidence suggests that CHIP plays a vital role in the regulation of multiple physiological and pathological processes (7–9). Consistently, CHIP-deficient mice (chip−/−) exhibit reduced life span with accelerated aging phenotypes including osteoporosis, cellular senescence, and accumulation of oxidized lipids (10). Importantly, several ubiquitylation substrates of CHIP are key proteins whose activity/level is dysregulated in a number of malignancies and/or neurodegenerative diseases. These include p53, PTEN, HIF1-α, c-Myc, phospho-tau, and NQO1 (11–15).
CHIP is also a key modulator of cellular response to stress and chip−/− mice are highly sensitive to hyperthermia and ischemia or reperfusion injury (16, 17), exhibit decreased cardiac function, and suffer from pressure overload-induced cardiac hypertrophy (7). Hypoxia-induced down regulation of CHIP causes cardiomyocyte apoptosis after myocardial infraction (18). The role of CHIP in mediating stress response may also depend on its ability to induce heat shock factor 1 (HSF1), which regulates the expression of chaperone proteins like HSP70 (16).
Here, we report that CHIP knockdown radiosensitizes lung cancer cells to ionizing radiation (IR) through enhancing p21 levels and IR-induced senescence. We show that p21 is a bona fide ubiquitylation substrate of CHIP. CRISPR/Cas9-mediated deletion of CDKN1A restored radioresistance of lung cancer cells following CHIP knockdown, defining a novel ubiquitylation axis for regulating radiation sensitivity in lung cancer cells.
Methods and Materials
Cell culture
Lung cancer cell lines A549, H1299, and H460 were purchased from the American Type Culture Collection (ATCC, Manassa, VA, USA) and were maintained in RPMI media (Gibco-Life Technologies, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. All cells were maintained in a 37 °C, 5% CO2 humidified atmosphere.
Western blotting and antibodies
Cells were lysed in modified RIPA lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 5 mM EDTA, 5 mM EGTA, 0.5% NP-40, 0.1% SDS, 50 mM NaF, 2 mM sodium orthovanadate) supplemented with a protease inhibitor mix (Thermo Scientific, Rockford, IL, USA). Unless otherwise described, 30 μg of protein were resolved by SDS–polyacrylamide gel electrophoresis (PAGE), transferred, and immunoblotted with various antibodies. The antibodies used were anti-p21 (sc-397) and anti-p53 (sc-126) from Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti-CHIP (C3B6) and anti-γH2AX (2577) from Cell Signaling Technology (Danvers, MA, USA); anti-ubiquitin (ab7780) and anti-ub-48-linked (ab140601) from Abcam (Cambridge, United Kingdom); mouse monoclonal anti-tubulin (Sigma-Aldrich); and anti-KAP1 and anti-p-KAP1 from Bethyl laboratories (Montgomery, TX, USA).
Clonogenic survival assay
Clonogenic survival assays were performed as described previously (19). Briefly, exponentially growing cells were trypsinized, rinsed, and counted, and appropriate numbers of cells were treated with or without various doses of radiation and plated immediately for colony formation. After 10 to 14 days of incubation, colonies measuring >50 cells were counted to determine the survival fraction (SF) (SF = number of colonies formed after IR/number of cells seeded x PE, where PE is the number of colonies formed/number of cells seeded × 100%). The colony numbers were fitted to standard linear dose–response curves. All data points were the average of at least three independent experiments.
Senescence-associated β-galactosidase activity assay
Senescence-associated β-galactosidase (SA-β-gal) activity was determined in a formaldehyde-fixed histochemical staining kit according to the manufacturer’s instructions (Cell Signaling Technology, Danvers, CA). Briefly, cells were grown in 6-well plates at a density of 5 × 104 cells/well and then treated with or without IR for 48 h. Cells were stained overnight with SA-β-gal staining solution at pH 6.0 in 37 °C incubator. Blue staining was observed and photographed under a bright-field microscope (AMG EvosXL Core Imager/Camera microscope, USA), counting 100 cells from at least 3 different fields.
Stable cell lines and siRNA transfection
Lung cancer cell lines A549, H1299, and H460 were stably depleted of CHIP using MISSION TRC shRNA lentiviral particles from Sigma according to the manufacturer’s protocol. Briefly, cells were seeded in 12-well plates, infected with 30 μL of virus particle solution in 1 mL of complete growth medium containing polybrene (4 μg/mL). Cells were selected by puromycin treatment and CHIP depletion was confirmed by Western blot analysis. siRNA transfections were performed using RNAi-MAX (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s protocol.
CDKN1A deletion by CRISPR/Cas9
Two single guide RNAs (sg-RNAs) targeting exon 2 of the human CDKN1A gene (encoding p21, sg-CDKN1A-1, and sg-CDKN1A-2) were cloned into a pX330 vector containing a human codon-optimized SpCas9 endonuclease and co-transfected with GFP-blasticidin vector in A549 cells. After selection with blasticidin, cells were seeded to obtain single colonies. Genomic DNA was extracted from individual colonies, and PCR reactions were performed using the following primers (Forward: 5′-TCACCTGAGGTGACACAGCAAAGC-3′ and Reverse: 5′-GGCCCCGTGGGAAGGTAGAGCTT-3′). The following genomic sequences of the human CDKN1A gene are targeted by sg-CDKN1A-1 and sg-CDKN1A-2 respectively: 5′-GCGCCATGTCAGAACCGGCTGGG-3′, and 5′-CCGCGACTGTGATGCGCTAATGG-3′. Successful targeting results in the removal of the intervening 97 bp in exon 2, resulting in frame-shift mutation. CDKN1A deletion was confirmed by the size and sequence of the PCR product.
In vivo and in vitro ubiquitylation assay
Cells were irradiated with 10 Gy as described above, incubated overnight, treated with or without the proteasome inhibitor MG132 for 4 h, harvested, and lysed using RIPA buffer. Ubiquitylated proteins were affinity purified using Tandem Ubiquitin Binding Entities (TUBEs)-conjugated agarose or control beads (LifeSensors, USA). Protein samples were resolved by SDS–PAGE, and immunoblotted for p21. In vitro ubiquitylation assays were performed using the CHIP Ubiquitin Ligase Kit (K-280, Boston Biochem, Cambridge, MA, USA) according to the manufacturer’s instructions. Recombinant p21 protein was purchased from Abcam (Cambridge, United Kingdom).
RNA isolation and RT–PCR
Cells were treated with the indicated doses of IR, and total RNA was isolated using TRIzol reagent (Life Technologies) according to the manufacturer’s instructions. Complementary DNA synthesis was carried out using the SuperScript III first-strand synthesis system for RT–PCR (Life Technologies). mRNA was detected by qRT-PCR using SYBR Green PCR master mix (Bio-Rad) in a Bio-Rad CFX96 cycler and quantified with Bio-Rad CFX manager software (Bio-Rad, Hercules, CA, USA).
FACS analysis
Cells were harvested and fixed with 70% ethanol for 24 h at 4 °C. Fixed cells were stained in 1 mL of propidium iodide (PI) solution (0.05% NP-40, 50 mg/mL PI, and 10 mg/mL RNase A) overnight at 4 °C. Stained cells were analyzed with a flow cytometer using CellQuest software (both from BD Biosciences, San Jose, CA, USA), and cell cycle phases were analyzed by ModFit LT V3.3.11 (Mac, Verity Software House, Topshan, ME, USA).
Cell proliferation assays
The indicated human lung cancer cells were plated (2,000 cells/well) in 96-well plates and treated with or without the indicated doses of IR. Cell growth was monitored at different time points with the Alamar Blue assay (Life Technologies) according to the manufacturer’s instructions.
Statistical analysis
Statistical analyses were conducted using Prism (GraphPad Software Inc., CA, USA), and results are presented as means ± SD. All statistical results were performed using unpaired two-tailed t-test to compare two groups (n≥3) to determine statistical significance.
Results
CHIP knockdown Sensitizes lung cancer cells to IR
In response to various cellular insults, such as thermal, hypoxic, or ischemic stress, CHIP has a protective role therefore we investigated the role of CHIP in response to IR in lung cancer cell lines. We used A549 and H460 lung cancer cells and two different lentiviral shRNAs to knockdown CHIP and examined the response to IR. As controls A549 or H460 cells were transduced with lentivirus expressing scrambled sh-RNA (non target shRNA that does not target any known gene from any species). Both shRNAs targeting CHIP (sh-CHIP-1 and sh-CHIP -2) effectively depleted endogenous CHIP protein in both cell lines (Fig. 1A, B). We exposed control or CHIP-knockdown A549 and H460 cells to increasing doses of IR and monitored responses with clonogenic survival assays. As shown in Fig. 1C, D, and consistent with the literature (20), A549 cells were relatively more radioresistant, while the H460 line was more radiosensitive. Although CHIP knockdown did not impact the proliferation of either of these lines (Supplementary Fig. S2), there was significantly increased lethality of IR in both knockdown cell lines (Fig. 1C, 1D and Supplementary Fig. S1A, B). Conversely, cells stably overexpressing CHIP were more radio-resistant compared with parental A549 cells or cells expressing empty expression vector, (Fig. 1E and Supplementary Fig. S1C). Similar results were observed in other lung cancer cell lines (data not shown). These data demonstrate that radiation resistance in lung cancer cells depends on CHIP.
Figure 1.

CHIP dependence of lung cancer cells response to ionizing radiation (IR). A, A549 and B, H460 cells were depleted of CHIP using two different shRNAs (shCHIP-1 & shCHIP-2). Cell lysates were immunoblotted for CHIP and tubulin as loading control. Clonogenic survival of C, A549 and D, H460 cells depleted of CHIP treated with different doses of IR. Survival fractions were fitted to linear-quadratic model. E, Clonogenic survival of A549 cells comparing ectopically expressed WT-CHIP or empty Vector treated and plotted as C or D. Immunoblot of ectopically expressed CHIP (inset). F, FACS analysis of control (sh-NT) and knockdown (sh-CHIP-2) A549 cells harvested at indicated time points after IR (10 Gy). G, CHIP proficient and deficient cells (as F) were treated with or without 10 Gy and cellular senescence was measured 24h after IR by β-gal staining. H, Quantification of senescence cells. Error bars for C–F and H represents the mean ±SD of at least three independent experiments. *, P<0.05; **, P<0.01.
We next asked whether the radiosensitizing effect associated with CHIP knockdown was due to its actions on sensors or effectors of the DNA damage response (DDR) and/or repair proteins. Using Western blotting, we found no significant differences in the levels of p-DNA-PK (pS2056), γH2AX, p-Kap1 and p-ATM (pS1981) in control shRNA- vs CHIP-knockdown A549 cells (Fig. Supplementary S3A, B). We have recently shown that knockdown of CHIP arrests prostate cancer cells in the G1 phase of the cell cycle (21). We used flow cytometry to assay whether CHIP knockdown arrested A549 lung cancer cells. Fig. 1F shows that CHIP knockdown did not alter cell cycle distribution before IR treatment. However, treatment of these cells with IR changed the cell cycle distribution. At 12h post-IR more cells were in G1 in CHIP knockdown cells demonstrating the impact on the cell cycle in response to IR. Interestingly we observed that, compared with control cells, IR caused more CHIP- knockdown A549 cells to undergo senescence than we observed in irradiated control cells (Fig. 1G, H). This result suggests that enhanced senescence in CHIP knockdown cells may account for the radiosensitization of lung cancer cells.
CHIP depletion enhances radiosensitivity through p53-independent stabilization of the p21 protein
The tumor suppressor p53 protein plays a vital role in the stress-induced DNA damage response (DDR) pathway (22, 23). Because the p53 protein is known as an ubiquitylation substrate for CHIP (12, 24), it was possible that failure to degrade p53 in CHIP depleted cells contributed to the radiosensitization of lung cancer cells. To test this hypothesis, we western blotted for p53 in control and in CHIP depleted A549 (with wt p53) before and at various time points after exposure to IR. CHIP knockdown in A549 cells slightly, but reproducibly, increased the levels of p53 protein in the absence of IR (Fig. 2A, compare lane 1 and lane 7), but we observed no potentiation of this effect upon IR treatment. Interestingly, the level of the p53 transcriptional target p21 paralleled the increase of p53 in the absence of IR, but increased significantly following IR treatment (Fig. 2A). Other cell cycle regulatory proteins, such as MDM2 (a regulator of p53), cyclin D1 and Cdk6 (G1 cell cycle regulators) were unaffected by these treatments (Fig. 2A).
Figure 2.
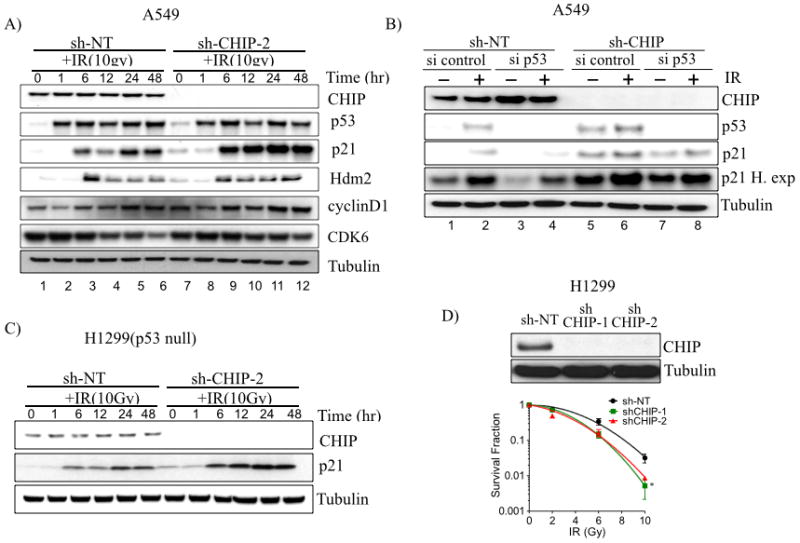
Depletion of CHIP enhances radiosensitivity independent of p53 status. A, A549 cells were treated with sh-NT or shCHIP-2 to stably deplete CHIP. Cells were treated with or without 10 Gy, harvested and immunoblotted for indicated proteins at indicated time. B, A549 cells (sh-NT or shCHIP-2) were treated with either control siRNA or p53 siRNA to deplete p53 for 48 h, irradiated (10Gy), and harvested after 24 h. Cell lysates were immunoblotted for indicated proteins. C, H1299 cells were treated with sh-NT or shCHIP-2 to stably deplete CHIP. Cells were treated with or without 10 Gy and harvested at indicated time points, Lysates were immunoblotted for CHIP and p21and tubulin as control. D, H1299 cells depleted of CHIP (shCHIP-1 and shCHIP-2) treated with different doses of IR. Immunoblot showing knockdown of CHIP (top) and clonogenic survival (bottom). *, P<0.05.
To test whether the increase of p21 in CHIP-depleted A549 cells exposed to radiation was p53-dependent, we carried out siRNA-mediated knockdown of p53 in control shRNA and CHIP-depleted A549 cells. The p21 protein levels were reduced following p53 knockdown, as expected (Fig. 2B-compare lane 1 and lane 3), however IR exposure still induced p21 protein both in control and in p53 -knockdown cells (Fig. 2B- lane 2 and lane 4). The higher level in p21 protein in CHIP depleted cells following p53 knockdown compared to control cells in both untreated (Fig 2B lane 3 and lane 7) and following IR (Fig 2B lane 4 and lane 8) cannot be explained by increased p21 mRNA expression due to residual p53 protein, since there were similar levels of p21 mRNA in either control or CHIP depleted cells following IR (Fig. 3A). These results indicate that CHIP plays a role in controlling the steady state levels of p21 independent of transcription in IR-treated cells. To rule out the possibility that p21 levels can still be induced by residual p53 in the p53 knockdown cells, we stably depleted CHIP in the p53-null H1299 lung cancer cells and monitored p21 protein levels with and without IR. Re-affirming our results above, the steady state levels of the p21protein was increased in CHIP-depleted H1299 cells in both the absence and presence of IR (Fig. 2C), and this was not accompanied by increased p21 transcription (Fig. 3B). As shown in Fig. 1C and 1D, knockdown of CHIP radiosensitized A549 and H460 cells (each wt for p53), we therefore tested the effect of CHIP knockdown on the radiosensitivity of the p53-null H1299 cell line, and found enhanced radiosensitivity, similar to that observed in A549 and H460 cells (Fig. 2D). These results support the concept that radiosensitization of lung cancer cells induced by CHIP knockdown is associated with increased steady-state level of p21 protein but is independent of the p53 tumor suppressor protein.
Figure 3.
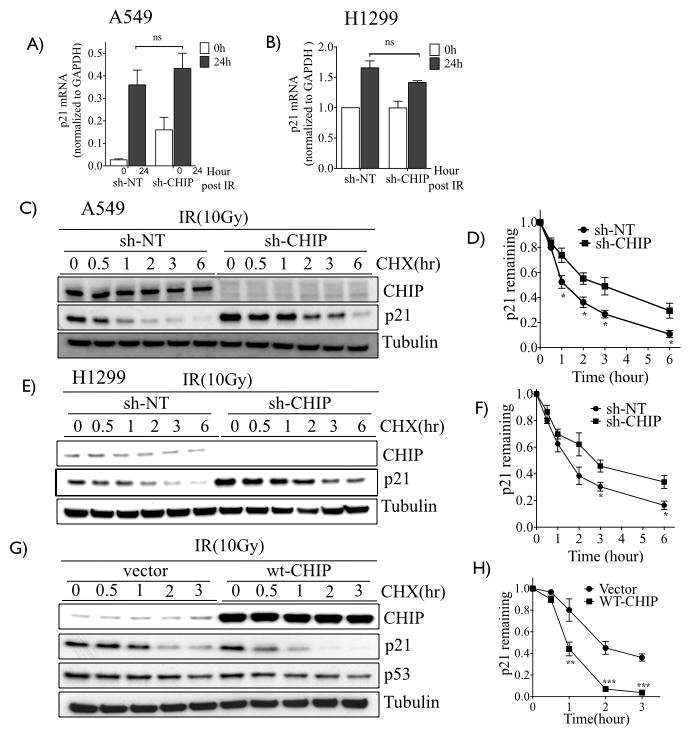
Depletion of CHIP enhances stability of p21. A–B, RT-qPCR of p21 of CHIP proficient (sh-NT) and knockdown (shCHIP-2) of A549 and H1299 cells treated with or without 10 Gy. C, A549 and E, H1299 cells (sh-NT or shCHIP-2), were subjected to 0 or 10 Gy and 24h later cells were treated with 100 μg/mL cycloheximide (CHX) and harvested as indicated time points. Lysates were immunoblotted for CHIP, p21 and tubulin as loading control. D, F, Remaining amount of p21 (normalized with corresponding 0h) was plotted for A549 (D) and H1299 (F). G, A549 cells were infected with retrovirus containing empty vector or wt-CHIP, treated as C. H, Remaining amount of p21 from G calculated as in D. Experiments D, F and H represents the mean ±SD of at least three independent experiments. *, P<0.05; **, P<0.01; ***, P< 0.001.
P21 is a bona fide ubiquitylation substrate for CHIP
To investigate the mechanistic basis for the p53-independent increase of the steady-state levels of p21 protein in CHIP-depleted and irradiated cells, we treated CHIP-expressing and -knockdown cells with 10 Gy. This radiation dose gave a maximal difference for viability between control and CHIP knockdown cells. Cells were treated with or without cycloheximide to prevent new protein synthesis and cell lysates were collected at different time points. Western blots showed there was a significant increase in the half-life of the p21 protein in irradiated CHIP-depleted A549 (Fig. 3C) and H1299 (Fig. 3E) cells compared to the matched controls. The quantified results from multiple independent experiments are shown in Fig. 3D and Fig. 3F for A549 and H1299 cells, respectively. Reciprocally, the p21 protein half-life was significantly shortened in irradiated A549 cells overexpressing wt CHIP (Fig. 3G and 3H). These results suggest that CHIP promotes the degradation of p21 protein in lung cancer cells exposed to IR. To test this hypothesis, we first transiently overexpressed wt CHIP in A549 cells and exposed the cells to IR. Four hours before harvesting, cells were treated with MG132 to inhibit the proteasome. Under these conditions, the endogenous p21 protein co-immunoprecipitated with ectopically expressed CHIP (Fig. 4A). Reciprocally, CHIP was detected in the p21 immunoprecipitate (Fig. 4B). The physical interaction of p21 with the E3 ligase CHIP prompted us to test whether p21 is ubiquitylated by CHIP. For this, we affinity purified total poly-ubiquitylated proteins from IR-irradiated A549 cells in the presence or absence of MG132 and immunoblotted for p21. Fig. 4C demonstrates that, compared with control cells, CHIP-depleted A549 cells have significantly reduced levels of poly-ubiquitylated p21 protein (compare lane 1 and lane 3). This effect was potentiated by MG132 inhibition of proteasome (Fig. 4C-compare lane 2 and 4). Similar results were obtained in H460 cells (Fig. 4D). Importantly, in vitro incubation of recombinant CHIP along with recombinant p21, HSP70, ATP and ubiquitin resulted in robust polyubiquitylation of p21 (Fig. 5A- compare lane 5 to lane 4). Collectively, these results demonstrated that p21 is an ubiquitylation substrate for CHIP.
Figure 4.
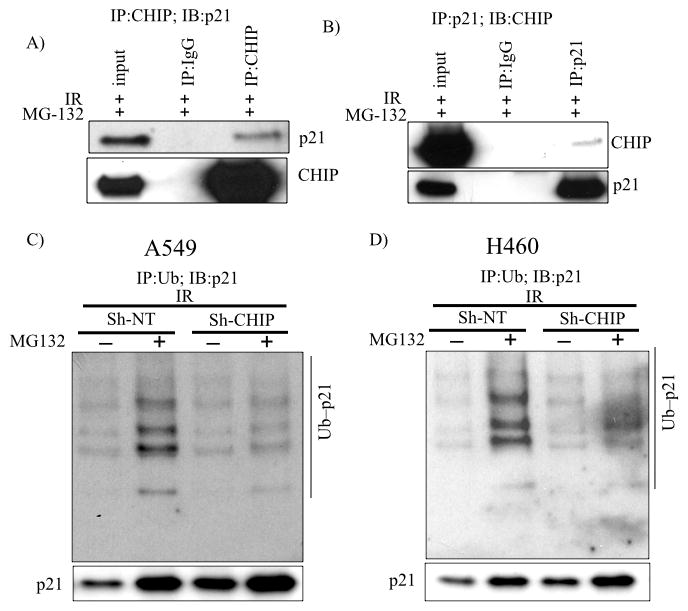
CHIP interacts with p21 and promotes polyubiquitination and degradation in living cells. A, A549 cells stably expressing WT-CHIP were treated with 10 Gy, incubated for 24 h at which point MG132 was added for 4 h. Lysates were prepared and CHIP was immunoprecipitated and immunoblot analysis performed. B, Reciprocally, p21 was immunoprecipitated from lysate A and blotted for CHIP. C, A549 and D, H460 cells (sh-NT or sh-CHIP-2) were treated as A and ubiquitinated proteins were affinity purified using TUBE (see Methods and Materials) and immunoblotted for p21. 5% input shown in the bottom.
Figure 5.
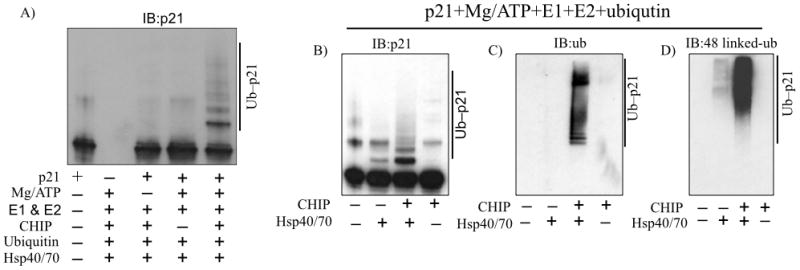
CHIP promotes p21 ubiquitination in vitro. A, Purified recombinant CHIP was incubated with recombinant p21 (purified) in the indicated reaction mixtures at 37 °C for 1 h and immunoblotted for p21. B–D, CHIP and p21 were incubated as in A in the presence or absence of HSP70/HSP40 and immunoblotted for p21 (B), ubiquitin (C) and 48-linked ubiquitin (D).
CHIP-mediated Lysine 48-linked polyubiquitylation of p21 is dependent on HSP40/70
CHIP serves as a bridge linking the HSP70/90 chaperone and the 26S proteasome (3, 25). We next asked whether CHIP mediated poly-ubiquitylation of p21 requires HSP40/70. We employed the in vitro ubiquitylation assay and found that the relatively modest levels of p21 poly-ubiquitylation seen with CHIP alone were dramatically increased by the addition of the Hsp40/70 complex (Fig. 5B). The results were verified by re-probing the blot with an anti-ubiquitin antibody (Fig. 5C). Lys-48-linked ubiquitin conjugation is the most common ubiquitylation leading to proteasomal degradation (26). This ubiquitylation of p21 by CHIP is mediated through Lys-48 linkage, and indeed the poly-ubiquitylated p21 species generated in the in vitro assay were reactive with the Lys-48 specific antibodies (5D). These results support that CHIP-mediated polyubiquitylation of p21 directs it for proteasomal degradation.
CDKNA1 deletion rescues CHIP-induced radiosensitivity and senescence in lung cancer cells
Our results correlate the radiation sensitivity of lung cancer cells with failure of CHIP to ubiquitylate and degrade p21. To test whether p21 is sufficient for the increased radiosensitivity of lung cancer cells depleted of CHIP, we engineered A549 cells with homozygous deletion of CDKN1A gene encoding p21 using CRISPR/Cas9, and isolated single clones as described in the materials and methods section. PCR analysis and Western blotting confirmed the intended bi-allelic deletion (frame-shift) and loss of p21 protein expression (Fig. 6A and Supplementary FIG. S4). Surprisingly, the homozygous deletion of CDKN1A by itself made A549 cells more radioresistant compared to wt control cells (Fig. 6B; black vs blue). As previously observed CHIP knockdown alone enhanced radiosensitivity compared with wt controls, (Fig. 6B; black vs red). Importantly, knockdown of CHIP in the CDKN1A homozygous deletion background failed to convert those radioresistant cells to radiosensitive cells (Fig. 6B). These results establish that the radiosensitivity of lung cancer cells induced by knockdown of the E3 ligase CHIP is mediated through p21. We also show in Fig. 1G that knockdown of CHIP enhanced IR-induced cellular senescence of A549 cells, a phenotype controlled by p21 (27, 28). Therefore, we sought to determine whether the induction of senescence in CHIP-depleted cells would be affected by the loss of p21. As shown in Figures 6C and 6D, CHIP knockdown enhanced senescence only in p21-proficient cells but not in cells with knockout of p21. These results demonstrate that p21 promotes senescence and radiation sensitivity in lung cancer cells.
Figure 6.
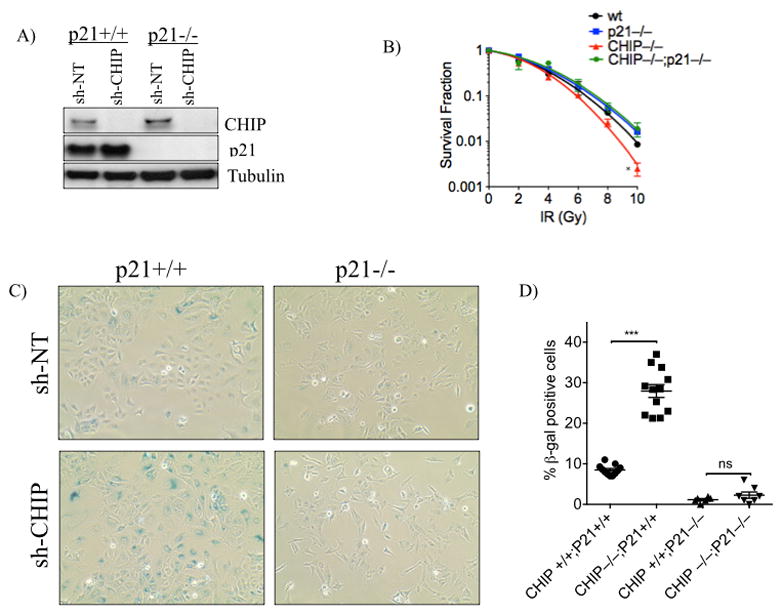
Deletion of CDKN1A rescues radiosensitivity of CHIP-depleted cells. A, Immunoblot demonstrating homozygous deletion of CDKN1A in A549 cells as well as knockdown of CHIP using shCHIP-2 or sh-NT. B, A549 (control), A549 p21−/−, A549 CHIP−/− and A549 p21−/−/CHIP−/− cells were treated with indicated doses of IR and survival fractions were plotted as in Fig 1. C–D, Cells mentioned in (B) were treated with 10 Gy and cellular senescence was measured using β-gal staining and quantified (D). Error bars represent the mean ±SD of at least three independent experiments. *, P<0.05; ***, P<0.001.
Discussion
Knockdown of CHIP with the consequent increase of p21 protein results in enhanced cellular senescence and increased sensitivity of lung cancer cells to IR. Elevated levels of p21 in CHIP-depleted cells result from deficiency in p21 ubiquitylation and degradation, and occur in p53-proficient and -deficient cells. This in turn promotes cellular senescence and contributes to the enhanced radiosensitivity of lung cancer cells. The identification of p21 as a direct ubiquitylation substrate for E3 ligase CHIP has several implication for how cells respond to cellular stress, such as that induced by IR.. On one hand, p21 is important for cellular senescence, so the ability to induce p21 and senescence in the absence of functional p53 would be critical to the therapeutic efficacy of IR in lung tumors where p53 mutations are prevalent. On the other hand, as a CDK inhibitor, p21 is an important regulator of cell cycle progression and its stabilization contributes to the ability of cells to enforce cell cycle arrest to promote DNA repair, which is critical for cellular responses to single and double strand DNA breaks, such as those induced by IR. The p21 protein also acts in response to various forms of cellular stresses (29), and thus, our identification of a new axis regulating p21 stability is important for understanding how cells respond to cellular stresses. CHIP regulates p21 polyubiquitylation and degradation not only in cells exposed to IR, but also in non-irradiated cells expanding possible role of CHIP in modulating cellular stresses.
The mechanism by which elevated p21 leads to enhanced radiosensitivity is not entirely clear, but the p21-dependent senescence phenotype in CHIP depleted cells suggests that senescence may contribute to the observed radiosensitivity phenotype. The role of senescence in response to therapeutic IR may involve the secretion of inflammatory cytokines, a phenomenon known as the senescence-associated secretory phenotype (SASP). Nevertheless, in vitro studies have demonstrated that the induction of senescence by IR is generally correlated with radiation sensitivity. Modulation of DNA repair by increased p21 levels (directly or indirectly) may contribute to the radiation sensitivity phenotype observed in CHIP- depleted cells. A transient cell cycle arrest in G1 phase of the cell cycle by increased p21 protein and inhibition of CDK2 activity can in principle promote DNA repair by extending the time available to cells to repair DNA damage. Elevated p21 expression can inhibit CDK1 activity with a cell cycle block in G2/M phase when cells are most susceptible to DNA damage. Indeed, while the majority of CHIP knockdown cells underwent senescence in response to IR, we detected a significant increase in the percentage of cells accumulating in G1 at the 12h post-radiation, compared to control lung cancer cells (see Fig. 1F). These observations raise the possibility that p21 may promote radiation sensitivity through mechanisms other than senescence.
CHIP is not the only regulator of p21 protein stability (29). Newly synthetized p21 is stabilized by WIS39, an adaptor that recruits HSP90 to p21 (30) while p21 ubiquitylation and degradation is promoted by several E3 ubiquitin ligase complexes during various phases of the cell cycle and following exposure of cells to UV irradiation, These include the SCFSKP2 ubiquitin ligase, the CRL4CDT2 ligase, and the APC/CCDC20 ubiquitin ligase (31–33). Even though p21 is polyubiquitylated in cells, ubiquitylation is not always required for p21 degradation, because a p21 mutant with all lysine residues mutated to arginine (p21K0) can still be degraded (34, 35). However, other groups reported that the p21K0 mutant is ubiquitylated at its N-terminus both in vivo and in vitro (36, 37). Our study adds CHIP to the growing list of E3 ubiquitin ligases controlling p21 protein abundance and highlights the complex network involved in regulating the abundance (and activity) of this important protein.
The expression of CHIP ligase is altered in many solid tumors, including those of breast, gastric, gallbladder and gliomas. Increased CHP expression has been correlated with poor prognosis in gallbladder and glioma while in the case of breast cancer increased CHIP expression has been associated with favorable prognosis (38). CHIP is well know to be a regulator of oxidative stress as well as has been shown to be a regulator of oncogenic pathways such as those involved in tumorigenesis, proliferation, and invasion in several malignancies, particularly breast cancer. The tissue specific basis for opposing effect of CHIP is unknown but likely related to the different role substrates play in various contexts. For example in breast cancer CHIP inhibits progression through ubiquitination and degradation of ERα. In case of lung adenocarcinoma, although CHIP is amplified, deleted or mutated in 1–2% of cases (cBio portal data) it remains to be determined whether CHIP levels correlate with patient outcome.
In summary, our results demonstrate that p21 is a bona fide ubiquitylation substrate for CHIP, and that CHIP knockdown induces radiation sensitivity through p21 accumulation. CHIP mediated degradation of p21 requires the HSP70 chaperone protein. Inactivation of the CHIP-HSP70-p21 ubiquitylation/degradation axis represents a novel strategy to enhance the efficacy of radiotherapy in patients with non-small cell lung cancer.
Supplementary Material
Acknowledgments
We thank Thomas L. Genetta for his critical comments and discussion during preparation of the manuscript. This work was supported by grant NIH-CA192669 and the Charles Burnett fund.
Footnotes
“The authors declare no conflicts of interest”
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Hohfeld J, et al. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol. 2001;3(1):93–6. doi: 10.1038/35050618. [DOI] [PubMed] [Google Scholar]
- 3.Ballinger CA, Connell P, Wu Y, Hu Z, Thompson LJ, Yin LY, et al. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol. 1999;19(6):4535–45. doi: 10.1128/mcb.19.6.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parsons JL, Tait PS, Finch D, Dianova II, Allinson SL, Dianov GL. CHIP-mediated degradation and DNA damage-dependent stabilization regulate base excision repair proteins. Mol Cell. 2008;29(4):477–87. doi: 10.1016/j.molcel.2007.12.027. [DOI] [PubMed] [Google Scholar]
- 5.Almeida KH, Sobol RW. A unified view of base excision repair: lesion-dependent protein complexes regulated by post-translational modification. DNA Repair (Amst) 2007;6(6):695–711. doi: 10.1016/j.dnarep.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ronnebaum SM, Wu Y, McDonough H, Patterson C. The ubiquitin ligase CHIP prevents SirT6 degradation through noncanonical ubiquitination. Mol Cell Biol. 2013;33(22):4461–72. doi: 10.1128/MCB.00480-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schisler JC, Rubel CE, Zhang C, Lockyer P, Cyr DM, Patterson C. CHIP protects against cardiac pressure overload through regulation of AMPK. J Clin Invest. 2013;123(8):3588–99. doi: 10.1172/JCI69080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie P, Fan Y, Zhang H, Zhang Y, She M, Gu D, et al. CHIP represses myocardin-induced smooth muscle cell differentiation via ubiquitin-mediated proteasomal degradation. Mol Cell Biol. 2009;29(9):2398–408. doi: 10.1128/MCB.01737-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li F, Xie P, Fan Y, Zhang H, Zheng L, Gu D, et al. C terminus of Hsc70-interacting protein promotes smooth muscle cell proliferation and survival through ubiquitin-mediated degradation of FoxO1. J Biol Chem. 2009;284(30):20090–8. doi: 10.1074/jbc.M109.017046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Min JN, Whaley RA, Sharpless NE, Lockyer P, Portbury AL, Patterson C. CHIP deficiency decreases longevity, with accelerated aging phenotypes accompanied by altered protein quality control. Mol Cell Biol. 2008;28(12):4018–25. doi: 10.1128/MCB.00296-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kajiro M, Hirota R, Nakajima Y, Kawanowa K, So-ma K, Ito I, et al. The ubiquitin ligase CHIP acts as an upstream regulator of oncogenic pathways. Nat Cell Biol. 2009;11(3):312–9. doi: 10.1038/ncb1839. [DOI] [PubMed] [Google Scholar]
- 12.Tripathi V, Ali A, Bhat R, Pati U. CHIP chaperones wild type p53 tumor suppressor protein. J Biol Chem. 2007;282(39):28441–54. doi: 10.1074/jbc.M703698200. [DOI] [PubMed] [Google Scholar]
- 13.Ahmed SF, Deb S, Paul I, Chatterjee A, Mandal T, Chatterjee U, et al. The chaperone-assisted E3 ligase C terminus of Hsc70-interacting protein (CHIP) targets PTEN for proteasomal degradation. J Biol Chem. 2012;287(19):15996–6006. doi: 10.1074/jbc.M111.321083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo W, Zhong J, Chang R, Hu H, Pandey A, Semenza GL. Hsp70 and CHIP selectively mediate ubiquitination and degradation of hypoxia-inducible factor (HIF)-1alpha but Not HIF-2alpha. J Biol Chem. 2010;285(6):3651–63. doi: 10.1074/jbc.M109.068577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paul I, Ahmed SF, Bhowmik A, Deb S, Ghosh MK. The ubiquitin ligase CHIP regulates c-Myc stability and transcriptional activity. Oncogene. 2013;32(10):1284–95. doi: 10.1038/onc.2012.144. [DOI] [PubMed] [Google Scholar]
- 16.Dai Q, Zhang C, Wu Y, McDonough H, Whaley RA, Godfrey V, et al. CHIP activates HSF1 and confers protection against apoptosis and cellular stress. EMBO J. 2003;22(20):5446–58. doi: 10.1093/emboj/cdg529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang C, Xu Z, He XR, Michael LH, Patterson C. CHIP, a cochaperone/ubiquitin ligase that regulates protein quality control, is required for maximal cardioprotection after myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2005;288(6):H2836–42. doi: 10.1152/ajpheart.01122.2004. [DOI] [PubMed] [Google Scholar]
- 18.Nishitani H, Shiomi Y, Iida H, Michishita M, Takami T, Tsurimoto T. CDK inhibitor p21 is degraded by a proliferating cell nuclear antigen-coupled Cul4-DDB1Cdt2 pathway during S phase and after UV irradiation. J Biol Chem. 2008;283(43):29045–52. doi: 10.1074/jbc.M806045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dziegielewski J, Baulch JE, Goetz W, Coleman MC, Spitz DR, Murley JS, et al. WR-1065, the active metabolite of amifostine, mitigates radiation-induced delayed genomic instability. Free Radic Biol Med. 2008;45(12):1674–81. doi: 10.1016/j.freeradbiomed.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tumati V, Kumar S, Yu L, Chen B, Choy H, Saha D. Effect of PF-02341066 and radiation on non-small cell lung cancer cells. Oncol Rep. 2013;29(3):1094–100. doi: 10.3892/or.2012.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sarkar S, Brautigan DL, Parsons SJ, Larner JM. Androgen receptor degradation by the E3 ligase CHIP modulates mitotic arrest in prostate cancer cells. Oncogene. 2014;33(1):26–33. doi: 10.1038/onc.2012.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reinhardt HC, Schumacher B. The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012;28(3):128–36. doi: 10.1016/j.tig.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meek DW. Tumour suppression by p53: a role for the DNA damage response? Nat Rev Cancer. 2009;9(10):714–23. doi: 10.1038/nrc2716. [DOI] [PubMed] [Google Scholar]
- 24.Esser C, Scheffner M, Hohfeld J. The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degradation. J Biol Chem. 2005;280(29):27443–8. doi: 10.1074/jbc.M501574200. [DOI] [PubMed] [Google Scholar]
- 25.McDonough H, Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones. 2003;8(4):303–8. doi: 10.1379/1466-1268(2003)008<0303:calbtc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243(4898):1576–83. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- 27.Noda A, Ning Y, Venable SF, Pereira-Smith OM, Smith JR. Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Exp Cell Res. 1994;211(1):90–8. doi: 10.1006/excr.1994.1063. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Blandino G, Givol D. Induced p21waf expression in H1299 cell line promotes cell senescence and protects against cytotoxic effect of radiation and doxorubicin. Oncogene. 1999;18(16):2643–9. doi: 10.1038/sj.onc.1202632. [DOI] [PubMed] [Google Scholar]
- 29.Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9(6):400–14. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jascur T, Brickner H, Salles-Passador I, Barbier V, El Khissiin A, Smith B, et al. Regulation of p21(WAF1/CIP1) stability by WISp39, a Hsp90 binding TPR protein. Mol Cell. 2005;17(2):237–49. doi: 10.1016/j.molcel.2004.11.049. [DOI] [PubMed] [Google Scholar]
- 31.Bornstein G, Bloom J, Sitry-Shevah D, Nakayama K, Pagano M, Hershko A. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J Biol Chem. 2003;278(28):25752–7. doi: 10.1074/jbc.M301774200. [DOI] [PubMed] [Google Scholar]
- 32.Abbas T, Sivaprasad U, Terai K, Amador V, Pagano M, Dutta A. PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. 2008;22(18):2496–506. doi: 10.1101/gad.1676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amador V, Ge S, Santamaria PG, Guardavaccaro D, Pagano M. APC/C(Cdc20) controls the ubiquitin-mediated degradation of p21 in prometaphase. Mol Cell. 2007;27(3):462–73. doi: 10.1016/j.molcel.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen X, Chi Y, Bloecher A, Aebersold R, Clurman BE, Roberts JM. N-acetylation and ubiquitin-independent proteasomal degradation of p21(Cip1) Mol Cell. 2004;16(5):839–47. doi: 10.1016/j.molcel.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 35.Sheaff RJ, Singer JD, Swanger J, Smitherman M, Roberts JM, Clurman BE. Proteasomal turnover of p21Cip1 does not require p21Cip1 ubiquitination. Mol Cell. 2000;5(2):403–10. doi: 10.1016/s1097-2765(00)80435-9. [DOI] [PubMed] [Google Scholar]
- 36.Bloom J, Amador V, Bartolini F, DeMartino G, Pagano M. Proteasome-mediated degradation of p21 via N-terminal ubiquitinylation. Cell. 2003;115(1):71–82. doi: 10.1016/s0092-8674(03)00755-4. [DOI] [PubMed] [Google Scholar]
- 37.Coulombe P, Rodier G, Bonneil E, Thibault P, Meloche S. N-Terminal ubiquitination of extracellular signal-regulated kinase 3 and p21 directs their degradation by the proteasome. Mol Cell Biol. 2004;24(14):6140–50. doi: 10.1128/MCB.24.14.6140-6150.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun C, Li HL, Shi ML, Liu QH, Bai J, Zheng JN. Diverse roles of C-terminal Hsp70-interacting protein (CHIP) in tumorigenesis. J Cancer Res Clin Oncol. 140(2):189–97. doi: 10.1007/s00432-013-1571-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.