

Abstract
Silk sericin, a water-soluble glue-like protein, is extensively used as a biomaterial due to its biocompatibility, hydrophilicity, biodegradability, and adequate resource. In addition, hydroxyapatite-based drug carriers are functionally efficient for drug or gene delivery due to their biodegradability, biocompatibility and easy metabolism in vivo. Herein, for the first time, this study used sericin, from a wild silkworm called Antheraea pernyi (A. pernyi), as a template to nucleate hydroxylapatite (HAp) nano-needles and form porous sericin-HAp nanocomposite microspheres as an anticancer drug carrier. Specifically, A. pernyi sericin (AS) was incubated in 1.5× simulated body fluid to induce the formation of porous AS/HAp microspheres in situ. Doxorubicin (DOX) loading and release assays proved that the microspheres exhibited pH-dependent controlled and sustained release of DOX. In particular, the microspheres can selectively release DOX at a higher rate at the acidic conditions typical for tumor microenvironment than at the physiological conditions typical for normal tissues, which will potentially reduce the side effect of the cancer drugs in normal tissues. Cancer cell toxicity assay, cancer cell imaging and intracellular DOX distribution assay provided further evidence to support the pH-dependent controlled and sustained release of DOX to cancer cells from the microspheres. Our work has demonstrated a biomimetic strategy for the design and synthesis of silk protein-based drug carriers that can be potentially employed in drug delivery and regenerative medicine.
TOC image
Protein-hydroxylapatite microspheres with a porous structure and a pH-dependent drug release profile are fabricated by incubating sericin in 1.5 SBF solution.
1. Introduction
Drug delivery carriers have important therapeutic applications in both pharmaceuticals and regenerative medicine.1, 2 To achieve the ability of drug carriers in controlling cancer cells, most of the previous studies have focused on developing cancer targeting technology. 3–5 In fact, it is also very important to avoid or decrease the side effect resulting from burst release of drug from drug carriers in designing drug delivery as the burst released drug can be absorbed within a short time in vivo and accumulated in adjacent tissues and metabolic organs (i.e., liver and spleen).6 Therefore, controlled drug delivery is increasingly demanded to sustain the drug release within the expected time and with a suitable dosage.7–9 Currently, the microspheres have been widely used as controlled drug release carrier by having superiority in bioavailability, high drug-loading, and penetrability to tumor tissues instead of normal tissue.10–13 Especially, the diameter of microspheres ranged from 0.1 μm to 4 μm seems an ideal dimension for drug carries because larger diameters of microspheres may be trapped or withheld in the blood capillary or lungs.12 In addition, the morphology and physico-chemical characteristics of the porous microspheres are also important factors for drug delivery.14, 15 Microspheres can be produced by methods such as spray drying, microemulsion, and phase separation, etc.16–18 Currently, there is an increasing concern to exploit suitable materials and corresponding technology for developing microspheres used in controlled drug delivery.
Hydroxylapatites (HAp) are the inorganic component of bone and teeth. It has been one of the popular substrate materials for bone tissue engineering scaffolds.19, 20 Collagen, phages and silk proteins have been used as a template to mediate the nucleation and growth of HAp.21–23 Our previous results have indicated that one of silk proteins, sericin spun from the domesticated silkworm, Bombyx mori (B. mori), can induce the aggregation of porous composite microspheres composed of HAp nanoneedles.24 Specifically, the anionic carboxyl groups of B. mori sericin electrostatically attract calcium ions to trigger the nucleation of HAp. In the meantime, two factors favor the assembly of the HAp and the protein itself into a microsphere.25,26 One is the hydrogen bonding between hydroxyl groups of HAp and carbonyl groups of B. mori sericin. Another is the change of the sericin conformation from random coils to β-sheets. Hence, we speculated that sericin extracted from a wild silkworm, Antheraea pernyi (A. pernyi), would also be a template mediating the successful nucleation of porous HAp microspheres as both A. pernyi sericin (AS) and B. mori sericin share similar amino acid composition and contain acidic amino acids.27 Although there are reports that the individual HAp microspheres without proteins28 or B.mori sericin particles29,30 can be used as drug carriers for cancer treatment, there is no report on the use of AS to template the formation of AS/HAp composite microspheres and demonstrating their further use in achieving sustained drug delivery. We assumed that the porous AS/HAp microspheres (ASM) mediated by AS would be capable of controlled drug delivery because the ASM contains two biocompatible components (HAp and sericin protein) that form unique porous structures.
AS is a soluble protein and has abundant acidic amino acids with carboxyl groups, which will serve as sites for binding the Ca2+ ions to promote the nucleation of HAp. Hence, we hypothesize that AS will experience a biomineralization process by incubating AS in a modified simulated body fluid (1.5 SBF), which contains Ca2+ and PO43− ions. We believe that the negatively charged -COOH groups on the AS in random coil ensure that the AS will firstly attract the Ca2+ by electrostatic interaction. Then the Ca2+ binding will trigger the HAp nucleation and growth in the form of nano-needles, and the change of AS conformation from random coils to β-sheets24, 31 will simultaneously assemble the mineralized HAp into an ASM (Scheme 1A). Doxorubicin (DOX) is an ideal anti-cancer drug, which can induce cytotoxic effects by oxidative DNA damage and preventing DNA replication inside the nuclei. 9 We hope that the obtained ASM will have a porous structure with a negative zeta-potential, favoring the loading of the DOX with a positive zeta-potential (Scheme 1B). Moreover, the DOX loaded ASM are expected to have a pH-responsive characteristic due to the presence of pH-responsive HAp, enabling the DOX to be selectively released into the acidic microenvironment around cancer cells (Scheme 1C). Finally, cancer cells will take up DOX and be killed by continuous incubation of DOX-loaded ASMs (Scheme 1D).
Scheme 1.
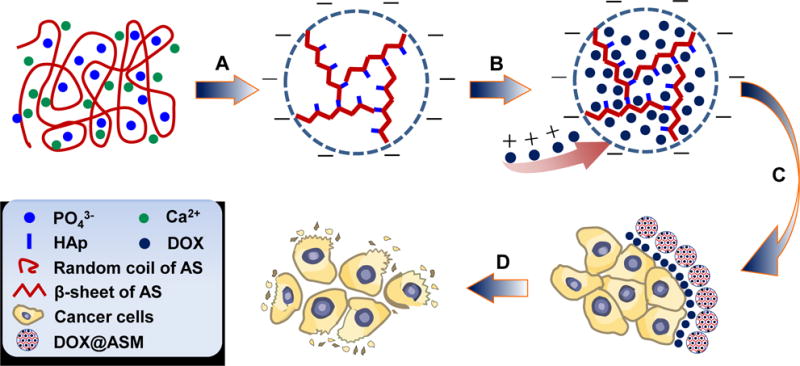
Schematic illustration of the preparation of ASM and the mechanism of pH-triggered DOX release from the DOX@ASM to cancer cells: (A) AS-mediated self-assembly and biomineralization when AS is incubated in 1.5 SBF containing Ca2+, PO43− and OH−; After 5 days of incubation, the nano-needlelike HAp crystals on the AS matrix are further assembled into AS/HAp microspheres (ASM). (B) DOX is loaded in the ASM to form DOX@ASM by electrostatic adsorption and nano-porous structure. (C) DOX@ASM is mixed with cell culture medium and then DOX is released to the acidic extracellular environment surrounding the cancer cells. (D) Cancer cells take up the DOX and, then are killed with the extended duration of DOX@ASM incubation.
2. Results and discussion
2.1. Protein-templated mineralization for preparation of ASM
Fig. 1 showed that ASM was successfully prepared by incubating AS in 1.5 SBF by 5 days. SEM images indicated that ASM was a porous sphere having a diameter at 1–3 μm compared to control, pure AS (Fig. 1 A–B). DLS analysis showed that ASM was distributed uniformly with a diameter at about 1.2 μm (Fig. S1). TEM images confirmed that ASM was composed of nano-needles of HAp (Fig. 1C). The electron diffraction pattern further verified the nucleation of nano-needled HAp due to the appearance of (002) and (211) planes (Fig. 1C, inset). XRD (Fig. 1D) and FTIR (Fig. S2) also confirmed the AS-templated nucleation of HAp. These results are consistent with our previous findings.24,32
Fig. 1.
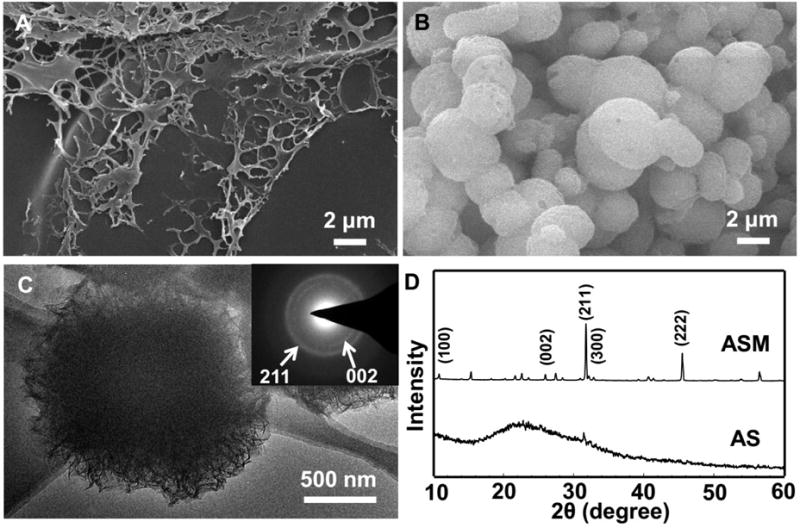
SEM images, TEM image (C) and XRD patterns (D) of AS incubated in 1.5 SBF at 37.2 °C for 5 h (A) and 5 d (B, C). The inset in (C) is an electron diffraction pattern of the corresponding microsphere shown in C. White arrows indicate the (002) and (211) planes of the HAp crystals. (D) Two prominent and sharp diffraction peaks at 25.9 and 31.5° assigned to (002) and (211) plane of apatite emerged, indicating that apatite crystals were nucleated on the AS after incubation in 1.5 SBF by day 5.
2.2. DOX loading ratio and encapsulation efficiency of ASM
Both of the time of incubating carriers in DOX solution and the amount of the carriers can affect the drug encapsulation efficiency.33, 34 We found that increasing the ratio of ASM can increase the DOX loading by ASM (DOX@ASM) (Fig. 2A). Decreasing the DOX loading content can increase the encapsulation efficiency (Fig. 2B). The point that the curve of encapsulation efficiency crossed the curve of DOX loading content (Fig. 2B) means the balance of encapsulation efficiency and loading amounts of DOX by ASM. This ratio is about at 2:1. Therefore, we used this ratio in our following experiments. After incubating ASM in DOX for 66 h, the cumulative encapsulation efficiency of ASM reached up to 62.6% (Fig. 2C), suggesting a high encapsulation efficiency of ASM. FTIR spectra indicated the successful adsorption of DOX by ASM (Fig. S2). The confocal microscopy indicated that the fluorescence intensity tended to be weaker from the edge region than from the center of ASM, implying that DOX was mainly distributed inside the porous structure of ASM (Fig. S3). Fig. 2D verified the zeta potential of the DOX and ASM to be 5.44 ±1.39 and −2.74 ±0.91mV, respectively. In contrast to ASM, silk microspheres with smooth surface were hardly loaded with DOX (Fig. S4). This explained that the negative zeta-potential and porosity of the ASM drive it to load and carry the DOX.
Fig. 2.
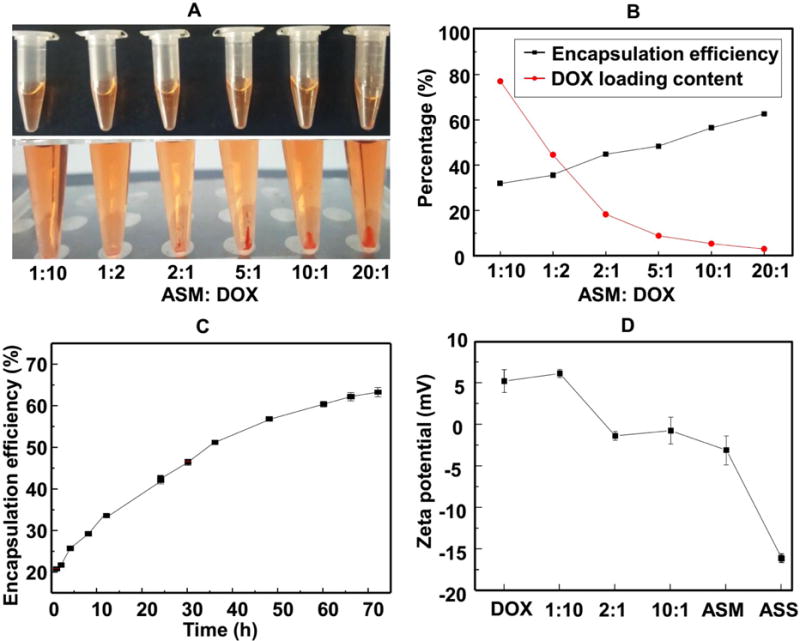
The DOX loading by ASM and encapsulation efficiency of ASM. (A) shows DOX/ASM mixed solutions with different ASM/DOX mass ratio before centrifugation (top, homogenous solution) and after centrifugation (bottom, DOX/ASM precipitation). (B) The DOX loading content and encapsulation efficiency under different ASM/DOX mass ratio after 36 h. The mass ratio of 2:1 was chosen as the subsequent experiment ratio. (C) Encapsulation efficiency of ASM at different adsorption times. (D) Zeta potential change of DOX@ASM with different mass ratios at pH 7.4.
2.3. Release of DOX@ASM in vitro
The DOX cumulative release profiles from DOX@ASM were estimated at pH 7.4 and pH 6.2, which are similar to the pH values of the human blood and the tumor microenvironment, respectively. As shown in Fig. 3, the DOX@ASM had an almost linear diffuse curve at pH 7.4, and no significant release of DOX was observed over 100 h. In contrast, the DOX release from DOX@ASM at pH 6.2 was much faster than that at pH 7.4 in a sustained manner. The maximum release of the DOX reached 90% at pH 6.2 within 72 h, indicating that the rate of DOX release from DOX@ASM is higher at a more acidic condition (Fig. 3A). The pH value can serve as a stimulus for driving functional changes of drug carriers because adjusting pH value can result in changes in protein conformation, solubility, surface potential and hydrophilicity/hydrophobicity, which can enable controlled drug release 35,36. Therefore, we believe that the faster-sustained release at the acidic pH value arose from the fact that the HAp contained in the ASM became less stable at the acidic condition, making the porous structure of the ASM become more open and favor the DOX release. In contrast, the free DOX release from a dialysis membrane was completed within 6 h by having a DOX cumulative release reaching up to about 88% (Fig. 3B), suggesting DOX@ASM displayed a slower drug release than DOX under the same pH condition. Therefore, the release characteristic of DOX@ASM was pH-dependent, and this high release characteristic at slightly low pH would allow the DOX to be released in a short time. Taken together, the faster DOX release DOX@ASM at the acidic pH achieved a sustained release pattern. Hence, DOX@ASM is expected to selectively release drug at tumor sites (with a pH close to 6.2) at a higher rate and thus avoid burst release of DOX in normal tissue (with a pH close to 7.4), leading to a reduced side effect of DOX to normal tissues.
Fig. 3.
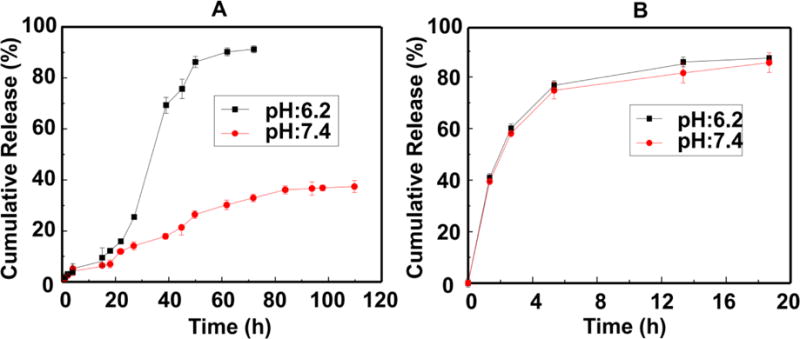
Cumulative release profiles of DOX@ASM (A) and free DOX (B) from dialysis membrane against pH 7.4 PBS buffer and pH 6.2 buffer at 37 °C, respectively. Data were presented as mean ±standard deviation (n=4).
2.4. Cytotoxicity of DOX@ASM
The cytocompatibility of ASM before loading DOX was investigated. The results of cytotoxicity test (Fig. 4A) indicate that ASM is suitable to be drug carriers because it does not bring any toxic effects to the cells. To assess the cytotoxicity of DOX-loaded ASM, the cells were treated by DOX@ASM for 12 h by using PBS and DOX as the positive and negative control, respectively. The DOX concentration in DOX@ASM was 2, 10 and 50 μg mL−1, respectively. As shown in Fig. 4A, DOX and DOX@ASM might result in cell apoptosis, which seemed to follow a dose-dependent manner after 12 h. Regardless of DOX concentration set at 2, 10 or 50 μg mL−1, DOX tended to have a relatively lower Bcap-37 cell viability than DOX@ASM at same DOX concentration. Furthermore, the half-maximal inhibitory concentration (IC50) value for DOX was 50 μg mg−1, which was distinctively lower than the DOX@ASM after a 12 h treatment.
Fig. 4.
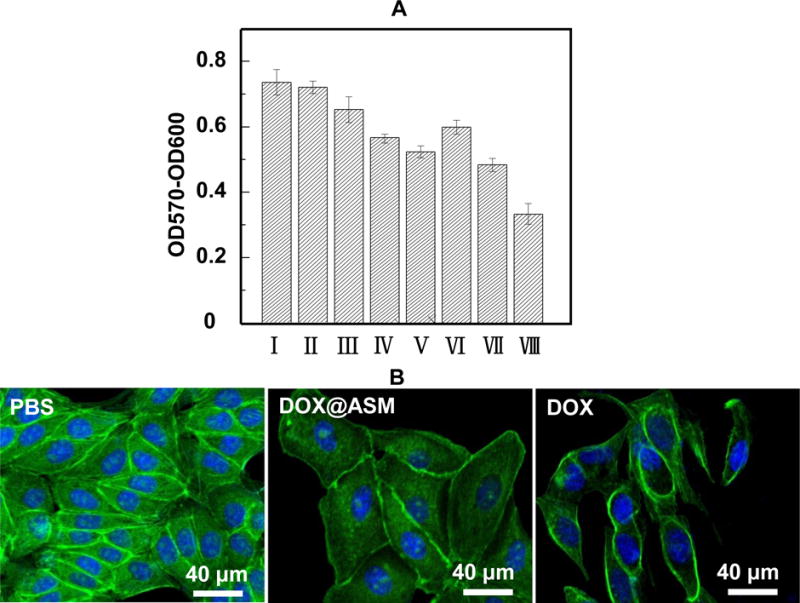
Cytotoxicity and morphology of ASM and DOX@ASM after incubation with Bcap-37 cells: (A) Alamar Blue® cell viability reagent is used to assess Bcap-37 cell viability. The absorbance is monitored at 570 nm with 600 nm as a reference wavelength; (B) Morphology of Bcap-37 cells after 12 h incubation with PBS, DOX@ASM (10 μg mL−1) and free DOX (10 μg mL−1); *p < 0.05, **p < 0.01. ASM: AS/HAp microspheres; DOX@ASM: DOX loaded by ASM.I: PBS; II: ASM; III: DOX@ASM (2 μg mL−1); IV: DOX@ASM (10 μg mL−1); V: DOX@ASM (50 μg mL−1); VI: DOX (2 μg mL−1); VII: DOX (10 μg mL−1); VIII: DOX (50 μg mL−1).
The morphology of Bcap-37 cells incubated with DOX@ASM after 12 h was observed by staining Alexa Fluor® 488 and DAPI. Fig. 4B showed that the cells in the control group (PBS) had a well-defined actin cytoskeleton. However, the cellular morphology from DOX@ASM and the free DOX group dramatically changed, and many cells were floated away from the cell culture plate. Besides, the free DOX group had the lightest green fluorescence staining, indicating the free DOX group could reach a higher level of cytotoxicity than DOX@ASM carriers within a short time.
To further explore the effect of duration time on cancer cells, the morphology of Bcap-37 cells (Fig. 5 and Fig. S5) was examined by a phase-contrast microscope at 3, 24 and 60 h. Fig. 5 showed the presence of regular Bcap-37 cell outline on the DOX@ASM after 3 h of incubation which was similar to the PBS control group. However, a lot of apoptotic cells or necrotic cells could be observed in the free DOX group, demonstrating that the cellar uptake of the drug in the free DOX group was more rapid. After 24 h, Bcap-37 cells on the DOX@ASM group were distorted, implying that the DOX was efficient and slowly released from DOX@ASM. In contrast to Bcap-37 cells, C2C12 cells showed apoptosis until incubation of 60 h (Fig. S6). This indicates that cancer cells are subjected to more apoptosis than normal cells, proving that DOX@ASM prefers to release DOX in the tumor microenvironment. Our data indicated that the drug release from the DOX@ASM was more favorable for delaying the cellular uptakes by Bcap-37 cells.
Fig. 5.
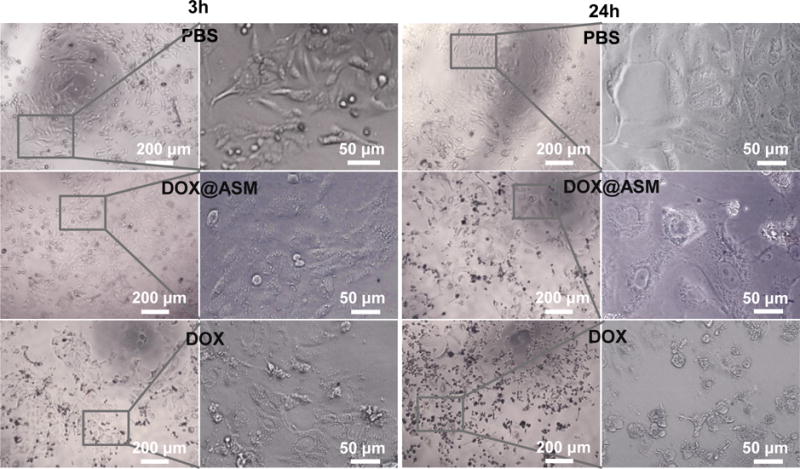
Morphologies of Bcap-37 cells incubation with PBS, DOX@ASM (DOX:20 μg mL−1) and free DOX (20 μg mL−1) after cultured for 3 h and 24 h. Left images are low magnification images and right images are high magnification images in each group. ASM: AS/HAp microspheres; DOX@ASM: DOX loaded by ASM.
2.5. Distribution of DOX inside of cells
To further evaluate the dynamic process of DOX release from cell membrane to nucleus controlled by ASM in vitro, HeLa cells were stained with Hoechst 33342 (blue) and the spontaneous red fluorescence in relation to the intracellular localized DOX was observed by CLSM. Fig. 6 showed that the cells treated by the free DOX group had a brighter red- fluorescence than those treated by the DOX@ASM group after incubation for 3 h, suggesting that the DOX was rapidly localized inside the HeLa cells in the free DOX group. There was a slight accumulation of DOX in the cells treated with the DOX@ASM group according to the dark red fluorescence, suggesting the cellular uptakes of the DOX were considerably reduced after incubation for 3 h.
Fig. 6.
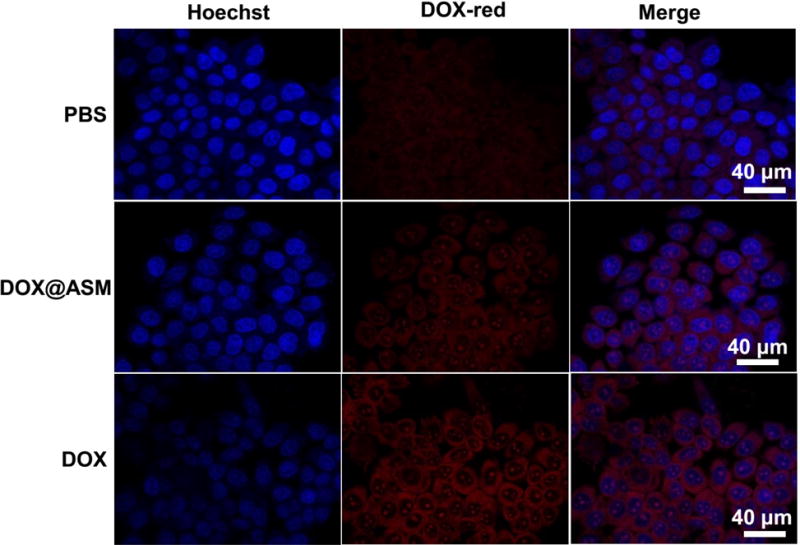
Fluorescence images of cells incubated with PBS, DOX@ASM, and free DOX, showing the cellular uptake and distribution of DOX after incubation with HeLa cells for 3 h. Hoechst 33342 (blue) was used to stain cell nuclei. The red fluorescence was spontaneously emitted by DOX. ASM: AS/HAp microspheres; DOX@ASM: DOX loaded by ASM.
After one day of incubation, the cells treated by the DOX@ASM had the brightest red fluorescence from the cytoplasm compared to controls (Fig. 7), indicating the more efficient and controlled release of DOX by ASM. In addition, the DOX@ASM group had brighter blue fluorescence than the free DOX group, suggesting that the cells cultured with free DOX had been dead for a while. In addition, the blue fluorescence from the free DOX tended to become weaker and the red fluorescence was mostly concentrated on the nuclei. This suggested that the DOX was accumulated in the cell nuclei. According to the intracellular localization profiles of DOX, the drug release from the ASM was more effective than the free DOX, which is consistent with the selectively more effective DOX release profile (Fig. 3) from ASM at an acidic pH value typical for the tumor microenvironment. Cancer cells are known to secrete lactic acid. Hence, the tumor tissue has an acidic microenvironment, different from the normal tissue which has a normal physiological pH.37, 38 Therefore, the DOX@ASM is expected to be more effective in DOX release at the tumor microenvironment that at the normal tissue.
Fig. 7.
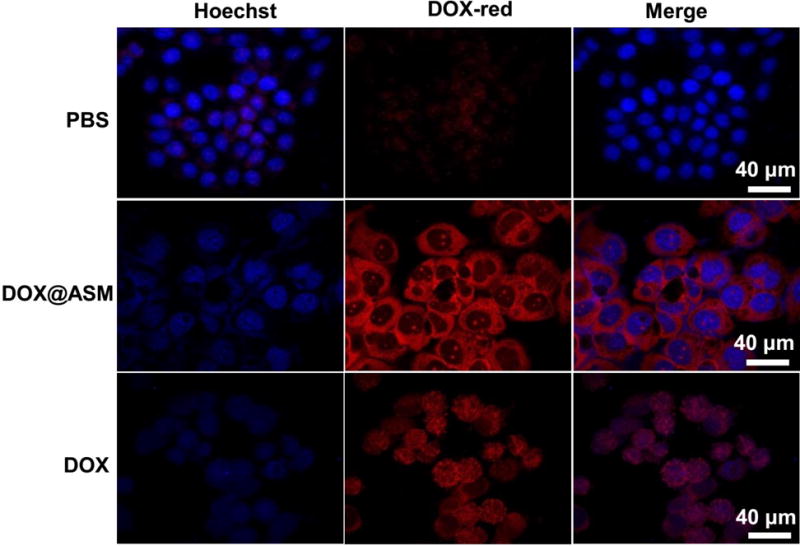
Fluorescence images of cells incubated with PBS, DOX@ASM, and free DOX, showing the cellular uptake and distribution of DOX (red) after incubation with HeLa cells for 24 h. Hoechst 33342 was used to stain cell nuclei. The red fluorescence was spontaneously emitted by DOX. ASM: AS/HAp microspheres; DOX@ASM: DOX loaded by ASM.
The above observations were further confirmed by quantitative analysis in vitro cytotoxicity using the Alamar blue cell viability analysis (Fig. 8). Quantitative analysis showed that HeLa cells treated with the free DOX had the lowest growth rate than other groups after 3 h, suggesting a rapid accumulation of DOX in the cytoplasm (Fig. 8A). However, the DOX@ASM group presented fewer HeLa cells than the free DOX group after 36 and 60 h, suggesting the DOX release from DOX@ASM was more sustained. This was consistent with the CLSM observation results (Fig. 7). In order to further investigate the sustained release efficiency of ASM, the data were further analyzed by dividing the quantitative results of Alamar blue into different periods (Fig. 8B–D). Fig. 8B showed that the DOX@ASM resulted in the minimum growth rate of HeLa cells between 3 to 36 h, indicating that the DOX were continuously released from the DOX@ASM. Fig. 8C shows that the cells treated with the free DOX group showed a higher growth rate than those treated with the DOX@ASM from 36 to 60 h, indicating that the release of DOX into the HeLa cells was much faster in the DOX@ASM group. Fig. 8 D showed the relative growth of HeLa cells between 3 and 60 h in the ASM group was the lowest among all of the testing groups. Collectively, these results were consistent with the results of intracellular DOX distribution, implying that the ASM carrier system could efficiently release DOX into cancer cells for a long time in a sustained manner.
Fig. 8.
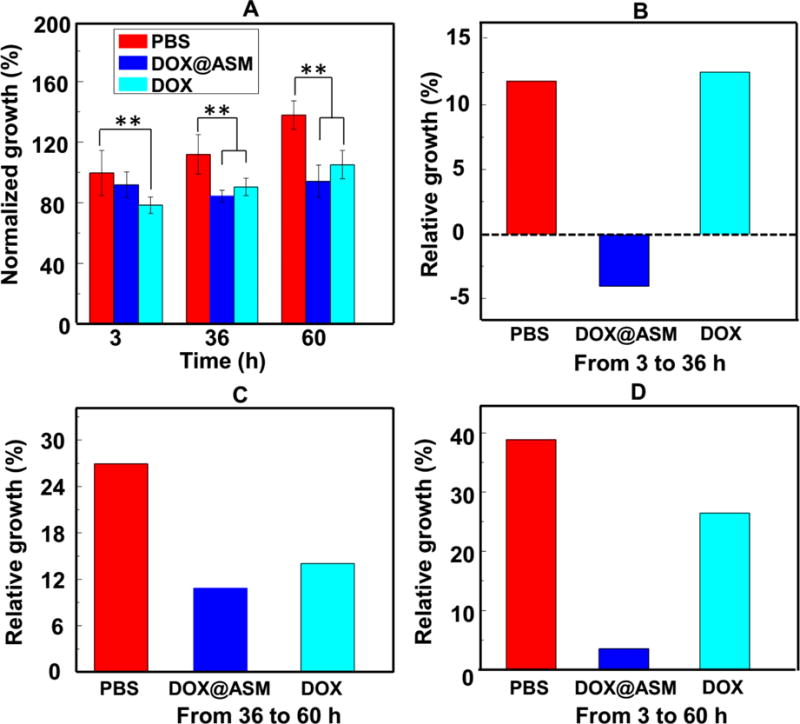
Inhibition of cancer cell growth by DOX@ASM. (A) Alamar Blue® cell viability reagent was used to assess HeLa cell viability at different time points. (B, C, D) Relative growth of HeLa cells for three different durations (A, 3–36 h; B, 36–60 h; C: 3–60 h). *p < 0.05, **p < 0.01. ASM: AS/HAp microspheres; DOX@ASM: DOX loaded by ASM.
3. Experimental
3.1 Preparation of aqueous AS solution
The AS solution was prepared following our reported protocol.27 The cocoons of A. pernyi were cut into films of 5×5 mm2. The films were immersed in deionized water. For extracting AS, this container was firstly heated at 120 °C for 30 min. The resultant suspension was then centrifuged at a speed of 8000 rpm for a period of 10 min. Finally, the supernatant was collected to form the AS aqueous solution. The solution was dried and weighed in order to determine the AS concentration in the collected solution.
3.2 Preparation of ASM by incubating AS in 1.5 SBF
To make 1.5 SBF solution, NaCl, CaCl2, NaHCO3, KCl, K2PO4·3H2O, MgCl2·6H2O, and Na2SO4 were dissolved in deionized water. Tris-hydroxymethyl-aminomethane and hydrochloric acid were then added to allow the solution to be buffered at pH=7.27 The nucleation and growth of HAp induced by AS template were performed according to our previous procedure.23, 24 Briefly, AS aqueous solution (2.0 mg mL−1) was placed into a dialysis bag. The subsequent dialysis was performed against 1.5 SBF, which was replaced with fresh one every 12 h, at the body temperature. After 5 days, the solution in the dialysis bag was centrifuged for collecting the precipitate. The ASM was obtained after rinsing the precipitate with deionized water followed by freeze-drying.
3.3 Characterization of ASM
To characterize the ASM, the freeze dried powder obtained above was observed with field emission scanning electron microscopy (SEM) and Fourier Transform Infrared Spectroscopy (FTIR) and X-ray diffractometry spectra (XRD). For SEM, gold was deposited on the samples prior to imaging. For XRD, the scanning was performed from 10° to 60° at 6° min−1. For FTIR, the samples mixed with KBr were examined over 4000–400 cm−1. In addition, the samples’ surface charge was calculated through a zeta potential analyzer (ZetaSizer Nano series) at pH 7.4 and a fixed scattering angle of 90°. For DLS measurements, the size distribution of ASM aqueous solution (1mg/mL) was obtained by using Zetasizer (Nano-ZS90, Malvern instruments, UK).
3.4 DOX loading into and release from ASM
Here, DOX (Zhejiang Hisun Chemical Co., Ltd, China), an anticancer drug, was selected to investigate the encapsulation efficiency of ASM. The mixture was prepared by placing 10 mg of the ASM and 4 mg of DOX powder into 6 mL of PBS (Phosphate Buffer Solution). For preparing DOX-loaded carriers, this mixture was completely shaken by placing it on a shaking table at the speed of 100 rpm at 37 °C. Then the mixture was centrifuged for 10 min at a speed of 6000 rpm. The supernatants were saved for measuring the amount of the residual DOX that was not absorbed by ASM. The concentration of the residual DOX in supernatants was quantitatively monitored using a DOX calibration curve by employing a microplate reader to detect optical density at 490 nm. The DOX loading content (termed DLC) and encapsulation efficiency (termed EE) adsorbed by ASM were calculated according to the following equations, where, Wt and Wr indicate the total weight of DOX dissolved in a buffer (phosphate-buffered saline, PBS), and residual DOX in the same buffer, respectively, while Wm is defined as the total amount of ASM in PBS:
The powders were isolated through centrifugation, followed by freeze-drying to form the DOX-loaded ASM (DOX@ASM). The signal intensities of the DOX from DOX@ASM were determined using a confocal laser scanning microscope (CLSM, ZEISS LSM780, Germany). Red fluorescence from DOX was excited at 490 nm and emitted light over a wavelength range of 535 to 625 nm. In addition, the release of DOX from DOX@ASM was performed in vitro. 5 mg of the DOX@ASM powder was suspended in 5 mL of PBS. Then, this suspension was put in a cellulose membrane bag (MWCO=8 kDa), followed by dialysis against 15 mL of buffer solution at 37 °C. During dialysis, the bag was shaken at 100 rpm. Here, two media, PBS with a pH of 7.4 as well as a buffered solution of potassium dihydrogen phosphate and sodium hydroxide (KH2PO4/NaOH) with a pH of 6.2, were used as the release buffer, respectively. At different time points, 100 μL of a sample was collected from the corresponding release buffer, and 100 μL of the fresh buffer solution was placed to keep the volume of the release medium buffer to be 15 mL. To determine the amount of the released DOX, the absorbance at 490 nm was measured to form a DOX calibration curve. Silk fibroin microspheres having a smooth surface and a diameter of about 2 μm (similar to ASM) were as a control group.
3.5 Cell toxicity assay, cell morphology and intracellular distribution of DOX@ASM
Cell culture was performed by using two cancer cell lines (Bcap-37 cells and HeLa cells) and a normal cell line (C2C2 cells). Both cell lines were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in Dulbecco’s Modified Eagle Media (DMEM, Gibco, USA) with 10% fetal bovine serum (FBS, Gibco, USA). All cells were incubated at 37°C in a humidified, 5% CO2 incubator. For passaging, cells were detached by incubating with 0.25 % trypsin-EDTA solution (Invitrogen, USA) and centrifuged at 1000 rpm for 5 min. The number of cells was determined by a Scepter™ handheld automated cell counter (Millipore, USA). Then, the cells were seeded in 96-well plates with 1×104 cells in each well. After incubation for one day, the culture medium was exchanged with 100 μL of fresh DMEM supplemented with DOX@ASM. The DOX concentrations from DOX@ASM were set at 2, 10 and 50 μg mL−1, respectively. Free DOX served as a control sample.
3.5.1 Cytotoxicity assay
For evaluating the cell toxicity of ASM and DOX@ASM, 10 μL of Alamar blue solution (Life Technologies, USA) was placed into the testing wells, followed by incubation at 37 °C for 3 h. Cell toxicity was monitored using a microplate reader, which was operated at an excitation wavelength of 570 nm and a reference wavelength of 600 nm. The cell viability was represented as a relative value by OD570–OD600 nm.
3.5.2 Cell morphology
For observing the fluorescence images of the cancer cells, Bcap-37 cells were placed with 1×104 cells in each well by adding DOX-loaded microspheres into the culture medium in 5% CO2 at 37°C. After cultured for 12 h, the cells were fixed in 4% paraformaldehyde in PBS for 1 h, followed by washing 3 times by 0.2 % Triton X-100 in PBS. After fixation, the cytoskeletons were stained with F-actin using Alexa Fluor® 488 phalloidin (Invitrogen), and cell nuclei were stained with DAPI at room temperature. Cell morphology was acquired using an excitation wavelength of 488 nm and an emission wavelength of 520 nm by a confocal laser scanning microscope (CLSM, ZEISS LSM780, Germany).
3.5.3 Cellular uptake of DOX
Fluorescent imaging is commonly used to explore the subcellular localization of drug absorption. Hoechst 33342 is used for specifically staining the nuclei of living cells as it can pass the cell membrane and bind DNA.39 Cellular uptake was observed by CLSM. HeLa cells (1×104 cells dish−1) were grown on cover glasses (35 mm×10 mm) and interacted with the DOX@ASM at the final concentration of 20 μg mL−1 or PBS. After the cells were incubated at 3 and 24 h, they were washed twice with PBS. Then they were fixed in 5% glutaraldehyde at room temperature, followed by rinsing with PBS to remove the residual ASM. After that, Hoechst 33342 solution (0.1 mL) was added and incubated for 15 min in order to achieve the cell nuclei staining. The cells were eventually washed twice and characterized by CLSM imaging. Blue fluorescence from Hoechst 33342 was excited at the wavelength of 346 nm and emitted a light at around 461 nm. Red fluorescence from DOX was excited at 490 nm and emitted a light over the wavelength range of 535 to 625 nm.
4. Conclusions
We report the development of AS/HAp microspheres by AS-templated biomineralization of HAp. These AS/HAp microspheres are uniform and porous. The porous AS/HAp microspheres have a good DOX encapsulation efficiency, and a controllable and sustained release rate. The sustained release of the DOX from the porous AS/HAp microspheres is both time dependent and pH responsive. In particular, the microspheres can selectively release DOX at a higher rate at the acidic conditions (pH=6.2) typical for tumor microenvironment than at the physiological conditions (pH=7.4) typical for normal tissues, which will potentially reduce the side effect of the cancer drugs in normal tissues. Given the fact that silk proteins and HAp can find applications as scaffolds in tissue regeneration, the controlled and sustained release behavior implies that the AS/HAp microspheres have a potential application in drug delivery and regenerative medicine.
Supplementary Material
Acknowledgments
We acknowledge the support of Zhejiang Provincial Natural Science Foundation of China (LZ17C170002 and LZ16E030001), State of Sericulture Industry Technology System (CARS-22-ZJ0402), National Natural Science Foundation of China (51673168), National High Technology Research and Development Program 863 (2013AA102507), Zhejiang Provincial Science and Technology Plans (2016C02054). CBM would also like to thank the financial support from National Institutes of Health (CA200504), Oklahoma Center for Adult Stem Cell Research (434003), and Oklahoma Center for the Advancement of Science and Technology (HR14-160).
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Cao B, Yang M, Zhu Y, Qu X, Mao C. Adv Mater. 2014;26:4627–4631. doi: 10.1002/adma.201401550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gandra N, Abbineni G, Qu X, Huai Y, Wang L, Mao C. Small. 2013;9:215–221. doi: 10.1002/smll.201202090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Savla R, Taratula O, Garbuzenko O, Minko T. J Controlled Release. 2011;153:16–22. doi: 10.1016/j.jconrel.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 4.Du C, Deng D, Shan L, Wan S, Cao J, Tian J, Achilefu S, Gu Y. Biomaterials. 2013;34:3087–3097. doi: 10.1016/j.biomaterials.2013.01.041. [DOI] [PubMed] [Google Scholar]
- 5.Ang C, Tan SY, Wang X, Zhang Q, Khan M, Bai L, Selvan ST, Ma X, Zhu L, Nguyen KT. J Mater Chem B. 2014;2:1879–1890. doi: 10.1039/c3tb21325k. [DOI] [PubMed] [Google Scholar]
- 6.Sun T, Zhang YS, Pang B, Hyun DC, Yang M, Xia Y. Angew Chem, Int Ed. 2014;53:12320–12364. doi: 10.1002/anie.201403036. [DOI] [PubMed] [Google Scholar]
- 7.Zhang X, Meng L, Lu Q, Fei Z, Dyson PJ. Biomaterials. 2009;30:6041–6047. doi: 10.1016/j.biomaterials.2009.07.025. [DOI] [PubMed] [Google Scholar]
- 8.Uhrich KE, Cannizzaro SM, Langer RS, Shakesheff KM. Chem Rev. 1999;99:3181–3198. doi: 10.1021/cr940351u. [DOI] [PubMed] [Google Scholar]
- 9.Pan L, Liu J, He Q, Shi J. Adv Mater. 2014;26:6742–6748. doi: 10.1002/adma.201402752. [DOI] [PubMed] [Google Scholar]
- 10.Freiberg S, Zhu X. Int J Pharm. 2004;282:1–18. doi: 10.1016/j.ijpharm.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 11.Bao W, Zhou J, Luo J, Wu D. J Microencapsulation. 2006;23:471–479. doi: 10.1080/02652040600687613. [DOI] [PubMed] [Google Scholar]
- 12.Neuberger T, Schöpf B, Hofmann H, Hofmann M, Von Rechenberg B. J Magn Magn Mater. 2005;293:483–496. [Google Scholar]
- 13.Wolinsky JB, Colson YL, Grinstaff MW. J Controlled Release. 2012;159:14–26. doi: 10.1016/j.jconrel.2011.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gref R, Domb A, Quellec P, Blunk T, Müller R, Verbavatz J, Langer R. Adv Drug Delivery Rev. 2012;64:316–326. doi: 10.1016/0169-409X(95)00026-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng S. Expert Rev Med Devices. 2004;1:115–125. doi: 10.1586/17434440.1.1.115. [DOI] [PubMed] [Google Scholar]
- 16.Sollohub K, Cal K. Spray drying technique: II. J Pharm Sci. 2010;99:587–597. doi: 10.1002/jps.21963. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Zhou A, Yang Z. Mater Lett. 2008;62:1930–1932. [Google Scholar]
- 18.Elbert DL. Acta Biomater. 2011;7:31–56. doi: 10.1016/j.actbio.2010.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cox SC, Thornby JA, Gibbons GJ, Williams MA, Mallick KK. Mater Sci Eng, C. 2015;47:237–247. doi: 10.1016/j.msec.2014.11.024. [DOI] [PubMed] [Google Scholar]
- 20.Wang JL, Yang MY, Zhu Y, Wang L, Tomsia AP, Mao CB. Adv Mater. 2014;26:4961–4966. doi: 10.1002/adma.201400154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao BR, Yang MY, Mao CB. Materials and Medicine. Acc Chem Res. 2016;49:1111–1120. doi: 10.1021/acs.accounts.5b00557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Almora-Barrios N, De Leeuw NH. Cryst Growth Des. 2012;12:756–763. [Google Scholar]
- 23.Yang MY, He W, Shuai YJ, Min SJ, Zhu LJ. J Polym Sci, Part B: Polym Phys. 2013;51:742–748. [Google Scholar]
- 24.Yang M, Zhou G, Shuai Y, Wang J, Zhu L, Mao C. J Mater Chem B. 2015;3:2455–2462. doi: 10.1039/C4TB01944J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li W, Cai Y, Zhong Q, Yang Y, Kundu SC, Yao J. J Mater Chem B. 2016;4:340–347. doi: 10.1039/c5tb02328a. [DOI] [PubMed] [Google Scholar]
- 26.Liu J, Liu Y, Kong Y, Yao J, Cai Y. Mater Lett. 2013;110:221–224. [Google Scholar]
- 27.Yang M, Shuai Y, Zhang C, Chen Y, Zhu L, Mao C, OuYang H. Biomacromolecules. 2014;15:1185–1193. doi: 10.1021/bm401740x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang KW, Zhu YJ, Chen XY, Zhai WY, Wang Q, Chen F, Chang J, Duan YR. Chem -Asian J. 2010;5:2477–2482. doi: 10.1002/asia.201000463. [DOI] [PubMed] [Google Scholar]
- 29.Mandal BB, Kundu S. Nanotechnology. 2009;20:355101. doi: 10.1088/0957-4484/20/35/355101. [DOI] [PubMed] [Google Scholar]
- 30.Huang L, Tao K, Liu J, Qi C, Xu L, Chang P, Gao J, Shuai X, Wang G, Wang Z. ACS Appl Mater Interfaces. 2016;8:6577–6585. doi: 10.1021/acsami.5b11617. [DOI] [PubMed] [Google Scholar]
- 31.Melcher M, Facey SJ, Henkes TM, Subkowski T, Hauer B. Biomacromolecules. 2016;17:1716–1726. doi: 10.1021/acs.biomac.6b00135. [DOI] [PubMed] [Google Scholar]
- 32.Yang M, He W, Shuai Y, Min S, Zhu L. J Polym Sci, Part B: Polym Phys. 2013;51:742–748. [Google Scholar]
- 33.Yang X, Zhang X, Liu Z, Ma Y, Huang Y, Chen Y. J Phys Chem C. 2008;112:17554–17558. [Google Scholar]
- 34.Peng C, Zhao Q, Gao C. Colloids Surf, A. 2010;353:132–139. [Google Scholar]
- 35.Schmaljohann D. Adv Drug Delivery Rev. 2006;58:1655–1670. doi: 10.1016/j.addr.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 36.Lee ES, Gao Z, Bae YH. J Controlled Release. 2008;132:164–170. doi: 10.1016/j.jconrel.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S. Blood. 2007;109:3812–3819. doi: 10.1182/blood-2006-07-035972. [DOI] [PubMed] [Google Scholar]
- 38.Choi SYC, Collins CC, Gout PW, Wang Y. J Pathol. 2013;230:350–355. doi: 10.1002/path.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin RM, Leonhardt H, Cardoso MC. Cytometry, Part A. 2005;67:45–52. doi: 10.1002/cyto.a.20172. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.