

Abstract
Objective
Adrenocortical carcinoma is an aggressive endocrine malignancy with an annual incidence of 0.5–2 cases per million. The most important factors that determine prognosis are tumor stage at the time of diagnosis and the success of surgery. However, advanced age, large tumor size, hormone secretion, high Ki-67 index (>10%), tumor necrosis and high mitotic activity are other factors associated with poor prognosis. In the present study, we aimed to evaluate the contribution of the patient and treatment- related factors to the prognosis in adrenocortical carcinoma.
Material and methods
We included 15 adrenocortical carcinoma patients who were followed in our center between 2005 and 2015. The effects of age, gender, tumor size, type of operation, postoperative resection status and adjuvant treatment on disease-free survival and overall survival were analyzed.
Results
Disease-free survival was 23.32±3.69 months and overall survival was 36.60±10.78 months. Gender, tumor size, tumor stage, type of operation, hormonal activity, presence of necrosis, recurrence and development of metastasis were not found to be associated with disease-free survival and overall survival (p>0.05). Postoperatively applied adjuvant treatments including mitotane, chemotherapy and radiotherapy did not significantly affect disease-free survival in our study, but statistically significant increase in overall survival was observed in patients getting adjuvant treatments (p=0.006).
Conclusion
Adrenocortical carcinoma has poor prognosis and short overall survival, and in its clinical course, recurrence and development of metastasis can be commonly observed even after complete resection of the tumor. Therefore, the patients should be evaluated carefully while determining the surgical procedure during the preoperative period, and the operation and post-operative follow-up should be performed in experienced centers. However, due to the positive effects of adjuvant treatments on survival, all patients should be evaluated postoperatively for the necessity of adjuvant treatments, especially mitotane.
Keywords: Adrenocortical carcinoma, chemotherapy, mitotane, surgery
Introduction
Adrenocortical carcinoma (ACC) is a rarely seen endocrone malignancy with an annual incidence of 0.5–2 million cases.[1] It is more frequently seen in women (female/male ratio, 1.5/1).[2] Diagnosis is generally established based on symptoms due to hypersecretion from tumor or local effects of large tumoral mass lesions. However, they can be detected incidentally thanks to realization of increasingly frequent radiological imaging modalities.[3,4] In ACC computed tomography (CT), and magnetic resonance imaging (MRI) are preferred imaging modalities for the localization of the tumor, and determination of metastases. When incidentally an adrenal mass is detected, for further evaluation priorly CT imaging is preferred. In non-contrasted measurements tissue densities more than 10 HU (Hounsfield unit) indicates the presence of ACC. However in contrasted-CT measurements wash-out rate of >50% (clearance rate of the mass from the contrast agent) is an indicator of malignancy. However in MRI evaluation of signal intensity facilitates the discrimination between benign, and malignant lesions.[5] In the differential diagnosis between adrenal adenoma, and adrenal carcinoma biopsy is not useful. Also, in the presence of suspect ACC, taking biopsy material from the mass is not recommended because of the risk of implantation of tumor cells into surrounding tissues. Since serious complications can be seen, pheochromocytoma should be ruled out preoperatively in patients for whom adrenal biopsy is planned.[6]
When staging system of European Network for the Study of Adrenal Tumors (ENSAT) is taken as a base, 5-year survival rates are 82% in Stage I, 61% in Stage II, 50% in Stage III, and in Stage IV it dramatically drops down to 13 percent.[7] Although it is a tumor with an aggressive course, the most important prognostic factors are stage at the time of diagnosis, and success of the first surgery. However advanced age, tumor size, functional state of the tumor, higher Ki-67 index (>10%), increased mitotic activity, and necrotic areas in the tumor are indicators of poor prognosis.[2,4,7,8] German Adrenocortical Carcinoma Registry reported mean diameter of tumor in patients with ACC at the time of diagnosis as 11.5±4.7 cm.[9] According to National Institutes of Health consensus conference tumors with a diameter of >6 cm have a higher potential for malignancy, and surgery is recommended.[10]
Surgery is the first-line treatment in ACC. Tumor should be completely resected, and integrity of the capsule should be preserved.[9] In invasive Stage III tumors open adrenalectomy is indisputable recommended. While for Stage I, and II noninvasive tumors with a diameter of <10 cm, laparoscopic surgery is also recommended as a selectable method.[11–13] However peritoneal recurrence, and development of carcinomatosis are more frequently seen following laparoscopic surgery when compared with open surgery. Therefore open adrenalectomy is the the general treatment approach in ACC.[14,15] Since complete resection of the tumor is an important prognostic factor, invasive tumors should be resected en bloc with the surrounding tissue or organs.[2]
Even in non-metastatic tumors where complete resection is achieved, in most of the cases adjuvant treatment is recommended because of higher recurrence rates.[4] Mitotane which is a derivative of an insecticide dichlorodiphenyl trichloroethane, blocks synthesis of cortisole, and exerts adrenolytic effects.[16] Use of mitotane is recommended in adjuvant treatment where postoperatively microscopic residual tissue (R1) is detected, when presence of residual tissue is not known for sure (Rx), in patients with Ki-67 index is above 10%, and in cases with increased risk where Ki-67 index is >10% in whom R0 resection is achieved (Stage III or Ki-67 >10%).[2] Treatment algorithm of ACCs is given in Figure 1.[17] In the present study, we aimed to evaluate the contribution of the patient and treatment- related factors to the prognosis in adrenocortical carcinoma.
Figure 1.
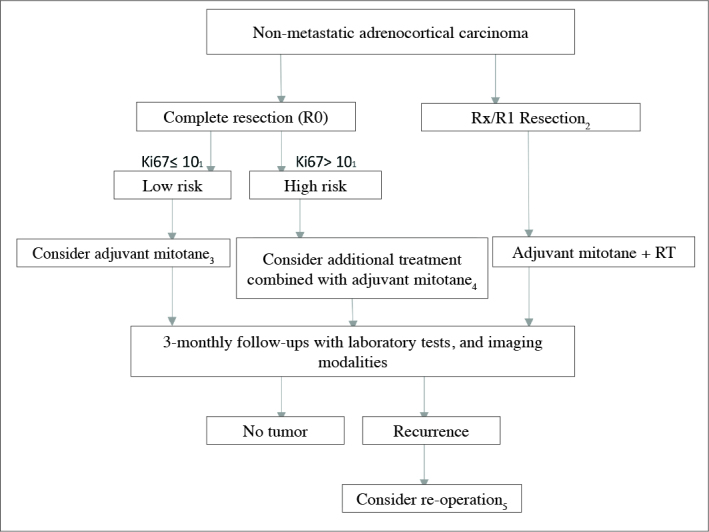
Treatment algorithm of non-metastatic adrenal carcinoma
1) If Ki67 index can not be measured, then higher proliferative index (>mitoses/5 large field) can be used in the evaluation of risk. 2) In patients who had undergone R2 resection, re-operation should be considered by an experienced surgeon. 3) Tumor diameter less than 8 cm, and lack of evidence of microscopic invasion into tumor capsule are indicators of decreased risk for recurrence.In cases which meet these conditions follow-up may be planned. 4) Individualized treatment (especially in patients who demand aggressive treatment): Ki67 ≥20, in the presence of microscopic invasion to vessels and tumor capsule decision favoring additional radiotherapy directed to tumor bed, if tumor thrombus is seen in vena cava, then decision favoring administration of additional dose of streptozotocin. 5) Re-operation is recommended if complete resection is feasible, and a time interval of less than 12 months passed between primary surgery and recurrence. If this time interval is less than 6 months, since most of the patients would not benefit from resection of the recurrent lesion, systemic treatment is initiated as soon as possible.
Material and methods
A total of 15 patients were included in the study who had been operated between January 2005, and August 2015 because of an adrenal mass, and histopathological examination established the diagnosis of ACC which resulted in continuation of their monitorization. Written consent of the study participants, and ethics committee approval from our center were obtained. Archival files of the patients were reviewed, and data about their age, gender, preoperative biochemical test, and radiological imaging results, type of surgery, surgical resection status, and postoperative follow-ups were studied. Similarly, location, and stage of the tumor were evaluated based on the information about histopathological data found in patients’ files. The presence of recognized poor prognostic factors as Ki-67 index, mitotic activity index, and necrosis was investigated. Ki-67 index above 10%, and mitotic activity index >5 mitoses per 50 large field of magnification were considered as abnormally high. ACCs were staged based on ENSAT TNM staging system (Table 1).
Table 1.
TNM staging recommended by European Network for the Study of Adrenal Tumors (ENSAT)
| Stage | ENSAT 2008 |
|---|---|
| I | T1, N0, M0 |
| II | T2, N0, M0 |
| III | T1-2, N1, M0 | T3-4, N0-N1, M0 |
| IV | T1-4, N0-N1, M1 |
T1, tumor size ≤5 cm; T2, tumor size >5 cm; T3, tumoral infiltration into surrounding tissue; T4, tumor invasion into adjacent organ or venous tumor trombus in vena cava or renal vein. N0, absence of positive lymph node; N1, positive lymph node(s). M0, absence of distant metastasis; M1, distant metastasis
In patients preoperative presence of humoral activation of the tumor was investigated. Excess amounts of cortisole release were examined based on the results of 1 mg dexamethasone suppression test (DST), and basal ACTH levels were analyzed by screening the data in patients’ files. Cushing syndrome was ruled out in patients whose cortisol levels were below 1.8 μg/dL when 1 mg DST suppression tests were performed. Patients whose serum cortisol levels were > 1.8 μg/dL after 1 mg DST which were maintained after confirmatory tests with 2 mg DST for 2 days, and those with baseline ACTH levels were below 5 pg/mL were evaluated as Cushing syndrome of adrenal origin. The patients whose data in their medical files indicated the presence of Cushing symptoms and signs, and their biochemical tests revealed hypercortisolemia were evaluated as having Cushing syndrome, while cases with subtle or lack of symptoms and signs were assessed as subclinical Cushing syndrome. In patients with preoperative dihydroepiandrosterone sulphate (DHEAS) levels were within normal reference ranges, hyperandrogenemia was discarded. Similarly, plasma aldosterone concentration/plasma renin activity <30 which is examined to evaluate release of excess amounts of mineralocorticoid preoperatively was considered to be normal. Pheochromocytoma was discarded in patients whose 24-hour urinary metanephrine, and normetanephrine levels were within reference ranges.
Postoperative data contained in the patients’ files about clinical, and biochemical test results, abdominal CT, MRI or fluorodeoxyglucose-positron emission tomograms (FDG-PET) were examined in detail to search for the development of recurrence, and metastases. In patients who developed recurrences or metastases, locations of metastatic foci, and time to development of recurrences or metastases were investigated. Besides applications of adjuvant mitotane, systemic chemotherapy, and radiotherapy, and responses to these treatments were assessed.
Statistical analysis
Descriptive statistical data were expressed as median (min–max) for numerical variables, and for categorical variables as frequencies, and percentages. Survival times were analyzed using Kaplan-Meier curves. Factors were indicated using log-rank tests. P<0.05 was accepted as statistically significant. For statistical analysis IBM SPSS (IBM Statistical Package for the Social Sciences Statistics; Armonk, NY, ABD) version 20 was used.
Results
Five (33.3%) female, and 10 (66.7%) male patients were included in our study. Median age of the patients at the time of diagnosis was 52.45 (29.52–72.13) years. Tumors were detected in the right (n=7; 46.7%), and left (n=8; 53.3%) adrenal glands. Demographic characteristics, results of clinical, and histopathological evaluations, treatments and follow-ups of the patients are demonstrated in Table 2.
Table 2.
Data related to demographic characteristics, treatment, histopathological evaluation, and follow-up of the patients
| Gender | Female | 5 (33.3%) |
| Male | 10 (66.7%) | |
| Age at diagnosis | (years) | 52.45 (29.52–72.13) |
| Stage | I | 3 (20%) |
| II | 1 (6.7%) | |
| III | 11 (73.3%) | |
| IV | 0 | |
| Localization | Right | 7 (46.7%) |
| Left | 8 (53.3%) | |
| Hormonal Activity | No | 11 (73.3%) |
| Yes | 4 (26.7%) | |
| • Cortisole | 1 (6.7%) | |
| • Cortisole + DHEAS | 3 (20%) | |
| Surgery | Open | 12 (80%) |
| Laparoscopic | 3 (20%) | |
| Adrenal/En bloc | Adrenalectomy | 6 (40%) |
| En bloc | 9 (60%) | |
| Resection | Rx | 2 (13.3%) |
| R0 | 7 (46.7%) | |
| R1 | 4 (26.7%) | |
| R2 | 2 (13.3%) | |
| Mitotic Activity | High | 4 (26.7%) |
| Low | 1 (6.7%) | |
| Necrosis | Yes | 9 (60%) |
| No | 6 (40%) | |
| Ki67 | High | 7 (77.8%) |
| Low | 2 (22.2%) | |
| Treatment | None | 7 (46.7%) |
| Mitotan | 3 (20%) | |
| Mitotane+RT | 2 (13.3%) | |
| Mitotane+Systemic CT | 2 (13.3%) | |
| RT+Systemic CT | 1 (6.7%) | |
| Ketoconazole | 2 (13.3%) | |
| Recurrence and Metastasis | Development of recurrence | 7 (46.7%) |
| Recurrent operation | 3 (20%) | |
| Time to recurrence | 8 (2–24) mos | |
| Development of metastasis | 6 (40%) | |
| Metastasectomy | 2 (13.3%) |
DHEAS: dehydroepiandrosteron sulphate; RT: radiotherapy; CT: chemotherapy
Postoperatively 7 (46.7%) patients did not receive adjuvant therapy, while 3 (20%) patients received only mitotane treatment, 2 (13.3%) patients were given systemic combined treatment with mitotane, and radiotherapy and mitotane treatment were administered in 2 (13.3%) cases. Adjuvant radiotherapy was applied on tumor bed of one (6.7%) patient, and also systemic chemotherapy was administered. None of the patients required palliative radiotherapy.
In the adjuvant therapy, mitotane was initiated at daily dose of 1000 mg, and with weekly increases of 500 mg, its dosage was adjusted so as to reach target blood level of 14–20 mg/L at follow-up visits. Blood levels of mitotane were measured at 2–3-month- intervals for the first 3 months. When serum mitotane levels reached to targeted range, the follow-up periods were prolonged. Follow-up was maintained all along the treatment period. Mitotane treatment was initiated simultaneously with hydrocortisone treatment, and drug dosages were regulated based on clinical findings observed. During the follow-up period, blood pressure, serum potassium, plasma renin activity, and aldosteron levels were monitored, and for patients who developed mineralocorticoid deficiency, fludrocortisone treatment was initiated.
Three of 15 patients received adjuvant systemic chemotherapy. As a chemotherapy, 2 patients were given etoposide, doxorubisin, and cisplatin EDP regimen. During follow-up period after application of EDP protocol, and concurrent mitotane treatment, paclitaxel chemotherapy which was initiated by the oncology department was maintained in one patient due to disease progression. Within the first three postoperative months 3 patients received radiotherapy directed to tumor bed.
The patients were followed up for an average of 21 (1–42) months after surgery. During follow-up period local recurrence was observed in 70% of the patients. Average time to the development of recurrence was 8 (2–24) months. Three patients were operated due to development of recurrences. During the follow-up period metastases developed in 6 (40%) patients. One patient underwent metastasectomy in the form of adrenalectomy, and the other partial hepatectomy. During follow-up period nine patients (60%) died.
Mean overall survival times of our female, and male study participants were 30.48±9.53, and 27.16±4.74 months, respectively. While corresponding mean disease-free survival times were 15±6.1, and 26.17±4.03 months, respectively. A statistically significant difference was not detected between gender of the patients, overall-, and disease-free survival times.
In our study, in Stage I, and II ACC patients average overall-, and mean disease-free survival times were 24.35, and 24.35±4.07 months, respectively, while in Stage III ACC patients mean overall survival time was calculated as 27.77±5.31 months. Mean disease-free survival time in Stage I, and II patients was 18.5±6.83 months, while in Stage III patients it was 24.51±3.99 months. In our study a significant effect of disease stage on overall, and disease-free survival was not detected.
Overall, and disease-free survival times in hormone inactive patients were 29.69±5.2, and 19.79±3.77 months, while in hormone-active patients the corresponding survival times were 24.95±9.13, and 30.3±4.46 months, respectively. As an outcome of our study any significant effect of hormonal activation on survival was not observed.
In our study Ki-67 expression was histopathologically evaluated in 9 patients. So the impact of tumor Ki-67 index on survival could not be evaluated because of scarce number of patients. Similarly, statistical evaluation of mitotic activity index could not be made. Presence of necrosis in tumor as detected during histopathological examination did not exert a significant effect on overall-, and disease-free survival rates.
Disease-free survival, and overall survival curves are shown in Figure 2. Mean. disease-free survival was calculated as 23.32±3.69 months (95% CI; 16.10;30.55) Factors effecting disease-free survival were analyzed. The effects of stage at the time of diagnosis, hormonal activity, gender, type of the operation, treatment, and presence of necrosis on survival were examined. A significant impact of these variables on disease-free survival was not detected (Table 3). Our present results were associated with the scarce number of patients included in our study. Overall mean survival was detected as 36.60±10.78 months (95% CI 15.47–57.74). Factors effective on overall survival were analyzed, and any effect of gender, hormonal activity, recurrence, and development of metastases on survival were not observed (Table 4). However adjuvant treatment prolonged survival significantly (p=0.006). Overall survival curves of the treated, and untreated patients are demonstrated in Figure 3. Excluding adjuvant therapy, inability to observe the effects of other variables on survival was associated with scarce number of patients.
Figure 2. a, b.
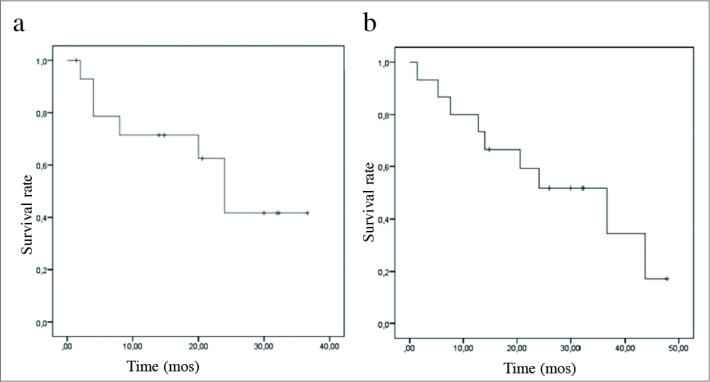
Kaplan-Meier curves demonstrating disease-free survival (a) and total survival (b) rates
Table 3.
Factors effecting disease-free survival
| Disease-free survival mean ± Standard error | 95%CI | p | |
|---|---|---|---|
| Gender | |||
| Female | 15±6.1 | 3.04;26.92 | 0.296 |
| Male | 26.17±4.03 | 18.27;34.06 | |
|
| |||
| Stage | |||
| Stage I+II | 18.5±6.83 | 5.11;31.89 | 0.710 |
| Stage III | 24.51±3.99 | 16.7;32.32 | |
|
| |||
| Hormonal activity | |||
| Yes | 30.3±4.46 | 21.6;39.03 | 0.436 |
| No | 19.79±3.77 | 12.4;27.17 | |
|
| |||
| Surgery | |||
| Adrenalectomy | 20.33±4.86 | 10.8;29.87 | 0.804 |
| En bloc | 24.07±4.77 | 14.73;33.41 | |
|
| |||
| Necrosis | |||
| Yes | 24.16±4.52 | 15.3;33.02 | 0.813 |
| No | 19.72±5.61 | 8.73;30.71 | |
|
| |||
| Treatment | |||
| Yes | 24.92±4.59 | 15.92;33.92 | 0.625 |
| No | 20±5.11 | 9.99;30.01 | |
Table 4.
Factors effecting overall survival
| Disease-free survival mean ± Standard error | 95%CI | p | |
|---|---|---|---|
| Gender | |||
| Female | 30.48±9.53 | 11.81;49.15 | 0.478 |
| Male | 27.16±4.74 | 17.87;36.45 | |
|
| |||
| Stage | |||
| Stages I+II | 24.35±4.07 | 16.38;32.32 | 0.967 |
| StageIII | 27.77±5.31 | 17.37;38.18 | |
|
| |||
| Hormonal activity | |||
| Yes | 24.95±9.13 | 7.06;42.84 | 0.924 |
| No | 29.69±5.2 | 19.49;39.88 | |
|
| |||
| Surgery | |||
| Adrenalectomy | 36.3±6.52 | 23.52;49.08 | 0.199 |
| En bloc | 23.67±5.85 | 12.21;35.12 | |
|
| |||
| Necrosis | |||
| Yes | 25.01±5.14 | 14.93;35.09 | 0.246 |
| No | 34.23±7.95 | 18.66;49.81 | |
|
| |||
| Treatment | |||
| Yes | 38.96±4.21 | 30.72;47.21 | 0.006 |
| No | 15±3.88 | 7.39;22.61 | |
|
| |||
| Development of recurrence | |||
| Yes | 26.74±7.46 | 12.11;41.37 | 0.988 |
| No | 26.05±4.77 | 16.71;35.4 | |
|
| |||
| Development of metastasis | |||
| Yes | 30.29±6.56 | 17.25;43.34 | 0.623 |
| No | 24.57±5.25 | 14.29;34.35 | |
Figure 3.
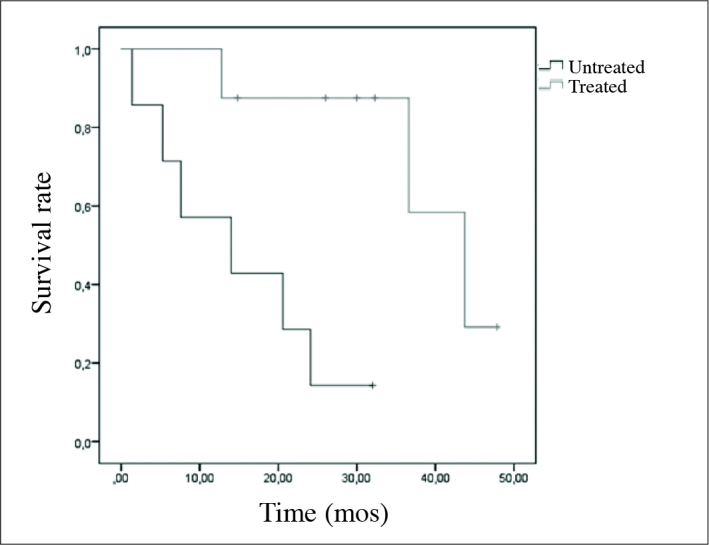
Total survival curve in treated, and untreated patients
Discussion
Adrenocortical syndrome is a rarely seen endocrine malignancy with an aggressive course. Five-year overall survival rate is less than 35 percent. In our study our mean overall survival time was 36.60±10.78 months. Since complete surgical resection is one of the most important factors which prolong survival, the success of the first surgical intervention is very important.[3] Since our study participants were limited in number, the impact of complete resection on disease-free, and overall survival could not be evaluated statistically. However, rates of development of recurrence in patients in whom complete surgical resection was achieved were not statistically significantly different from those with incompletely resected tumors. While development of metastasis was found to be statistically significantly lower in patients in whom complete resection was accomplished. One of the important prognostic parametres is stage of the disease at diagnosis. In our study disease-free survival, and overall survival rates were slightly lower in Stage I+II patients relative to Stage III patients without any statistically significant intergroup difference. Available results were presumably attributed to scarce number patients in our study population.
Open surgery is recommended for mass lesions in invasive ACC, while in localized stage I, and II tumors where tumor diameter is less than 10 cm without invasion into surrounding tissue, a consensus on application of laparoscopic or open surgery does not exist. Beneficial effects of laparoscopic adrenalectomy on decreasing morbidity, and hospital stay are already known. However, previous studies have indicated increase in locoregional recurrence following laparoscopic surgery.[14,15] Therefore for ACCs as a general approach open surgery is recommended in tumors less than 8 cm without any evidence of preoperative invasion, however for these tumors laparoscopic surgery should be performed by experienced hands.[2] We performed open adrenalectomy in 12 (80%) of our 15 patients, while 3 (20%) patients underwent laparoscopic adrenalectomy. As a result of our study any statistically significant difference could not be found between the laparoscopic and open surgery groups as for mean survival, local recurrence, and development of metastasis.
In the literature a slightly higher incidence rates of ACCs have been reported among women. In our center we reviewed patient series within a 10 year period and observed higher number of male patients with ACC. In our study gender of the patients was compared with recurrence, time to development of metastases, average survival, and disease-free survival times, and any statistically significant intergroup difference was not detected as for these prognostic parametres.
In nearly 60% of ACCs hypersecretion adrenal hormones is detected. In case of excess amounts of glucocorticoid release clinical symptoms of Cushing syndrome including central obesity, moon face, facial plethora, hirsutism, proximal myopathy, easy bruising, red-purple stria, hypertension, and diabetes can be observed. In aldostreone secreting ACCs hypertension, and marked hypokalemia are found. In the presence of excessive androgen release signs of virilization as hirsutism, deep voice in women, androgenic male-type alopecia, and oligomenorrhea may be detected. While estrogen-secreting tumors may lead to the development of gynecomastia, and testicular atrophy.[4] Most frequently, increases in hormone levels (hypercortisolemia, and hypercortisolemia plus hyperandrogenemia) have been reported.[4,18] In our study hypercortisolemia was detected in 4 (26.6%) of our 15 patients. In one patient Cushing syndrome was detected at the time of diagnosis. In one patient during preoperative examination primary tumor did not secrete hormones, while postoperative local recurrence led to development of manifest Cushing clinic. In the other 2 patients subclinical Cushing syndrome was detected. In other two patients subclinical Cushing syndrome was found. In 3 of our 15 patients androgen–secreting tumor was observed. These patients also had hypercortisolemia. In none of our patients solely androgen or aldosterone release was seen. As an outcome of our study, any statistically significant unfavourable contribution of excess release of hormone to survival has not been demonstrated.
Mitotane occupies an important place in adjuvant medical treatment. Nearly 60% of oral mitotane is absorbed. It is stored in adipose tissue, liver, brain, and adrenal glands. Therefore higher doses as 4–6 g are needed for the treatment. Treatment is started at a lower dose, blood mitotane levels should be maintained between its therapeutic range in blood (ie. 14–20 mg/L).[8,19] Because of its adrenolytic effects, all patients under mitotane therapy should receive glucocorticoids, and in case of need mineralocorticoid treatment at higher than standard doses to curtail development of adrenal failure should be given. Glucocorticoid dosages are regulated in consideration of clinical symptoms of the patients. Need for mineralocorticoids is determined by monitorization of aldosterone levels, and evaluating symptoms suggesting mineralocorticoid deficiency as postural hypotension.[2] Four out of 15 patients included in our study received adjuvant mitotane treatment. The patients received prednisolone at average daily doses of 15 mg concurrently with mitotane treatment, and during follow-up if required the patients were given fludrocortisone at daily doses of 0.1–0.2 mg.
When mitotane blood levels exceed 20 mg/dL, mitotane-related side effects mainly gastrointestinal, and neurological system symptoms are seen.[4] In our study one patient who received adjuvant mitotane treatment was hospitalized because of the presence of complaints as nausea, vomiting, and tendency to sleep. In patients with mitotane level of 27 mg/dL, the treatment was discontinued for a while, and when symptomatic improvement was achieved after palliative treatments, mitotane treatment was re-initiated at a lower dose.
In patients with ACC receiving adjuvant mitotane treatment, during the treatment periodic follow-ups with liver function tests, lipid levels, and whole blood cell count have been recommended.[2] During monitorization of our patients, any change in available tests was not observed.
In a multicenter study performed with a total of 117 patients with diagnosis of ACC, who did and did not receive mitotane treatment, the risks of recurrence, and disease-related mortality were lower in mitotane group relative to mitotane-naive patients, while median and overall survival rates were significantly higher.[20] Scarce number of patients in our study did not allow statistical evaluation of the group which received only mitotane as an adjuvant therapy.
Adrenocortical carcinomas most frequently metastasize into liver, lungs, bone, and abdominal lymph nodes.[21] However in our study most frequently metastases were observed in liver, and bone followed by lungs, contralateral adrenal gland, and orbita. Cytotoxic chemotherapy is recommended for patients whose disease progressed under mitotane therapy or in rapidly progressive ACCs or life-threatening metastatic disease.[2] In ACCs various chemotherapy protocols have been tried, nowadays EDP-Mitotane (EDP-M), and Streptozotocin combined with mitotane have been recommended thanks to its highest effectiveness. The efficacies of these two treatment protocols have been compared in the First International Randomized trial in advanced or Metastatic Adrenocortical Carcinoma Treatment (FIRM-ACT) study.[22] Response rate was higher (23.2 vs. 9.2%), and progression-free survival was longer (5.0 vs. 2.1 mos) in the group which received EDP-M. Based on available data nowadays EDP-M is recommended as the first-line chemotherapy.[2] In our study survival, and remission of the patients who received cytotoxic chemotherapy could not be evaluated due to scarce number of study participants.
Cases with ACC where excessive amounts of hormones were secreted (specifically, Cushing syndrome) have a poor prognosis, and their quality of life is dramatically impaired. Mitotane fails to control hypersecretion of ACC, and so adrenostatic drugs should not be used Ketoconazole (400–1200 mg/d) is the most frequently used drug, and it can be combined with mitotane.[3] In our study ketoconazole treatment was used for 2 patients with manifest Cushing syndrome to keep hypercortisolemia under control.
Though ACCs are known to be radiotherapy-resistant tumors, as another method of adjuvant therapy, radiotherapy is applied postoperatively on the tumor bed.[23] Some publications in the literature have indicated that radiotherapy increased or at least did not alter tumor recurrence rates, while radiotherapy of tumor bed is recommended postoperatively in cases with residual microscopic (R1), and macroscopic (R2) tumors which can not be reoperated, and in cases where presence of residual tumor tissue is not known (RX).[20,23–25]
In ACCs radiotherapy can be used with an intent of palliative treatment apart from adjuvant treatment. Radiotherapy has been found to be effective in the palliative treatment of pain, and neurological symptoms due to metastases. Radiotherapy is recommended in metastases into skeletal system, and brain.[26] Palliative radiotherapy was not required for the patients included in our study.
In our study various adjuvant treatments were applied for scarce number of patients. Because of limited number of patients the contribution of each adjuvant treatment to prognosis could not be statistically evaluated. However, statistical evaluation of the patients who did or did not receive adjuvant treatment revealed significant increase in overall survival of the patients under adjuvant treatment. Available data are in compliance with those of the literature. Nevertheless, significant effect of adjuvant treatment on disease-free survival was not detected. We have thought that results of our study might be related to scarce number of patients included in our study.
According to the guideline of European Society for Medical Oncology, in patients in whom complete postoperative resection was achieved, abdominal CT (or MRI), thoracic CT, and in patients with baseline hormone release hormonal evaluation is recommended. Dependent on the treatment applied for patients with locally advanced or metastatic disease follow-up intervals can be shortened. The role of PET imaging is not clear-cut yet.[2] Montorization of the patient may be performed using only CT or CT plus PET.[3]
Important limitations of our study are its retrospective design, and limited number of patients. Small sample size could not allow statistical evaluation of all parametres which might effect prognosis. Despite inadequate number of our patients, statistically significant positive effects of adjuvant treatment on overall survival have been demonstrated. Therefore patients with ACC should be evaluated for the need for postoperative adjuvant treatment, their follow-up, and treatment should be maintained by experienced centers.
In conclusion, ACC is an endocrine malignancy with an aggressive course. The most important prognostic factors are disease stage at diagnosis, and achievement of complete resection. However despite complete resection, since higher recurrence rates in ACCs are detected, adjuvant treatments as mitotane, radiotherapy or systemic chemotherapy may be needed for these patients. In our study important contribution of adjuvant treatment on prognosis has been found, other important prognostic factors did not make any significant difference. We think that in more comprehensive studies containing increased number of of patient series, additional contributory prognostic factors in ACC may be revealed more clearly.
Footnotes
Ethics Committee Approval: Ethics committee approval was received for this study from the ethics committee of Uludağ University School of Medicine.
Informed Consent: Written informed consent was obtained from patients who participated in this study.
Peer-review: Externally peer-reviewed.
Author Contributions: Concept – P.Ş.; Design – S.C.; Supervision – C.E.; Resources – Ö.Ö.G., C.E.; Materials – A.B.Ş.; Data Collection and/or Processing – A.B.Ş. H.P.; Analysis and/or Interpretation – E.E., S.C.; Literature Search – P.Ş., Ö.Ö.G., E.E.; Writing Manuscript – P.Ş., S.C.; Critical Review – S.C., E.E., C.E.; Other – A.B.Ş., P.Ş., H.P.
Conflict of Interest: No conflict of interest was declared by the authors.
Financial Disclosure: The authors declared that this study has received no financial support.
References
- 1.Michalkiewicz E, Sandrini R, Figueiredo B, Miranda EC, Caran E, Oliveira-Filho AG, et al. Clinical and outcome characteristics of children with adrenocortical tumors: a report from the International Pediatric Adrenocortical Tumor Registry. J Clin Oncol. 2004;22:838–45. doi: 10.1200/JCO.2004.08.085. https://doi.org/10.1200/JCO.2004.08.085. [DOI] [PubMed] [Google Scholar]
- 2.Berruti A, Baudin E, Gelderblom H, Haak HR, Porpiglia F, Fassnacht M, et al. Adrenal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(Suppl 7):vii131–8. doi: 10.1093/annonc/mds231. https://doi.org/10.1093/annonc/mds231. [DOI] [PubMed] [Google Scholar]
- 3.Glover AR, Ip JC, Zhao JT, Soon PS, Robinson BG, Sidhu SB. Current management options for recurrent adrenocortical carcinoma. Onco Targets Ther. 2013;6:635–43. doi: 10.2147/OTT.S34956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allolio B, Fassnacht M. Clinical review: Adrenocortical carcinoma: clinical update. J Clin Endocrinol Metab. 2006;91:2027–37. doi: 10.1210/jc.2005-2639. https://doi.org/10.1210/jc.2005-2639. [DOI] [PubMed] [Google Scholar]
- 5.Elsayes KM, Emad-Eldin S, Morani AC, Jensen CT. Practical approach to adrenal imaging. Radiol Clin North Am. 2017;55:279–301. doi: 10.1016/j.rcl.2016.10.005. https://doi.org/10.1016/j.rcl.2016.10.005. [DOI] [PubMed] [Google Scholar]
- 6.Bancos I, Tamhane S, Shah M, Delivanis DA, Alahdab F, Arlt W, et al. The diagnostic performance of adrenal biopsy: a systematic review and meta-analysis. Eur J Endocrinol. 2016;157:65–80. doi: 10.1530/EJE-16-0297. https://doi.org/10.1530/EJE-16-0297. [DOI] [PubMed] [Google Scholar]
- 7.Fassnacht M, Johanssen S, Quinkler M, Bucsky P, Willenberg HS, Beuschlein F, et al. Limited prognostic value of the 2004 International Union Against Cancer staging classification for adrenocortical carcinoma: proposal for a Revised TNM Classification. Cancer. 2009;115:243–50. doi: 10.1002/cncr.24030. https://doi.org/10.1002/cncr.24698. [DOI] [PubMed] [Google Scholar]
- 8.Sisman P, Ersoy C. The Treatment and Follow up of Adrenocortical Carcinoma. Med-Science. 2015;4:2098–108. https://doi.org/10.5455/medscience.2014.03.8213. [Google Scholar]
- 9.Dackiw AP, Lee JE, Gagel RF, Evans DB. Adrenal cortical carcinoma. World J Surg. 2001;25:914–26. doi: 10.1007/s00268-001-0030-7. https://doi.org/10.1007/s00268-001-0030-7. [DOI] [PubMed] [Google Scholar]
- 10.National Institutes of Health 2002 NIH state of the science statement on management of the clinically inapparent adrenal mass (‘incidentaloma’) NIH Consens State Sci Statements. 19:1–25. [PubMed] [Google Scholar]
- 11.Brix D, Allolio B, Fenske W, Agha A, Dralle H, Jurowich C, et al. Laparoscopic versus open adrenalectomy for adrenocortical carcinoma: surgical and oncologic outcome in 152 patients. Eur Urol. 2010;58:609–15. doi: 10.1016/j.eururo.2010.06.024. https://doi.org/10.1016/j.eururo.2010.06.024. [DOI] [PubMed] [Google Scholar]
- 12.Porpiglia F, Fiori C, Daffara F, Zaggia B, Bollito E, Volante M, et al. Retrospective evaluation of the outcome of open versus laparoscopic adrenalectomy for stage I and II adrenocortical cancer. Eur Urol. 2010;57:873–8. doi: 10.1016/j.eururo.2010.01.036. https://doi.org/10.1016/j.eururo.2010.01.036. [DOI] [PubMed] [Google Scholar]
- 13.Donatini G, Caiazzo R, Do Cao C, Aubert S, Zerrweck C, El-Kathib Z, et al. Long-term survival after adrenalectomy for stage I/II adrenocortical carcinoma (ACC): a retrospective comparative cohort study of laparoscopic versus open approach. Ann Surg Oncol. 2014;21:284–91. doi: 10.1245/s10434-013-3164-6. https://doi.org/10.1245/s10434-013-3164-6. [DOI] [PubMed] [Google Scholar]
- 14.Leboulleux S, Deandreis D, Al Ghuzlan A, Aupérin A, Goéré D, Dromain C, et al. Adrenocortical carcinoma: is the surgical approach a risk factor of peritoneal carcinomatosis? Eur J Endocrinol. 2010;162:1147–53. doi: 10.1530/EJE-09-1096. https://doi.org/10.1530/EJE-09-1096. [DOI] [PubMed] [Google Scholar]
- 15.Cooper AB, Habra MA, Grubbs EG, Bednarski BK, Ying AK, Perrier ND, et al. Does laparoscopic adrenalectomy jeopardize oncologic outcomes for patients with adrenocortical carcinoma? Surg Endosc. 2013;27:4026–32. doi: 10.1007/s00464-013-3034-0. https://doi.org/10.1007/s00464-013-3034-0. [DOI] [PubMed] [Google Scholar]
- 16.Touitou Y, Bogdan A, Luton JP. Changes in corticosteroid synthesis of the human adrenal cortex in vitro, induced by treatment with o,p’-DDD for Cushing’s syndrome: evidence for the sites of action of the drug. J Steroid Biochem. 1978;9:1217–24. doi: 10.1016/0022-4731(78)90015-8. https://doi.org/10.1016/0022-4731(78)90015-8. [DOI] [PubMed] [Google Scholar]
- 17.Fassnacht M, Allolio B. What is the best approach to apparently nonmetastatic adrenocortical carcinoma? Clin Endocrinol. 2010;73:561–5. doi: 10.1111/j.1365-2265.2010.03867.x. https://doi.org/10.1111/j.1365-2265.2010.03867.x. [DOI] [PubMed] [Google Scholar]
- 18.Koschker AC, Fassnacht M, Hahner S, Weismann D, Allolio B. Adrenocortical carcinoma -- improving patient care by establishing new structures. Exp Clin Endocrinol Diabetes. 2006;114:45–51. doi: 10.1055/s-2006-923808. https://doi.org/10.1055/s-2006-923808. [DOI] [PubMed] [Google Scholar]
- 19.Heilmann P, Wagner P, Nawroth PP, Ziegler R. Therapy of the adrenocortical carcinoma with Lysodren (o,p’-DDD). Therapeutic management by monitoring o,p’-DDD blood levels. Med Clin. 2001;96:371–7. doi: 10.1007/pl00002218. [DOI] [PubMed] [Google Scholar]
- 20.Terzolo M, Angeli A, Fassnacht M, Daffara F, Tauchmanova L, Conton PA, et al. Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med. 2007;356:2372–80. doi: 10.1056/NEJMoa063360. https://doi.org/10.1056/NEJMoa063360. [DOI] [PubMed] [Google Scholar]
- 21.Abiven G, Coste J, Groussin L, Anract P, Tissier F, Legmann P, et al. Clinical and biological features in the prognosis of adrenocortical cancer: poor outcome of cortisol-secreting tumors in a series of 202 consecutive patients. J Clin Endocrinol Metab. 2006;91:2650–5. doi: 10.1210/jc.2005-2730. https://doi.org/10.1210/jc.2005-2730. [DOI] [PubMed] [Google Scholar]
- 22.Fassnacht M, Terzolo M, Allolio B, Baudin E, Haak H, Berruti A, et al. Combination chemotherapy in advanced adrenocortical carcinoma. N Engl J Med. 2012;366:2189–97. doi: 10.1056/NEJMoa1200966. https://doi.org/10.1056/NEJMoa1200966. [DOI] [PubMed] [Google Scholar]
- 23.Polat B, Fassnacht M, Pfreundner L, Guckenberger M, Bratengeier K, Johanssen S, et al. Radiotherapy in adrenocortical carcinoma. Cancer. 2009;115:2816–23. doi: 10.1002/cncr.24331. https://doi.org/10.1002/cncr.24331. [DOI] [PubMed] [Google Scholar]
- 24.Fassnacht M, Hahner S, Polat B, Koschker AC, Kenn W, Flentje M, et al. Efficacy of adjuvant radiotherapy of the tumor bed on local recurrence of adrenocortical carcinoma. J Clin Endocrinol Metab. 2006;91:4501–4. doi: 10.1210/jc.2006-1007. https://doi.org/10.1210/jc.2006-1007. [DOI] [PubMed] [Google Scholar]
- 25.Pommier RF, Brennan MF. An eleven-year experience with adrenocortical carcinoma. Surgery. 1992;112:963–70. [PubMed] [Google Scholar]
- 26.Percarpio B, Knowlton AH. Radiation therapy of adrenal cortical carcinoma. Acta Radiol Ther Phys Biol. 1976;15:288–92. doi: 10.3109/02841867609131965. https://doi.org/10.3109/02841867609131965. [DOI] [PubMed] [Google Scholar]