

Abstract
“pHlash” is a novel bioluminescence-based pH sensor for measuring intracellular pH, which is developed based on Bioluminescence Resonance Energy Transfer (BRET). pHlash is a fusion protein between a mutant of Renilla luciferase (RLuc) and a Venus fluorophore. The spectral emission of purified pHlash protein exhibits pH dependence in vitro. When expressed in either yeast or mammalian cells, pHlash reports basal pH and cytosolic acidification. In this chapter, we describe an in vitro characterization of pHlash, and also in vivo assays including in yeast cells and in HeLa cells using pHlash as a cytoplasmic pH indicator.
Keywords: pH, BRET, Luminescence, resonance transfer, ratiometric
1. Introduction
Intracellular pH (pHi) is a very important modulator for cells to maintain proper function. Most proteins, especially enzymes, are affected by even subtle pH changes. To maintain stable intracellular pH values, all cells have mechanisms to pump protons in or out across cellular membranes. Moreover, the distribution of protons is not uniform within a cell and depends on the nature of subcellular compartments [1]. Current methods for measuring intracellular pH include: (I) a classical microelectrode technique that relies on a specific apparatus that is not present in most labs; (II) fluorescent dyes, such as BCECF(2′7′-bis-2-carboxyethyl-5- (and -6)-carboxyfluorescein) and SNARF (seminaphthorhodafluor) [2], which are popular because of their convenience and accessibility for many labs; (III) genetically encodable fluorescent pH-sensitive GFPs (e.g., “pHluorin” or “Pt-GFP”) [3,4,5], which overcome some of the limitations of fluorescent dyes (such as being able to measure the pH within subcellular compartments).
We are developing luminescence-based pH probes. Since luciferases are enzymes, many luciferases show the significant pH dependency of activity that is common to most enzymes. Some luciferases like firefly luciferase show both changes in activity and shifts in spectra with pH change [6], while other luciferases show activity changes but have little spectral shift, e.g. RLuc [7], Gaussia luciferase (GLuc) [8], PpyRE8 [9] and NanoLuc [10]. The activity of most of these luciferases peaks near neutral pH (e.g., pH 7.0-7.5).
By fusing a bright mutant of Renilla luciferase (“RLuc8”) [11] via an Ala-Glu-Leu linker to the circularly permuted Venus fluorophore (“cpVenus”) [12], we developed a fusion protein, called “pHlash,” that shows a dramatic pH dependent change in vitro in its spectrum of emission–as the pH increases, the spectral ratio shifts increasingly to the yellow (see Fig. 1). pHlash utilizes Bioluminescence Resonance Energy Transfer (BRET) [13], which makes its response ratiometric like that of pHluorins [3,4,5]; however, pHlash avoids fluorescence excitation since it is luminescent and BRET-based. In the absence of fluorescence excitation, problems associated with this irradiation are avoided, such as autofluorescence, photobleaching, and photo-response/toxicity. The spectral response of pHlash is specific for pH–neither CaCl2, MgCl2, NaCl, nor KCl had a significant effect on the spectra or pH dependency of pHlash [14]. In this chapter, we describe the in vitro pH calibration assay of purified pHlash protein; as well as in vivo assays to monitor the intracellular pH changes in yeast cells and HeLa cells using pHlash as the pH indicator.
Fig. 1. pH response of purified pHlash protein in vitro.
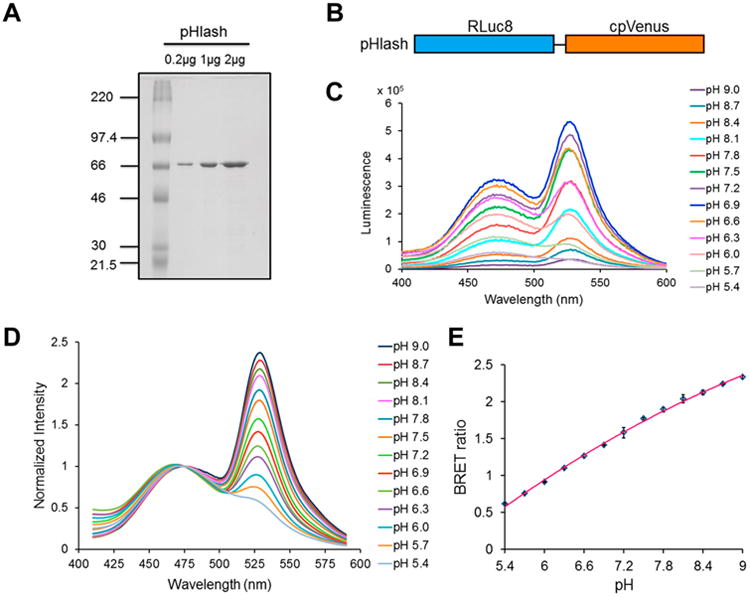
(A) SDS-PAGE gel of purified His-tagged pHlash protein stained with Coomassie Blue dye. Leftmost lane is molecular weight standards with KDa indicated, while the other lanes are the purified pHlash protein loaded at 0.2, 1, and 2 μg per lane. (B) Construct of the pHlash fusion protein. Rluc8 was linked to cpVenus by the sequence Ala-Glu-Leu. (C) Raw data (not normalized) of luminescence emission spectra of purified pHlash protein with 10 μM native coelenterazine at different pH values (pH 5.4-9.0) in 50 mM BIS-Tris-propane, 100 mM KCl, and 100 mM NaCl. (D) Normalized luminescence emission spectra of pHlash measured as in panel C. Luminescence intensity was normalized to the peak at 475 nm. (E) The BRET ratio (luminescence at 525nm:475 nm) as a function of pH is shown for pHlash in vitro. Error bars are +/- S.D., but in most cases the error bars are so small that they are obscured by the symbols (n = 3).
2. Material
2.1 Constructs information
The pHlash construct encodes a fusion protein of RLuc8 [11] and cpVenus (cp173Venus) [12] linked by the sequence GCCGAGCTC encoding the amino acids Ala-Glu-Leu.
Plasmid used for protein expression in E. coli : pRSETb-pHlash (Addgene, Plasmid #61550), built from pRSETb (Invitrogen).
Plasmid used for expression in yeast cells (Saccharomyces cerevisiae strain CEN.PK113-7D): pRS305-hph-PACT1pHlash (see Note 1)
Plasmid used for expression in HeLa cells: pcDNA3.1+-pHlash (Addgene, Plasmid#61552), built from pcDNA3.1+ (Invitrogen).
2.2 Protein expression and purification
To express His-tagged fusion proteins (N-terminal fusions of His6) of pHlash, E. coli strain BL21 (DE3) cells were used to transform those constructs.
50-mL flask and 2-L flask
LB medium with ampicillin (60 μg/mL)
TALON metal affinity resin (Clontech)
Equilibration/Wash buffer: 50 mM sodium phosphate, 300 mM NaCl, pH 7.0
Elution buffer: 50 mM sodium phosphate, 300 mM NaCl, 150 mM imidazole, pH 7.0
10% SDS-PAGE gel
Dialysis cassette (20K MWCO) (Thermo Scientific)
Dialysis buffer: 30 mM MOPS, 5% Glycerol, pH 7.2
BSA standard solution and Protein Assay Dye Reagent Concentrate (Bio-Rad)
2.3 In vitro characterization of pHlash
QuantaMaster QM-7/SE spectrofluorometer (Photon Technology International,Birmingham NJ)
Calibration buffer: 50 mM BIS-Tris-propane, 10 mM KCl, and 100 mM NaCl (see Note 2). pH was adjusted with 1N HCl to 13 different pH values ranging from 5.4 to 9.0 (see Note 3).
Substrates for luciferase: native coelenterazine (NanoLight, Pinetop, AZ) (see Note 4).
1.5-mL disposable plastic cuvettes
2.4 In vivo pH calibration of yeast strain expressing pHlash
Yeast strain: Saccharomyces cerevisiae strain CEN.PK113-7D
Yeast complete synthetic medium (CSM) (100 mL): 650 mg Nitrogen base (without amino acids), 11 mg complete amino acids mix, 2 g glucose.
Yeast permeabilization buffer: 50 mM MES, 50 mM HEPES, 50 mM KCl, 50 mM NaCl, 0.2 M ammonium acetate, 10 mM NaN3, 10 mM 2-deoxyglucose, 75 μM monensin, and 10 μM nigericin, titrated to 8 different pH values adjusted with 1M NaOH from 5.5 to 9.0 [15]. (see Note 5)
BCECF-AM (Molecular Probes, Eugene OR)
YPD medium: 1% yeast extract, 2% peptone, and 2% D-glucose
QuantaMaster QM-7/SE spectrofluorometer (Photon Technology International,Birmingham NJ)
1.5-mL disposable plastic cuvettes
4.5-mL glass cuvette
Magnetic micro stir bar (∼7 mm length)
2.5 In vivo experiments with HeLa cells expressing pHlash
HeLa cells (obtained from ATCC, ATCC® Number: CCL-2™)
Culture medium: Dulbecco's Modified Eagle's Medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Invitrogen)
FuGene6 (Invitrogen, Carlsbad, CA)
Recording media: DMEM medium without phenol red (Invitrogen) supplemented with 10% FBS
ViviRen™ (a serum-insensitive version of coelenterazine-h; Promega, Madison WI) (see Note 6)
pH calibration buffer: 100 mM KCl, 100 mM NaCl, 1.36 mM CaCl2, 4.5 g/L glucose, 20 μM nigericin, and 50 mM BIS-Tris-propane (pH 5.5-9.0).
QuantaMaster QM-7/SE spectrofluorometer (Photon Technology International,Birmingham NJ)
4.5-mL glass cuvette
Magnetic micro stir bar (∼7 mm length)
MES buffer: 50mM MES, 130 mM KCl, 1mM MgCl2, 25 mM NaCl, pH 6.1
2.6 BRET imaging
Dual-View™ micro-imager (Optical Insights, Tucson AZ, USA)
Modified electron bombardment-charge coupled device (EB-CCD) camera (Hamamatsu Photonic Systems, Bridgewater NJ, USA) [16].
IX-71 inverted microscope (Olympus America Inc., Melville NY, USA) with an Apo N 60× objective, NA 1.49 (oil immersion, Olympus)
Temperature controlled (22-37°C) light-tight box
35-mm Glass Bottom Cell Culture Dish (MatTek Corporation)
HeLa cells transfected with pcDNA3.1+/pHlash
3. Methods
3.1 Protein expression and purification
In a 50-mL flask, inoculate E. coli strain BL21 (DE3) cells bearing pRSETb/pHlash in 10 mL LB medium with ampicillin (60 μg/mL). Grow the starter culture overnight at 37°C with agitation.
In a 2-L flask, dilute the overnight culture into 500 mL fresh LB medium with ampicillin (60 μg/mL).
Grow the culture at 37°C with agitation until an OD600 of 0.6 is reached. Add 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) to induce the expression at 25°C with gentle agitation.
After incubation with IPTG for 5 h at 25°C, harvest the cells by centrifugation at 6000 g for 15 min. Suspend the pellet with 25 mL Equilibration buffer.
Disrupt the cells by sonication (1 min for 8 times, with 30 sec interval, keep the sample on ice).
To remove the cell debris, centrifuge the sample at 22000 g for 40 min. Transfer the supernatant to a new 50-mL tube, and then add 5 mM imidazole to the supernatant.
Incubate the supernatant with balanced TALON metal affinity resin with gentle shaking at 4°C for 1 h.
Load the sample into one empty column and let the resin settle down. Collect the flow-through and then wash the resin with at least 20 bed volumes of Equilibration/Wash buffer containing 10 mM imidazole.
Elute the protein according to the manufacturer's protocol. (see Note 7)
After the gradient elution, 10 μL samples from each eluted fraction should be assayed by SDS-PAGE to confirm which fraction(s) contains pHlash. Fractions with high yield and purity can be combined. (see Note 8)
Dialyse eluted pHlash protein in 1 L 30 mM MOPS buffer (pH 7.2) (containing 5% glycerol) at 4°C to remove imidazole (see Note 8). After 12 h dialysis, transfer the dialysis cassette (or dialysis bag) to fresh MOPS buffer and incubate for another 12 h.
Quantify the purified pHlash protein using the Bio-Rad protein assay, then flash-freeze it with liquid nitrogen. Store the samples at -80°C for later use.
3.2 In vitro pH calibration of pHlash
For measurement of BRET emission, use a QuantaMaster QM-7/SE spectrofluorometer with the slit width set to 16 nm. The excitation beam of the QuantaMaster is not needed and can be turned off.
Set the following parameters for pH calibration. Select “Emission Scan” mode (see Note 9) and set the wavelength from 400 nm to 600 nm in 1 nm steps with 0.1 sec integration for each step.
For each individual measurement, prepare a 1.5 mL disposable cuvette. Dilute purified pHlash protein (1 μg in 2 μL of the MOPS storage buffer) 250-fold to achieve a final concentration of 1 μg purified protein per 500 μL calibration buffer. Mix the sample well with a pipette.
Add fresh coelenterazine to the sample to a final concentration of 10 μM before each measurement. (see Note 10) Mix the sample quickly, then insert the cuvette and start the measurement immediately.
Repeat the measurements with calibration buffers at different pH.
To analyze data from Emission Scan mode, smooth the curve using a ± 10 nm running average and then normalize the luminescence intensity from each measurement curve to each curve's peak at 475 nm (see Fig. 1D). The BRET ratio is obtained by dividing the luminescence intensity at 525 nm by the luminescence intensity at 475 nm.
Plot the BRET ratio from each curve as a function of the pH to generate a standard curve (see Fig. 1E) that can be used to estimate pH of experimental samples when their BRET ratios are determined.
3.3 In vivo pH calibration of yeast strain expressing pHlash
3.3.1 Genetic construct for yeast expression
Transform the desired strain of yeast with the LEU2 integrating plasmid pRS305-hph-PACT1pHlash and select for resistance on YPD medium that includes 200 μg/mL hygromycin (see Note 1 for how this plasmid was constructed). Once transformed and selected, the resulting yeast cells have a stable integration of the construct, which allows pHlash experiments to be conducted in a medium or buffer of choice without the need for antibiotics or auxotrophic requirements.
3.3.2 In vivo pH calibration
Prepare a 2 mL starter culture of yeast strain expressing pHlash and let it grow overnight at 28–30°C with agitation. Dilute the starter culture 10-fold with fresh Yeast complete synthetic medium and incubate for 5 h.
Harvest the yeast cells by centrifugation at 1000 g for 5 min, wash with distilled water.
In vivo pH calibration should be performed in yeast cells expressing pHlash that have been slightly permeabilized to protons by incubating in yeast permeabilization buffer [15]. For each pH value to be tested, use yeast cells harvested from 1 mL culture (see Note 11), and resuspend the cells with 1 mL yeast permeabilization buffer. Incubate at room temperature with gentle rotation for 30 min.
Use the QuantaMaster QM-7/SE spectrofluorometer to measure the BRET ratio of the in vivo calibration samples in “Emission Scan” mode as previously described in step 2 in section 3.2 and step 6 in section 3.2. Or alternatively the “Two-Wavelength Switching” mode (described hereafter) can be used to ensure that the in vivo BRET ratios are stable (see Note 9). To use Two-Wavelength Switching mode, shut off the excitation beam and set the slit width to 16 nm. Select Two-Wavelength Switching mode, and set the wavelength to record at 475 nm and 525 nm, with 0.5 sec integration. The QuantaMaster will selectively monitor these two wavelengths continuously or for a set amount of time.
Transfer each sample into a 4.5-mL glass cuvette. A stir bar placed on the bottom of the cuvette allows gentle stirring to maintain the cells in suspension. Add fresh coelenterazine to the sample to a final concentration of 10 μM before each measurement. Insert the cuvette and start the measurement in Two-Wavelength Switching mode. Continue to record until a stable BRET ratio is achieved. Fig. 2B shows examples of stable BRET ratios recorded in Two-Wavelength Switching mode.
Plot the BRET ratio of each pH unit tested to generate a standard curve like that shown in (see Fig. 2A). This standard curve can be used to estimate the intracellular pH of experimental samples of yeast when their BRET ratios are determined.
If desired, the standard curve and in vivo experiments can be corroborated by BCECF-AM (Molecular Probes, Eugene OR) in permeabilized nontransformed yeast cells as shown in (see Fig. 2A and C). For in vivo calibration using BCECF-AM, incubate yeast cultures in YPD medium (at pH 7.5) with 50 μM BCECF-AM at 30°C for 30 min, wash and suspend in yeast permeabilization buffer. Incubate at room temperature with gentle rotation for 30 min. Measure fluorescence with dual excitation at 440 nm and 490 nm and record emission at 535 nm with the QuantaMaster QM-7/SE spectrophotometer.
Fig. 2. Response of yeast pH to weak acid treatment.
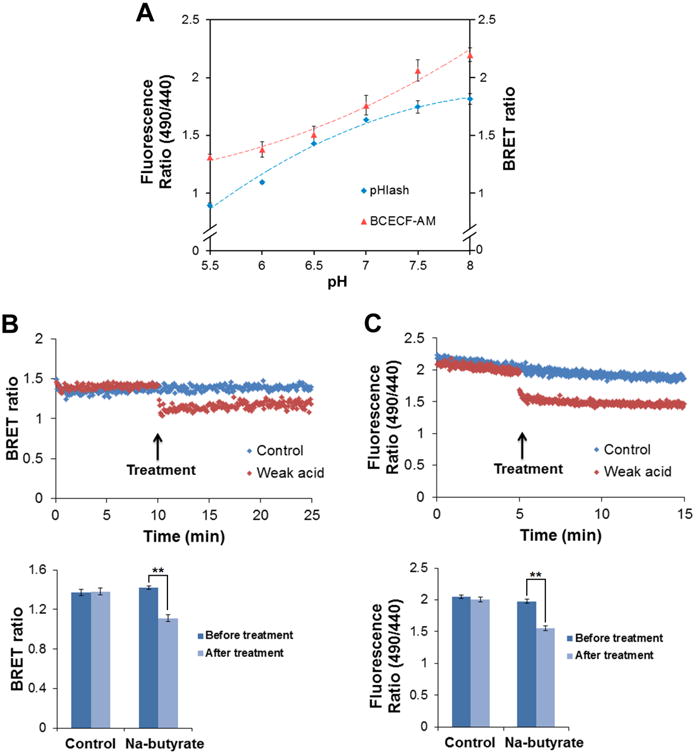
(A) pH response of BCECF-AM loaded yeast cells (red) compared with pHlash transformed yeast cells (blue). (B) Response of pHlash-expressing yeast cells to weak acid. Yeast cells were suspended in 20 mM MES buffer (pH 5.0) with 10 μM native coelenterazine. After 10 min baseline recording, 20 μM final concentration of Na-butyrate (pH 5.0) was added (upper portion of the panel). An equal volume of pH 5.0 buffer was added as control. The change in BRET ratio of pHlash reports the intracellular acidification after treatment with Na-butyrate, and is quantified in the lower portion. (C) Response of BCECF-AM loaded yeast cells to weak acid (20 μM sodium butyrate, upper portion). Yeast cells were suspended in 20 mM MES buffer (pH 5.0). After 10 min baseline recording, 20 μM final concentration of Na-butyrate (pH 5.0) was added (total added volume was 20 μL, which is the same volume as that for the pH 5.0 buffer that was used as a control). In panels A, B, and C, error bars are +/- S.D., but in some cases the error bars are so small that they are obscured by the symbols (n = 3 for panel A, n = 5 for panels D and E). ** p < 0.01.
3.3.3
If desired to confirm that yeast cytosolic pH as indicated by pHlash is responding in real time to pH perturbation, pHlash-expressing yeast cells can be suspended in 20 mM MES buffer (pH 5.0) with 10 μM native coelenterazine. Using the Two -Wavelength Switching mode to monitor dual emission at 475 nm and 525 nm with 0.5 sec integration, add 20 μM final concentration of Na-butyrate (pH 5.0) (see Fig. 2B, red points). For a control recording, add a volume of 20 mM MES buffer (pH 5.0) equal to that of the Na-butyrate solution used above (but without Na-butyrate; see Fig. 2B, blue points). The change in BRET ratio of pHlash reports the intracellular acidification after treatment with Na-butyrate (see Fig. 2B, lower portion).
3.4 In vivo experiments with HeLa cells expressing pHlash
3.4.1 HeLa Cell Culture and Transfection
Grow HeLa Cells in 35-mm cell culture dishes in DMEM medium (Invitrogen) with 10% FBS at 37°C with 5% CO2 until they reach 80% confluence.
Dilute 3 μL FuGene6 with serum-free DMEM medium. Mix gently and keep at room temperature for 5 min.
Add 1 μg of plasmid DNA to the mixture. Mix gently and then spin down shortly. Incubate at room temperature for 30 min.
Add the mixture to cells in 35-mm dishes. Swirl gently to mix. Return the dish to the incubator.
24-48 h after transfection, cells can be used for BRET measurement or imaging (see Note 12).
3.4.2 In vivo pH calibration
24-48 h after transfection, wash the cells with Dulbecco's Phosphate-Buffered Saline (DPBS) and then trypsinize the cells for 5-10 min. Transfer the cells to 1.5 mL tube and centrifuge at 900 g for 5 min.
Resuspend the cells with pH calibration buffers such that extracellular pH values cover a range of 5.5 to 9.0.
Incubate the cells in the calibration buffer for 10 min. Add ViviRen™ (substrate) to the assay buffer to a final concentration of 2.5 μM.
Use a QuantaMaster QM-7/SE spectrophotometer to measure dual emission at 475 nm and 525 nm for 10 min. A stir bar placed on the bottom of the cuvette gently stirs the cells to maintain them in suspension.
Generate a standard curve by plotting the BRET ratio as a function of pH to cover the range of buffers tested. This standard curve can be used to estimate intracellular pH of other pHlash-expressing HeLa cells from experimental conditions.
3.4.3 Imaging of BRET in HeLa cells
Prepare apparatus for BRET imaging (see Note 13 for detailed information). Set the temperature controller of the light-tight box to 37°C.
Prepare pHlash-expressing HeLa cells for imaging (see Note 12).
In brightfield, focus on a group of cells using the inverted microscope using the Dual-View™ in“Bypass Mode.”
Switch to excitation mode and look for cells with bright fluorescence (of the cpVenus moeity) (see Note 14). Move the target cell to the center view, focus quickly and turn off the excitation light.
Drop in ViviRen™-containing medium directly to the dish without touching the dish. Close the box and switch the Dual-View™ out of “Bypass Mode” by pushing the filter holder inside the Dual-View™. Close the box for imaging.
For single HeLa cell imaging, collect and integrate 10 sequential 2 sec exposures by choosing the median value for each pixel over the sequence of 10 exposures. Then subtract background with ImageJ by using a single pixel from the non-sample region of the image as a background value.
Calculation of BRET ratio: A pixel-by-pixel BRET ratio can be calculated with ImageJ, and the numerical ratios visualized with a pseudocolor look-up-table (LUT) (see Fig. 3A).
Fig. 3. Response of pHlash to acidification of the cytoplasm in HeLa cells.
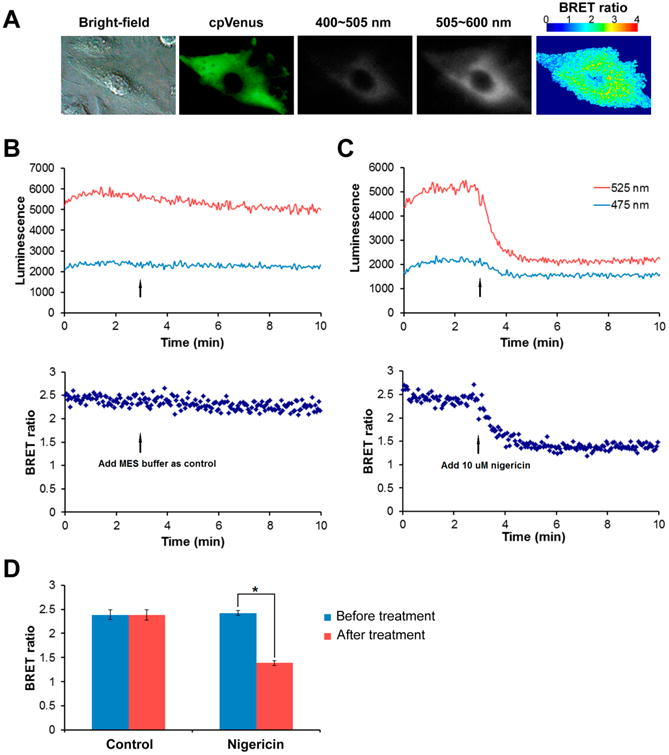
(A) BRET imaging of single pHlash-expressing HeLa cell. Panels from left to right are bright-field, fluorescence from cpVenus, 400-505 nm luminescence, 505-600 nm luminescence, and BRET ratio image. (B) & (C) Time-course recording of total luminescence from pHlash-transfected HeLa cells with 2.5 μM ViviRen™ as substrate in pH 6.1 MES buffer (50mM MES, 130mM KCl, 1mM MgCl2, 25mM NaCl). Luminescent signals were recorded at 475 nm and 525 nm using the QM-7/SE spectrofluorometer. (B) Control recording. After 3 min baseline recording, 100 μL MES buffer was added as control. (C) Response of pHlash to acidification. After 3 min baseline recording, 10 μM final concentration of nigericin (in 100 μL MES buffer) was added. (D) Histogram shows change in BRET ratio change of pHlash-expressing HeLa cells treated with nigericin compared with control. Error bars are +/- S.D.
3.4.4 Response of pHlash to acidification of the cytoplasm in HeLa cells
In a 4.5 mL glass cuvette, initiate a time-course recording from pHlash transfected HeLa cells with 2.5 μM ViviRen™ in pH 6.1 MES buffer. A stir bar placed on the bottom of the cuvette provides gentle stirring to maintain the cells in suspension.
Record the luminescence at 475 nm and 525 nm using the QM-7/SE spectrofluorometer with the excitation beam turned off. After recording for 3 min, add nigericin to a final concentration of 10 μM in 5 μl ethanol (see Fig. 3C).
For control, prepare another batch of cells and record as before. After recording for 3 minutes, add the same volume of pH 6.1 MES buffer (including 5 μL ethanol) as that for the nigericin solution described in step 3.4.4.2 (see Fig. 3B).
4. Notes
A yeast integrating vector that includes pHlash driven by a constitutively expressed promoter (pRS305-hph-PACT1pHlash) was constructed from the pRS305-hph-PGAL1CBG99 backbone [17] as follows. The yeast ACT1 promoter was amplified by PCR from S.cerevisiae genomic DNA (strain S288C) using the 5′ primert tcactCCCGGGTAAGTAAATAAGACACACGCGAG that contained an XmaI overhang, and 3′ primer atgattAGATCTTGTTAATTCAGTAAATTTTCGATC that contained a BglII overhang. This PCR product was used to replace PGAL1 of pRS305-hph-PGAL1CBG99. A PCR product containing the pHlash CDS was then added in place of the CBG99 CDS of the backbone. This pHlash PCR product was generated from the template plasmid pcDNA3.1+-pHlash (Addgene) using the 5′ primer atgattAGATCTATGGCTTCCAAGGTGTACGAC that contained a BglII overhang, and the 3′ primer ttcactTCTAGATTACTCGATGTTGTGGCGG that contained an XbaI overhang. The result was pRS305-hph-PACT1pHlash, a plasmid that integrates into the native yeast LEU2 gene when the plasmid is linearized with AflII and transformed by the standard lithium acetate method. Yeast transformants can be selected on YPD containing 200 μg/mL hygromycin.
BIS-Tris-propane has two pKa values at 25°C, 6.8 and 9.0, which give it a wide buffering range from approximately pH 6 to 9.5.
For in vitro pH calibration, we chose the pH range from 5.4 to 9.0 because the activity of Rluc8 luciferase peaks at neutral pH (∼pH 7.0), and decreases at both low and high pH. Beyond the pH 5.4 to 9.0 range, luminescence is very dim.
Native Coelenterazine is very sensitive to light and not stable in solution. Stocks are stored at -80°C. We prepare fresh working solutions before each experiment, and wrap the tubes with aluminum foil to protect coelenterazine from light. Keep the working solution on ice. We usually use 10 μM coelenterazine for both in vitro and in vivo experiments.
Monensin and nigericin are dissolved in ethanol. The stock solutions are stored at -20°C and added to the permeabilization buffer before use.
ViviRen™ is very sensitive to light. We make a 10 mM stock solution in DMSO. Aliquots are stored at -80°C. We prepare a fresh working solution before each experiment and wrap the tube with aluminum foil to protect the solution from light. During the experiment, the substrate is mantained on ice. For single cell BRET imaging, we use 10 μM ViviRen™ as substrate; and when recording with the QuantaMaster QM-7/SE spectrofluorometer, we reduced the concentration to 2.5 μM.
For 500 mL E. coli culture, we used 2 mL TALON metal affinity resin (1 mL bed volume). For protein elution, we used an imidazole gradient from 20 mM to 150 mM.
The elution buffer we used contained 150 mM imidazole. To remove imidazole, the buffer was exchanged to 30 mM MOPS buffer by dialysis at 4°C.
Calibration curves were derived from measurements of the BRET by two different methods: (I) Emission Scan Mode in the QM-7/SE spectrofluorometer, where the spectrum was continuously scanned from 400-600 nm; (II) Two-Wavelength Switching Mode in the QM-7/SE spectrofluorometer, where the monitoring of luminescence emission was alternately switched between 475 nm and 525 nm for measurement of time courses. In Two-Wavelength Switching Mode in the QM-7/SE spectrofluorometer, the switching requires additional time, so the average time for one cycle to measure luminescence at both 475 nm and 525 nm is about 1.5 sec. The calibration curve obtained with the Emission Scan Mode is more linear than that obtained with the Two-Wavelength Switching Mode. However, the Two-Wavelength Switching Mode was needed for the time-course measurements.
After adding coelenterazine to the sample, mix quickly by swirling the cuvette for 3 sec and then start the measurement immediately. Perform each measurement in the same time so that the results obtained can be directly compared.
We perform yeast in vivo pH calibration with 8 different pH values from pH 5.5 to 9.0. For each pH value to be tested, we harvest yeast cells from 1 mL yeast culture, wash with distilled water and resusped with1 mL yeast permeabilization buffer.
24-48 h after transfection, we trypsinize and transfer the cells to 35-mm glass bottom dishes. Culture the cells in DMEM medium without phenol red supplemented with 10% FBS until the cells adhere to the bottom of the dishes.
BRET imaging was accomplished using a Dual-View™ micro-imager and a modified EB-CCD camera as described previously [16, 18]. The Dual-View™ consists of a dichroic mirror, which was here selected to split the incoming image at 505 nm (dichroic Q505LPxr) and interference filters to further refine wavelengths below 505 nm (HQ505SP) and above 505 nm (HQ505LP). The use of the Dual-View™ therefore allows the simultaneous acquisition of luminescence images at two wavelengths. A “BRET ratio” image of emission in the two ranges can be calculated thereafter without the complication that the total luminescence intensity may change over the time course of the exposure. Our EB-CCD camera had a GaAsP photocathode with low ion feedback and cooling to -25°C (Hamamatsu Photonic Systems, Bridgewater NJ, USA). This EB-CCD camera model C7190-13W has a resolution of 512 × 512 pixels with a pixel size of 24 × 24 μm. The acquisition software was Photonics-WASABI (Hamamatsu). The Dual-View™ and EB-CCD were attached to the bottom port of an IX-71 inverted microscope (Olympus America Inc., Melville NY, USA). This setup allows the measurement of fluorescence using an epifluorescence attachment (EX 500/20 nm, EM 520 LP). The entire IX-71 microscope was enclosed in a temperature controlled (22-37°C) light-tight box. The IX-71 microscope was used with an Apo N 60× objective, NA 1.49 (oil immersion, Olympus).
Here we use a Light-Emitting Diode flashlight as an excitation source for fluorescence microscopy [19] so that the light source can be rapidly switched on and off without bulb afterglow. This allows one to immediately begin acquisition of bioluminescent images following fluorescent identification of target samples, and it offers dimer excitation with safer wavelengths for protecting the samples.
References
- 1.Roos A, Boron WF. Intracellular pH. Physiol Rev. 1981;61:296–434. doi: 10.1152/physrev.1981.61.2.296. [DOI] [PubMed] [Google Scholar]
- 2.Fricker MD, Plieth C, Knight H, Blancaflor E, Knight MR, White NS, Gilroy S. Fluorescence and luminescence techniques to probe ion activities in living plant cells. In: Mayson WT, editor. Fluorescent and Luminescent Probes for Biological Activity. San Diego: Academic Press; 1999. pp. 569–596. [Google Scholar]
- 3.Miesenbock G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- 4.Kneen M, Farinas J, Li Y, Verkman AS. Green fluorescent protein as a noninvasive intracellular pH indicator. Biophys J. 1998;74:1591–1599. doi: 10.1016/S0006-3495(98)77870-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schulte A, Lorenzen I, Böttcher M, Plieth C. A novel fluorescent pH probe for expression in plants. Plant Methods. 2006;2:1–13. doi: 10.1186/1746-4811-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ugarova NN, Maloshenok LG, Uporov IV, Koksharov MI. Bioluminescence Spectra of Native and Mutant Firefly Luciferases as a Function of pH. Biochemistry (Moscow) 2005;70:1534–1540. doi: 10.1007/s10541-005-0257-2. [DOI] [PubMed] [Google Scholar]
- 7.Matthews JC, Hori K, Cormier MJ. Purification and properties of Renilla reniformis luciferase. Biochemistry. 1977;16:85–91. doi: 10.1021/bi00620a014. [DOI] [PubMed] [Google Scholar]
- 8.Verhaegen M, Christopoulos TK. Recombinant Gaussia luciferase. Overexpression, purification and analytical application of a bioluminescent reporter for DNA hybridization. Anal Chem. 2002;74:4378–4385. doi: 10.1021/ac025742k. [DOI] [PubMed] [Google Scholar]
- 9.Branchini BR, Ablamsky DM, Rosenberg JC. Chemically Modified Firefly Luciferase Is an Efficient Source of Near-Infrared Light. Bioconjugate Chem. 2010;21:2023–2030. doi: 10.1021/bc100256d. [DOI] [PubMed] [Google Scholar]
- 10.Hall MP, Unch J, Binkowski BF, Valley MP, Butler BL, Wood MG, Otto P, Zimmerman K, Vidugiris G, Machleidt T, Robers MB, Benink HA, Eggers CT, Slater MR, Meisenheimer PL, Klaubert DH, Fan F, Encell LP, Wood KV. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem Biol. 2012;7:1848–1857. doi: 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loening AM, Fenn TD, Wu AM, Gambhir SS. Consensus guided mutagenesis of Renilla luciferase yields enhanced stability and light output. Protein Eng Des Sel. 2006;19:391–400. doi: 10.1093/protein/gzl023. [DOI] [PubMed] [Google Scholar]
- 12.Nagai T, Yamada S, Tominaga T, Ichikawa M, Miyawaki A. Expanded dynamic range of fluorescent indicators for Ca2+ by circularly permuted yellow fluorescent proteins. Proc Natl Acad Sci USA. 2004;101:10554–10559. doi: 10.1073/pnas.0400417101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu Y, Piston D, Johnson CH. A bioluminescence resonance energy transfer (BRET) system: Application to interacting circadian clock proteins. Proc Natl Acad Sci USA. 1999;96:151–156. doi: 10.1073/pnas.96.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, Xie Q, Roberson JB, Johnson CH. pHlash: A New Genetically Encoded and Ratiometric Luminescence Sensor of Intracellular pH. PLOS ONE. 2012;7:e43072. doi: 10.1371/journal.pone.0043072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brett CL, Tukaye DN, Mukherjee S, Rao R. The Yeast Endosomal Na+(K+)/H+ Exchanger Nhx1 Regulates Cellular pH to Control Vesicle Trafficking. Mol Biol Cell. 2005;16:1396–1405. doi: 10.1091/mbc.E04-11-0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu X, Soutto M, Xie Q, Servick S, Subramanian C, et al. Imaging protein interactions with bioluminescence resonance energy transfer (BRET) in plant and mammalian cells and tissues. Proc Natl Acad Sci USA. 2007;104:10264–10269. doi: 10.1073/pnas.0701987104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krishnamoorthy Archana, Robertson J Brian. Dual color monitoring overcomes limitations of single bioluminescent reporters in fast growing microbes and reveals phase-dependent protein productivity during metabolic rhythms of yeast. Appl Environ Microbiol. 2015 doi: 10.1128/AEM.01631-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie Q, Soutto M, Xu X, Zhang Y, Johnson CH. Bioluminescence Resonance Energy Transfer (BRET) Imaging in Plant Seedlings and Mammalian Cells. Methods Mol Biol. 2011;680:3–28. doi: 10.1007/978-1-60761-901-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberson JB, Y Zhang Y, Johnson CH. Light-emitting diode flashlights as effective and inexpensive light sources for fluorescence microscopy. J Microsc. 2009;236:1–4. doi: 10.1111/j.1365-2818.2009.03208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]