

Summary
Cdk7, the CDK-activating kinase and transcription factor IIH component, is a target of inhibitors that kill cancer cells by exploiting tumor-specific transcriptional dependencies. However, whereas selective inhibition of analog-sensitive (AS) Cdk7 in colon cancer-derived cells arrests division and disrupts transcription, it does not by itself trigger apoptosis efficiently. Here we show that p53 activation by 5-fluorouracil or nutlin-3 synergizes with a reversible Cdk7as inhibitor to induce cell death. Synthetic lethality was recapitulated with covalent inhibitors of wild-type Cdk7, THZ1 or the more selective YKL-1-116. The effects were allele-specific; a CDK7as mutation conferred both sensitivity to bulky adenine analogs and resistance to covalent inhibitors. Non-transformed colon epithelial cells were resistant to these combinations, as were cancer-derived cells with p53-inactivating mutations. Apoptosis was dependent on death receptor DR5, a p53 transcriptional target whose expression was refractory to Cdk7 inhibition. Therefore, p53 activation induces transcriptional dependency to sensitize cancer cells to Cdk7 inhibition.
Keywords: Cdk7, p53, colon cancer, synthetic lethality, transcription, 5-fluorouracil (5-FU), nutlin-3, apoptosis, RNA polymerase II, chemical genetics, TFIIH, CDK inhibitor
eTOC Blurb
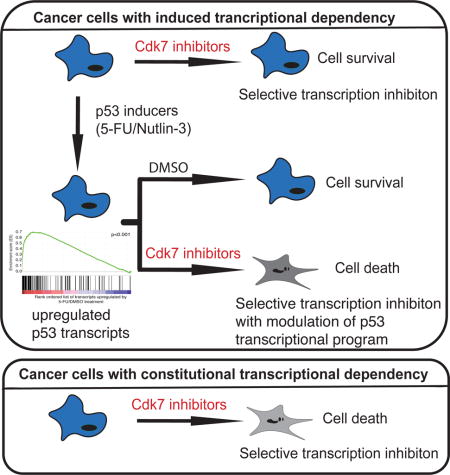
Kalan et al. find that activation of the p53 tumor suppressor protein in human colon cancer-derived cells can induce transcriptional dependency on Cdk7, analogous to constitutive dependencies described in other tumors driven by oncogenic transcription factors. This work provides a proof of concept for combining p53-activating agents with Cdk7 inhibitors to elicit synthetic lethality.
Introduction
Cyclin-dependent kinases (CDKs) regulate eukaryotic cell division and RNA polymerase II (Pol II)-dependent transcription [reviewed in (Morgan, 2007; Sansó and Fisher, 2013)]. Cdk7 plays essential, direct roles in both cell-division and transcription cycles, as a CDK-activating kinase (CAK) that phosphorylates CDKs on the activation segment (T loop), and as part of general transcription factor (TF) IIH [reviewed in (Fisher, 2005)]. Physiologic functions and targets of Cdk7 have been identified by a chemical-genetic, “bumped-hole” approach—ATP-binding site expansion by mutation of the “gatekeeper” residue to accommodate bulky inhibitors or substrate analogs that cannot bind wild-type kinases (Bishop et al., 1998). Gene-targeting to replace Cdk7WT with an analog-sensitive (AS) mutant version allowed selective inhibition of the endogenous kinase in human colon cancer-derived HCT116 cells (Larochelle et al., 2007).
As a CAK, Cdk7 is inherently able to influence both cell division and gene expression; T-loop phosphorylation is required for the functions of both cell-cycle and transcriptional CDKs (Larochelle et al., 2012; Schachter et al., 2013). Apart from its CAK function, Cdk7, working in the context of TFIIH, phosphorylates the carboxy-terminal domain (CTD) of the Pol II large subunit Rpb1, with a preference for Ser5 and Ser7 positions of the heptad repeat consensus sequence, Y1S2P3T4S5P6S7 (Ramanathan et al., 2001; Glover-Cutter et al., 2009). Cdk7 activity is needed for the promoter-proximal pause, a rate-limiting step in expression of genes transcribed by Pol II (Adelman and Lis, 2012). Cdk7 promotes recruitment of the DRB-sensitivity inducing factor (DSIF) and negative elongation factor (NELF) to the transcription complex (Glover-Cutter et al., 2009; Larochelle et al., 2012), which throws up a block to elongation that is relieved by positive transcription elongation factor b (P-TEFb) (Peterlin and Price, 2006). P-TEFb is itself a CDK, consisting of Cdk9 and cyclin T1, activation of which depends on phosphorylation by Cdk7 (Larochelle et al., 2012). Therefore, Cdk7 acts both to establish and to overcome the pause imposed by DSIF and NELF; loss of this regulation diminishes Pol II occupancy in gene bodies and is likely to uncouple RNA synthesis and co-transcriptional processing (Glover-Cutter et al., 2009; Larochelle et al., 2012).
Despite extensive efforts to target cell-cycle CDKs for anti-cancer therapy (Malumbres and Barbacid, 2009), parallel approaches aimed at transcriptional CDKs only recently gained traction. A covalent inhibitor of Cdk7, THZ1, induced global transcriptional shutdown at high doses, but had gene-selective repressive effects at lower doses (Kwiatkowski et al., 2014). In vitro, THZ1 recapitulated effects of Cdk7 inhibition on Pol II pausing, and impaired co-transcriptional RNA 5′-end capping (Nilson et al., 2015). Moreover, THZ1 triggered apoptosis of sensitive cells in culture, and limited or reversed growth of specific tumors—T-cell acute lymphoblastic leukemia (T-ALL), neuroblastoma, small cell lung cancer (SCLC) and triple-negative breast cancer (TNBC)—with minimal toxicity in human xenograft mouse models (Chipumuro et al., 2014; Christensen et al., 2014; Kwiatkowski et al., 2014; Wang et al., 2015).
Here we show that selective inhibition of Cdk7 in HCT116 cells does not by itself trigger apoptosis, but potentiates p53-dependent cell-killing by the antimetabolite 5-fluorouracil (5-FU), and switches cell fate from division arrest to death upon p53 stabilization by nutlin-3. These synthetic-lethal effects were 1) recapitulated with covalent inhibitors of transcriptional CDKs, such as THZ1 or the more Cdk7-selective YKL-1-116, in wild-type cells; 2) dependent on wild-type p53 function and ongoing transcription; and 3) accompanied by caspase 8 activation and suppressed by depletion of the death receptor (and p53 target) DR5, suggesting engagement of the extrinsic apoptotic pathway (Henry et al., 2012). Previous studies reported lethality due to combined inhibition of poly(ADP- ribose) polymerase (PARP) and CDKs implicated in DNA damage responses (Johnson et al., 2011; Johnson et al., 2016). We provide proof of concept for synthetic-lethal strategies targeting CDK functions in p53-responsive transcription.
Results
Inhibition of multiple CDKs promotes apoptosis
To dissect Cdk7 functions in vivo, we previously replaced both wild-type copies of CDK7 with CDK7F91G/D92E (CDK7as) in HCT116 cells (Larochelle et al., 2007). Treatment of these cells with bulky adenine analogs such as 3-MB-PP1, which potently inhibits Cdk7as in vitro, prevented T-loop phosphorylation of Cdk1, -2, -4, -6 and -9, and led to cell-cycle arrests and transcription defects (Larochelle et al., 2007; Glover-Cutter et al., 2009; Larochelle et al., 2012; Schachter et al., 2013). However, it did not efficiently induce apoptosis, measured by PARP cleavage, accumulation of annexin V-positive cells or caspase activation (Figure S1A–C). This is in contrast to the pro-apoptotic effects of THZ1 in T-ALL and other sensitive cell types (Kwiatkowski et al., 2014).
We considered two possible, but not mutually exclusive, explanations for the difference. First, THZ1 might elicit apoptosis through inhibition of Cdk7 and additional targets in the same pathway, such as Cdk12 and/or Cdk13 (Kwiatkowski et al., 2014). Second, vulnerability to selective CDK inhibitors might be based on transcriptional dependencies unique to certain cancers but lacking in HCT116 cells. To explore the first possibility, we treated CDK7as/as cells with increasing doses of 3-MB-PP1 in the absence or presence of flavopiridol (FP), a pan-CDK inhibitor that is most potent towards Cdk9, but which also inhibits Cdk12 at higher doses (Chao and Price, 2001; Bosken et al., 2014; Bartkowiak and Greenleaf, 2015). Addition of sublethal doses of FP (10 or 50 nM) sensitized cells to killing by Cdk7 inhibition. However, 3-MB-PP1 doses >2 μM suppressed PARP cleavage in the presence of 50 nM FP and, at 150 nM FP (a lethal dose on its own in CDK7as/as cells), 3-MB-PP1 suppressed PARP cleavage at doses >100 nM (Figure S1A).
Similarly, the response of CDK7as/as cells to FP alone was biphasic, with a maximum at ~125 nM and suppression at ≥250 nM (Figure 1A). Expression of p53 increased after FP treatment with half-maximal induction at ~150 nM, within the pro-apoptotic range. Addition of 1 μM 3-MB-PP1 shifted the FP dose needed for maximal PARP cleavage to ~50 nM. At higher doses of FP apoptosis was suppressed; PARP cleavage returned to background levels at 125 nM, the optimal dose in the absence of 3-MB-PP1. Cdk7 inhibition similarly potentiated FP effects on p53 expression, which remained elevated as the FP dose was raised. In contrast, a p53 transcriptional target—the CDK inhibitor p21—was induced over a narrow FP dose range, which was roughly co-extensive with the pro-apoptotic range and likewise shifted to lower doses by 3-MB-PP1 addition. Therefore, simultaneous inhibition of multiple CDKs can induce apoptosis in HCT116 cells, but biphasic responses imply a limitation on the ability of broad-specificity CDK inhibitors to trigger cell death; at higher concentrations these drugs lose efficacy, possibly because they also block transcription of pro-apoptotic p53 targets.
Figure 1. Synthetic lethal effects of Cdk7 inhibition combined with p53 activation.
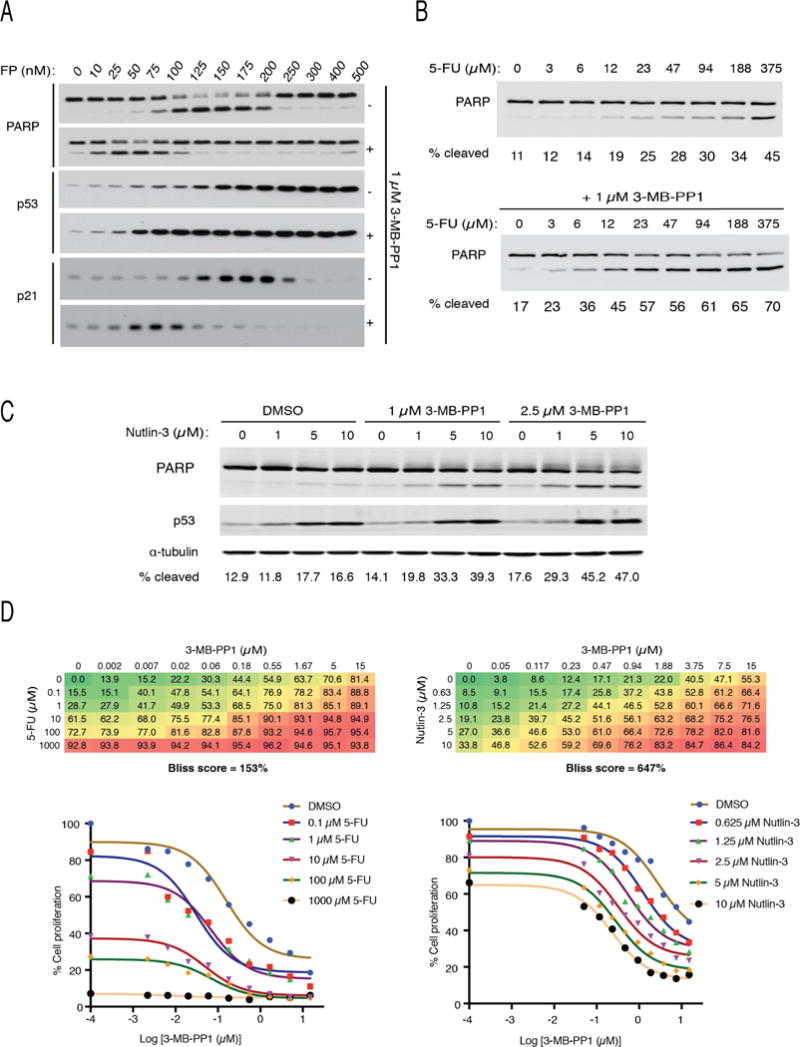
(A) CDK7as/as cells were treated with indicated doses of flavopiridol (FP), without (−) or with (+) addition of 1 μM 3-MB-PP1 for 14 hr prior to extract preparation and immunoblot detection of PARP, p53 and p21.
(B) CDK7as/as cells were treated with indicated doses of 5-FU, without (top) or with (bottom) addition of 1 μM 3-MB-PP1 for 14 hr prior to extract preparation and immunoblot detection of PARP. Signals were quantified by densitometry and expressed as a percentage of cleaved PARP (cleaved/uncleaved + cleaved) below each lane.
(C) CDK7as/as cells were treated with indicated doses of nutlin-3, without or with addition of 3-MB-PP1 at indicated doses for 14 hr prior to extract preparation and immunoblot detection of PARP, p53 and α-tubulin. Signals were quantified by densitometry and expressed as a percentage of cleaved PARP (cleaved/uncleaved + cleaved) below each lane. In (A–C), results are representative of multiple (n ≥ 2) biological replicates.
(D) Bliss independence analysis in CDK7as/as cells for 3-MB-PP1 and 5-FU (left) or nutlin-3 (right). Bliss scores were calculated from mean values of full concentration matrices performed in triplicate. Numbers in matrices indicate % reduction in metabolic activity as measured by resazurin staining, relative to DMSO-treated cells. Growth inhibition curves derived from these data are shown below each matrix.
See also Figure S1
Cdk7 inhibition potentiates cell killing by p53-activating agents
FP elicited a p53 response and cell death, which were potentiated by a Cdk7-selective drug that by itself did not activate p53. At lower doses, where it is more likely to be Cdk7-selective, THZ1 triggered apoptosis in vulnerable tumor cells with fixed dependencies on oncogenic transcription factors (Kwiatkowski et al., 2014). We reasoned that p53-activating agents might induce a similar dependency and, because they do not directly target the CDK network, do so without the dose limitation seen with FP. We tested 5-FU and nutlin-3, which elicit different p53-dependent phenotypes—death or arrested division, respectively—in HCT116 cells (Donner et al., 2007). In CDK7as/as cells, Cdk7 inhibition potentiated the effect of 5-FU by ~20-fold; half-maximal PARP cleavage occurred at ~20 μM 5-FU in the presence of 1 μM 3-MB-PP1, compared to >300 μM in its absence (Figure 1B). Similarly, treatment of CDK7as/as cells with 1–10 μM nutlin-3 alone did not trigger apoptosis, but did so when combined with 3-MB-PP1 (Figure 1C). Combination of either 5-FU or nutlin-3 with 3-MB-PP1 led to greater-than-additive accumulation of annexin V-positive cells, and both PARP cleavage and annexin V staining could be blocked by addition of the caspase inhibitor Z-VAD (Figure S1B, C). There was no difference in levels of p53 induced by nutlin-3 in the presence or absence of 3-MB-PP1, indicating that Cdk7 inhibition works downstream of p53 to switch cell fate from division arrest to death in response to nutlin-3.
We tested for synergy between 3-MB-PP1 and p53-activating agents by combination analysis over full concentration matrices (Figure 1D). The analog acted synergistically to shift dose responses to 5-FU and, to a greater extent, nutlin-3, as indicated by strongly positive Bliss independence scores (Zhao et al., 2014). Cdk7 inhibition therefore lowers 5-FU doses needed to kill tumor cells that are normally responsive to the drug, and confers nutlin-3-sensitivity on normally resistant cells. These results suggest that p53 stabilization might induce transcriptional dependency, sensitizing cells to killing by single-CDK inhibition.
Synthetic lethality of p53 activators with covalent inhibitors of wild-type Cdk7
We next asked if synthetic-lethal effects of 3-MB-PP1 in CDK7as/as cells could be recapitulated with THZ1. Modeling of THZ1 docked with an X-ray crystal structure of human Cdk7 suggested close proximity of the drug to the side chain of gatekeeper residue Phe91 (Kwiatkowski et al., 2014). In vitro, Cdk7as complexes were resistant to THZ1 at concentrations up to 10 μM (Figure 2A). In contrast, Cdk7WT was inhibited by THZ1 with an IC50 of ~100 nM. Conversely, Cdk7WT was insensitive to 3-MB-PP1, which inhibited Cdk7as with an IC50 of ~10 nM, as reported (Merrick et al., 2008).
Figure 2. Synthetic lethality of 5-FU and Cdk7 inhibitors is CDK7 allele-specific and p53-dependent.
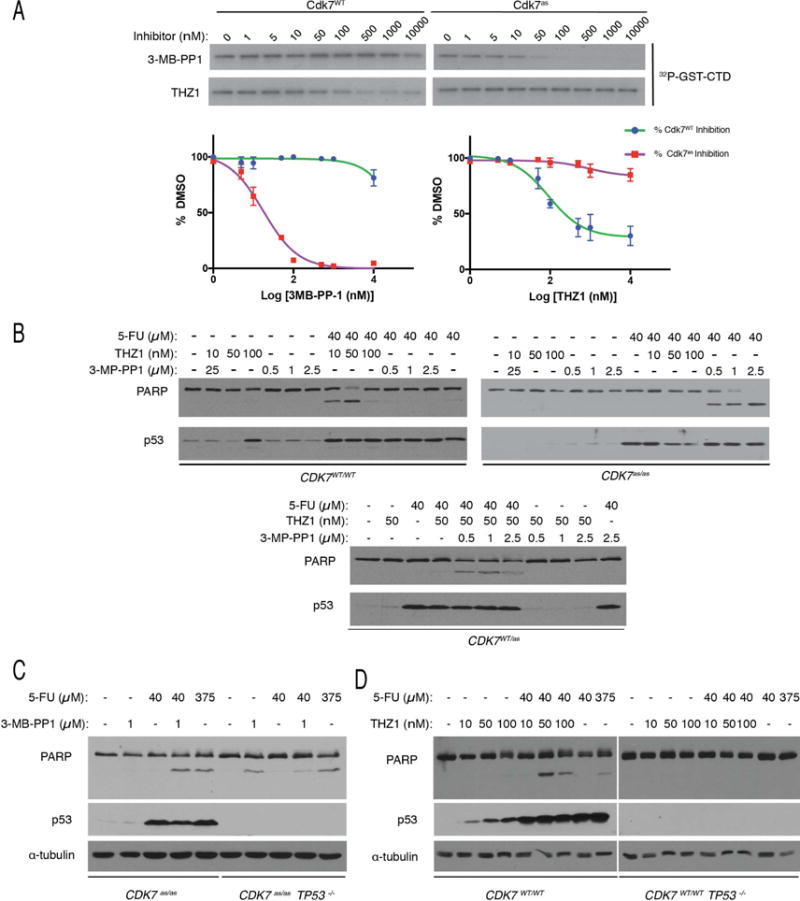
(A) Allele-specific inhibition of Cdk7 in vitro. T-loop-phosphorylated, trimeric complexes containing Cdk7 (WT or as), cyclin H and Mat1 (~5 nM each) were incubated with indicated concentrations of 3-MB-PP1 or THZ1 and tested for kinase activity towards a fusion protein containing the Pol II CTD (GST-CTD), with results shown by representative autoradiograms (top) and quantified by Phosphorimager (bottom). Error bars indicate ± S.E.M. of triplicate samples.
(B) CDK7WT/WT, CDK7as/as or CDK7WT/as HCT116 cells, as indicated, were treated with indicated combinations and doses of drugs for 14 hr prior to extract preparation and immunoblot detection of PARP and p53.
(C) CDK7as/as TP53−/− HCT116 cells were treated with indicated drugs, at indicated doses, for 14 hr prior to lysis and immunoblot detection of PARP, p53 and α-tubulin.
(D) Wild-type or TP53−/− HCT116 cells were treated with indicated drugs, at indicated doses, for 14 hr prior to lysis and immunoblot detection of PARP, p53 and α-tubulin.
See also Figure S2
We tested the allele-specificity of each drug in wild-type and CDK7as/as cells (Figure 2B). In the absence of other drugs, neither 10–100 nM THZ1 nor 0.5–2.5 μM 3-MB-PP1 induced PARP cleavage in either genetic background (although 100 nM THZ1 induced p53 expression in wild-type cells). When combined with 40 μM 5-FU, THZ1 induced PARP cleavage in CDK7WT/WT cells, but only at doses of 10 and 50 nM, not at 100 nM. In CDK7as/as cells, however, there was no synthetic lethal effect of combining 5-FU and THZ1. As expected, 3-MB-PP1 had the opposite allele-specificity—synthetic lethality with 5-FU in CDK7as/as but not CDK7WT/WT cells—with no loss of efficacy at higher doses.
In deriving CDK7as/as cells, we generated heterozygous, CDK7WT/as intermediates (Larochelle et al., 2007), which express wild-type and AS Cdk7 at approximately equal levels and therefore should retain Cdk7 activity in the presence of either 3-MB-PP1 or THZ1, but not both drugs. In accordance with this prediction, CDK7WT/as cells did not undergo apoptosis when treated with pairwise combinations of 5-FU and either 3-MB-PP1 or THZ1 but did so when all three drugs were added (Figure 2B). Therefore, a single copy of either CDK7 allele—WT or as—can complement inactivation of the enzyme encoded by the other. This further validates Cdk7 as the unique, common target of 3-MB-PP1 and THZ1 relevant for synthetic lethality.
Synthetic lethality of 5-FU and CDK inhibitors is p53-dependent
We hypothesized that p53 stabilization establishes dependency on Cdk7, such that Cdk7 inhibition modifies the downstream transcriptional response to favor pro-apoptotic over pro-survival pathways. It follows that synthetic lethality should depend on a functional p53. To test that prediction, we used recombinant adeno-associated virus (rAAV) vectors (Topaloglu et al., 2005) to disrupt both copies of TP53 (encoding p53) in CDK7as/as HCT116 cells (Figure S2A). In CDK7as/as TP53−/− cells, neither p53 nor p21 was detectable after treatment with the DNA-damaging agent doxorubicin, whereas both were induced in CDK7as TP53+ cells (Figure S2B). Loss of p53 led to slight sensitization to 3-MB-PP1, measured either by PARP cleavage or cell viability (Figures 2C, S2C). The effects of combined 3-MB-PP1 and 5-FU treatment were less than additive, however, in CDK7as/as TP53−/− cells (Figures 2C, S2D). (The CDK7as mutation also appeared to sensitize TP53−/− cells to treatment with 375 μM 5-FU [Figure S2E].) Similarly, we observed greater-than-additive effects of combining 5-FU with THZ1 in TP53+/+ but not TP53−/− HCT116 cells with wild-type Cdk7 (Figure 2D), and in TP53+ colorectal cancer-derived RKO (Figure S2F) or LoVo cells (data not shown), but not in the TP53− mutant lines HT29 or DLD1 (Figure S2G). Therefore, the synthetic lethality of 5-FU + Cdk7 inhibition in colon cancer-derived cells depends on an intact p53 pathway.
A Cdk7-selective covalent inhibitor synergizes with 5-FU and nutlin-3
By analogy with the biphasic response to FP (Figure 1A), we suspected that attenuated synthetic lethality at THZ1 doses >50 nM (Figure 2B) might be due to inhibition of additional THZ1 targets, such as Cdk12 and Cdk13 (Kwiatkowski et al., 2014). The response of CDK7as/as cells to 3-MB-PP1 was not similarly biphasic, so we reasoned that a more selective inhibitor of wild-type Cdk7 might work over a broader dose range. As part of efforts to develop more specific kinase inhibitors, we synthesized YKL-1-116 (Figure 3A), a covalent inhibitor of Cdk7 (Figure S3A, B) that does not target Cdk9, Cdk12 or Cdk13 (Figure 3B, Supplemental Tables 1, 2). In vitro, YKL-1-116 was more potent than THZ1 towards both Cdk7WT and Cdk7as, although Cdk7as was relatively resistant to this compound as well (Figure 3C). In wild-type cells, YKL-1-116 alone elicited minimal amounts of PARP cleavage only at the highest doses tested, reminiscent of the effects of 3-MB-PP1 on CDK7as/as cells. In combination with 40 μM 5-FU, however, YKL-1-116 induced PARP cleavage in dose-dependent fashion, with little loss of efficacy at higher concentrations (Figure 3D). Similarly, combining 5 μM nutlin-3 with 100–800 nM YKL-1-116 produced dose-dependent increases in PARP cleavage (Figure 3E). As with THZ1, synthetic lethality depended on potency towards Cdk7: CDK7as/as cells were resistant to combinations of YKL-1-116 and either 5-FU (Figure 3D) or nutlin-3 (Figure 3E). Moreover, both 5-FU and nutlin-3 shifted dose responses of wild-type cells to YKL-1-116, with strongly positive Bliss scores indicating drug synergy (Figure 3F). Similar independence analysis of THZ1 yielded weakly positive or even negative scores due to loss of efficacy at higher doses (data not shown). Combinations of YKL-1-116 with either 5-FU or nutlin-3 were effective against a variety of p53-positive cells derived from different tumor types (Figure S3C–F). Finally, combinations of THZ1 or YKL-1-116 with 5-FU did not trigger PARP cleavage in a non-transformed colorectal epithelial cell line, CCD 841 CoN, despite induction of p53 (Figure S3G). Taken together, the results with 3-MB-PP1, THZ1 and YKL-1-116 indicate that increasing the specificity of Cdk7 inhibition can enhance synergistic, synthetic-lethal effects of combinatorial treatments in cancer-derived cells with functional p53.
Figure 3. YKL-1-116, a Cdk7-selective covalent inhibitor, synergizes with 5-FU or nutlin-3 to kill HCT116 cells.
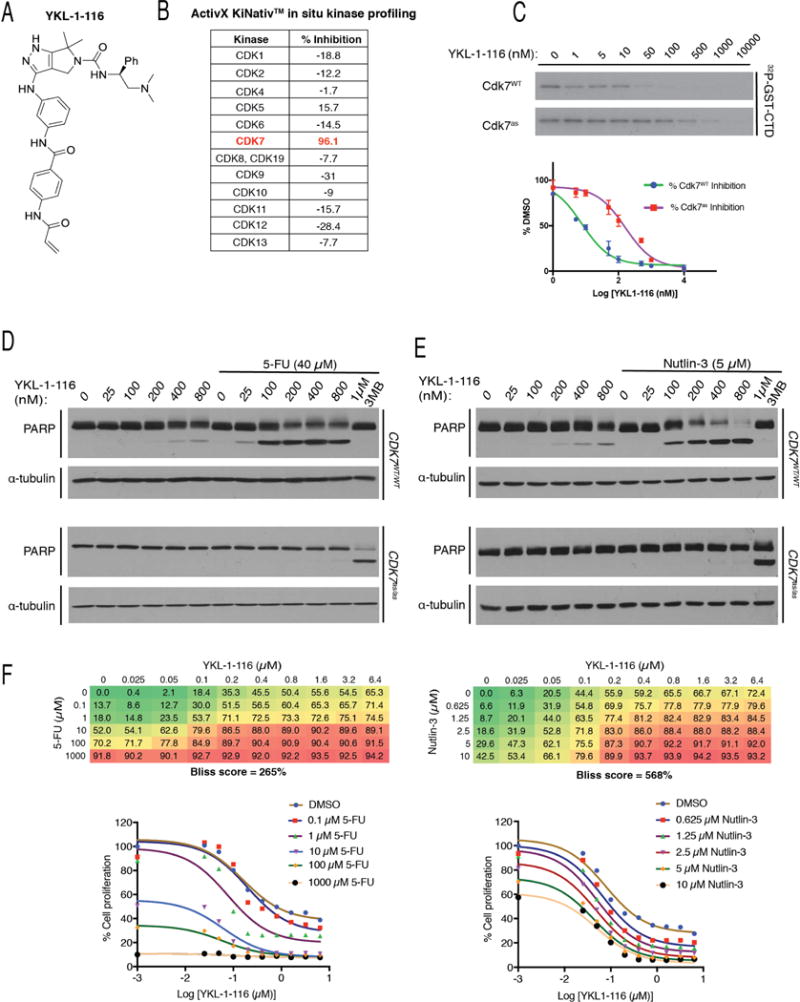
(A) YKL-1-116 structure.
(B) Selectivity of YKL-1-116 for Cdk7 over other CDKs, as determined by KiNativ™ kinome profiling. For each CDK, the percent inhibition of labeling by a desthiobiotin-ATP probe after exposure in vivo to YKL-1-116 is indicated.
(C) Inhibition of Cdk7 by YKL-1-116 in vitro. As in Figure 2A, complexes containing Cdk7 (WT or as) were incubated with indicated concentrations of YKL-1-116 and tested for kinase activity towards GST-CTD, with results shown by representative autoradiograms (top) and quantified by Phosphorimager (bottom). Error bars indicate ± S.E.M. of triplicate samples.
(D) CDK7WT/WT or CDK7as/as HCT116 cells were treated with indicated Cdk7 inhibitors, at indicated doses, with or without 40 μM 5-FU, as indicated, for 14 hr prior to lysis and immunoblot detection of PARP and α-tubulin.
(E) CDK7WT/WT or CDK7as/as HCT116 cells were treated with indicated Cdk7 inhibitors, at indicated doses, with or without 5 μM nutlin-3, as indicated, for 14 hr prior to lysis and immunoblot detection of PARP and α-tubulin.
(F) Bliss independence analysis in CDK7WT/WT cells for YKL-1-116 and 5-FU (left) or nutlin-3 (right). Bliss scores represent the mean of triplicate concentration matrices. Numbers in matrices indicate % reduction in metabolic activity as measured by resazurin staining, relative to DMSO-treated cells. Growth inhibition curves derived from these data are shown below each matrix.
Transcriptional disruption underlies synthetic lethality of 5-FU and Cdk7 inhibition
To unravel the mechanism(s) of this synthetic lethality, we first compared effects of FP, which at low doses primarily affects transcriptional CDKs, and purvalanol A, which inhibits cell-cycle CDKs activated by Cdk7, with selectivity for Cdk1 (Gray et al., 1998). In wild-type HCT116 cells, which are less FP-sensitive than CDK7as/as cells (Larochelle et al., 2012), 150 nM FP did not induce PARP cleavage on its own, but did so in combination with sublethal doses of 5-FU (Figure S4A). In contrast, there was no detectable PARP cleavage after treatment with 5-FU and purvalanol A. There was likewise no PARP cleavage in 5-FU-exposed CDK2as/as HCT116 cells treated with 3-MB-PP1 (Figure S4B), which impedes G1/S progression by inhibiting Cdk2as in these cells (Merrick et al., 2011). We infer that CDK functions in transcription are needed to avoid apoptosis after 5-FU exposure. Also suggestive of a transcription-dependent mechanism, synthetic lethality could be suppressed by simultaneous treatment with triptolide (Figure 4A), which covalently inhibits XPB, a TFIIH subunit required for transcription by Pol II (Titov et al., 2011). In contrast, specific inhibitors of Pol I or Pol III—CX5461 (Drygin et al., 2011) or ML60218 (Wu et al., 2003), respectively—were unable to suppress synthetic lethality, even when added together. Therefore, Pol II activity is necessary and sufficient for the transcription-dependent lethality of combined 5-FU treatment and Cdk7 inhibition.
Figure 4. Synthetic lethality of 5-FU + CDK inhibition is transcription-dependent.
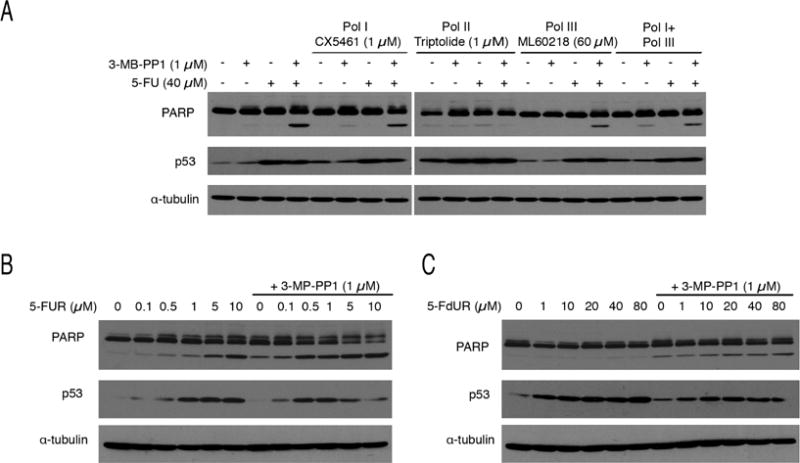
(A) CDK7as/as cells were treated with 5-FU (40 μM) and/or 3-MB-PP1 (1 μM), as indicated, with addition of inhibitors of Pol I (1 μM CX5461), Pol II (1 μM triptolide) or Pol III (60 μM ML60218), as indicated, for 14 hr prior to extract preparation and immunoblot detection of PARP, p53 and α-tubulin.
(B) CDK7as/as cells were treated with indicated doses of 5-fluorouridine (5-FUR), without or with addition of 1 μM 3-MB-PP1, as indicated, for 14 hr prior to extract preparation and immunoblot detection of PARP, p53 and α-tubulin.
(C) CDK7as/as cells were treated with indicated doses of 5-fluorodeoxyuridine (5-FUdR), without or with addition of 1 μM 3-MB-PP1, as indicated, for 14 hr prior to extract preparation and immunoblot detection of PARP, p53 and α-tubulin.
See also Figure S4
5-FU can be incorporated into DNA or RNA (Longley et al., 2003). Therefore, to test if synthetic lethality depended on RNA or DNA metabolism, we compared effects of ribose or deoxyribose derivatives of 5-FU—5-fluorouridine (5-FUR) or 5-fluoro-2′-deoxyuridine (5-FdUR), respectively (Figure 4B, C). Even in the absence of 3-MB-PP1, CDK7as/as HCT116 cells were more sensitive to 5-FUR than to 5-FU or 5-FdUR. Addition of 1 μM 3-MB-PP1 potentiated pro-apoptotic effects of 5-FUR but not 5-FdUR, suggesting that the relevant toxic metabolite is 5-FU-containing RNA, rather than DNA.
Prior exposure to 5-FU sensitizes cells to Cdk7 inhibition
To dissect these responses further, we first determined when CDK7as/as HCT116 cells exposed to various treatments became committed to apoptosis. In cells treated with 5 μM nutlin-3 + 2.5 μM 3-MB-PP1, or with 375 μM 5-FU alone, PARP cleavage measured at 24 hr was largely but not completely prevented if drugs were removed as late as 12 hr after addition. In contrast, combined treatment with 40 μM 5-FU and 2.5 μM 3-MB-PP1 accelerated passage of a point of no return; cells became irreversibly committed to apoptosis 6–9 hr after drug addition (Figure 5A).
Figure 5. Cdk7 inhibition after 5-FU exposure triggers apoptosis dependent on transcription.
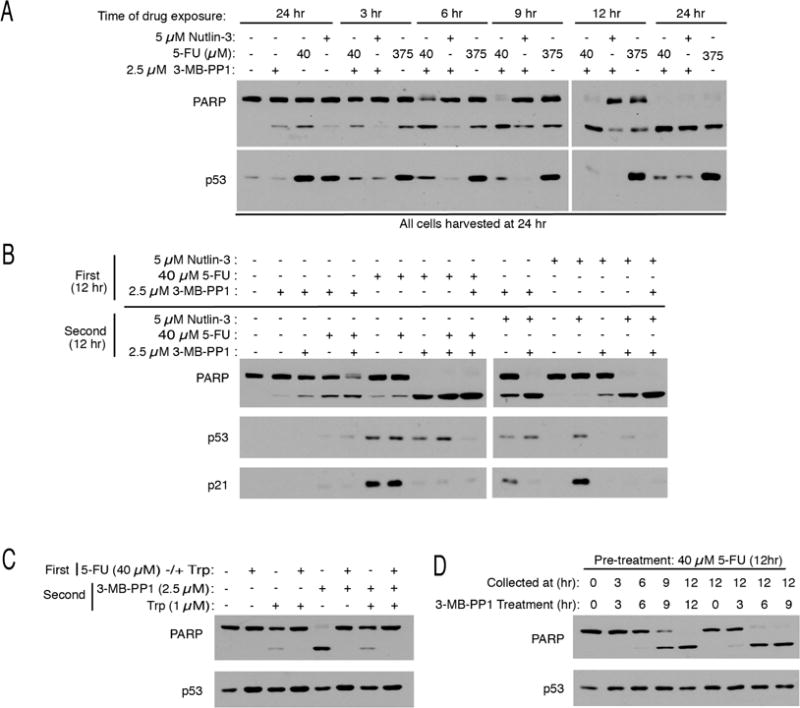
(A) CDK7as/as cells were treated with 5-FU, nutlin-3 and/or 3-MB-PP1, as indicated, for indicated times. Where indicated, drug-containing media were removed and replaced with fresh, drug-free medium. All cells were harvested after 24 hr for extract preparation and immunoblot detection of PARP and p53.
(B) CDK7as/as cells were first incubated with the indicated drugs for 12 hr (First), washed and incubated for an additional 12 hr with indicated drugs (Second), prior to extract preparation and immunoblot detection of PARP, p53 and p21.
(C) As in (B) but with addition of 1 μM triptolide (Trp) to indicated cell populations, during either first or second incubation, as indicated.
(D) CDK7as/as cells were pretreated with 40 μM 5-FU for 12 hr, washed and treated with 2.5 μM 3-MB-PP1 for indicated times and collected at indicated times for extract preparation and immunoblot detection of PARP and p53.
Nutlin-3 acts directly to inhibit ubiquitylation and proteasomal degradation of p53 (Vassilev et al., 2004). The mechanism of 5-FU action is indirect, requiring a toxic intermediate that, once it accumulates, can sustain p53 activation in the absence of the drug. Consistent with this scenario, we achieved similar cell-killing efficacy by simultaneous or sequential treatment with 5-FU and 3-MB-PP1, whereas nutlin-3 and 3-MB-PP1 needed to be administered together (Figure 5B). Moreover, the lethality of sequential treatment depended on the order of drug addition: 3-MB-PP1 was equally effective whether it was given with or after 5-FU treatment, and either regimen was more effective than 5-FU given only after 3-MB-PP1 removal.
Addition of 1 μM triptolide during the first (5-FU) or second (3-MB-PP1) treatment suppressed PARP cleavage (Figure 5C). Rescue by triptolide after removal of 5-FU indicates that ongoing Pol II transcription is required to activate caspases when Cdk7 is inhibited. Taken together with the relative toxicities of 5-FUR and 5-FUdR (Figure 4B, C), the results suggest that transcription is needed for both steps in the pathway leading to cell death, with a specific requirement for Pol II at the second, 3-MB-PP1-dependent step. Moreover, a time course of 3-MB-PP1 treatment after 5-FU washout indicated that the relevant differences in gene expression caused by Cdk7 inhibition would be detectable by 3–6 hr after addition of 3-MB-PP1, when cells have committed to apoptosis but before effector caspases are activated (Figure 5D).
Cdk7 inhibition modulates the transcriptional response to p53 activation
To detect alterations in transcription that might favor cell death when Cdk7 is inhibited, we performed RNA sequencing analysis (RNA-seq) of CDK7as/as cells that were pretreated with 40 μM 5-FU or DMSO for 12 hr, washed, and incubated in fresh medium containing 2.5 μM 3-MB-PP1 or DMSO for an additional 4 hr (Figure 6A). Treatment with 5-FU increased expression of 145 transcripts, while repressing 420 genes (Figure 6B, left column). We asked if either of these transcript pools was enriched for known p53 targets included in the “p53 Hallmark Pathway” gene set (Broad Institute data base MSigDB). Gene set enrichment analysis [GSEA (Subramanian et al., 2005)] revealed that members of this set were enriched among transcripts induced by 5-FU (Figure 6C). Expression of p53 targets was significantly elevated upon 5-FU treatment in comparison to changes in global mRNA levels (Figure 6D top panel, 6E top panel). Addition of 3-MB-PP1 to 5-FU-treated cells blunted induction of p53 targets relative to 5-FU treatment alone (Figure 6B right column, 6D bottom panel, 6E bottom panel), although it did not restore expression of these genes to their pre-5-FU levels (Figure 6B middle column, Figure S5A–C). Expression of p53-regulated genes such as p21 and MDM2 was reduced, suggesting attenuation of pro-survival transcriptional signaling induced by 40 μM 5-FU (Figure 6F). We validated the decrease in MDM2 transcripts by RT-qPCR analysis of cells treated simultaneously with 3-MB-PP1 and 5-FU (Figure 6G) or nutlin-3 (Figure 6H), and verified that this change was dependent on the CDK7as mutation (i.e., it did not occur in wild-type HCT116 cells, Figure S5D, E). Expression of Mdm2 and p21 proteins was also attenuated by Cdk7 inhibition, in response to sequential treatment with 5-FU and 3-MB-PP1 (Figures 6I and S5F), or simultaneous treatment with nutlin-3 and 3-MB-PP1 (Figures 6J and S5G).
Figure 6. Cdk7 inhibition modulates the transcriptional response to p53 activation.
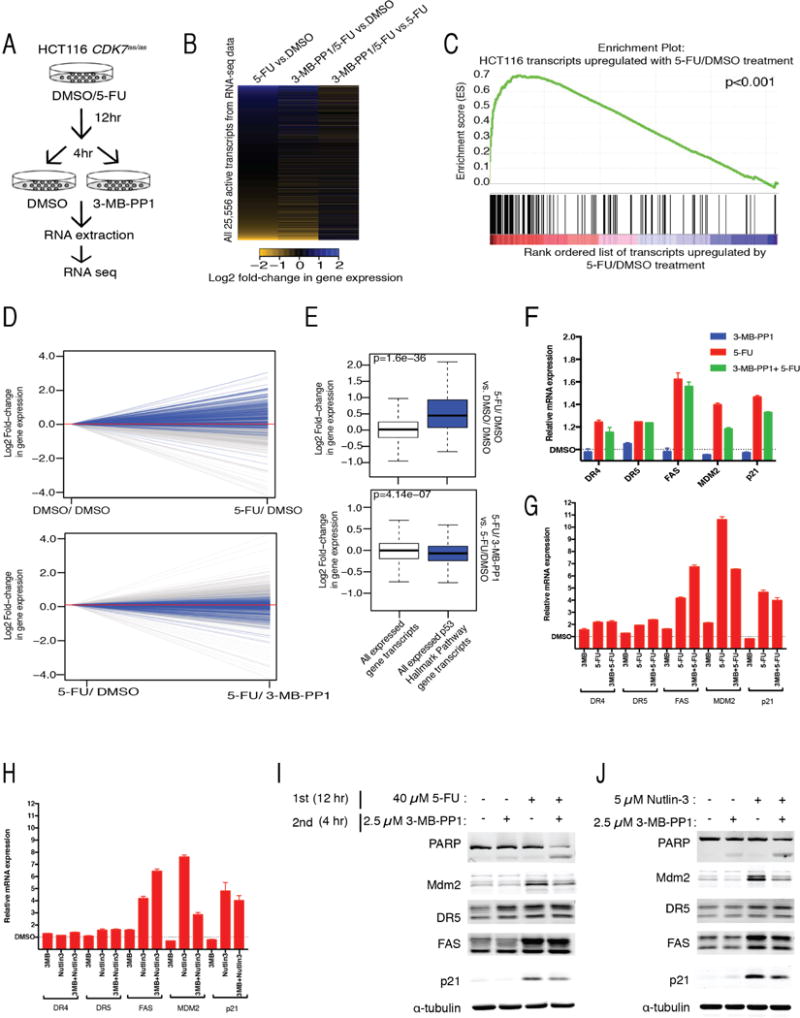
(A) Schematic of sequential treatments of CDK7as/as cells with 5-FU (40 μM) and 3-MB-PP1 (2.5 μM) used for RNA-seq analysis.
(B) 5-FU treatment changes steady-state mRNA levels. HCT116 cells were treated as indicated in Figure 6A. Heatmaps display the Log2 fold change in gene expression between indicated conditions for the 25,556 transcripts expressed in DMSO.
(C) Transcripts upregulated by 5-FU are enriched for p53 target genes. GSEA of all expressed transcripts rank-ordered from upregulated to downregulated following treatment with 5-FU in comparison to transcripts associated with the “p53 Hallmark Pathway Genes” from MSigDB. GSEA-supplied P-value < 0.001.
(D) Per-transcript line plots showing Log2 fold-change in expression following 5-FU/DMSO in comparison to DMSO/DMSO treatment (top panel) and following 5-FU/3MB-PP1 in comparison to 5-FU/DMSO treatment (bottom panel). Gray lines indicate top 10% of most highly expressed gene transcripts in DMSO. Blue lines indicate “p53 Hallmark Pathway Gene” transcripts. Red line indicates no change in gene expression.
(E) Box plots showing distribution of Log2 fold-changes in expression for the indicated transcript sets following 5-FU/DMSO in comparison to DMSO/DMSO treatment (top panel) and following 5-FU/3MB-PP1 in comparison to 5-FU/DMSO treatment (bottom panel).
(F) RNA-seq data for individual pro-apoptotic (DR4, DR5 and FAS) and pro-survival (MDM2 and p21) p53 transcriptional targets, after indicated drug treatments, relative to levels in DMSO (indicated by dashed horizontal line and defined as 1.0). Error bars indicate range of values obtained in two biological replicates.
(G) RT-qPCR analysis of selected p53 target gene expression in CDK7as/as cells after simultaneous exposure to 5-FU and/or 3-MB-PP1, relative to levels in DMSO-treated cells. Error bars indicate range of values obtained in two biological replicates.
(H) RT-qPCR analysis of selected p53 target gene expression in CDK7as/as cells after simultaneous treatment with nutlin-3 and/or 3-MB-PP1, relative to levels in DMSO-treated cells. Error bars indicate range of values obtained in two biological replicates.
(I) Immunoblot analysis of selected p53 target gene products in CDK7as/as cells after sequential treatment with 5-FU and 3-MB-PP1. Cells were harvested 12 hr after exposure to second drug (8 hr after second drug removal) for extract preparation and immunoblot detection of PARP, Mdm2, DR5, FAS, p21 and α-tubulin.
(J) Immunoblot analysis of selected p53 target gene products in CDK7as/as cells after simultaneous treatment with nutlin-3 and/or 3-MB-PP1. Cells were harvested after 12 hr drug exposure for extract preparation and immunoblot detection of PARP, Mdm2, DR5, FAS, p21 and α-tubulin.
See also Figure S5 and Supplemental Table 3
In contrast to these effects on MDM2 and p21, transcription levels of two pro-apoptotic p53 targets induced by 5-FU or nutlin-3—death receptor genes DR5 and FAS—were sustained in the presence of 3-MB-PP1 (Figure 6F–H). DR5 and FAS proteins were also induced to similar levels by 5-FU or nutlin-3 in the absence or presence of 3-MB-PP1 (Figures 6I, J and S5F, G). In cells pre-treated with DMSO, 3-MB-PP1 did not selectively affect expression of p53-regulated genes, indicating that it specifically modulated transcriptional responses to p53 stabilization (Figure S5A–C). These results suggest that Cdk7 inhibitors might synergize with 5-FU or nutlin-3 by tipping the balance of p53-dependent transcription towards death-promoting targets such as DR5 and FAS.
A role for the extrinsic pathway and a requirement for DR5 in synthetic lethality
The possible involvement of DR5 suggested engagement of the extrinsic pathway, which is implicated in HCT116 cell-killing by higher doses of 5-FU (Henry et al., 2012). Effectors specific to the extrinsic pathway include caspase 8, which was activated by treating CDK7as/as cells with 3-MB-PP1 in combination with either 5-FU (Figure 7A) or nutlin-3 (Figure 7B).
Figure 7. Activation of the extrinsic pathway of apoptosis by synthetic lethal combinations of p53 activators and Cdk7 inhibitors, dependent on DR5.
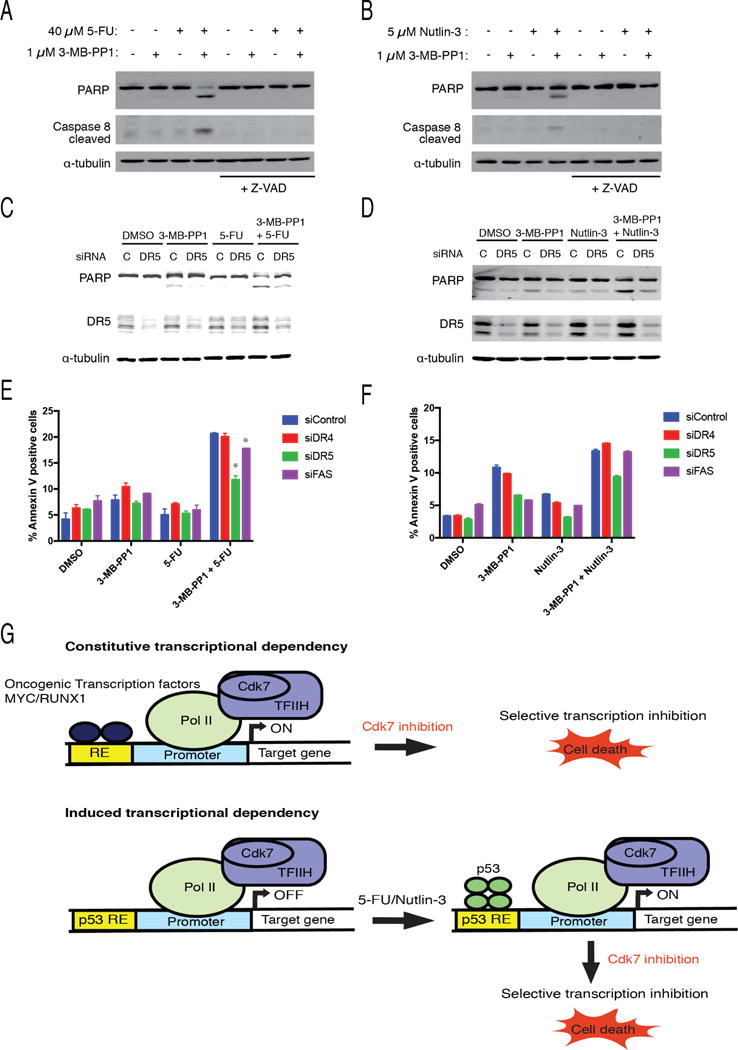
(A) CDK7as/as cells were treated with indicated dose of 5-FU, with or without 1 μM 3-MB-PP1 as indicated for 14 hr prior to extract preparation and immunoblot detection of PARP, activated caspase 8 (p18 isoform) and α-tubulin.
(B) CDK7as/as cells were treated with indicated dose of 3-MB-PP1, with or without 5 μM nutlin-3 as indicated for 14 hr prior to extract preparation and immunoblot detection of PARP, activated caspase 8 (p18 isoform) and α-tubulin.
(C) CDK7as/as cells were transfected with siRNA targeting DR5 or control (scrambled) siRNA (C) and treated with indicated drugs or drug combinations (doses: 40 μM 5-FU, 1 μM 3-MB-PP1) prior to lysis and immunoblot detection of PARP, DR5 and α-tubulin.
(D) CDK7as/as cells were transfected with siRNA targeting DR5 or control (scrambled) siRNA (C), and treated with indicated drugs or drug combinations (doses: 5 μM nutlin-3, 1 μM 3-MB-PP1) prior to lysis and immunoblot detection of PARP, DR5 and α-tubulin.
(E) Quantification of annexin V-positive populations measured by flow cytometry in CDK7as/as cells treated as in (C) (with addition of cells transfected with siRNA targeting DR4 or FAS). Error bars indicate ± S.E.M. of three biological replicates. Statistically significant differences between knockdowns and controls are indicated by (*).
(F) Quantification of annexin V-positive populations measured by flow cytometry in CDK7as/as cells treated as in (D) (with addition of cells transfected with siRNA targeting DR4 or FAS). Error bars indicate range of values obtained in two biological replicates.
(G) Transcriptional dependency on Cdk7 activity induced by p53 stabilization mimics constitutive dependency due to constitutive transcriptional activation by oncogenic transcription factors such as MYC or RUNX1.
See also Figure S6
In HCT116 cells exposed to high-dose 5-FU, DR5 and FAS were previously shown to translocate to the plasma membrane, and DR5 silencing impeded caspase activation (Can et al., 2013). To assess the involvement of DR5, FAS and DR4 (another death receptor) in synthetic lethality, we silenced expression of each with small interfering RNA (siRNA), treated cells with 3-MB-PP1 and either 5-FU or nutlin-3, and measured markers of apoptosis. We achieved similar degrees of knockdown of each of the three proteins (Figures 7C, D, S6A, B). Depletion of DR5 by ~50% diminished PARP cleavage and led to a statistically significant reduction in the annexin V-positive fraction (P ≈ 0.012, two- tailed t-test) in cells treated with 3-MB-PP1 + 5-FU (Figure 7C, E), and a reproducible reduction in cells treated with 3-MB-PP1 + nutlin-3 (Figure 7D, F). Knockdown of FAS also caused a significant reduction in annexin V staining upon treatment with 5-FU + 3-MB-PP1 (P ≈ 0.0015) but not nutlin-3 + 3-MB-PP1, whereas DR4 depletion did not significantly affect annexin V staining or PARP cleavage in response to either drug combination (Figures 7E, F, S6A, B). Taken together, the results suggest repression of pro-survival p53 targets, with sparing of DR5 and activation of the extrinsic pathway, as a common mechanism through which Cdk7 inhibition can potentiate pro-apoptotic effects of 5-FU, or switch cell fate from arrest to death in response to nutlin-3.
Discussion
Protein kinases are attractive targets for drug discovery, but conservation of their active sites has hindered development of selective small-molecule inhibitors. Chemical genetics circumvents this limitation to facilitate mechanistic studies. Homozygous replacement of wild-type CDKs with AS variants in human cells enabled dissection of complex biochemical pathways and identification of substrates of specific CDKs (Larochelle et al., 2007; Merrick et al., 2008; Merrick et al., 2011; Wohlbold et al., 2012). Here we demonstrate the utility of AS kinase-expressing human cell lines as platforms for discovery of synthetic-lethal drug combinations.
Genetically sensitized cells also provide a benchmark by which to gauge drug specificity; the ability of THZ1 to recapitulate effects of 3-MB-PP1 in CDK7as cells helped validate it as a Cdk7 inhibitor (Kwiatkowski et al., 2014). THZ1 treatment diminished bulk Ser2, Ser5 and Ser7 phosphorylation of the Pol II CTD, however, whereas allele-specific inhibition of Cdk7as did not (Larochelle et al., 2007; Glover-Cutter et al., 2009), suggesting that THZ1 had additional targets. THZ1 effects on both CTD phosphorylation and cell survival were nevertheless rescued by expression of a THZ1-refractory Cdk7 variant (Kwiatkowski et al., 2014). To reconcile these results, we propose that Cdk7 inactivation is necessary to elicit the observed effects in vivo but not sufficient; engagement of secondary targets such as Cdk12 and Cdk13—CTD kinases that can be selectively inhibited to trigger apoptosis (Zhang et al., 2016)—might contribute.
Irreversible inactivation of Cdk7 by THZ1 depends on covalent modification of Cys312, a residue outside the kinase domain conserved in Cdk12 and Cdk13 (Kwiatkowski et al., 2014). We now report THZ1-insensitivity of a Cdk7 variant with an active-site mutation. The resistance conferred by different mutations—F91G/D92E or C312S—indicates that specificity is determined by both the contours of the ATP-binding site and accessibility of a Cys residue for covalent modification (Kwiatkowski et al., 2014). Similar resistance to inhibitors of polo-like kinase 1 (Plk1) arose in human cells expressing an AS variant, due not to the gatekeeper mutation per se but rather to a second-site substitution needed for mutant kinase activity (Burkard et al., 2012). The resistance of CDK7as cells to THZ1 and YKL-1-116 provides a means to test whether phenotypes induced by these drugs depend on inhibition of their primary target, and suggests a potential path to acquired drug-resistance in tumors.
As anti-cancer therapeutic targets, transcriptional CDKs did not offer the obvious advantage of being needed only in proliferating cells, and seemed to entail a risk of toxicity to normal tissues. However, chemical-genetic ablation of Cdk7 catalytic function caused gene-specific rather than global repression of transcription in fission yeast (Viladevall et al., 2009) and human cells (this report). Although post-transcriptional mRNA stabilization might have masked more widespread effects on synthesis in budding yeast (Rodriguez-Molina et al., 2016), measurements of Pol II occupancy likewise indicate differential requirements for Cdk7 at different genes (Glover-Cutter et al., 2009; Viladevall et al., 2009; Larochelle et al., 2012). Similarly, different mRNAs exhibit a range of sensitivities to THZ1 in human cells (Kwiatkowski et al., 2014). A common feature of several THZ1-hypersensitive tumors is dependence on super-enhancers—regulatory elements that nucleate high-density assembly of Pol II machinery and histone modifications permissive for high levels of transcription (Hnisz et al., 2013; Loven et al., 2013). Reliance on oncogenic transcription factors—MYC in SCLC or neuroblastoma, or RUNX1 in T-ALL (Chipumuro et al., 2014; Christensen et al., 2014; Kwiatkowski et al., 2014)—might make these tumors especially vulnerable to perturbation of the Pol II cycle. Hypersensitivity to THZ1 need not be based on addiction to a single factor, but can be a cumulative effect of activating multiple, independent transcription programs, for example in TNBC (Wang et al., 2015). Here we show that p53 activation in cancer cells can also induce dependence on Cdk7 function, and might form the basis for a synthetic-lethal therapy (Figure 7G).
The balance between pro-apoptotic and pro-survival gene expression induced by p53— and thus the outcome of p53 activation—depends on the nature and strength of the p53-activating signal and varies among cell types exposed to the same stimulus (Donner et al., 2007), but is not clearly correlated with levels of pro- versus anti-apoptotic gene transcription. Comparative analyses of responses to 5-FU or nutlin-3 implicated post-transcriptional stabilization of DR4, leading to caspase 8 activation and BID cleavage, in the killing of HCT116 cells by 5-FU (Henry et al., 2012). Conversely, RNA interference-based screens identified signaling pathways that promote survival when p53 is activated by nutlin-3; depletion or inhibition of the kinases ATM or MET converted the response of HCT116 cells from division arrest to death without gross alterations in gene expression patterns (Sullivan et al., 2012). Cdk7 inhibition achieves a similar cell fate switch through a transcriptional mechanism, which depends on expression of DR5. The same p53 target contributes to Cdk7 inhibitor-mediated, transcription-dependent potentiation of cell-killing by 5-FU. Mechanistic studies will be needed to determine why DR5 transcription is refractory to Cdk7 inhibition. Moreover, depletion of DR5 only partially suppressed synthetic lethality; although this might reflect incomplete knockdown, further analyses are warranted to reveal other pro-apoptotic p53 targets expressed (or anti-apoptotic ones repressed) in the presence of Cdk7 inhibitors.
Finally, comparison between THZ1 and YKL-1-116 suggests a rationale for enhancing selectivity of clinically important kinase inhibitors. Although the ability to inhibit multiple kinases might potentiate cell-killing effects of drugs such as THZ1 (Knight et al., 2010), it increases the risk of toxicity in normal tissues and, as we have shown, can limit efficacy in combinations with other agents. Whereas simultaneous inhibition of closely related targets can have additive or synergistic effects, other “anti-targets” might need to remain active to elicit a therapeutically desirable outcome such as death of a cancer cell (Dar et al., 2012). We suggest that the organization of the transcription cycle, with precise coordination dependent on specialized functions of closely-related CDKs, makes it uniquely susceptible to “surgical” inactivation of individual kinases that leaves others active to trigger maladaptive stress responses and death in vulnerable cell populations.
Experimental Procedures
Cell culture, drug treatment and extract preparation
HCT116 cells were grown and lysates prepared as described (Larochelle et al., 2012). Cells were plated 24 hr prior to drug treatments. Synthesis and characterization of YKL-1-116 are described, and other drugs and antibodies used are listed, in Supplemental Experimental Procedures.
Drug synergy analysis
Drug synergy was measured by Bliss independence analysis (Zhao et al., 2014) as described (Dhawan et al., 2016). Details of drug treatments and analysis are described in Supplemental Experimental Procedures.
Kinase assays
Kinase assays were performed with complexes of phosphorylated Cdk7 (WT or AS), cyclin H and Mat1 generated by adding purified His-Mat1 to Cdk7-cyclin H dimer, as previously described (Larochelle et al., 2001), with or without treatment with various inhibitors, as described in Supplemental Experimental Procedures.
TP53 knockout in CDK7as/as cells
The TP53 gene was disrupted in HCT116 CDK7as/as cells with an rAAV vector as described (Topaloglu et al., 2005).
RNAseq
RNA-seq reads were aligned to a version of the hg19 human reference genome with ERCC spike-in reference sequences added using tophat (Trapnell et al., 2009) v.2.0.13 with parameters –no-novel-juncs and –G set to the human RefSeq transcript list downloaded in May 2013. Reads overlapping to the ENCODE list of blacklist regions (Consortium, 2012) were filtered using bedtools (Quinlan, 2014) intersect. Per-transcript expression values were created using RPKM_count.py from the RSeQC package (Wang et al., 2012) using –e, the RefSeq transcript list and ERCC probe regions. RPKM values between 0 and 0.1 were set to 0.1, and a pseudocount of 0.1 was added to all transcripts. For each transcript, replicates of each condition were averaged. For displays, expressed transcripts are those with RPKM >1 in the average of the DMSO replicates. Gene set enrichment analysis (Subramanian et al., 2005) was performed with GSEA software, using the “p53 Hallmark Pathway” gene set from Broad Institute data base MSigDB. This same gene set was used in all expression plots to indicate p53 pathway genes.
RNA interference
Cells were transfected with short interfering RNA (siRNA) oligonucleotides as described in Supplemental Experimental Procedures.
Supplementary Material
Supplemental Table S1 YKL-1-116 selectivity measured by KiNativ™ kinome profiling (see accompanying Excel file)
Supplemental Table S3 RNAseq data (see accompanying Excel file)
Highlights.
Cdk7 inhibition plus p53 activation causes synthetic lethality in cancer cells.
Chemical genetics leads to discovery of synthetic lethal drug combinations.
A Cdk7-selective covalent inhibitor produces similar, synthetic-lethal effects.
p53 activation induces transcriptional dependency targeted by Cdk7 inhibitors.
Acknowledgments
We thank K.A. Merrick, M.B. Yaffe and members of the Fisher lab for helpful discussions; C. Zhang and K. Shokat for 3-MB-PP1; F. Bunz for rAAV vector to disrupt TP53; N. Dhawan and A. Dar for advice on drug synergy assays; S. Mungamuri and S. Aaronson for help with flow cytometry; J. M. Espinosa for advice during early stages of this work; A. Dar and K. Merrick for critical review of the manuscript. R.A. was supported by a Beatriu de Pinos fellowship of the Generalitat de Catalunya. M.M.S. was supported by a fellowship from the NIH (T32 CA78207). B.J.A. is the Hope Funds for Cancer Research Grillo-Marxuach Family Fellow. This work was supported by NIH grants GM105773 and GM104291 to R.P.F., CA179483 to N.S.G, HG002668 to R.A.Y., and by a Koch Institute-Dana Farber/Harvard Cancer Center bridge project grant to N.S.G. Flow cytometry was supported by support grant P30 CA196521 to the Tisch Cancer Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
S.K., R.A., M.M.S., N.K., B.J.A., S.L., R.A.Y., N.S.G. and R.P.F. designed experiments. S.K., R.A., N.K., B.J.A., Y.L., T.Z., C.M.O. and S.L. conducted experiments. S.K., R.A., N.K., B.J.A and R.P.F. wrote the paper.
Accession Number
The sequencing files have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) (Accession number GSE99794).
Competing Interests
N.S.G, T.Z., N.K. are inventors on a patent application covering THZ1 which is licensed to a company co-founded by N.S.G and R.A.Y.
References
- Adelman K, Lis JT. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet. 2012;13:720–731. doi: 10.1038/nrg3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkowiak B, Greenleaf AL. Expression, purification, and identification of associated proteins of the full-length hCDK12/CyclinK complex. J Biol Chem. 2015;290:1786–1795. doi: 10.1074/jbc.M114.612226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop AC, Shah K, Liu Y, Witucki L, Kung C-y, Shokat KM. Design of allele-specific inhibitors to probe protein kinase signaling. Curr Biol. 1998;8:257–266. doi: 10.1016/s0960-9822(98)70198-8. [DOI] [PubMed] [Google Scholar]
- Bosken CA, Farnung L, Hintermair C, Merzel Schachter M, Vogel-Bachmayr K, Blazek D, Anand K, Fisher RP, Eick D, Geyer M. The structure and substrate specificity of human Cdk12/Cyclin K. Nat Commun. 2014;5:3505. doi: 10.1038/ncomms4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkard ME, Santamaria A, Jallepalli PV. Enabling and disabling polo-like kinase 1 inhibition through chemical genetics. ACS Chem Biol. 2012;7:978–981. doi: 10.1021/cb200551p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Can G, Akpinar B, Baran Y, Zhivotovsky B, Olsson M. 5-Fluorouracil signaling through a calcium-calmodulin-dependent pathway is required for p53 activation and apoptosis in colon carcinoma cells. Oncogene. 2013;32:4529–4538. doi: 10.1038/onc.2012.467. [DOI] [PubMed] [Google Scholar]
- Chao SH, Price DH. Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J Biol Chem. 2001;276:31793–31799. doi: 10.1074/jbc.M102306200. [DOI] [PubMed] [Google Scholar]
- Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, Abraham BJ, Sharma B, Yeung C, Altabef A, et al. CDK7 Inhibition Suppresses Super-Enhancer-Linked Oncogenic Transcription in MYCN-Driven Cancer. Cell. 2014;159:1126–1139. doi: 10.1016/j.cell.2014.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T, Chipumuro E, Herter-Sprie GS, Akbay EA, Altabef A, et al. Targeting Transcriptional Addictions in Small Cell Lung Cancer with a Covalent CDK7 Inhibitor. Cancer Cell. 2014;26:909–922. doi: 10.1016/j.ccell.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar AC, Das TK, Shokat KM, Cagan RL. Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature. 2012;486:80–84. doi: 10.1038/nature11127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhawan NS, Scopton AP, Dar AC. Small molecule stabilization of the KSR inactive state antagonizes oncogenic Ras signalling. Nature. 2016;537:112–116. doi: 10.1038/nature19327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donner AJ, Hoover JM, Szostek SA, Espinosa JM. Stimulus-specific transcriptional regulation within the p53 network. Cell Cycle. 2007;6:2594–2598. doi: 10.4161/cc.6.21.4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drygin D, Lin A, Bliesath J, Ho CB, O’Brien SE, Proffitt C, Omori M, Haddach M, Schwaebe MK, Siddiqui-Jain A, et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011;71:1418–1430. doi: 10.1158/0008-5472.CAN-10-1728. [DOI] [PubMed] [Google Scholar]
- Fisher RP. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J Cell Sci. 2005;118:5171–5180. doi: 10.1242/jcs.02718. [DOI] [PubMed] [Google Scholar]
- Glover-Cutter K, Larochelle S, Erickson B, Zhang C, Shokat K, Fisher RP, Bentley DL. TFIIH-associated Cdk7 kinase functions in phosphorylation of C-terminal domain Ser7 residues, promoter-proximal pausing, and termination by RNA polymerase II. Mol Cell Biol. 2009;29:5455–5464. doi: 10.1128/MCB.00637-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray NS, Wodicka L, Thunnissen AM, Norman TC, Kwon S, Espinoza FH, Morgan DO, Barnes G, LeClerc S, Meijer L, et al. Exploiting chemical libraries, structure, and genomics in the search for kinase inhibitors. Science. 1998;281:533–538. doi: 10.1126/science.281.5376.533. [DOI] [PubMed] [Google Scholar]
- Henry RE, Andrysik Z, Paris R, Galbraith MD, Espinosa JM. A DR4:tBID axis drives the p53 apoptotic response by promoting oligomerization of poised BAX. EMBO J. 2012;31:1266–1278. doi: 10.1038/emboj.2011.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson N, Li YC, Walton ZE, Cheng KA, Li D, Rodig SJ, Moreau LA, Unitt C, Bronson RT, Thomas HD, et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat Med. 2011;17:875–882. doi: 10.1038/nm.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SF, Cruz C, Greifenberg AK, Dust S, Stover DG, Chi D, Primack B, Cao S, Bernhardy AJ, Coulson R, et al. CDK12 Inhibition Reverses De Novo and Acquired PARP Inhibitor Resistance in BRCA Wild-Type and Mutated Models of Triple-Negative Breast Cancer. Cell Rep. 2016;17:2367–2381. doi: 10.1016/j.celrep.2016.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight ZA, Lin H, Shokat KM. Targeting the cancer kinome through polypharmacology. Nat Rev Cancer. 2010;10:130–137. doi: 10.1038/nrc2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511:616–620. doi: 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larochelle S, Amat R, Glover-Cutter K, Sanso M, Zhang C, Allen JJ, Shokat KM, Bentley DL, Fisher RP. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat Struct Mol Biol. 2012;19:1108–1115. doi: 10.1038/nsmb.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larochelle S, Chen J, Knights R, Pandur J, Morcillo P, Erdjument-Bromage H, Tempst P, Suter B, Fisher RP. T-loop phosphorylation stabilizes the CDK7-cyclin H-MAT1 complex in vivo and regulates its CTD kinase activity. EMBO J. 2001;20:3749–3759. doi: 10.1093/emboj/20.14.3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larochelle S, Merrick KA, Terret ME, Wohlbold L, Barboza NM, Zhang C, Shokat KM, Jallepalli PV, Fisher RP. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol Cell. 2007;25:839–850. doi: 10.1016/j.molcel.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- Merrick KA, Larochelle S, Zhang C, Allen JJ, Shokat KM, Fisher RP. Distinct activation pathways confer cyclin-binding specificity on Cdk1 and Cdk2 in human cells. Mol Cell. 2008;32:662–672. doi: 10.1016/j.molcel.2008.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick KA, Wohlbold L, Zhang C, Allen JJ, Horiuchi D, Huskey NE, Goga A, Shokat KM, Fisher RP. Switching Cdk2 on or off with small molecules to reveal requirements in human cell proliferation. Mol Cell. 2011;42:624–636. doi: 10.1016/j.molcel.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan DO. The Cell Cycle: Principles of Control. London: New Science Press Ltd; 2007. [Google Scholar]
- Nilson KA, Guo J, Turek ME, Brogie JE, Delaney E, Luse DS, Price DH. THZ1 Reveals Roles for Cdk7 in Co-transcriptional Capping and Pausing. Mol Cell. 2015;59:576–587. doi: 10.1016/j.molcel.2015.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol Cell. 2006;23:297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Quinlan AR. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr Protoc Bioinformatics. 2014;47:11.12.11–34. doi: 10.1002/0471250953.bi1112s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan Y, Rajpara SM, Reza SM, Lees E, Shuman S, Mathews MB, Pe’ery T. Three RNA polymerase II carboxyl-terminal domain kinases display distinct substrate preferences. J Biol Chem. 2001;276:10913–10920. doi: 10.1074/jbc.M010975200. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Molina JB, Tseng SC, Simonett SP, Taunton J, Ansari AZ. Engineered Covalent Inactivation of TFIIH-Kinase Reveals an Elongation Checkpoint and Results in Widespread mRNA Stabilization. Mol Cell. 2016;63:433–444. doi: 10.1016/j.molcel.2016.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansó M, Fisher RP. Pause, Play, Repeat: CDKs Push RNAP II’s Buttons. Transcription. 2013;4 doi: 10.4161/trns.25146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachter MM, Merrick KA, Larochelle S, Hirschi A, Zhang C, Shokat KM, Rubin SM, Fisher RP. A cdk7-cdk4 T-loop phosphorylation cascade promotes g1 progression. Mol Cell. 2013;50:250–260. doi: 10.1016/j.molcel.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KD, Padilla-Just N, Henry RE, Porter CC, Kim J, Tentler JJ, Eckhardt SG, Tan AC, DeGregori J, Espinosa JM. ATM and MET kinases are synthetic lethal with nongenotoxic activation of p53. Nat Chem Biol. 2012;8:646–654. doi: 10.1038/nchembio.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titov DV, Gilman B, He QL, Bhat S, Low WK, Dang Y, Smeaton M, Demain AL, Miller PS, Kugel JF, et al. XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nat Chem Biol. 2011;7:182–188. doi: 10.1038/nchembio.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topaloglu O, Hurley PJ, Yildirim O, Civin CI, Bunz F. Improved methods for the generation of human gene knockout and knockin cell lines. Nucleic Acids Res. 2005;33:e158. doi: 10.1093/nar/gni160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Viladevall L, St Amour CV, Rosebrock A, Schneider S, Zhang C, Allen JJ, Shokat KM, Schwer B, Leatherwood JK, Fisher RP. TFIIH and P-TEFb coordinate transcription with capping enzyme recruitment at specific genes in fission yeast. Mol Cell. 2009;33:738–751. doi: 10.1016/j.molcel.2009.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang S, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012;28:2184–2185. doi: 10.1093/bioinformatics/bts356. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang T, Kwiatkowski N, Abraham BJ, Lee TI, Xie S, Yuzugullu H, Von T, Li H, Lin Z, et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell. 2015;163:174–186. doi: 10.1016/j.cell.2015.08.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlbold L, Merrick KA, De S, Amat R, Kim JH, Larochelle S, Allen JJ, Zhang C, Shokat KM, Petrini JH, et al. Chemical genetics reveals a specific requirement for cdk2 activity in the DNA damage response and identifies nbs1 as a cdk2 substrate in human cells. PLoS Genet. 2012;8:e1002935. doi: 10.1371/journal.pgen.1002935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Pan J, Thoroddsen V, Wysong DR, Blackman RK, Bulawa CE, Gould AE, Ocain TD, Dick LR, Errada P, et al. Novel small-molecule inhibitors of RNA polymerase III. Eukaryot Cell. 2003;2:256–264. doi: 10.1128/EC.2.2.256-264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Kwiatkowski N, Olson CM, Dixon-Clarke SE, Abraham BJ, Greifenberg AK, Ficarro SB, Elkins JM, Liang Y, Hannett NM, et al. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat Chem Biol. 2016;12:876–884. doi: 10.1038/nchembio.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Sachsenmeier K, Zhang L, Sult E, Hollingsworth RE, Yang H. A New Bliss Independence Model to Analyze Drug Combination Data. J Biomol Screen. 2014;19:817–821. doi: 10.1177/1087057114521867. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1 YKL-1-116 selectivity measured by KiNativ™ kinome profiling (see accompanying Excel file)
Supplemental Table S3 RNAseq data (see accompanying Excel file)