

Abstract
As the catalogue of sequenced genomes and metagenomes continues to grow, massively parallel approaches for the comprehensive and functional analysis of gene products and regulatory elements are becoming increasingly valuable. Current strategies to synthesize or clone complex libraries of DNA sequences are limited by the length of the DNA targets, throughput and cost. Here, we show that long-adapter single-strand oligonucleotide (LASSO) probes can capture and clone thousands of kilobase DNA fragments in a single reaction. As a proof-of-principle, we simultaneously cloned >3,000 bacterial open reading frames (ORFs) from E. coli genomic DNA (spanning 400–5,000 bp targets). Targets were enriched up to a median of ~60-fold compared to non-targeted genomic regions. At a cutoff of 3 times the median non-target reads per kilobase of genetic element per million reads, ~75% of the targeted ORFs were successfully captured. We also show that LASSO probes can clone human ORFs from complementary DNA, and an ORF library from a human-microbiome sample. LASSO probes could be used for the preparation of long-read sequencing libraries and for massively multiplexed cloning.
Introduction
DNA sequencing has created an unprecedented wealth of biological information1. With dramatic advances in parallelization, ‘reading’ DNA sequences has now become orders of magnitude more efficient and cost-effective than ‘writing’ them (i.e. synthesizing or cloning sequences of interest). Consequently, a bottleneck has formed between our knowledge of DNA sequences and our understanding of their functional significance. Massively parallel technologies enabling the synthesis and cloning of long DNA sequences are therefore required to bridge the widening gap from sequence to significance.
Highly multiplexed cloning of long target sequences has not been previously demonstrated. Traditional mutliplexed polymerase chain reaction (PCR) is generally not feasible for this purpose, due to the unpredictable interactions among large numbers of primer sets. The parallel amplification of hundreds of DNA targets (~200 base pairs; bp) in a single reaction using short selector oligonucleotides (~70bp) that act as a template for the circularization of specific target sequences has been shown2.. Despite its usefulness for highly multiplex PCR based enrichment3, this technique cannot be used for the amplification of full length DNA sequences because it relies on the presence of specific restriction enzymes sites in the target sequences. Molecular inversion probes (MIPs) have proven to be a useful tool for short DNA target capture and enrichment, since they exhibit high specificity and can be massively multiplexed.4 MIPs are short single-stranded DNA molecules (~150 bp) that become circularized by gap filling after annealing to target sequences that flank a desired DNA fragment.5, 6 However, traditional MIPs are inefficient at capturing larger target sequences (greater than ~200 bp) due to the persistence length (“stiffness”) of double stranded DNA7 (Supplementary Fig 1). This constraint has prevented their use for the capture of larger fragments and for the cloning of open reading frames (ORFs) that encode full-length proteins or large protein domains. In an attempt to address this target size limitation, increasing the length of the MIP linker backbone has been shown to permit capture of somewhat longer targets (up to ~400 bp).8–10 However, the method used to construct these probes required a separate PCR reaction for each individual probe, thus limiting its scalability. Furthermore, these long MIPs were constructed as double stranded DNA (dsDNA) probes, resulting in the capture of both sense and anti-sense DNA strands9, which is an undesirable feature for certain applications, such as the cloning of ORFs from genomic DNA.
Here, we report the construction and use of Long Adapter Single Strand Oligonucleotide (LASSO) probe libraries (Fig 1), which enable the massively multiplexed capture of kilobase-sized fragments for downstream sequencing or expression. Our approach was developed to permit the assembly of LASSO probes from a complex pool of shorter, synthetic oligonucleotides, which can be readily obtained using programmable DNA microarray synthesis technology.11 Using LASSO probe libraries we show the simultaneous capture of thousands of target ORFs, including those >4 kilobases (kb) in length (10-fold longer than previously reported). LASSO-based ORFeome cloning captured 95% of targeted bacterial ORFs (~3,000 ORFs; over 3 megabases in total) enriching targeted regions over non-targeted regions by an average of up to 300-fold. The functional utility of our cloned ORFs is illustrated by the LASSO-based cloning of a kanamycin gene, which conferred antibiotic resistance to host cells. LASSO cloning can also be used on human cDNA libraries. Finally, the capture and cloning of Escherichia coli (E. Coli) ORFs using human stool derived DNA as input demonstrate the application of LASSO cloning to studies of the human microbiome at the molecular level.
Figure 1. Synthesis of DNA LASSO Probe Components.
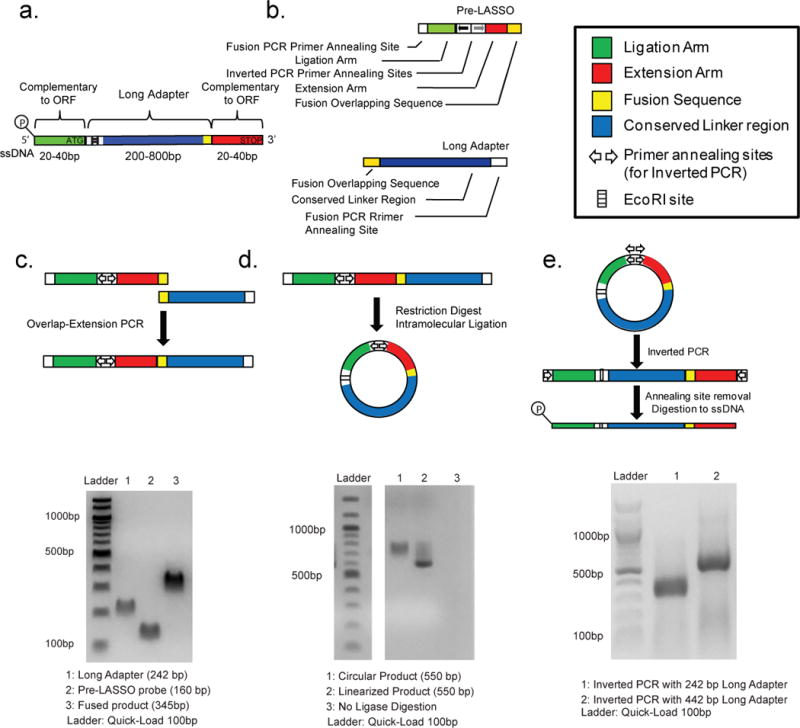
(a) Schematic of a final ssDNA LASSO probe. Two sequences complementary to regions that flank a target are linked to a universal adapter by a series of processing reactions. (b) Schematic of starting components for LASSO probe synthesis, consisting of pre-LASSO probe and a Long Adapter. (c) Schematic of PCR reaction used to fuse the Long Adapter and pre-LASSO probe. Gel electrophoresis results illustrate successful fusion. (d) Schematic of the intramolecular circularization reaction of the fusion PCR product. Gel electrophoresis results illustrate successful, ligation-dependent circularization. (e) Inverted PCR is used to create linear probe precursors. Gel electrophoresis results confirm the product of inverse PCR. A 125 bp pre-LASSO probe was used with either a 220 bp adapter or a 440 bp adapter in the example shown.
LASSO Construction and Single Target ORF Cloning
LASSO probe construction begins with the fusion of a precursor probe (pre-LASSO probe), designed to hybridize with sequences that flank the targeted region, with a common long adapter sequence (Fig 1a–b). The fusion of a long adaptor and a pre-LASSO probe (Fig 1c) occurred with better specificity if the hybridized complex was extended prior to amplification and was robust to varying concentrations of adapter and pre-LASSO probe lengths (Supplementary Fig. 2 a–b). The resulting pre-LASSO fusion product is then circularized (Fig 1d; Supplementary Fig. 3) and subjected to inverse PCR, so that the LASSO annealing arms are made to flank the long adapter sequence in the final configuration (Fig 1e, Supplementary Fig. 4). The external primer sites are next removed and the final single stranded DNA (ssDNA) LASSO probe is produced by exonuclease digestion (Materials and Methods). The final LASSO probe pool is then purified and ready to use in massively parallel target capture reactions.
LASSO probes were first evaluated individually for their ability to clone long DNA targets. The capture reaction involves a multi-step process of annealing, extension, ligation, digestion, and amplification of the probe-target complex (Fig 2a). Starting with a 100bp target, we used single target reactions to determine the optimal conditions for gap filling and ligation (Supplementary Fig. 5). Four LASSO probes (fused with a 442bp Long Adapter) were designed to capture four different target DNA sequences of approximately 0.6 kb, 1 kb, 2 kb, and 4 kb in size, located within the ssDNA genome of the M13 bacteriophage. All four probes were able to capture their targets with high specificity (Fig 2b). Notably, the LASSO probe successfully captured a 4 kb fragment, which is tenfold longer than any previously reported MIP-captured target.
Figure 2. Single ORF target capture with LASSO probes.
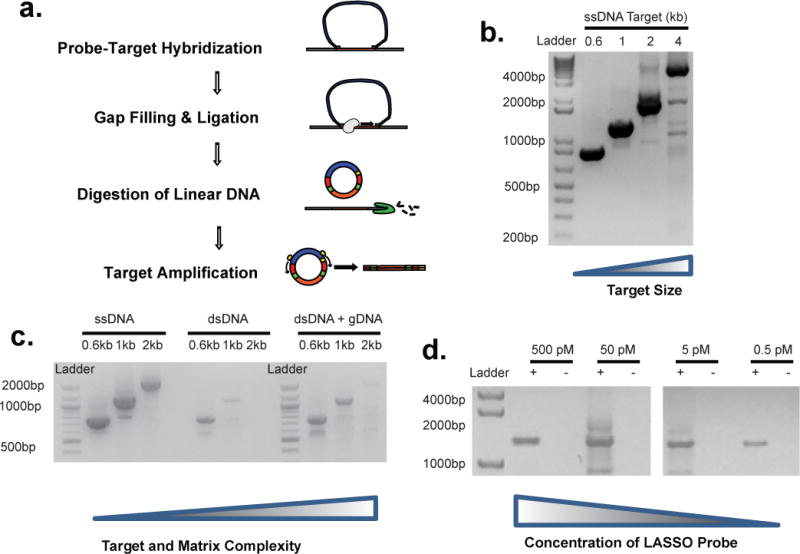
(a) Schematic of single target capture, purification, and amplification. (b) Post capture PCR of circles obtained from the capture of 620 bp, 1 kb, 2 kb, 4 kb target sequences within the M13Mp18 ssDNA genome using 4 different pre-LASSO probes assembled with a 445 bp adapter. (c) Post capture PCR of circles obtained from the capture of 620bp, 1kb, or 2kb sequences using as template ssDNA M13Mp18, dsDNA M13Mp18 amplicon alone, or dsDNA M13Mp18 amplicon in a background of 10 pM sheared E. coli K12 genomic DNA. (d) Post capture PCR of circles obtained by capturing a 1,038 bp target sequence within the M13Mp18 dsDNA (~500 fM) in presence of an equimolar (~500 fM) background of total genomic DNA of E. coli, using serial dilution of a LASSO probes. Negative controls contain sheared gDNA but no target.
To mimic target capture within a complex background of unrelated DNA, we assessed the influence of target DNA strandedness and background matrix complexity. The same concentration of LASSO probe was applied to M13 ssDNA, the corresponding M13 dsDNA target sequence produced by PCR, and M13 dsDNA, in the presence or absence of sheared E. coli whole genomic DNA (gDNA). In the absence of background gDNA, dsDNA target capture was less efficient compared to ssDNA target capture. Efficiency was recovered, however, when the dsDNA template was first melted within a complex background of sheared gDNA (Fig 2c). This finding is consistent with dsDNA target re-hybridization, which is expected to compete with LASSO probe annealing. Next, capture was performed using a dilution series of LASSO probe to test the sensitivity of the reaction, and the feasibility of performing massively multiplexed reactions that include thousands of LASSO probes (individually at low concentration) in the same reaction. A 1 kb dsDNA target sequence (500 fM) was spiked into an equimolar background of E. coli gDNA in order to simulate capture of a single copy target gene. We detected captured product even at the lowest dilution of the LASSO probe tested (500 fM) (Fig 2d, Supplementary Fig. 6). Importantly, “off target” products were not observed when the target sequence was absent from the reaction (but with background gDNA), thus highlighting the specificity of the capture reaction.
We assessed the fidelity of LASSO probe-based cloning, using the kanamycin resistance gene (KanR2, 815bp) as a model target. The KanR2 gene was captured successfully from total gDNA or purified plasmid DNA template (Fig 3a), and cloned via Gibson Assembly into the pET- 21(+) vector. Dual selection for ampicillin resistance (present in the pET- 21(+) backbone) and kanamycin resistance demonstrated that 94% of the captured KanR2 genes were functionally expressed (Fig 3b–c, Supplementary Fig. 7).
Figure 3. Functional assessment of a LASSO-captured ORF target.
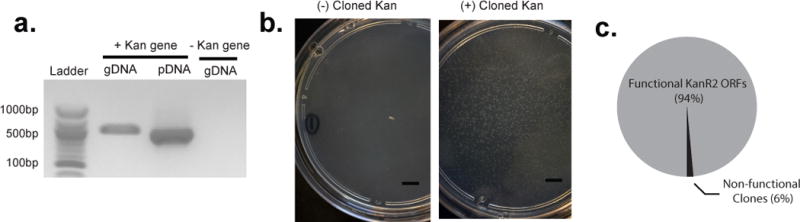
(a) PCR of circles obtained from the capture of Kanamycin resistance determinant (KanR2) from total DNA (gDNA) or plasmid DNA (pDNA). Negative control for capture was total genomic DNA extracted from an E. coli clone without vector. (b) Kanamycin resistant E. coli colonies obtained by cloning the post-capture PCR of KanR2 into a pET21 expression vector and transformation into BL21 Kanamycin susceptible E. coli cells by electroporation. (c) The percentage of functional KanR2 ORFs present in the Kanamycin resistant E. coli colonies.
Multiplex LASSO Cloning of the E. coli ORFeome
We next evaluated the ability of LASSO probes to capture a library of kilobase-sized ORFs from E. coli genomic DNA, using two different adapter lengths (~350 bp and ~550 bp). These results were additionally compared to capture using traditional MIP probes (~120 bp in length). A schematic of the workflow is presented in Fig. 4a. ORFeome cloning is a particularly stringent test of multiplexed long sequence capture, since the design of probes is highly constrained by the target sequences downstream and upstream of each ORF’s start and stop codons, respectively. Using parameters defined by our optimization experiments, we developed a LASSO probe design algorithm, which we used to generate thousands of pre-LASSO probe sequences. Of the 3,999 annotated E. coli K12 (ATCC 27325) ORFs, the algorithm produced 3,664 pre-LASSO probe sequences that satisfied our algorithm’s probe design requirements (~92% of targets). Adjusting the thresholds for target length, melting temperature, or the length of the ligation/extension arms determines the number of acceptable probes. Of the 3,664 acceptable probes, we removed those corresponding to ORF targets smaller than 400bp, as a precaution to avoid potentially skewing our capture library during its subsequent PCR amplification. Approximately 20% of the E. coli K12 ORFeome was thus left untargeted (835 ORFs) and used as an internal, negative control for our experiments. A programmable DNA microarray was used to synthesize the pool of 3,108 × 160 bp pre-LASSO probes (sequences available in Supplementary File ‘pre-LASSO Library’). These precursor probes were then converted into mature LASSO probe libraries of two different lengths (~350 bp or ~550 bp) by fusion with two different length adapters (242bp or 442bp in length, respectively; sequences available in Supplementary Table 1) during probe assembly. A series of library capture optimization experiments were performed using a single adapter and a partial ORFeome LASSO probe library (Supplementary Fig. 8a–c).
Figure 4. Comparison of ORFeome capture using LASSO or MIP probe libraries.
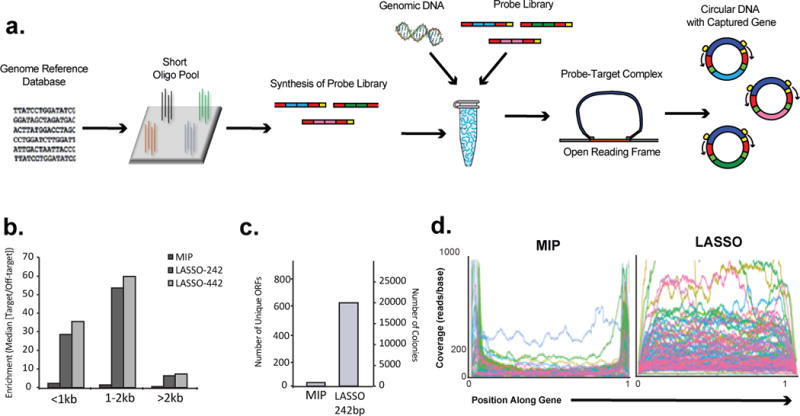
(a) Schematic of workflow of ORFeome capture using LASSO or MIP probe libraries. A genomic database was used to guide the design of the probe library, which was synthesized as pre-LASSO or pre-MIP DNA oligonucleotides on a programmable array. The pre-probe pools were converted into the mature probe pools in pooled format. The libraries of probes were hybridized on target DNA. Closed DNA circles containing captured ORFs were selected by exonuclease digestion, and then PCR amplified using universal primers. (b) Median RPKM enrichment ratios of targeted ORFs versus non-targeted genetic elements for LASSO-242bp, LASSO-442bp and MIP captures. When comparing the enrichment ratios of LASSO probes to those of MIP probes, 100 bases on either end of the ORFs were omitted for computational purposes as described further in Methods. (c) Quantification of unique ORFs cloned and sequenced from MIP and LASSO-242bp capture transformations. (d) Positions of captured reads mapped across the length-normalized target ORFs for LASSO-242bp and MIP captures. All ORFs having size > than 1kb were included in the graphs.
A corresponding library of 3,108 pre-MIPs (~160 bp in length) was obtained from a second programmable DNA microarray and amplified with common primers. The MIPs of this library targeted the same ORFs and were prepared to be identical to the mature LASSO probe libraries described above, aside from the shorter conserved linker (sequences available in Supplementary File ‘pre-MIP Library’). The amplicon corresponded to the expected size (data not shown), and was converted to mature MIPs as described in Materials and Methods.
The E. coli ORFeome capture was performed using the two LASSO probe sets and corresponding MIP probes. The post-capture PCR amplicons were sheared and sequenced on an Illumina NextSeq instrument to obtain 50 nucleotide single end reads. When captures were performed using LASSO probes libraries, more than half of the sequencing reads aligned with the E. coli genome (65% for LASSO-242bp and 50% for LASSO-442bp). In comparison, only 20% of the reads from the MIP capture libraries could be aligned to the E. coli genome; most of the remaining 80% arose from self-circularization reactions (ligation of an extension arm to a ligation arm without an intervening sequence). For reads mapping to the E. coli genome, we calculated target enrichment factors, which we defined as the reads per kilobase of genetic element per million reads (RPKM), which were mapped to the targeted ORFs versus non-targeted ORFs or other genetic elements. Further, RPKM targeted/non-targeted ratios were analyzed for different length genetic elements by binning (Fig. 4b). In this experiment, LASSO targeted ORFs were enriched in all bins (up to ~60x for 1–2 Kb ORFs). Standard MIPs exhibited little to no target enrichment (≤ 2x), regardless of ORF length.
We performed recombination-based cloning on post-capture PCR product in order to move the captured E. coli ORFeome library into the pDONR221 entry vector. We obtained far more colonies when using LASSO capture material compared to MIP capture material (~20,000 versus hundreds); the same was true for complete coverage of unique target ORFs observed by sequencing (640 versus 14; Fig. 4c).
For the LASSO-242bp and MIP capture libraries, we plotted the frequency of mapped sequence reads according to their normalized positions within the corresponding target for all ORFs >1kb in length in the LASSO-242bp capture library (Fig. 4d). LASSO captured sequences were uniformly distributed across the full-length of target ORF. In contrast, MIP captured sequences were strongly enriched at the end of the ORFs, suggesting that MIP capture products were largely derived from incomplete or misprimed polymerase extensions that were then able to ligate to the ligation arm and form closed circular products. Importantly, the size distribution of the LASSO capture amplicon corresponded well with that of the targeted ORFs (Supplementary Figure 9). In summary, our data (lack of FPKM target enrichment, poor fragment distribution, and low cloning yield) indicate that traditional MIPs cannot be used for cloning of kilobase-sized DNA libraries.
Using optimized capture conditions, we repeated the capture of the E. coli ORFeome using the LASSO-242bp probe library. The products of post-capture amplification are shown in Fig. 5a. Their apparent size distribution corresponded well with that of the targeted ORFs. These PCR amplicons were sheared and sequenced on an Illumina HiSeq instrument (50bp single-end reads). Fig. 5b illustrates the distribution of read counts per kilobase for each targeted ORF, each untargeted ORF >400 bp, and each intergenic region >400 bp. Targeted ORFs were significantly enriched compared to non-targeted ORFs and intergenic regions (p < 2.2 × 10−16 by Welch two sample t-test; no significant difference was observed between non-targeted ORFs and intergenic regions). The mean and median RPKM of the targets were 465.3 and 50.3, respectively, whereas the mean and median RPKM of the non-targeted genomic regions were 2.9 and 0, respectively; fold-enrichment of targets was calculated to be between 17.5- and 160-fold (by median or mean of target RPKM, respectively, over the mean non-target RPKM). At a cutoff of 3x the median non-target RPKM, ~75% of targeted ORFs were successfully captured. There was an excellent positive predicted value (AUC = 0.959) for LASSO targets as a function of normalized read depth by ROC analysis (Fig. 5c). In terms of absolute sequence coverage, a majority of the targeted ORF sequences were fully covered by the mapped reads, whereas close to none of the non-targeted sequences were fully covered (Fig. 5d). We observed a negative correlation between the normalized abundance of each target ORF and its length; ORF representation was observed to decline by 60% with each doubling of length (Fig. 5e). This bias may reflect target length-dependent capture efficiency, post capture PCR bias, or a combination of the two effects. Importantly, however, 89.4% of the cloned library was present within 10-fold normalized abundance of the median, indicating a relatively uniform representation of the captured ORFs. Using a >2 kb target probe subpool from the original pre-LASSO library, we observed a much more homogeneous distribution of ORF abundance, as expected (Fig 5f). Target size-based subpooling may be a useful strategy for creating more uniform capture libraries. The integrity of several ORFs was additionally confirmed by Sanger sequencing of capture library clones. An abridged sequence of the start and stop regions of a representative cloned ORF is shown in Fig 5g. As shown, the sequence contains the long adapter between the primer used for post capture PCR and the ligation arm, the ATG start codon followed by the complete captured ORF, and the sequence of the long adapter between the STOP codon and the primer used for PCR. These data provide additional evidence that the cloned sequences are indeed derived from the desired LASSO capture product.
Figure 5. Multiplex capture and sequencing of an E. coli ORFeome library.
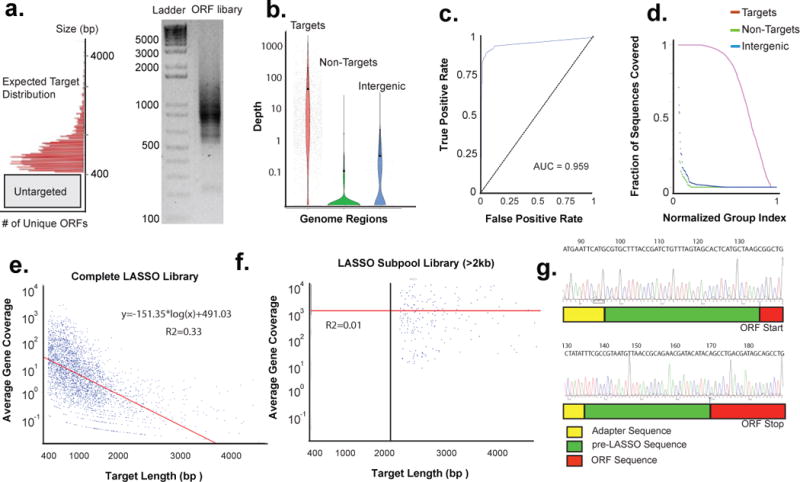
(a) Post capture PCR of circles obtained from the capture of 3,164 ORFs of E. coli K12 performed by using the LASSO probe library assembled with a 242 bp adapter. The inset is a histogram denoting the size distribution of the targeted ORFs split into bin size of 40 bp. Targeted ORFs will have an increase in 140 bp of residual LASSO sequences once captured and run on a gel. (b) Average depth of sequencing per kilobase for each targeted ORF, non-targeted ORF >400bp, and intergenic region >400bp. All sequences with depth <0.01 are displayed on the 0.01 floor of the graph. (c) Receiver operating characteristic (ROC) of the normalized read depth data shown in 3c as a function of read depth. (d) Fractional base coverage across the targeted ORFs, non-targeted ORFs >400bp, and intergenic regions >400bp from the E. coli genome. Normalized read depth as a function of the length of the ORF is shown in (e) for the complete LASSO probe library or in (f) for the >2kb LASSO probe subpool library. Red line illustrates a linear regression of the logged sequencing depth as a function of target length. (g) Sanger sequencing analysis of a randomly selected E.coli clone obtained from the capture library (NP_414738.1). The top inset shows the long adapter sequence, the ligation arm of the LASSO probe, and the start codon of the ORF. The bottom inset shows the end of the long adapter sequence, the extension arm of the LASSO probe, and the stop codon of the ORF.
LASSO Cloning of Human and Commensal ORFs
Finally we evaluated the utility of LASSO cloning in the context of two important biomedical applications: (i) the cloning of a human ORFeome from cDNA and (ii) the capture of bacterial ORF libraries from human gut microbiome samples. Using capture conditions optimal for massively multiplexed cloning, we evaluated the ability of LASSO probes to capture two individual full length ORFs from a mammalian cell-derived cDNA library. TP53 and PRLP0 were successfully captured in this manner (Fig. 6a), as confirmed by Sanger sequencing, thus demonstrating the potential utility of our method for the multiplexed construction of human protein expression libraries. We also applied the E. coli LASSO-242bp probe library (designed using a K12 reference strain) to DNA extracted from a human stool sample. Given the extreme complexity of this DNA sample, which likely includes hundreds of bacterial species and host gDNA, one might expect increased off-target capture. To control for this, we performed a parallel capture from the same microbiome sample, but using the MIP probe library instead. The capture amplicons obtained from LASSO-242bp and MIP libraries showed band patterns consistent with a successful and unsuccessful capture, respectively (Supplementary Figure 10). The amplicons were cloned in pDONR.221 and electroporated into E. coli cells as described before. MIP based cloning produced hundreds of colonies, while with LASSO produced several thousands (Fig 6b). Quantification of the colonies and NGS analysis of the pDONR plasmids from E. coli colonies recovered from agar plates revealed that 1,129 ORFs from E. coli K12 were captured with various degrees of coverage (Fig 6c). The top 500 ORFs sequenced from pDONR plasmids showed an approximate 2x enrichment (Fig 6d, median RPKM target = 11.41, median RPKM non-target = 5.23) suggesting that while the LASSO cloning worked, this experiment was likely impacted by the extreme complexity of the sample. Using the MIP library in parallel, we only detected a few ORFs from E. coli, all at very low coverage, indicating that the LASSO library was effectively able to capture and clone ORFs from a microbiome sample.
Figure 6. LASSO-based cloning and characterization of full-length ORFs from human cDNA or DNA isolated from a human microbiome sample.
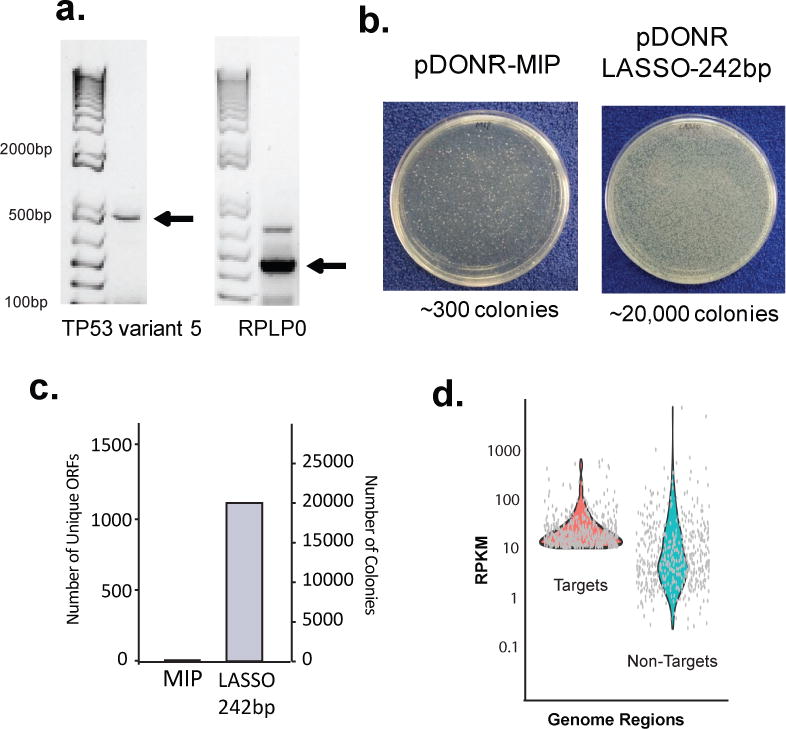
(a) Capture of Human ORFs from a mammalian cDNA library. Post capture PCR of tumor suppressor TP53 variant 5 (783 bp) (left), and housekeeping RPLP0 internal fragment (323 bp) (right) captured from Jurkat E6-1 cDNA. Note there is the addition of 142 bp from linker for each cDNA captured. (b) E. coli colonies transformed with captured ORFs using LASSO-242bp or MIP libraries and cloned into the pDONR211 vector. (c) Number of detected ORFs from LASSO-242bp-pDONR211 and MIP-pDONR211 captured from DNA isolated from a human stool sample. Number of colonies from the transformations as in (b). (d) Average depth of sequencing, analyzed as target reads per kilobase per million reads, for each targeted ORF, or non-targeted genomic region >400 bp. Sequencing depth of top 500 targeted and non-targeted ORFs in the LASSO-242bp-pDONR211 stool DNA-captured library.
Outlook
We have demonstrated that LASSO probes can be used to clone thousands of kilobase-sized DNA fragments from a prokaryotic genome in a single reaction (over 3 megabases in this study), and that this technique can be adapted to cDNA-derived expression libraries, as well as ORFeome cloning from microbiome samples. Conventional MIPs, by comparison, were unable to demonstrably capture ORF targets >400 nt in length. LASSO cloned ORFs include their native start and stop codons, while maintaining their intended reading frames; resulting libraries can thus be expressed using standard vectors for functional biomedical screening applications. By design, new libraries of protein domains (e.g. extracellular, catalytic, DNA binding, etc.) may be produced in this way. LASSO probe-based cloning may also be used to construct libraries of promoters, enhancers, lncRNAs, untranslated regions of mRNAs, etc., for use in high throughput studies of gene expression.12 We expect that the ability to produce inexpensive large-fragment DNA libraries will find many additional applications, including the targeted construction of long-read sequencing libraries, or the assembly of chromosome-scale synthetic DNA fragments.13
Methods
Pre-LASSO Probes and Long Adapters
Pre-LASSO probes were obtained as double-stranded DNA oligonucleotides (IDT GBlocks) or as pools of single stranded DNA oligonucleotides derived from programmable DNA microarray (Custom Array Inc.). The pre-LASSO probes were approximately 160 bp long. For single LASSO probes and for the 3,108 E. coli K12 ORF LASSO library subpool the design was: 5′-GAGTATTACCGCGGCGAATTC, ligation arm (variable), AACACTTCTTGCGGCGATGGTTCCTGGCTCTTCGATC, extension arm (variable), AGAGAAGTCCTAGCACGGTAACC-3′. For the E. coli LASSO library sub pool (ORFs>2kb) the design was: 5′-CGGTGCTGACGATGCCGAATTC, ligation arm (variable), AACACTTCTTGCGGCGATGGTTCCTGGCTCTTCGATC, extension arm (variable), TGCTGCTTGGATGCGTTAAATGG-3′.
The ORFs of the E. coli K12 genome that are longer than 400 nucleotides were targeted with ligation and extension arms positioned at the beginning and end of the sequences respectively and extended until the desired melting temperature was reached. Specifically, the algorithm first selected the ORF’ leading and trailing 32-mer sequences for the two arms and that the melting temperature for the ligation and extension arms were between 65 °C and 85 °C and 55 °C and 80 °C respectively. If at least one of these conditions were not satisfied, the algorithm increased the length of the arms by one nucleotide and re-tested the conditions until they are satisfied or the end of the ORF is reached. Since an EcoRI digestion step was used to assemble the LASSO probes, the algorithm discarded the design of pre-LASSO probes where an EcoR1 restriction site was present in the ligation or extension arm. The full table of the ORFs with valid ligation and extension arms and the corresponding pre-LASSO probes (subpool > 400bp) and for the pre-LASSO subpool that only included only the ORFs > 2kb (subpool > 2kb) are reported in Supplementary File 1
The Long Adapters (242 bp and 442 bp) were obtained by PCR performed by using tailed primers and the plasmid pCDH-CMV-MCS-EF1-Puro (System Bioscience) as template. The forward primer used for PCR was FusionBlaF and was the same for Long Adapter 242bp and Long Adapter 442bp. The reverse primers were RFP200EcoR1 for Long Adapter 242 bp and RFP400EcoR1 for Long Adapter 442 bp. For the E. coli K12 LASSO library subpool that included only the ORFs >2kb the forward primer used for PCR was Fusion2kbF and was the same for Long Adapter 242 bp and Long Adapter 442 bp, The reverse primers were RFP200EcoR1 for Long Adapter 242 bp RFP400EcoR1 for Long Adapter 442 bp. PCR reactions were performed In 25 μl of 1X Klentaq Mutant Buffer containing 0.2 μl of Omni Klentaq LA (DNA Polymerase Technology), 0.4 μM of each primer, dNTPs 200 μM and 10ng of pCDH-CMV-MCS-EF1-Puro plasmids. The PCR program was initiated for 5 min at 95°C; thirty cycles of 15 sec at 95°C, 20 sec at 55°C, and 40 sec at 72°C; and 5 min at 72°C. The PCR products was loaded in an 1% agarose gel and DNA band correspondent to the expected size of the Long Adapters were cut and purified from the gel using Wizard SV Gel and PCR Clean-Up System (Promega, USA). The sequences of the 242 bp, 442 bp, Long Adapters are listed in Supplementary Table 1 all DNA sequences of primers are listed in Supplementary Table 2
LASSO probe assembly
Fusion PCR
The fusion PCR reactions was performed in 25 μl containing 2.5 μl of Klentaq Mutant Buffer 10X, 0.6 μl of dNTPs 10 mM, 0.2 μl of Omni Klentaq LA (DNA Polymerase Technology), ~20 ng of pre-LASSO probe (a single dsDNA pre-LASSO probe or a pool of ssDNA pre-LASSO probes), ~20 ng of Long Adapter. The solution was denatured 4 min at 95 °C and subjected to 10 thermal cycles as follow; 15 sec at 95 °C, 20 sec at 50 °C, 40 sec at 72 °C. After the 10 cycles the PCR was stopped and 1 μl of 10 μM Fusion forward primer BLAF and 1 μl of 10 μM Fusion reverse primer RFPR200EcoR1 for Long Adapter 242bp or RFPR400EcoR1 for Long Adapter 442bp, were added. To obtain the fusion of the Long Adapter with the subpool for ORF > 2Kb we added 1 μl of 10 μM Fusion forward primer SubPool2kbF and 1 μl of 10 μM Fusion reverse primer RFPR200EcoR1for Long Adapter 242bp or RFPR400EcoR1 for Long Adapter 442bp. The PCR was continued for 30 more cycles: 15 sec at 95 °C, 20 sec at 50 °C, 40 sec at 72°C. Fusion PCR products were subjected to agarose gel electrophoresis (1.1% agarose). Amplicons corresponding to the expected sizes of the fusion PCR products were purified using QIAquick Gel Extraction Kit (Quiagen) or Wizard SV Gel and PCR Clean-Up System (Promega) and eluted in 50 μl of water.
Self-circularization
The approximately 45 μl solution containing gel purified LASSO fusion PCR products as described above were digested by adding 5 μl of EcoRI 10X buffer and 1μl (20 units/μl) of EcoRI restriction enzyme (NEB) for 1h at 37°C followed by 10′ at 80°C. The digested DNA was purified using AMPure beads (1.4X and washed twice with 70% ethanol) and eluted in 40 μl of water. Self-circularization was performed in a total volume of 2 ml of 1X T4 Ligase Buffer (NEB) containing approximately 5 ng of EcoRI digested fusion PCR product and 400 units of T4 DNA ligase; DNA ligase was added last. Ligation was performed in a 15 ml conical tube in a cold water bath (16 °C) for 16 hr and then concentrated to a volume ~ 20μl in a Savant SpeedVac concentrator (Thermo Scientific). The concentrated DNA was adjusted to 100 μl final volume by adding water, purified with AMPure beads (1.4X and washed twice with 70% ethanol), and finally eluted into 50 μl water. Uncircularized linear DNA was digested by adding 2 μl of solution containing 1 μl of Lambda Exonuclease (5U/μl) and 1 μl of Exonuclease I (20 U/μl) (both from NEB) directly into the 50 μl volume containing the self-circularized DNA. Digestion was performed at 37 °C for 30 min followed by 20 min at 80°C.
Inverted PCR
Inverted PCR was performed in a 25 μl total volume containing 10 μl of the circularized LASSO precursors as described above, 2.5 μl of Klentaq Mutant Buffer 10 X, 0.2 μl of Omni Klentaq LA (DNA Polymerase Technology), 0.6 μl of dNTPs (NEB), 1 μl of 0.4 μM reverse primer TiolNew and forward primer SapINew. Both SapI and TiolNew anneal with opposite orientations in the conserved central section of the pre-LASSO probe (AACACTTCTTGCGGCGATGGTTCCTGGCTCTTCGATC). TiolNew includes a xxxx base to prevent digestion during subsequent ExoI treatment, and a 3′-terminal uracil base for subsequent primer removal using Uracil-DNA Glycosylase (USER enzyme); SapINew includes the SapI (Type IIS side cutting restriction enzyme) site for primer removal via digest with the isoschizomer BspQI. The PCR thermal profile was 4 min at 95 °C; thirty cycles of 10 sec at 95 °C, 20 sec at 55 °C, 40 sec at 72 °C; 4 min at 72 °C.
The inverted PCR product was subsequently purified by using AMPure beads (1.4 ×), washed with 70% ethanol twice and eluted with 40 μl of nuclease free water. The concentration of purified inverted PCR product was measured by Nanodrop.
Production of mature LASSO probes
Approximately 1 μg of purified Inverted PCR product were digested by adding 4 μl of CutSmart buffer 10 × (NEB) and 1 μl of BspQI restriction enzyme (NEB). Digestion was performed at 50 °C for 1h followed by 20 min at 80 °C. After digestion, 1 μl (5 units) of Lambda exonuclease (NEB) was added directly to the BspQI digested DNA and incubated for 30 min at 37 °C followed by 10 min at 80 °C. At this point, 2 μl (1 unit/μl) of USER enzyme (NEB) were added in solution and incubated for 30 min at 37 °C. The mature ssDNA form of LASSO probes were purified using AMPure beads (1.4 × and washed twice with 70% ethanol) and eluted in 40 μl of water. The final concentration of mature ssDNA LASSO probes was determined by Nanodrop.
MIP probe library assembly
The MIP library was designed and synthesized with flanking adapters for PCR amplification and primer removal as above. The pre-MIP probes were approximately 160bp long and had this design: 3′-ATCGCCGCAAGAAGTGTT, ligation arm (variable), TGAGATTTAAGGTCAAGATGGAGGGAAGCGCTCCCCTTCTCCTGGGATATTCTG, extension arm (variable), GATCGAAGAGCCAGGAACC-5′. The ligation and extension arms of MIP probes were identical to ligation and extension of LASSO probes. The sequences of the pre-MIP probes are available in Supplementary File 1.
The pre-MIP library was PCR amplified with primers TiolNew and SapINew. PCR reaction was performed in 25 μl of 1X Klentaq Mutant Buffer containing 0.2 μl of Omni Klentaq LA (DNA Polymerase Technology), 0.4 μM of each primer, dNTPs 200 μM and 130 ng of ssDNA pre-MIP probes. The PCR program was initiated for 5 min at 95°C; thirty cycles of 15 sec at 95°C, 20 sec at 55°C, and 40 sec at 72°C; and 5 min final extension at 72°C. PCR product was purified using Agencourt® AMPure XP (Beckman Coulter and eluted with 40 μl of nuclease free water. The concentration of the purified PCR product was measured by Nanodrop (Thermo Fisher Scientific). Approximately 350 ng of purified PCR product above were digested by adding 4 μl of CutSmart buffer 10 X (NEB) and 1 μl of BspQI (10 units) restriction enzyme (NEB). Digestion was performed at 50 °C for 1h followed by 20 min at 80 °C. After digestion, 1 μl (5 units) of Lambda exonuclease (NEB) was added directly to the BspQI digested DNA and incubated at 37 °C for 30 min followed by 10 min at 80 °C. Finally, 2 μl (1 unit/μl) of USER enzyme (NEB) were added and incubated at 37 °C for 30 min. The mature ssDNA MIP probes were purified using AMPure beads (1.4 X), washed twice with 70% ethanol and eluted in 40 μl of water. The concentration of the mature ssDNA MIP library was determined by Nanodrop.
DNA templates used in capture experiments
For LASSO probe capture optimization experiments, we used a 7,249 bp circular, single-stranded DNA isolated from the M13mp18 phage (NEB) or alternatively the double-stranded, covalently closed, circular form of DNA derived from bacteriophage M13 (NEB). For capture experiments of E.coli ORFeome by MIP or LASSO probes, total genomic DNA of the E.coli strain K12 substrain W3110, (Migula) Castellani and Chalmers (ATCC 27325) was extracted from 500 μl of LB broth (Sigma Aldrich) overnight culture using Charge Switch gDNA Mini Bacteria Kit (Life Technology). Sheared total genomic DNA of E.coli K12 was obtained by sonicating 1 μg of total DNA in a volume of 200 μl in a 1.5 ml Eppendorf tube on ice by using a Branson sonifier 450 (VWR scientific) at output control 2, duty cycle 50% for 40 sec. For the capture of the 815 bp long kanamycin resistance gene KanR2, we used total DNA of the E.coli clone n 29664 (Addgene) that contained the pET StrepII TEV LIC cloning vector harboring KanR2 gene.
E. coli ORFeome Capture
LASSO libraries or the MIP library were hybridized on E.coli gDNA. The hybridization was performed in 15 μl of 1X Ampligase DNA Ligase buffer (Epicentre) containing: 250 ng of unsheared or 250 ng of sheared E.coli K12 total genomic DNA and 5 ng LASSO-242bp or 9 ng of LASSO-442bp or 1.7ng of MIP library. In the hybridization volume the concentration of E. coli chromosomes was approximately 10 pM. The concentration of individual LASSO probes or MIP was approximately 14 pM (44 nM for the complete LASSO or MIP libraries). The solution (15 μl) containing the MIP or LASSO probe pool and the E. coli DNA was denatured for 5 min at 95 °C in a PCR thermocycler (Eppendorf Mastercycler), then incubated at 65 °C for 60 min. After hybridization, 5 μl of freshly prepared gap filling mix were added into the hybridization solution, while maintaining the reaction at 65 °C in the thermocycler. Gap filling and ligation was performed for 30 min at 65 °C. After capture, the DNA samples were denatured for 3 min at 95 °C, and the temperature reduced to 37 °C. 4 μl Linear DNA Digestion Solution was added immediately. Digestion was performed for 1 h at 37 °C, followed by 20 min at 80 °C. After digestion, the capture reaction was purified using AMPure beads (1.8 X and washed with 70% ethanol) and eluted in 25 μl of DNAse free water.
Gap Filling Mix was prepared fresh for each capture experiment and the composition for 50 μl of gap filling mix was: 2 μl of 1 mM dNTPs, 1 μl of Ampligase DNA Ligase (5 U/μl), 2 μl of Omni Klentaq LA that was previously diluted 1/10 in 1X Ampligase DNA Ligase Buffer, 5 μl of 10X Ampligase DNA ligase Buffer, 40 μl of DNAase free water. Linear DNA Digestion Solution (volume of 48 μl) was composed of 24μl of nuclease free water, 6 μl of Exonuclease I (20 units/μl), 6 μl of Exonuclease III (100 units/μl), 6 μl of Lambda Exonuclease (5 units/μl) and 6 μl Exonuclease VII (10 units/μl) (all from NEB).
Capture of DNA targets from phage M13 using single LASSO probes
The capture of the 620 bp, 1 kb, 2 kb and 4 kb target sequences located in the DNA of the phage M13 were performed with the same gap filling mix composition and the same thermal profile for hybridization and capture used for the LASSO probe libraries as described above. We used approximately 0.3 fmol of single LASSO probes, and 4 fmol of M13Mp18 dsDNA or ssDNA. The E.coli k12 total genomic DNA background was 10 pM (500 ng DNA in15 μl capture volume).
For the LASSO probe sensitivity test, E. coli K12 total genomic DNA background was ~500 fM (25 ng in15 μl capture volume). The concentration of M13Mp18 dsDNA was ~500 fM (0.03 ng in 15 μl). The serial dilution concentration of the LASSO 1kB probe were 500 pM, 50 pM, 5 pM and 500 fM.
Capture of KanR2 gene was performed by using 20 ng of total genomic DNA of E.coli clone n 29664 (Addgene) 3 fmol of LASSO probe KnaR2 (pre-LASSO KnaR2 assembled with 442 bp Long Adapter). Capture was performed using the same gap filling mix and thermal profile used for the LASSO probe pool. The DNA sequences of single pre-LASSO probes are in Supplementary Table 3.
Capture E.coli ORFs from human gut microbiome
Approval for the collection of stool from healthy volunteers was obtained from the Institutional Review Board of the University of Trento, Italy. Total gDNA was extracted from 200 mg of stool from a healthy human donor by using a QIAamp DNA Stool Mini Kit (Qiagen) as described by the vendor. For the captures, we used 500 ng of unsheared total gDNA recovered from stool and 5 ng of MIP or LASSO probes pools. Hybridization and capture were performed as described above.
Capture of human ORFs using single LASSO probes from human cDNA
To capture human ORFs, total RNA of Jurkat E6-1 (ATCC TIB-152) cells was extracted using miRNeasy Micro Kit (Qiagen). First-strand cDNA synthesis was performed using SuperScript VILO cDNA synthesis kit (Thermo Fisher Scientific). cDNA was treated with RNase H (New England Biolabs) and purified using Agencourt AMPure XP beads (Beckman Coulter). Capture of TP53 transcript variant 5 and RPLP0 internal fragment, respectively, were performed using 50 ng of Jurkat E6-1 total cDNA, 0.02 ng of LASSO probe TP53 Δ133α Anti or RPLP0 Anti (pre-LASSO probes assembled with 242 bp Long Adapter). Hybridization and capture were performed as described above and the sizes of the captured ORFs were verified by gel electrophoresis. To confirm the identities of those captured ORFs, the captured TP53 Δ133α and RPLP0 were cloned into pMiniT vector (NEB) by using NEB PCR cloning kit. The purified colony PCR products from the single transformants containing TP53 Δ133α or RPLP0 in pMiniT were analyzed by Sanger sequencing. The DNA sequences of single pre-LASSO probes are in Supplementary Table 3
Post Capture PCR
The captured ORFs were amplified using 5 μl of the capture reaction containing DNA circles in 25 μl of PCR master mix composed of 0.1 μl of Omni Klentaq LA, dNTPs 200 μM, and 0.4 μM of primers that annealed in the Long Adapter sequence or in the MIP linker region. For LASSO-242 bp and MIP library the primers for post capture PCR were: ICeuI200CaptF and PCR1kbCaptR200 For LASSO-442 bp the primers for post capture PCR were PCR1kbCaptF400 and PCR1kbCaptR200. The PCR thermal profile was 5 min at 95 °C; 30 cycles of 15 sec at 95 °C, 15 sec at 55 °C, and 2 min at 72 °C; final extension 5 min at 72°C. To visualize the amplicons derived from the circles, 3 μl of PCR products were loaded in a 1.1% agarose gel containing GelGreen Nucleic Acid Gel Stain (Biotium) (0.2 μg/ml).
Cloning into pDONR211
Post capture PCR amplicons obtained from LASSO or MIP captures, were subjected to a second PCR amplification using tailed primers containing Gateway attB1 and attB2 sequences. PCR was performed by using 5 ng of post capture PCR amplicons as template in 25 μl of PCR master mix composed of 0.1 μl of Omni Klentaq LA, dNTPs 200 μM, and 0.4 μM of attB-primers. For LASSO-242bp and MIP, the primers were: attB1Capt200F, attB2Capt200R. For LASSO-442bp, the primers were: attB1Capt400F, attB2Capt200R. Primers sequences available in Supplementary Table 2. The PCR amplification products were purified by using AMPure beads (1.8 X and washed with 70% ethanol) and eluted in 25 μl of DNAse free water. Approximately 60 ng of the PCR product was mixed with the Gateway “Donor vectors” (pDONR221) and the BP Clonase enzyme mix (Invitrogen) as described by the vendor and incubated at 25 °C overnight. After the overnight incubation, the BP reaction was purified using AMPure beads (1.8X and washed with 70% ETOH) and eluted in 10 μl of DNAse free water. Two μl of the purified BP reaction were added to 25 μl of electrocompetent 5-alpha E.coli cells (NEB) into a chilled 1mm cuvette (BioRad) and electroporated using Micro Pulser (BioRad). The transformed cells were recovered in 500 μl SOC medium and were all plated on a 150 mm petri dish containing 50 μg/ml Kanamycin. The numbers of single colonies were estimated by serial dilutions. All colonies from the petri dish were recovered, resuspended in 5 ml LB medium, and subjected to plasmid extraction (Miniprep Kit, Qiagen). The DNA concentration was measured by Nanodrop.
Expression cloning of KanR2 gene
Post capture PCR amplicons were cloned via Gibson Assembly in the vector pET- 21(+) (Novagen) that was previously linearized by PCR using tailed-primers pET21RGibson and pET21FGibson. Gibson Assembly reaction was performed as described by the vendor (NEB). Transformation of BL21 electrocompetent E.coli cells (Sigma) was performed using a 0.1 cm cuvette (Bio Rad) and a Bio Rad Micro Pulser. E. coli transformed clones were selected with agar plates containing ampicillin (100 μg/ml).
Sanger sequencing of colony PCR products for KanR2
Post capture PCR products were cloned into pMiniT (NEB) by using NEB PCR cloning kit and used to transform chemically competent NEB 10-beta E. coli cells (NEB) as described by the vendor. Sixty-six single colonies of transformed E. coli clones were picked from selective plate containing ampicillin (100 μg/ml). The presence of DNA inserts was determined by using the colony as DNA template for PCR with the primers provided with the kit. PCR product (5 μl) were visualized by agarose gel electrophoresis and purified using AMPure beads. Sanger sequencing of colony PCR amplicons was performed by capillary electrophoresis on the 96-well capillary matrix of an ABI3730XL DNA Analyzer.
NGS sequencing (NextSeq) of capture libraries
Illumina library construction
Post capture PCR products (25 μl) were purified using magnetic beads Agencourt AMPure XP system and eluted in 40 μl of water. The captures cloned into pDONR211 vector were minipreped and eluted into 50 μl water. The DNA concentration was measured by Nanodrop. Purified Post capture PCR (500 ng) or the captures in pDONR211 (500 ng) were collected, brought to 80 μl with nuclease free water and sonicated in an eppendorf tube on ice using a Branson sonifier 450 at output control 4.5, duty cycle 50% for 60 sec. The sheared DNA (55.5 μl) was subjected to end repair, 5′ phosphorylation, dA-tailing and Illumina adaptor ligation using the NEBNext Ultra DNA Library Prep Kit for Illumina (NEB) as described by the vendor. PCR enrichment of adaptor ligated DNA was performed using NEBNext Multiplex Oligos (NEB) with index primers. Thermal profile was: 30 sec at 98 °C, 8 cycles of 10 sec at 98 °C, 75 sec at 63 °C, and, 5 min at 72°C. PCR products were finally purified using Agencourt AMPure XP system as described in the NEB protocol. The quality of the Illumina library was verified by checking the size distribution on an Agilent Bioanalyzer using a high sensitivity DNA chip. The concentration of the Illumina library was measured by qPCR using the NEBNext Library Quant Kit for Illumina (NEB). DNA sequencing was performed by using the Illumina HiSeq or NextSeq instruments and standard reagents (Illumina).
NGS computational analyses
To check if the sequencing and sample preparation went well, we did the quality check on the raw data using FastQC tool (version 0.11.5). The low quality read trimming along with adapter clipping was performed using Trimmomatic version 0.36. Then the resulting fastq files from trimmomatic output were mapped against a reference genome sequence using BowTie2. The reference genome used was E. coli K12. For the human stool sample analyses, we did 2 different mappings, one with the original K12 and the other using the commensal strains in K12, HS, Nissle 1917, HS, CTF073, UTI189. Using the Samtools, we filtered the reads to include only those satisfying MAPQ of at least 30 and then sorted the resulting bam file. Since the probes were made for 3108 genes that satisfy the requirements of the current protocol, we considered these genes as targets. The rest of the genes, along with the intergenic regions are considered as non-targets.
When comparing the enrichment ratios of LASSO probes to those of MIP probes (Fig 4b), we trimmed 100 bases on either end of the ORFs during data pre-processing, as the MIP capture products were largely derived from incomplete or misprimed polymerase extensions that were then able to ligate to the ligation arm and form closed circular products. For ROC, we considered targets as true positives and non-targets as false positives. For each of these regions i.e., targets and non-targets, we created 2 bed files separately using the custom scripts. We then used BEDtools to estimate the depth of the regions. We then estimated the RPKM for each of these regions. Using RPKM allows us to easily compare the metrics across different samples that might have genes sequenced at different depths, and different number of total reads. We also calculated the fraction coverage i.e., fraction of the bases covered in the regions of interest. We repeated all the above steps for LASSO and MIP analyses of E. coli, pDONR and human stool sample. We then looked at various analyses i.e., ROC, plot fraction coverage for targets and non-targets, scatter plots of depth and RPKM vs length etc. We have prepared an R markdown for all the above analyses.
Statistical analysis
All data are presented as mean or median ± standard error of the mean (SEM), as stated in the figure legends. Statistical significance was assessed using Student’s t-test for pair-wise comparison, and 1-way ANOVA for comparison between multiple (≥3) conditions; p<0.05 was considered significant.
Supplementary Material
Acknowledgments
This work was supported in part by the Shriners Hospitals for Children (B.P, L.T.), a Prostate Cancer Foundation Young Investigator award (H.B.L.), National Institutes of Health Grants R01EB012521 (B.P.), K01DK087770 (B.P.), and 1U24AI118633 (H.B.L.).
Footnotes
Data Availability: The authors declare that all data supporting the findings of this study are available within the paper and its Supplementary Information. NGS data of the captures performed with LASSO and MIP libraries are available at https://www.ncbi.nlm.nih.gov/sra/?term=SRP079989
Code Availability: All the parameters used in trimmomatic, bowtie2, samtools along with custom based scripts used with BEDtools and the R markdown are provided in github: https://github.com/viswam78/LASSOprobes.
Author Contributions
LT, HBL, and BP conceived and designed the study. LT, VS, YY, DG, PS, NS performed experiments, analyzed, and interpreted the data. LT, HBL, and BP wrote the manuscript.
Competing interests
A patent application on the technology has been filed (PCT/US2016/035919). The authors declare no other competing financial interests.
Additional Information
Supplementary information accompanies this paper online.
References
- 1.van Dijk EL, Auger H, Jaszczyszyn Y, Thermes C. Ten years of next-generation sequencing technology. Trends Genet. 2014;30:418–426. doi: 10.1016/j.tig.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 2.Nilsson M, Dahl F, Larsson C, Gullberg M, Stenberg J. Analyzing genes using closing and replicating circles. Trends Biotechnol. 2006;24:83–88. doi: 10.1016/j.tibtech.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 3.Dahl F, et al. Multigene amplification and massively parallel sequencing for cancer mutation discovery. Proc Natl Acad Sci U S A. 2007;104:9387–9392. doi: 10.1073/pnas.0702165104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turner EH, Lee C, Ng SB, Nickerson DA, Shendure J. Massively parallel exon capture and library-free resequencing across 16 genomes. Nat Methods. 2009;6:315–316. doi: 10.1038/nmeth.f.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nilsson M, et al. Padlock probes: circularizing oligonucleotides for localized DNA detection. Science. 1994;265:2085–2088. doi: 10.1126/science.7522346. [DOI] [PubMed] [Google Scholar]
- 6.Landegren U, et al. Molecular tools for a molecular medicine: analyzing genes, transcripts and proteins using padlock and proximity probes. J Mol Recognit. 2004;17:194–197. doi: 10.1002/jmr.664. [DOI] [PubMed] [Google Scholar]
- 7.Hagerman PJ. Flexibility of DNA. Annu Rev Biophys Biophys Chem. 1988;17:265–286. doi: 10.1146/annurev.bb.17.060188.001405. [DOI] [PubMed] [Google Scholar]
- 8.Krishnakumar S, et al. A comprehensive assay for targeted multiplex amplification of human DNA sequences. Proc Natl Acad Sci U S A. 2008;105:9296–9301. doi: 10.1073/pnas.0803240105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shen P, et al. Multiplex target capture with double-stranded DNA probes. Genome Med. 2013;5:50. doi: 10.1186/gm454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen P, et al. High-quality DNA sequence capture of 524 disease candidate genes. Proc Natl Acad Sci U S A. 2011;108:6549–6554. doi: 10.1073/pnas.1018981108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kosuri S, Church GM. Large-scale de novo DNA synthesis: technologies and applications. Nat Methods. 2014;11:499–507. doi: 10.1038/nmeth.2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tewhey R, et al. Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell. 2016;165:1519–1529. doi: 10.1016/j.cell.2016.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boeke JD, et al. The Genome Project-Write. Science. 2016 doi: 10.1126/science.aaf6850. [DOI] [PubMed] [Google Scholar]
- 14.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.