

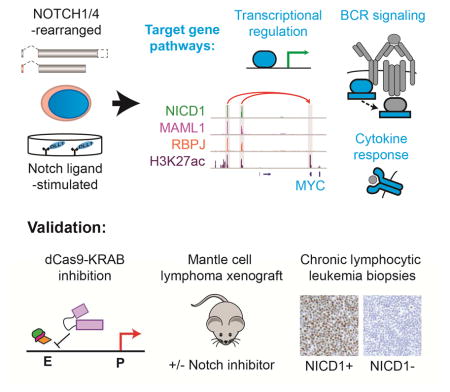
Summary
Gain-of-function Notch mutations are recurrent in mature small B-cell lymphomas such as mantle cell lymphoma (MCL) and chronic lymphocytic leukemia (CLL), but the Notch target genes that contribute to B-cell oncogenesis are largely unknown. We performed integrative analysis of Notch-regulated transcripts, genomic binding of Notch transcription complexes, and genome conformation data to identify direct Notch target genes in MCL cell lines. This unique B-cell Notch regulome is largely controlled through Notch-bound distal enhancers and includes genes involved in B-cell receptor and cytokine signaling and the oncogene MYC, which sustains proliferation of Notch-dependent MCL cell lines via a Notch-regulated lineage-restricted enhancer complex. Expression of direct Notch target genes is associated with Notch activity in an MCL xenograft model and in CLL lymph node biopsies. Our findings provide key insights into the role of Notch in MCL and other B-cell malignancies and have important implications for therapeutic targeting of Notch-dependent oncogenic pathways.
eTOC blurb
Ryan et al. reveal targets of Notch signaling in B-cell cancers associated with Notch gain-of-function mutations. Many Notch-responsive genes are part of pathways implicated in B-cell cancer pathogenesis. These findings provide insights into the role of Notch and a rationale for targeting Notch in B-cell cancers.
Introduction
Notch signaling controls development and tissue homeostasis in metazoan animals (reviewed in (Bray, 2016) and when dysregulated contributes to the pathogenesis of several hematologic malignancies and solid tumors (reviewed in (Aster et al., 2016)). Signaling relies on ligand-mediated proteolysis of Notch receptors by gamma-secretase, which releases the Notch intracellular domain (NICD), allowing it to translocate to the nucleus and form a Notch transcription complex (NTC) with the DNA-binding factor RBPJ and co-activators of the Mastermind-like (MAML) family. NTCs recruit factors such as p300 and Mediator and activate Notch target gene expression.
Outcomes produced by Notch signaling are cell context-specific, presumably because Notch drives distinct gene expression programs in different cell types. Both gain- and loss-of-function Notch mutations are observed in various human cancers, indicating that Notch can be oncogenic or tumor suppressive depending on cell context. However, detailed descriptions of Notch target genes and linked regulatory elements have been confined to a single cancer, T-cell acute lymphoblastic leukemia (T-ALL) (Wang et al., 2014), in which Notch has an oncogenic role.
Notch-mutated cancers include several subtypes of mature small B-cell lymphomas. NOTCH1 is the most frequently mutated gene in chronic lymphocytic leukemia (CLL, also known as small lymphocytic lymphoma) (Puente et al., 2011; Puente et al., 2015), NOTCH1 and NOTCH2 mutations occur in mantle cell lymphoma (MCL) (Bea et al., 2013; Kridel et al., 2012), and NOTCH2 is often mutated in splenic marginal zone B-cell lymphoma (Kiel et al., 2012; Rossi et al., 2012). Most Notch mutations in B-cell tumors are frameshift or nonsense mutations in a C-terminal PEST degron domain that increase NICD half-life, pointing to an oncogenic role for Notch in B-cell tumors. Such mutations are linked to disease progression and decreased survival in CLL and MCL (Fabbri et al., 2011; Rossi et al., 2012). In situ studies detected activated NOTCH1 (NICD1) in >80% of CLL lymph node biopsies (Kluk et al., 2013), suggesting a broad role for Notch signaling in CLL.
In this study, we used model cell lines and primary tumor samples to identify Notch target genes and associated regulatory elements in small B-cell lymphomas. The B-cell-specific Notch regulome revealed by these studies has broad implications for the role of Notch signaling in B-cell lymphomagenesis and lays the groundwork for developing novel therapeutic strategies involving the use of Notch pathway inhibitors in these cancers.
Results
Notch-addicted MCL cell lines bear activating Notch gene rearrangements
The growth of the MCL cell lines Rec-1 and SP-49 is suppressed by gamma-secretase inhibitors (GSI) (Figure S1A) and by dominant-negative MAML1 (Kridel et al., 2012), features that identify “Notch-addicted” cell lines (Weng et al., 2004). In Rec-1, a NOTCH1 allele with an intragenic deletion (Figures 1A and S1B) encodes a truncated NOTCH1 protein activated by gamma secretase in a ligand-independent fashion (Kluk et al., 2013). Sequencing of RNA from SP-49 cells identified a transcript consisting of the first exon of HLA-DMB spliced in-frame to exons 24-30 of NOTCH4 that is predicted to encode a truncated NOTCH4 protein (Figure 1A). This transcript is the product of an HLA-DMB—NOTCH4 fusion gene created by a 740 kb interstitial deletion (Figures S1B and S1C). Among MCL lines, high levels of NICD1 were seen only in Rec-1 cells and a truncated form of NOTCH4 was detected only in SP-49 cells (Figure 1B). GSI treatment reduced NICD1 in Rec-1 cells and slightly increased the size of the NOTCH4 polypeptide in SP-49 cells (Figure 1B), consistent with it being a gamma-secretase substrate. In line with prior work showing that MYC expression depends on Notch in Rec-1 cells (Stoeck et al., 2014), GSI treatment decreased MYC levels in Rec-1 and SP-49 cells but not in cell lines lacking Notch gene rearrangements (Figure 1B).
Figure 1. Identification of Notch-regulated transcripts in MCL cell lines. See also Figure S1.

(A) Wild-type NOTCH1 protein and Notch mutants in MCL cell lines. ‘S1’ indicates furin cleavage site. ’TM’ indicates transmembrane domain. Other abbreviations as in (Aster et al., 2016). (B) Western blot for Notch and MYC proteins in MCL cell lines treated for 3 d with GSI or DMSO. The NOTCH1 antibody recognizes NICD1, while the NOTCH4 antibody recognizes an epitope near the NOTCH4 C-terminus. Cell lines with Notch gene or MYC rearrangements are noted. (C) GSI-washout experiments in MCL lines with ligand-independent (top) and -dependent (bottom) Notch signaling. (D) Western blot showing modulation of NICD1 and MYC levels by GSI-washout in Mino and Rec-1 cells. (E) GSEA of genes ranked by differential expression following GSI-washout (WO) or mock washout Rec-1, SP-49, and DLL1ext-IgG-stimulated Mino cells for the Hallmark MYC targets V2 gene set. (F) Heatmaps showing transcripts significantly increased in GSI-washout versus mock-washout experiments in at least 2 of 3 MCL lines (Mino, Rec-1, SP-49). Heatmap clusters are defined and numbered as in the Venn diagram and sorted within clusters such that genes showing Notch-dependent activation in multiple cell types are at the top of each cluster and B-cell specific Notch targets are at bottom. Canonical Notch target genes are labeled in red, while other genes of interest are labeled in black.
Identification of Notch-activated genes in B-cell lymphoma cell lines
To model ligand-dependent Notch activation, MCL lines were grown on immobilized Notch ligand (DLL1ext-IgG), which generated NICD1 in Jeko-1 and Mino cells (Figure S1D); NICD1 levels were higher in Mino cells, which harbor a PEST domain mutation (NOTCH1 Q2487*). We selected Mino, Rec-1 and SP-49 cells for study using GSI-washout, which permits timed activation of Notch (Figures 1C and 1D). In both Rec-1 and DLL1-stimulated Mino cells, GSI-washout rapidly increased NICD1 levels. Notch activation increased MYC in Rec-1 cells but not in Mino cells (Figure 1D), which harbor a rearrangement placing MYC under the control of heterologous regulatory elements (Table S1; discussed below). Consistent with this, RNA-seq data revealed that Notch markedly increased MYC target gene expression only in Rec-1 and SP-49 cells (Figure 1E). Altogether, the transcript levels of 377 genes were increased and 203 genes were decreased by Notch activation in at least two MCL lines (Table S2). Most decreases in transcript abundance were modest (mean log2 fold change < 0.5) and we focused further analysis on up-regulated transcripts, which include potential direct Notch target genes.
To separate Notch and MYC target genes, transcripts increased by GSI-washout were divided into two groups (Figure 1F). Group 1 includes transcripts increased by Notch in Mino cells (in which MYC is not activated by Notch) and in one or both Notch-rearranged MCL lines, while Group 2 includes transcripts increased by Notch in Notch-rearranged lines, but not Mino cells. RNA-seq data obtained from GSI-washout in Notch-rearranged T-ALL and breast cancer cell lines also were analyzed to assess the lineage-specificity of Notch-activated genes (Stoeck et al., 2014). Group 1 genes (n=226) include known Notch target genes (e.g., HES1, HES4, NRARP, and DTX1) that are also upregulated by Notch in T-ALL and/or breast cancer cells and a larger number of genes that are only up-regulated in MCL lines, suggesting that these are B cell-specific Notch target genes. Consistent with this, Group 1 is enriched for genes associated with B-cell or lymphocyte biology, including B-cell receptor, interleukin, and interferon signaling, and NF-κB activation (Table S3). By contrast, Group 2 (n=151) are enriched for known MYC target genes and MYC-regulated biological processes (Table S3); indeed, MYC is one of the most significantly up-regulated transcripts in Group 2. Nearly all Group 2 transcripts also are increased by Notch in Notch-rearranged T-ALL and breast cancer cells (Figure 1F), suggesting that these genes belong to a MYC-driven expression module common to diverse Notch-related cancers.
Notch activates 5′ MYC enhancers in MCL cell lines
We next explored the mechanism by which Notch increases MYC expression in B cells. Lineage-specific enhancer elements are scattered across a 3 Mb gene desert surrounding the MYC gene body (Ahmadiyeh et al., 2010; Herranz et al., 2014; Shi et al., 2013; Yashiro-Ohtani et al., 2014). We previously identified two enhancer clusters on the 5′ side of MYC in primary MCL and CLL cells (Ryan et al., 2015) that are associated with polymorphisms linked to CLL risk. These candidate enhancers (E1 and E2, Figure 2A) are the most highly acetylated elements (based on H3K27ac ChIP-Seq) near MYC in SP-49 cells. By contrast, a Notch-responsive enhancer located ~1.4 Mb 3′ of MYC in T-ALL cells (Herranz et al., 2014; Yashiro-Ohtani et al., 2014) (termed the T-NDME) lacks H3K27ac in primary B-cell tumors and MCL lines. ChIP-Seq data from nine MCL lines and one CLL line showed that E1 and E2 are also acetylated in Rec-1 cells and in three Epstein-Barr virus (EBV)-positive CLL and MCL lines (Figure 2B). These EBV-positive lines express EBNA2 (Figure 2C), an EBV-encoded RBPJ-binding factor that mimics Notch activities (Henkel et al., 1994) and binds RBPJ within E1 and E2 in EBV-positive human B-lymphoblastoid cell lines (LCLs) (Zhao et al., 2011). We also detected EBNA2 and RBPJ binding to E1 and E2 in EBV-positive B-cell lymphoma lines (Figures 2D, 2E, S2A, and S2B) as well as binding of RBPJ to E1 and E2 in Notch-rearranged, EBV-negative MCL lines. Importantly, MYC gene rearrangements were detected in the five MCL lines that lack EBV and Notch gene rearrangements (Table S1), explaining why MYC levels are high in these lines (Figure 2C). Our data therefore suggest that CLL and MCL cell lines with an intact MYC locus require RBPJ-NICD (or RBPJ-EBNA2) binding to drive MYC expression via the E1/E2 enhancers.
Figure 2. Identification of B cell-restricted Notch-dependent 5′ MYC enhancers. See also Figure S2.

(A) H3K27ac ChIP-Seq data showing acetylation of 5′ MYC enhancer regions in a Notch-dependent MCL cell line and 3′ MYC enhancer regions in a Notch-dependent T-ALL cell line. Arrows indicate looping interactions with the MYC promoter in MCL (Ryan et al., 2015) and T-ALL (Herranz et al., 2014; Yashiro-Ohtani et al., 2014). (B) H3K27ac ChIP-Seq data for 5′ MYC enhancers (E1 and E2) and CD79A promoter (control) regions in CLL (blue) and MCL (green) cell lines. Cell lines are as follows: Me, Mec-1; Jv, JVM2; Gr, Granta-519; Re, Rec-1; Sp, SP-49; Mi, Mino; Je, Jeko-1; Z1, Z138; Ma, MAVER1; Hb, HBL-2. (C) Western blotting for EBNA2 and MYC in nuclear extracts from CLL and MCL lines. (D) ChIP-qPCR showing RBPJ binding at site E2 in CLL and MCL cell lines. Data represent mean ± SD. (E) ChIP-qPCR showing binding of EBNA2 at site E2 in CLL and MCL cell lines. Data represent mean ± SD. (F) ChIP-Seq in Rec-1 shows binding of ICN1, MAML1, and RBPJ at the E1 and E2 sites. Long-distance interactions involving the MYC promoter in EBV-transformed B cells are at top, with looping to E1 and E2 shown in red and other interactions shown in black. Loop thickness is proportional to the number of supporting paired-end tags. Position of SNPs associated with CLL and others in linkage disequilibrium are noted. (G) ChIP-Seq showing NTC factor binding and H3K27ac modification dynamics in MCL cell lines. Top: NICD1 and RBPJ binding and H3K27ac modification at the E1 and E2 sites in Rec-1 cells. ChIP-Seq data are shown after GSI-washout (‘Washout’) and mock-washout (‘GSI’, black overlay). Middle: Binding of MAML1 and RBPJ at the 5′ E1 and E2 sites in SP-49 cells. H3K27ac ChIP-Seq data is shown following 4 h GSI-washout (purple) or mock washout (black overlay). Bottom: Binding of NICD1 and RBPJ at the 5′ E1 and E2 sites in Mino cells stimulated for 48 h with DLL1extIgG. H3K27ac ChIP-Seq data is shown following 48 h of growth on DLL1extIgG (purple) or control IgG (black overlay). (H) 4C-Seq data for Rec-1 cells showing interactions between E1, E2, and the MYC promoter. Plots are aligned to genomic elements shown in Fig. 4F. A dotted red line marks the viewpoint for each experiment. The heatmap at bottom displays median normalized contact intensities at resolutions of 1kB to 50 kB, while the black trace at top shows median normalized contact intensity at a single, selected resolution. Top: MYC promoter viewpoint (15 kb resolution, y-axis scale 0–0.5) Middle: E1 viewpoint (15 kb resolution, y-axis scale 0–1). Bottom: E2 viewpoint (15 kb resolution, y-axis scale 0–0.3).
To confirm that E1 and E2 bind all NTC components in Notch-rearranged MCL lines, we performed ChIP-Seq for NICD1, RBPJ and MAML1, which revealed binding of all three proteins in Rec-1 cells (Figure 2F) and RBPJ and MAML1 binding in SP-49 cells (Figure 2G). MAML1 requires NICD to bind RBPJ (Nam et al., 2006), implying that NICD4 is present in NTCs associated with E1 and E2 in SP-49 cells. NTC loading onto E1 and E2 was dynamic, as NICD1 and RBPJ signals were increased by GSI washout in Rec-1 cells (Figure 2G) and H3K27ac was increased by GSI washout in both Rec-1 and SP-49 cells. We also noted increased NTC binding and H3K27ac signals in Mino cells after DLL1 stimulation, despite decoupling of MYC expression from Notch control, presumably due to the MYC-IGH rearrangement (Figure 2G). Importantly, RBPJ and NICD1 did not bind to the T-NDME in MCL lines, nor did RBPJ or NICD1 bind to E1 and E2 sites in T-ALL cells (Figure S2C), suggesting that access of NTCs to these sites requires additional lineage-specific factors.
Enhancers regulate transcription through looping interactions with gene promoters. To identify interactions between E1, E2 and the MYC promoter in MCL cells, we performed 4C-Seq on Rec-1 cells treated for 3 days with GSI or vehicle control (DMSO). Reproducible mutual interactions were noted between all three elements in datasets generated from viewpoints near the MYC promoter (Figures 2H and S2D) and E1 and E2 (Figure 2H and data not shown) in vehicle-treated cells. MYC-E1 interactions were maintained following GSI treatment (Figure S2D), albeit with lower contact intensity, while MYC-E2 interactions were no longer significant in the Notch-off state. Interestingly, interactions between the MYC promoter and elements 3′ of MYC, such as the non-coding RNA gene PVT1, were relatively increased in the GSI-treated cells (Figure S2D).
Direct Notch targets include regulators of B-cell signaling and differentiation
We next sought to identify other Notch target genes of potential importance in B-cell tumors. In Rec-1 cells, a large fraction of genomic sites bound by NICD1 showed RBPJ and MAML1 co-binding (Figure 3A), and essentially all were depleted of NICD1 following GSI treatment, confirming their specificity (Figure 3B). ChIP-Seq studies conducted in Rec-1, SP-49 and Mino cells identified 1,791 sites (Figure 3B) that were reproducibly bound by at least two NTC proteins in the same line, or by NICD1 in three independent experiments; these sites were termed “consensus NTC sites”. De novo analysis of consensus NTC sites revealed that the most enriched DNA sequence was the RBPJ binding motif. Motifs bound by transcription factors with important roles in B-cells were also enriched, including ETS-IRF composite element, E-box and NF-κB motifs (Figures 3C and 3D). MAML1 peaks were less enriched for RBPJ motifs than RBPJ or NICD1 peaks (Figure 3D), possibly because MAMLs associate with additional transcription complexes (Shen et al., 2006; Zhao et al., 2007). In all three MCL lines, the Notch-on state was associated with increased acetylation of NTC-bound distal elements (Figure 3E), consistent with recruitment of histone acetyltransferases, such as p300, by NTCs (Skalska et al., 2015).
Figure 3. Direct Notch target genes involved in signaling and transcriptional regulation.

(A) Euler diagram showing overlap between genome-wide binding peaks for NTC proteins in Rec-1 cells (FDR = 10−3). NICD1 GSI-washout data (wo; Notch-on state) is shown. (B) Heatmaps for the indicated ChIP-Seq datasets in a 2 kb window around each consensus NTC peak. Each peak achieved genome-wide significance (FDR = 0.001) for at least two NTC proteins in the same cell line, or in three independent NICD1 ChIP-Seq datasets produced in Rec-1 cells (two replicates) and DLL1-stimuated Mino cells (one dataset). Note the absence of NICD1 signal in mock washout (gsi) conditions in Rec-1 cells. (C) DNA sequence motifs detected in de novo analysis of consensus NTC binding peaks from three MCL cell lines (Rec-1, SP-49, and DLL1ext-IgG-stimulated Mino). (D) Significance of enrichment for transcription factor binding motifs (HOMER library) most closely matching the de novo motifs shown in (C). (E) Box plots showing differential H3K27ac signal at distal enhancers in GSI-washout (‘notch on’) versus mock washout (‘notch off’) experiments in MCL cell lines. Mino cells were stimulated with DLL1ext-IgG. Genome-wide distal enhancers were divided into two groups according whether they overlapped a consensus NTC binding site detected in any cell line (NTC+) or not (NTC−). (F) Linkage of Notch-activated genes identified in MCL cell lines to consensus NTC binding sites. Proximal, distal, and CTCF-defined chromatin contact domain (CCD) linkages were defined by proximity, GM12878 Pol2 ChIA-PET data, and GM12878 CTCF ChIA-PET data, respectively. Notch-activated genes were sub-grouped as in Fig. 1F (G1 = Group 1, G2 = Group 2).
Only a minority of Notch-activated genes in MCL had high-confidence NTC binding sites within 5 Kb of their transcriptional start sites, as in T-ALL cells (Wang et al., 2014), suggesting that most Notch-response elements reside in distal enhancers. Functional enhancer-promoter interactions occur within topologically isolated domains bounded by the DNA-binding protein CTCF (Rao et al., 2014; Tang et al., 2015), and we found that 95% of Group 1 genes contained at least one consensus NTC peak within the same CTCF-defined chromatin contact domain (CCD, (Tang et al., 2015)), compared to 69% of Notch-insensitive expressed genes. To more specifically link distal elements to Notch target genes, we used maps of looping interactions mediated by RNA polymerase II (Pol2 ChIA-PET) in GM12878 lymphoblastoid cells (Tang et al., 2015), revealing NTC binding site interactions (promoter-proximal or linked enhancer) for 67% of Group 1 genes (Figure 3F), compared to 10% of Notch-insensitive expressed genes. Genes linked to NTC-bound enhancers include MYC (the E1 and E2 elements, Figure 2F) and numerous genes regulated by EBNA2 (e.g. CR2, DNASE1L3, ABHD6, RHOH, CDK5R1, RUNX3, and BATF) (Johansen et al., 2003; Maier et al., 2006; Spender et al., 2002). By contrast, the sets of genes that appear to be up-regulated by Notch via its effects on MYC (Group 2) or that are down-regulated followed Notch activation showed no enrichment for linkage to NTC binding sites (Figure 3F).
Integrative analysis of RNA-seq, ChIP-seq, and ChIA-PET data sets identified 148 NTC-linked direct Notch target genes in MCL cell lines (Table S4). This list may be incomplete, as GM12878 LCLs may not fully recapitulate enhancer-promoter interactions in MCL cells. Approximately 60% of NTC target genes were not responsive (log2 fold change < 0.2) to Notch in T-ALL or breast cancer cell lines and appear to be B-cell-specific. By contrast, only 7% of Group 2 (MYC-associated) genes failed to increase upon Notch activation in T-ALL or breast cancer lines. The positions of Notch response elements in MCL lines are diverse (Figures 4 and S3). Proximal NTC binding sites are often 5′ of the transcriptional start site or within the proximal first intron. Distal sites can be intergenic or located within the target gene body or an adjacent gene. Complex multi-enhancer and/or multi-gene regulatory units also exist.
Figure 4. Examples of Notch target genes involved in signaling and transcriptional regulation. See also Figure S3.

(A) Representative Notch target genes with NTC binding to promoter-proximal sites. ChIP-Seq data are shown for the indicated factors in Rec-1 cells and for H3K27ac in lymph node biopsies MCL-003 and CLL-007. (B–C) Representative Notch target genes (red gene symbols) encoding signaling regulators (B) or transcription factors (C) associated with distal NTC-binding enhancers. GM12878 Pol2 ChIA-PET anchors are indicated by small green loops at top. Black or red loops indicate the presence of paired-end tags that connect the promoter of a Notch-regulated gene to a distal anchor containing (red) or lacking (black) a consensus NTC binding site. Loop thickness is proportional to the number of supporting paired-end tags for each interaction within each displayed region.
NTC-linked Notch target genes include genes involved in cytokine/interleukin signaling (IL6R, IL10RA, IL21R) and B-cell receptor activation (FYN, LYN, BLK, BLNK, PIK3AP1, SH2B2, NEDD9), and Ingenuity system analysis of Group 1 genes identified B-cell receptor signaling as the most enriched pathway (Table S3). Several transcription factor genes also are NTC-linked Notch target genes, suggesting that Notch activates or reinforces transcriptional regulatory programs in MCL cell lines. IRF8 and TLE3 are of particular interest, since these loci (like MYC) are near single nucleotide polymorphisms associated with risk of CLL (Crowther-Swanepoel et al., 2010).
Notch-dependent MYC enhancer function sustains MCL growth
To show that Notch-dependent enhancers have a role in lymphoma growth and survival, we generated SP-49 (NOTCH4-rearranged), Granta-519 (EBV+), and Jeko-1 (MYC-rearranged and amplified) cells stably expressing a KRAB-dCAS9-P2A-mCherry transgene, which encodes a nuclease-dead Cas9 fusion protein that mediates local epigenetic repression (Gilbert et al., 2014). To validate this approach, we transduced SP-49 and Granta-519 cells with sgRNAs targeting the promoter and 5′ distal enhancer complex of the Notch target gene CR2. As expected, this decreased levels of CD21 (encoded by CR2) in KRAB-dCAS9-expressing cells but not control cells (Figures S4A and S4B).
We next designed sgRNA constructs to target the MYC promoter and RBPJ motifs in the 5′ E1 and E2 elements, the T-NDME, and other intergenic sites near MYC (Figures 5A and S2C). In Granta-519 and SP-49 cells, sgRNAs targeting the MYC promoter, the E1 RBPJ motif, or the E2 RBPJ motifs decreased MYC expression, whereas sgRNAs targeting the T-NDME or intergenic regions had no effect (Figure S4C). E1- and MYC promoter-targeting sgRNAs also inhibited growth of Granta-519 (Figure S4D) but not SP-49 cells, possibly due to relatively mild MYC repression by single sgRNAs in that cell line. To test the effect of co-repressing the E1 and E2 modules, KRAB-dCAS9-P2A-mCherry lines were co-transduced with E1- and E2-targeting sgRNA lentiviruses. This led to decreased MYC expression (Figure 5B) and cell proliferation (Figure 5C) in SP-49 and Granta-519 cells, but not in MYC-rearranged Jeko-1 cells. Thus, Notch stimulates MYC expression in B cells through the E1 and E2 sites, referred to as the B-cell Notch-dependent MYC enhancer (B-NDME).
Figure 5. B-NDME elements associated with NTC-binding drive MYC expression and growth in Notch-addicted MCL lines. See also Figures S4 and S5.

(A) NICD1 and RBPJ binding at the E1 and E2 sites after GSI-washout in Rec-1 cells and Phastcons 46-vertebrate conservation score (‘conservation’). Consensus RBPJ logos are aligned with conserved RBPJ motifs in each enhancer. Positions of sgRNAs are indicated; sgRNAs used for dual guide experiments are labeled in red. (B) qRT-PCR measurement of MYC transcripts after transduction of KRAB-dCAS9-P2A-mCherry-expressing Granta-519 (EBV+), SP-49 (NOTCH4-rearranged), and Jeko-1 (MYC-rearranged/amplified) MCL cell lines with sgRNAs targeting the E1 and E2 sites (g005 and g009). CCND1 expression is the negative control. Data represent mean ± SEM of “n” independent experiments, as indicated. Significance calculated by t-test: * p < 0.05, *** p< 0.001. (C) Growth of KRAB-dCAS9-P2A-mCherry-expressing MCL cell lines after transduction with sgRNAs as in (B). Data represent mean ± SEM of “n” biological replicates, as indicated. Significance calculated by t-test on data from d 12: ** p < 0.01, *** p< 0.001. (D) Growth of pINDUCER-20-MYC transduced Rec-1 cells treated with vehicle, GSI, or GSI + doxycycline (3.3 ug/ml). Data represent mean ± SEM of 3 biological replicates. Significance calculated by t-test: * p < 0.05. (E) Growth of parental SP-49 and pINDUCER-22-MYC–transduced single-cell SP-49 clones treated with vehicle, GSI or GSI + doxycycline. Doxycycline doses shown produced optimal rescues in individual clones: Clones 3 & 7 – 33.6 ng/ml, Clone 4 (and parental) – 100 ng/ml. Data represent mean ± SEM of 3 biological replicates. Significance calculated by t-test: * p < 0.05, ** p< 0.01.
To test the role of MYC in the Notch-dependency of MCL cell lines, we transduced Rec-1 cells with a doxycycline-inducible MYC transgene, and observed doxycycline dose-dependent rescue of cell growth in the presence of GSI (Figures 5D, S5A and S5B). SP-49 cells were sensitive to MYC expression above baseline levels, so we generated single-cell SP-49 clones bearing the MYC transgene, which allowed us to identify clone-specific doxycycline doses that facilitated partial rescue from GSI-mediated growth suppression, (Figures 5E, S5C and S5D). These data show that the Notch/MYC signaling axis contributes to the growth of Notch-rearranged MCL lines.
Notch target genes show microenvironment-specific activation in MCL cells in vivo
To determine if Group 1 target genes are activated by Notch in MCL cells in vivo, we utilized an MCL patient-derived xenograft (PDX) model (PDX-98848, (Townsend et al., 2016)) that bears a NOTCH1 PEST domain mutation and homes to the spleens of NOD-Scid IL2Rgamma-null (NSG) mice. Immunohistochemistry (IHC) showed strong NICD1 staining in a subset of splenic MCL cells in PDX-98848-bearing animals, whereas three NOTCH1 wild-type MCL PDX models showed little NICD1 staining (Figure 6A). Analysis of PDX-98848 mice with high tumor burden treated for 5 days with vehicle or GSI (DBZ) revealed higher expression of the Notch-regulated proteins CD300A and SEMA7A in MCL cells from spleen versus blood or bone marrow, and that GSI lowered expression of these markers (Figure 6B). RNA-Seq analysis of splenic MCL cells revealed inhibition of additional Notch target genes in DBZ-treated mice (Figures 6C and S6A and Table S5), including MYC, cytokine receptors (IL21R, IL10RA), genes related to BCR signaling (FGR, SH2B2, FCRL2), and transcriptional regulators (IRF8, POU2AF1, and MYBL2) (FDR < 0.05).
Figure 6. Notch-dependent activation of target genes in an MCL xenograft system. See also Figure S6.

(A) IHC for NICD1 in the spleens of NOTCH1 wild-type MCL xenografts and an MCL xenograft with a NOTCH1 PEST truncating mutation (PDX 98848). NICD1 staining of endothelial cells (elongated nuclei, marked by arrows) is an internal control. (B) Flow cytometric analysis of Notch target gene products CD300A and SEMA7A in PDX 98848 MCL cells harvested from the indicated tissues in mice treated for five days with vehicle (n=5) or the GSI DBZ (n=4). Significance calculated by t-test. (C) Gene set enrichment analysis of RNA-seq data from PDX 98848 MCL cells harvested from the spleens of vehicle- or DBZ-treated mice, using the direct Notch target gene set. Genes were ranked according to fold-change in vehicle versus DBZ-treated mice.
Notch target expression is associated with Notch activation in CLL lymph nodes
We next investigated Notch target genes in CLL by performing RNA-Seq on NOTCH1 mutated and wild-type CLL lymph node biopsies with high and negligible amounts of NICD1 IHC staining, respectively (Figure 7A). Among the Notch target genes expressed at higher levels in NOTCH1-mutant CLL were SEMA7A, CD300A, IL6R, and the BCR signaling pathway genes FYN and NEDD9 (Figure 7B and Table S6). Gene set analysis using the direct Notch target genes identified in MCL cell lines showed higher expression of these transcripts in NOTCH1-mutant versus wild-type CLL biopsies (Figures 7C and S6B). To study Notch response elements in CLL, ChIP-Seq was performed for NICD1 and RBPJ in a NOTCH1 mutant CLL lymph node biopsy (CLL-M13). While the number and amplitude of NICD1 peaks were lower than in MCL cell lines, motif analysis revealed enrichment for the RBPJ-binding motif in both data sets (Figure S6C), with the NICD1 ChIP-seq peaks showing the most specific association with the RBPJ motif (40% of peaks, p=10−49). NICD1 and RBPJ-bound regulatory elements shared between CLL-M13 and MCL cell lines were associated with H3K27 acetylation in NOTCH1-mutant and wild-type CLL and primary MCL cells (Figures 7D and S7 and Table S7), while acetylation of most elements was substantially lower in diffuse large B-cell lymphoma. Sites of RBPJ and NICD1 binding in CLL-M13 included elements linked to IL6R, FYN, BLK, and other B-cell signaling pathway genes, transcription factor genes, and the E2 element of the B-NDME.
Figure 7. Notch-dependent activation of target genes in CLL lymph node biopsies. See also Figures S6 and S7.

(A) IHC staining for NICD1 in representative lymph node biopsies involved by NOTCH1-mutant/NICD1-high or NOTCH1-wild-type/NICD1-low CLL. (B) Heatmap of RNA-Seq data showing relative transcript abundance in NOTCH1-mutant versus NOTCH1-wild-type CLL of genes identified as Notch-regulated in MCL cell lines. Genes marked by the red bar (names in bold) showed significantly higher transcript abundance in the NOTCH1-mutant group at an FDR-adjusted p-value < 0.05. Other genes were significant by individual (non-FDR-adjusted) analysis at p < 0.05. (C) GSEA of CLL biopsy RNA-Seq data using the direct Notch target gene set. Genes were ranked according to fold-change in NOTCH1 mutant versus NOTCH1 wild-type CLL lymph node biopsies. (D) ChIP-Seq data showing NTC protein binding and H3K27 acetylation at enhancers linked to direct Notch target genes in Rec-1 cells and a NOTCH1-mutant CLL lymph node biopsy (CLL-M13). Where indicated, overlays demonstrate dynamic NICD1 factor binding and/or H3K27 acetylation in the GSI-washout versus the mock washout state (Rec-1 cells). All enhancers shown had significant NICD1 and RBPJ binding in Rec-1 cells and CLL-M13 at FDR < 0.001.
Discussion
Using integrated, genome-wide approaches, we identified a B-cell-specific Notch regulome in Notch-mutant MCL cell lines that is broadly associated with Notch activity in MCL cells in the splenic microenvironment of a PDX model and in NOTCH1-mutated CLL cells in lymph nodes. Our findings may be relevant to B-lymphoid cancers that show Notch activation in the absence of a Notch gene mutation, as has been observed in CLL (Fabbri et al., 2017; Kluk et al., 2013). Genomic profiling has identified CLL subgroups defined by mutations in specific pathways, including Notch, BCR signaling, RNA metabolism, and chromatin/transcriptional regulation (Puente et al., 2015). Our findings link Notch signaling to each of these pathways, either directly or secondarily via MYC, providing a potential basis for the selective drive for Notch gain-of-function mutations in small B cell lymphomas.
Target gene identification via GSI-washout and ligand-mediated activation of endogenous Notch genes offers an alternative approach to other recent work that relied on induction of an NICD1 transgene in an EBV-positive CLL cell line to identify Notch target genes (Fabbri et al., 2017). Only 41 target genes were shared between this signature (14% of 291 genes) and our signature (28% of 148 genes, Table S4), but these included many of the most strongly up-regulated direct target genes in our models, as well as the signaling component genes BLK, FGR, BLNK and IL10RA. Evaluation of NICD1 binding and activity in EBV+ systems are likely confounded in part by competition with EBNA2, which also activates transcription via RBPJ-bound regulatory elements (Henkel et al., 1994; Zhao et al., 2011). This may explain why many direct Notch target genes identified in our models, including MYC, FYN, IL6R, NEDD9, SH2B2, KLF13 and others, were not identified as functional Notch targets in EBNA2-expressing CLL cells. While our direct Notch target signature was identified in MCL cell lines, it showed greater enrichment for genes associated with Notch activity in MCL xenografts and CLL lymph nodes than did the NICD1-induced gene set from M01043 cells (Figures S6A and S6B). This result highlights the specificity of our signature and its generality across multiple Notch-driven B-cell lymphoma subtypes. Importantly, use of cell lines that are “addicted” to constitutive Notch signaling allowed us to show that MYC mediates the growth-sustaining effects of oncogenic Notch signaling in a B-cell cancer.
Direct Notch target genes identified in this study include transcription factor genes implicated in B-cell activation and proliferation (RUNX3, POU2AF1) (Brady et al., 2009; Kim et al., 1996), differentiation (PAX5) (Usui et al., 1997), or both (IRF8) (Carotta et al., 2014; Xu et al., 2015). Our identification of MYBL2 as a direct Notch target is intriguing, given the established role of MYB family transcription factors in cycle progression and oncogenesis in diverse cancer types (Gonda and Ramsay, 2016; Martinez and Dimaio, 2011; Ramsay and Gonda, 2008). Also notable is Notch-dependent regulation of ZMIZ1, which encodes a transcriptional co-regulator of Notch target genes in T-ALL (Pinnell et al., 2015). Notch also directly up-regulates cytokine receptors and regulators of B-cell receptor signaling. These include Src-family kinases (BLK, FYN, FGR, LYN), adaptor proteins (PIK3AP, BLNK, NEDD9, SH2B2), BCR signaling modulators (CD21, CD300A) and members of the Fc-receptor-like family (FCRL3, FCRL4, and FCRL5). Further study is needed to determine the possible functional and therapeutic significance of Notch/BCR signaling interactions in B cell malignancies.
Finally, our studies show that Notch-mediated up-regulation of MYC, a pivotal regulator of cellular growth that is frequently dysregulated in a wide spectrum of B-cell neoplasms, occurs through lineage-restricted enhancers lying 5′ of the MYC gene body. We previously suggested that these enhancers might contribute to the development of CLL (Ryan et al., 2015), based on recurrent genomic amplifications in CLL (Edelmann et al., 2012; Fabbri et al., 2017) and association with germline polymorphisms linked to CLL risk (Crowther-Swanepoel et al., 2010). MYC expression within CLL cells in lymph nodes is dynamic and high levels of MYC protein are restricted to a subset of cycling cells found mainly in “proliferation centers” (Gibson et al., 2016; Krysov et al., 2012). Study of this sub-population of cells may be necessary to fully define the factors that interact with Notch to regulate MYC expression in CLL. Our identification of MYC as a direct target of Notch also lends support to recent work linking Notch to MYC-dependent metabolic and ribosomal alterations in CLL (Jitschin et al., 2015; Pozzo et al., 2017).
MCL cell lines are highly proliferative, and resemble blastoid MCL, an aggressive variant associated with higher MYC protein levels and MYC gene rearrangements (Choe et al., 2016). MYC rearrangements in CLL are also associated with histological progression or large cell transformation (Richter syndrome) (Fabbri et al., 2011; Li et al., 2016; Rossi et al., 2011). NOTCH1 mutations and MYC rearrangements tend to be mutually exclusive in Richter syndrome (Fabbri et al., 2011), suggesting that both may serve to produce sustained MYC expression. Our finding that MYC rearrangements are associated with B-NDME- and Notch-independent MYC expression and growth suggests that MYC rearrangement status should be considered in the design of clinical trials targeting Notch in transformed CLL or MCL. Ligand-dependent Notch activation is thought to predominate in B-cell cancers, but the identity of the relevant ligand-presenting cell is unclear, and the cryptic nature of the Notch gene rearrangements we detected in MCL cell lines suggest that such lesions may be more common than currently appreciated. Understanding the mechanism of Notch activation in B-cell cancers will be critical for optimal testing of new Notch inhibitors, such as ligand- or receptor-blocking antibodies (Kuhnert et al., 2011; Wu et al., 2010) that may have a more favorable toxicity profile than GSI.
Experimental Procedures
Cell culture and GSI washout assay
Cell lines were grown in RPMI 1640 with 10% fetal calf serum, penicillin/streptomycin, nonessential amino acids, 1mM sodium pyruvate and 5μM 2-mercaptoethanol. For GSI washout studies, Rec-1 and SP-49 cells were treated with the GSI compound E (1μM) for 72 h, washed, and then cultured for 4 h in media containing GSI (mock washout) or DMSO (washout) as described (Weng et al., 2006). Mino cells were cultured on DLL1ext-IgG or control IgG for 48 h in the presence of 1μM compound E and then subjected to GSI washout or mock washout for 4 h. Following washout, cells were harvested from culture and either cross-linked for ChIP-Seq, frozen in liquid nitrogen for RNA extraction, or processed for protein extraction and Western blotting. ChIP-Seq of cell lines and biopsies was performed as described (Ryan et al., 2015) with antibodies recognizing NICD1 (Val1744 epitope, CST; #4147), RBPJ (CST, #5313), MAML1 (CST; #12166), and H3K27ac (Active Motif, #39133). RNA-Seq and ChIP-Seq protocols are provided in the Supplemental Experimental methods.
Western Blotting
Western blotting of whole-cell or nuclear extracts was performed by standard methods using antibodies specific for NICD1 (Val1744, CST; #4147), NOTCH4 (Cell Signaling, #2423), MYC (abcam, ab32072), TBP (Abcam, 1TBP18), α-tubulin (Abcam, ab7291) and β-actin (Sigma, A5316).
dCas9-KRAB analysis of MYC regulatory elements
SP-49, Granta-519 and Jeko-1 cells stably expressing SFFV-KRAB-dCas9-P2A-mCherry or pLX-304-GFP were used in these studies rather than Rec-1 cells, which suffered from a high rate of transgene inactivation and poor lentiviral transduction efficiency. To perform dual sgRNA targeting, SFFV-KRAB-dCas9-P2A-mCherry+ cells or LentiCas9-Blast+ cells were transduced with lentivirus produced with pLKO5.sgRNA.EFS.tRFP (Addgene #57823) encoding sgRNA g005 or g015 (non-targeting control) and/or with lentivirus produced with pLKO5.sgRNA.EFS.GFP (Addgene #57822) encoding sgRNA g009 or g015. Cells were sorted for expression of GFP, RFP and mCherry on d4 and harvested for RNA extraction or re-plated at equal density in 24-well plates. To measure growth, cells were counted every 3 d and re-plated at equal density for a total of 12 d. Details of sgRNA design and single-guide targeting of the CR2 and MYC loci are given in the Supplemental Experimental methods.
MYC transgene rescue of GSI-treated cell lines
Rec-1 cells stably transduced with pINDUCER-20-MYC were plated in 96-well plates in media containing 1μM Compound E or DMSO, with or without a range of doxycycline concentrations (three replicates per treatment). Cell growth was assessed by CellTiterGlo (Promega) on d7. To assess MYC protein levels, selected clones or parental Rec-1 cells were grown in the presence of DMSO, 1μM Compound E, or 1μM Compound E plus 3.3μg/mL doxycycline and harvested on d3. SP-49 cells were stably transduced with pINDUCER-22-MYC, sorted for GFP-positivity, and grown as single cell clones generated by limiting dilution in 96-well plates. Growth response to GSI and doxycycline treatment or MYC protein expression was studied as in Rec-1 cells.
Patient-derived xenograft (PDX) models
Murine studies were performed with approval of the Dana-Faber Cancer Institute IACUC. PDX DFBL-98848 was established through the Public Repository of Xenografts (PRoXe) program (Townsend et al., 2016). DFBL-98848 xenografts were established in NOD-scid-gamma mice via tail vein injection of 1.5×106 cells. Treatment was initiated upon appearance of MCL cells in the peripheral blood at d32 and consisted of IP injection of DBZ (10μMol/kg) in 0.5% CMC + 1% Tween 80 or vehicle alone every other day for 5 d. Details of xenograft treatment with DBZ are given in the Supplemental Experimental methods.
Human specimen collection
Studies on patient samples were conducted with approval of the Dana-Farber/Harvard Cancer Center IRB. Biopsy samples were obtained as excess surgical tissue from the Departments of Pathology at the Massachusetts General Hospital and Brigham and Women’s Hospital.
RNA-Seq, ChIP-Seq, and 4C-Seq data analysis
RNA-Seq data was aligned to the hg19 genome reference. Differential expression analysis was performed with DESeq2. For GSI-washout experiments, differentially expressed genes were defined as log2 fold change > 0.2 or < −0.2 and FDR-adjusted p-value <0.05. ChIP-Seq data was aligned to hg19 with BWA, filtered, and analyzed for significant peaks with Homer findPeaks for transcription factors (“factor” style, FDR 1E-3) or MACS2 for H3K27ac (FDR 1E-6). 4C-seq data was analyzed with the 4CSeq Pipe and FourCSeq pipelines. Additional details, including ChIA-PET linkage analysis, are described in the Supplemental Methods.
Supplementary Material
Table S1: Summary of evidence for presence or absence of MYC locus rearrangements in CLL and MCL cell lines. Related to Figures 1 and 2.
Tab 1: Genes increased in Notch-on state. Median transcript abundance (transcripts per million; TPM) for each gene was calculated with Kallisto; Tab 2: Genes decreased in Notch-on state.
Tab 1: Gene sets from the MSigDB Hallmark and Reactome databases enriched in Group 1 genes. FDR q-values are for combined analysis of both gene set collections. Tab 2: Gene sets from the Hallmark and Reactome databases enriched in Group 2 genes. Tab 3: Results of Ingenuity Canonical Pathways analysis on Group 1 genes. ‘Ratio’ corresponds to ratio of pathway members in the test gene set to total pathway members.
Tab 1: NTC-linked direct Notch target genes identified in MCL cell lines. The strongest proximal and/or distal Pol2 ChIA-PET-linked NTC peaks are listed for each gene, as well as total number of independently linked NTC peaks in each category (proxLinks and distLinks). Log2-fold-change (l2fc) and FDR-adjusted p-value (padj) are from DESeq2 analysis of gsi-washout RNA-Seq data in the indicated line. ChIP-Seq signal intensities are listed for each linked peak, normalized as fragments over peak per 10 million total aligned fragments. Tab 2: All NTC site – Notch-up-regulated gene links, defined by TSS proximity or Pol2 ChIA-PET. Tab 3: Direct Notch target genes identified in this analysis and shared with the Notch target gene signature identified by NICD1 transgene overexpression in the CLL cell line MO1043 (Fabbri et al., 2017).
Median transcript abundance (transcripts per million; TPM) for each gene in vehicle-treated mice was calculated with Kallisto.
Median transcript abundance (transcripts per million; TPM) for each gene in NOTCH1-mutant CLL biopsies was calculated with Kallisto.
Listed are all NTC binding sites identified in MCL cell lines with significant binding in both NICD and RBPJ ChIP-Seq performed on CLL-M13 lymph node biopsy material. ChIP-Seq signal intensities are listed for each linked peak, in MCL cell lines and CLL-M13 normalized as fragments over peak per 10 million total aligned fragments.
Highlights.
Genome-wide identification of Notch target enhancers and genes in B-cell lymphomas
Target genes regulate critical B-cell pathways
B-cell cancer growth relies on lineage-restricted Notch-activated MYC enhancers
Provides a rationale for targeting Notch in lymphoma
Acknowledgments
We thank Irwin Bernstein for recombinant DLL1ext-IgG and protocols. We thank Vivianna van Deerlin, Patrick McLaughlin, David Dombkowski, Scott Mordecai, and Will Flavahan for technical contributions. Contributing core facilities at the University of Pennsylvania included the Abramson Cancer Center Flow Cytometry Core (P30-CA016520), the AFCRI Core, the Functional Genomics Core, and the NIH/NIDDK P30 Center for Molecular Studies (P30-DK050306). Core facilities at Massachusetts General Hospital included the Pathology flow core (S10OD016372), and the HSCI flow core. This work was supported by fellowships from the Leukemia and Lymphoma Society to JP and RJHR; by grants from the National Institutes of Health to JCA, WSP, SCB (P01 CA119070), WSP (R01AI047833), BEB (ENCODE U54 HG004570), and RJHR (K08 CA208013); by a Leukemia and Lymphoma Society Specialized Center of Research grant (JCA, BEB); by a UPenn Epigenetics Pilot Grant (RBF, WSP), by donors to the MGH Center for Lymphoma Building Infrastructure for Future Discovery project (RJHR, EPH); and by the Loring Fund for Lymphoma Research (JCA, BEB).
Footnotes
The authors have declared that no conflict of interest exists.
Author Contributions
Authors made the following contributions to this work: Designed research studies (RR, JP, MK, AC, DT, BG, SB, DW, BB, JA, WP), conducted experiments (RR, JP, DR, MK, AC, WL, LD, SG, VN), acquired data (RR, JP, DR, MK, AC, WL, LD, SG, VN, JA), performed computational and statistical analysis (RR, YZ, CL, RF), provided reagents (BG, EH, SB, DW, BB, JA, WP), facilitated patient consenting (RR, EH), generated figures (RR, JP, YZ, CL, RF) and wrote the manuscript (RR, JP, SB, RF, BB, JA, WP)
Accession numbers
The GEO accession number for new datasets reported in this paper is GSE97541. ChIP-Seq data for lymphoma biopsies are available through dbGaP accession phs000939.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmadiyeh N, Pomerantz MM, Grisanzio C, Herman P, Jia L, Almendro V, He HH, Brown M, Liu XS, Davis M, et al. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:9742–9746. doi: 10.1073/pnas.0910668107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aster JC, Pear WS, Blacklow SC. The Varied Roles of Notch in Cancer. Annual review of pathology. 2016 doi: 10.1146/annurev-pathol-052016-100127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bea S, Valdes-Mas R, Navarro A, Salaverria I, Martin-Garcia D, Jares P, Gine E, Pinyol M, Royo C, Nadeu F, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:18250–18255. doi: 10.1073/pnas.1314608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady G, Whiteman HJ, Spender LC, Farrell PJ. Downregulation of RUNX1 by RUNX3 requires the RUNX3 VWRPY sequence and is essential for Epstein-Barr virus-driven B-cell proliferation. Journal of virology. 2009;83:6909–6916. doi: 10.1128/JVI.00216-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray SJ. Notch signalling in context. Nat Rev Mol Cell Biol. 2016 doi: 10.1038/nrm.2016.94. [DOI] [PubMed] [Google Scholar]
- Carotta S, Willis SN, Hasbold J, Inouye M, Pang SH, Emslie D, Light A, Chopin M, Shi W, Wang H, et al. The transcription factors IRF8 and PU.1 negatively regulate plasma cell differentiation. The Journal of experimental medicine. 2014;211:2169–2181. doi: 10.1084/jem.20140425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe JY, Yun JY, Na HY, Huh J, Shin SJ, Kim HJ, Paik JH, Kim YA, Nam SJ, Jeon YK, et al. MYC overexpression correlates with MYC amplification or translocation, and is associated with poor prognosis in mantle cell lymphoma. Histopathology. 2016;68:442–449. doi: 10.1111/his.12760. [DOI] [PubMed] [Google Scholar]
- Crowther-Swanepoel D, Broderick P, Di Bernardo MC, Dobbins SE, Torres M, Mansouri M, Ruiz-Ponte C, Enjuanes A, Rosenquist R, Carracedo A, et al. Common variants at 2q37.3, 8q24.21, 15q21.3 and 16q24.1 influence chronic lymphocytic leukemia risk. Nature genetics. 2010;42:132–136. doi: 10.1038/ng.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmann J, Holzmann K, Miller F, Winkler D, Buhler A, Zenz T, Bullinger L, Kuhn MW, Gerhardinger A, Bloehdorn J, et al. High-resolution genomic profiling of chronic lymphocytic leukemia reveals new recurrent genomic alterations. Blood. 2012;120:4783–4794. doi: 10.1182/blood-2012-04-423517. [DOI] [PubMed] [Google Scholar]
- Fabbri G, Rasi S, Rossi D, Trifonov V, Khiabanian H, Ma J, Grunn A, Fangazio M, Capello D, Monti S, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. The Journal of experimental medicine. 2011;208:1389–1401. doi: 10.1084/jem.20110921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri G, Holmes AB, Viganotti M, Scuoppo C, Belver L, Herranz D, Yan XJ, Kieso Y, Rossi D, Gaidano G, et al. Common nonmutational NOTCH1 activation in chronic lymphocytic leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:E2911–E2919. doi: 10.1073/pnas.1702564114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson SE, Leeman-Neill RJ, Jain S, Piao W, Cieply KM, Swerdlow SH. Proliferation centres of chronic lymphocytic leukaemia/small lymphocytic lymphoma have enhanced expression of MYC protein, which does not result from rearrangement or gain of the MYC gene. Br J Haematol. 2016;175:173–175. doi: 10.1111/bjh.13844. [DOI] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonda TJ, Ramsay RG. Adenoid Cystic Carcinoma Can Be Driven by MYB or MYBL1 Rearrangements: New Insights into MYB and Tumor Biology. Cancer discovery. 2016;6:125–127. doi: 10.1158/2159-8290.CD-15-1470. [DOI] [PubMed] [Google Scholar]
- Henkel T, Ling PD, Hayward SD, Peterson MG. Mediation of Epstein-Barr virus EBNA2 transactivation by recombination signal-binding protein J kappa. Science. 1994;265:92–95. doi: 10.1126/science.8016657. [DOI] [PubMed] [Google Scholar]
- Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, Xu L, Castillo-Martin M, Llobet-Navas D, Cordon-Cardo C, et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med. 2014;20:1130–1137. doi: 10.1038/nm.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jitschin R, Braun M, Qorraj M, Saul D, Le Blanc K, Zenz T, Mougiakakos D. Stromal cell-mediated glycolytic switch in CLL cells involves Notch-c-Myc signaling. Blood. 2015;125:3432–3436. doi: 10.1182/blood-2014-10-607036. [DOI] [PubMed] [Google Scholar]
- Johansen LM, Deppmann CD, Erickson KD, Coffin WF, 3rd, Thornton TM, Humphrey SE, Martin JM, Taparowsky EJ. EBNA2 and activated Notch induce expression of BATF. Journal of virology. 2003;77:6029–6040. doi: 10.1128/JVI.77.10.6029-6040.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel MJ, Velusamy T, Betz BL, Zhao L, Weigelin HG, Chiang MY, Huebner-Chan DR, Bailey NG, Yang DT, Bhagat G, et al. Whole-genome sequencing identifies recurrent somatic NOTCH2 mutations in splenic marginal zone lymphoma. The Journal of experimental medicine. 2012;209:1553–1565. doi: 10.1084/jem.20120910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim U, Qin XF, Gong S, Stevens S, Luo Y, Nussenzweig M, Roeder RG. The B-cell-specific transcription coactivator OCA-B/OBF-1/Bob-1 is essential for normal production of immunoglobulin isotypes. Nature. 1996;383:542–547. doi: 10.1038/383542a0. [DOI] [PubMed] [Google Scholar]
- Kluk MJ, Ashworth T, Wang H, Knoechel B, Mason EF, Morgan EA, Dorfman D, Pinkus G, Weigert O, Hornick JL, et al. Gauging NOTCH1 Activation in Cancer Using Immunohistochemistry. PLoS ONE. 2013;8:e67306. doi: 10.1371/journal.pone.0067306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kridel R, Meissner B, Rogic S, Boyle M, Telenius A, Woolcock B, Gunawardana J, Jenkins C, Cochrane C, Ben-Neriah S, et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood. 2012;119:1963–1971. doi: 10.1182/blood-2011-11-391474. [DOI] [PubMed] [Google Scholar]
- Krysov S, Dias S, Paterson A, Mockridge CI, Potter KN, Smith KA, Ashton-Key M, Stevenson FK, Packham G. Surface IgM stimulation induces MEK1/2-dependent MYC expression in chronic lymphocytic leukemia cells. Blood. 2012;119:170–179. doi: 10.1182/blood-2011-07-370403. [DOI] [PubMed] [Google Scholar]
- Kuhnert F, Kirshner JR, Thurston G. Dll4-Notch signaling as a therapeutic target in tumor angiogenesis. Vasc Cell. 2011;3:20. doi: 10.1186/2045-824X-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Hu S, Wang SA, Li S, Huh YO, Tang Z, Medeiros LJ, Tang G. The clinical significance of 8q24/MYC rearrangement in chronic lymphocytic leukemia. Mod Pathol. 2016;29:444–451. doi: 10.1038/modpathol.2016.35. [DOI] [PubMed] [Google Scholar]
- Maier S, Staffler G, Hartmann A, Hock J, Henning K, Grabusic K, Mailhammer R, Hoffmann R, Wilmanns M, Lang R, et al. Cellular target genes of Epstein-Barr virus nuclear antigen 2. Journal of virology. 2006;80:9761–9771. doi: 10.1128/JVI.00665-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez I, Dimaio D. B-Myb, cancer, senescence, and microRNAs. Cancer research. 2011;71:5370–5373. doi: 10.1158/0008-5472.CAN-11-1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam Y, Sliz P, Song L, Aster JC, Blacklow SC. Structural basis for cooperativity in recruitment of MAML coactivators to Notch transcription complexes. Cell. 2006;124:973–983. doi: 10.1016/j.cell.2005.12.037. [DOI] [PubMed] [Google Scholar]
- Pinnell N, Yan R, Cho HJ, Keeley T, Murai MJ, Liu Y, Alarcon AS, Qin J, Wang Q, Kuick R, et al. The PIAS-like Coactivator Zmiz1 Is a Direct and Selective Cofactor of Notch1 in T Cell Development and Leukemia. Immunity. 2015;43:870–883. doi: 10.1016/j.immuni.2015.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzo F, Bittolo T, Vendramini E, Bomben R, Bulian P, Rossi FM, Zucchetto A, Tissino E, Degan M, D’Arena G, et al. NOTCH1 mutated chronic lymphocytic leukemia cells are characterized by a MYC-related overexpression of nucleophosmin-1 and ribosome associated components. Leukemia. 2017 doi: 10.1038/leu.2017.90. in press (epub April 18, 2017) [DOI] [PubMed] [Google Scholar]
- Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, Escaramis G, Jares P, Bea S, Gonzalez-Diaz M, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101–105. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente XS, Bea S, Valdes-Mas R, Villamor N, Gutierrez-Abril J, Martin-Subero JI, Munar M, Rubio-Perez C, Jares P, Aymerich M, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526:519–524. doi: 10.1038/nature14666. [DOI] [PubMed] [Google Scholar]
- Ramsay RG, Gonda TJ. MYB function in normal and cancer cells. Nature reviews Cancer. 2008;8:523–534. doi: 10.1038/nrc2439. [DOI] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi D, Spina V, Deambrogi C, Rasi S, Laurenti L, Stamatopoulos K, Arcaini L, Lucioni M, Rocque GB, Xu-Monette ZY, et al. The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood. 2011;117:3391–3401. doi: 10.1182/blood-2010-09-302174. [DOI] [PubMed] [Google Scholar]
- Rossi D, Rasi S, Fabbri G, Spina V, Fangazio M, Forconi F, Marasca R, Laurenti L, Bruscaggin A, Cerri M, et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood. 2012;119:521–529. doi: 10.1182/blood-2011-09-379966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan RJ, Drier Y, Whitton H, Cotton MJ, Kaur J, Issner R, Gillespie S, Epstein CB, Nardi V, Sohani AR, et al. Detection of Enhancer-Associated Rearrangements Reveals Mechanisms of Oncogene Dysregulation in B-cell Lymphoma. Cancer discovery. 2015;5:1058–1071. doi: 10.1158/2159-8290.CD-15-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, McElhinny AS, Cao Y, Gao P, Liu J, Bronson R, Griffin JD, Wu L. The Notch coactivator, MAML1, functions as a novel coactivator for MEF2C-mediated transcription and is required for normal myogenesis. Genes Dev. 2006;20:675–688. doi: 10.1101/gad.1383706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Whyte WA, Zepeda-Mendoza CJ, Milazzo JP, Shen C, Roe JS, Minder JL, Mercan F, Wang E, Eckersley-Maslin MA, et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013;27:2648–2662. doi: 10.1101/gad.232710.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalska L, Stojnic R, Li J, Fischer B, Cerda-Moya G, Sakai H, Tajbakhsh S, Russell S, Adryan B, Bray SJ. Chromatin signatures at Notch-regulated enhancers reveal large-scale changes in H3K56ac upon activation. EMBO J. 2015;34:1889–1904. doi: 10.15252/embj.201489923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spender LC, Cornish GH, Sullivan A, Farrell PJ. Expression of transcription factor AML-2 (RUNX3, CBF(alpha)-3) is induced by Epstein-Barr virus EBNA-2 and correlates with the B-cell activation phenotype. Journal of virology. 2002;76:4919–4927. doi: 10.1128/JVI.76.10.4919-4927.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeck A, Lejnine S, Truong A, Pan L, Wang H, Zang C, Yuan J, Ware C, MacLean J, Garrett-Engele PW, et al. Discovery of biomarkers predictive of GSI response in triple-negative breast cancer and adenoid cystic carcinoma. Cancer discovery. 2014;4:1154–1167. doi: 10.1158/2159-8290.CD-13-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Luo OJ, Li X, Zheng M, Zhu JJ, Szalaj P, Trzaskoma P, Magalska A, Wlodarczyk J, Ruszczycki B, et al. CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription. Cell. 2015;163:1611–1627. doi: 10.1016/j.cell.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend EC, Murakami MA, Christodoulou A, Christie AL, Koster J, DeSouza TA, Morgan EA, Kallgren SP, Liu H, Wu SC, et al. The Public Repository of Xenografts Enables Discovery and Randomized Phase II-like Trials in Mice. Cancer Cell. 2016;30:183. doi: 10.1016/j.ccell.2016.06.008. [DOI] [PubMed] [Google Scholar]
- Usui T, Wakatsuki Y, Matsunaga Y, Kaneko S, Koseki H, Kita T. Overexpression of B cell-specific activator protein (BSAP/Pax-5) in a late B cell is sufficient to suppress differentiation to an Ig high producer cell with plasma cell phenotype. Journal of immunology. 1997;158:3197–3204. [PubMed] [Google Scholar]
- Wang H, Zang C, Taing L, Arnett KL, Wong YJ, Pear WS, Blacklow SC, Liu XS, Aster JC. NOTCH1-RBPJ complexes drive target gene expression through dynamic interactions with superenhancers. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:705–710. doi: 10.1073/pnas.1315023111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C, Del Bianco C, Rodriguez CG, Sai H, Tobias J, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006;20:2096–2109. doi: 10.1101/gad.1450406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, Finkle D, Venook R, Wu X, Ridgway J, et al. Therapeutic antibody targeting of individual Notch receptors. Nature. 2010;464:1052–1057. doi: 10.1038/nature08878. [DOI] [PubMed] [Google Scholar]
- Xu H, Chaudhri VK, Wu Z, Biliouris K, Dienger-Stambaugh K, Rochman Y, Singh H. Regulation of bifurcating B cell trajectories by mutual antagonism between transcription factors IRF4 and IRF8. Nature immunology. 2015;16:1274–1281. doi: 10.1038/ni.3287. [DOI] [PubMed] [Google Scholar]
- Yashiro-Ohtani Y, Wang H, Zang C, Arnett KL, Bailis W, Ho Y, Knoechel B, Lanauze C, Louis L, Forsyth KS, et al. Long-range enhancer activity determines Myc sensitivity to Notch inhibitors in T cell leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E4946–4953. doi: 10.1073/pnas.1407079111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Zou JY, Wang H, Johannsen E, Peng CW, Quackenbush J, Mar J, Morton CC, Freedman ML, Blacklow SC, et al. Epstein-Barr virus exploits intrinsic B-lymphocyte transcription programs to achieve immortal cell growth. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:14902–14907. doi: 10.1073/pnas.1108892108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Katzman RB, Delmolino LM, Bhat I, Zhang Y, Gurumurthy CB, Germaniuk-Kurowska A, Reddi HV, Solomon A, Zeng MS, et al. The notch regulator MAML1 interacts with p53 and functions as a coactivator. J Biol Chem. 2007;282:11969–11981. doi: 10.1074/jbc.M608974200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Summary of evidence for presence or absence of MYC locus rearrangements in CLL and MCL cell lines. Related to Figures 1 and 2.
Tab 1: Genes increased in Notch-on state. Median transcript abundance (transcripts per million; TPM) for each gene was calculated with Kallisto; Tab 2: Genes decreased in Notch-on state.
Tab 1: Gene sets from the MSigDB Hallmark and Reactome databases enriched in Group 1 genes. FDR q-values are for combined analysis of both gene set collections. Tab 2: Gene sets from the Hallmark and Reactome databases enriched in Group 2 genes. Tab 3: Results of Ingenuity Canonical Pathways analysis on Group 1 genes. ‘Ratio’ corresponds to ratio of pathway members in the test gene set to total pathway members.
Tab 1: NTC-linked direct Notch target genes identified in MCL cell lines. The strongest proximal and/or distal Pol2 ChIA-PET-linked NTC peaks are listed for each gene, as well as total number of independently linked NTC peaks in each category (proxLinks and distLinks). Log2-fold-change (l2fc) and FDR-adjusted p-value (padj) are from DESeq2 analysis of gsi-washout RNA-Seq data in the indicated line. ChIP-Seq signal intensities are listed for each linked peak, normalized as fragments over peak per 10 million total aligned fragments. Tab 2: All NTC site – Notch-up-regulated gene links, defined by TSS proximity or Pol2 ChIA-PET. Tab 3: Direct Notch target genes identified in this analysis and shared with the Notch target gene signature identified by NICD1 transgene overexpression in the CLL cell line MO1043 (Fabbri et al., 2017).
Median transcript abundance (transcripts per million; TPM) for each gene in vehicle-treated mice was calculated with Kallisto.
Median transcript abundance (transcripts per million; TPM) for each gene in NOTCH1-mutant CLL biopsies was calculated with Kallisto.
Listed are all NTC binding sites identified in MCL cell lines with significant binding in both NICD and RBPJ ChIP-Seq performed on CLL-M13 lymph node biopsy material. ChIP-Seq signal intensities are listed for each linked peak, in MCL cell lines and CLL-M13 normalized as fragments over peak per 10 million total aligned fragments.