

SUMMARY
The mechanistic target of rapamycin complex 1 (mTORC1) supports proliferation through parallel induction of key anabolic processes, including protein, lipid, and nucleotide synthesis. We hypothesized that these processes are coupled to maintain anabolic balance in cells with mTORC1 activation, a common event in human cancers. Loss of the tuberous sclerosis complex (TSC) tumor suppressors results in activation of mTORC1 and development of the tumor syndrome TSC. We find that pharmacological inhibitors of guanylate nucleotide synthesis have selective deleterious effects on TSC-deficient cells, including in mouse tumor models. This effect stems from replication stress and DNA damage caused by mTORC1-driven ribosomal RNA synthesis, which renders nucleotide pools limiting. These findings reveal a metabolic vulnerability downstream of mTORC1 triggered by anabolic imbalance.
Keywords: mTOR, rapamycin, tumor metabolism, tuberous sclerosis complex, TSC2, IMPDH, mizoribine, mycophenolic acid, nucleotide synthesis, lymphangioleiomyomatosis
eTOC Blurb/In Brief
mTORC1 drives anabolic tumor metabolism, including ribosome biogenesis and nucleotide synthesis. Valvezan et al. show that blocking synthesis of guanine nucleotides while sustaining mTORC1 activity depletes nucleotide pools, causing DNA replication stress and apoptosis in mTORC1-driven tumor models.
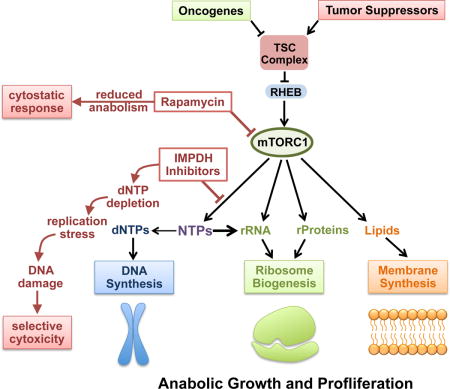
INTRODUCTION
Proliferating cells utilize coordinated anabolic pathways to synthesize essential macromolecules and generate new biomass to facilitate cell replication. Such large-scale anabolism creates a high demand for the requisite nutrients and building blocks. The mechanistic target of rapamycin complex I (mTORC1) is a central regulator of anabolic growth and proliferation that senses signals from nutrients, energy, and growth factors to control downstream metabolic outputs. Under favorable growth conditions, mTORC1 stimulates synthesis of the major macromolecules required for cell growth and proliferation, including proteins, nucleic acids, and lipids, while simultaneously inducing mechanisms to generate macromolecular precursors (Ben-Sahra and Manning, 2017). While the importance of coupling these anabolic processes downstream of mTORC1 is unknown, it is likely that cells need to maintain anabolic balance and prevent depletion of essential metabolites during growth and proliferation.
One established function of mTORC1 is to promote ribosome biogenesis as a key part of a larger program to increase the protein synthesis capacity of cells (Iadevaia et al., 2014; Ma and Blenis, 2009; Mayer and Grummt, 2006). mTORC1 induces translation of mRNAs which encode the ribosomal protein subunits, and also stimulates rRNA synthesis through coordinated regulation of 3 essential RNA Polymerase I (Pol I)-specific transcription factors, UBF, SL-1, and TIF1A, and by relieving MAF1-mediated inhibition of RNA Polymerase III (Pol III) transcription (Iadevaia et al., 2014; Mayer and Grummt, 2006; Michels et al., 2010; Shor et al., 2010). rRNA accounts for greater than 50% of ribosome mass and approximately 85% of total cellular RNA, and the rRNA of ribosomes is where the bulk of cellular nucleotides reside, indicating that ribosome biogenesis is accompanied by a significant increase in demand for nucleotides. To this end, mTORC1 signaling also stimulates de novo synthesis of purine and pyrimidine nucleotides, which are rapidly incorporated into RNA and, in proliferating cells, DNA (Ben-Sahra et al., 2013; Ben-Sahra et al., 2016; Robitaille et al., 2013). mTORC1 also stimulates flux through glycolysis and the pentose phosphate pathway, which provide essential precursors for de novo nucleotide, amino acid, and lipid synthesis (Duvel et al., 2010). Thus mTORC1 stimulates ribosome biogenesis, while simultaneously inducing metabolic changes to supply the essential precursors. This paradigm suggests that mTORC1 activation could confer unique dependence on the metabolic pathways that maintain anabolic balance during growth and proliferation, and that targeting these pathways could unveil a selective metabolic vulnerability. De novo nucleotide synthesis pathways are upregulated when cells are induced to proliferate by growth factors, cytokines, or oncogenic transformation, including in activated lymphocytes and cancer cells (Lane and Fan, 2015). Nucleotide synthesis pathways are attractive therapeutic targets, given that there are many well-tolerated clinicially approved inhibitors, that, like the mTORC1 inhibitor rapamycin, are generally used as immunosuppressants. A common target of pharmacological inhibitors of nucleotide synthesis is inosine monophosphate dehydrogenase (IMPDH), the rate limiting enzyme in guanylate nucleotide synthesis (Ishikawa, 1999). Within this class of compounds, mizoribine (Bredinin) is a natural purine analog used in Asia for preventing organ rejection after transplantation and treating autoimmune disorders, and mycophenolic acid (MPA) (Cellcept, Myfortic) is used in the USA for similar indications. Importantly, mizoribine and MPA are well tolerated, with safety profiles similar or superior to rapamycin (Ishikawa, 1999; Kalluri and Hardinger, 2012).
Rapamycin and its analogs (rapalogs) are also currently approved to treat specific manifestations of tuberous sclerosis complex (TSC), a tumor syndrome driven by uncontrolled mTORC1 signaling, and the related neoplastic progressive lung disease lymphangioleiomyomatosis (LAM) (Henske et al., 2016). Germline mutations in the TSC1 or TSC2 tumor suppressor genes give rise to TSC. TSC1 and TSC2 form a protein complex, referred to as the TSC complex, that is a critical negative regulator of mTORC1 (Dibble and Manning, 2013). Thus, loss of TSC1 or TSC2 results in constitutive activation of mTORC1 and its downstream anabolic program, including in sporadic tumors, such as renal angiomyolipomas, that arise in TSC patients due to spontaneous loss of the wild-type copy of the TSC gene (Henske et al., 2016; Kwiatkowski and Manning, 2014). Rapalogs cause tumor shrinkage of approximately 40% in these lesions, highlighting the essential role of mTORC1 in the growth of TSC-deficient tumor cells. However, rapalogs largely induce cytostatic, rather than cytotoxic, responses, and tumors rapidly regrow when treatment is stopped (Bissler et al., 2008; Franz et al., 2006; Fruman and Rommel, 2014). As mTORC1 activation stimulates parallel anabolic processes, mTORC1 inhibitors proportionally decrease these anabolic processes and, thus, likely maintain a balanced anabolic state. It seems possible that this feature of mTORC1 signaling might contribute to the lack of cytotoxicity with rapalog therapy. We hypothesized that loss of the TSC complex renders cells dependent on specific anabolic pathways activated downstream of mTORC1 and represents an excellent genetic and disease model to test the idea that anabolic balance is required for sustained cell growth and survival upon mTORC1 activation.
RESULTS
IMPDH inhibition selectively targets the viability of TSC2-deficient cells in an mTORC1-dependent manner
To determine whether the induction of purine nucleotide synthesis downstream of mTORC1 is essential for the proliferation of cells with aberrantly elevated mTORC1 signaling, we tested 4 clinically approved inhibitors of IMPDH on pairs of wild-type and TSC2-deficient cells. Mizoribine, MPA and ribavirin inhibited the proliferation of Tsc2−/−;Trp53−/− MEFs and TSC2 null human renal angiomyolipoma (AML)-derived cells (621-101) with varying degrees of selectivity relative to their TSC2-expressing counterparts, Tsc2+/+;Trp53−/− MEFs and the 621-101 cells stably reconstituted with wild-type TSC2 (Figures 1A and S1A, S1B). On the other hand, Azathioprine, which has several targets in addition to IMPDH (Maltzman and Koretzky, 2003), selectively inhibited TSC2-expressing cells (Figure S1A). Mizoribine demonstrated the highest degree of selectivity in targeting TSC2-deficient cells, and was therefore chosen for further characterization. The selective effects of mizoribine were confirmed by examining 4 additional isogenic pairs of cell lines with or without TSC2, including Tsc2−/−;Trp53+/+ MEFs and the Tsc2−/− 105K renal tumor-derived cell line expressing empty vector or reconstituted with wild-type TSC2 (Filippakis et al., 2017)(Figure 1B and 1C) and MCF10A or HeLa cells with stable shRNA-mediated knockdown of TSC2 relative to control knockdowns (Figure S1C), and by viable cell counts (Figure S1D and S1E). Thus, TSC2-deficient cells, with elevated and growth factor-independent activation of mTORC1, as scored by phosphorylation of S6K1 and phosphorylation-dependent mobility shifts in 4E-BP1 (Figures 1D–1F, S1B, S1C), displayed increased sensitivity to mizoribine in all 6 pairs of cell lines. Importantly, the selective effects of mizoribine are not due to differences in rates of proliferation, which were nearly identical within each pair of cell lines (Figure S1F). Consistent with mTORC1 signaling influencing this sensitivity, wild-type cells displayed a modest increase in sensitivity to mizoribine with increasing serum concentrations, which activates mTORC1, whereas TSC2-deficient cells, which exhibit serum-independent activation of mTORC1, were equally hypersensitive at all serum concentrations (Figure 1D–1F and S1G). Growth in dialyzed serum, which lacks exogenous nucleosides, did not affect sensitivity to mizoribine (Figure S1H). Importantly, mizoribine exerted selective effects on TSC2-deficient cells without inhibiting mTORC1 signaling (Figures 1D–1F) and exhibited similar or superior selectivity for these cells as rapamycin, with no additional effects from combining the two compounds (Figures 1G–1I). siRNA-mediated knockdown of IMPDH1 and 2 also selectively and strongly reduced the growth of TSC2-deficient cells (Figure 1J).
Figure 1. IMPDH inhibitors selectively induce cell death in TSC2-deficient cells due to elevated mTORC1 signaling.
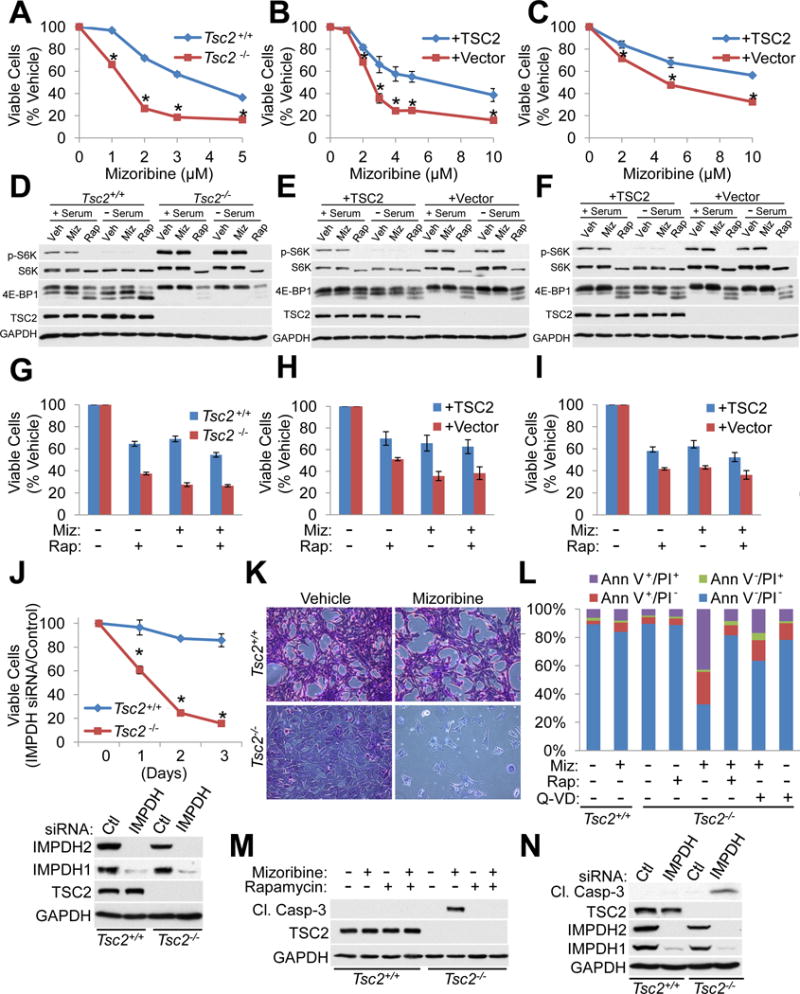
(A–C) Tsc2+/+ Trp53−/− and Tsc2−/− Trp53−/− MEFs (A), Tsc2−/− Trp53+/+ 3T3 MEFs with stable reconstitution of TSC2 or empty vector (B), and Tsc2−/− 105K renal tumor cells with stable reconstitution of TSC2 or empty vector (C) were treated with the indicated concentrations of mizoribine for 72 hr and viable cells are graphed as a percentage of vehicle-treated cells. n=12 biological replicates
(D–F) Tsc2+/+ Trp53−/− and Tsc2−/− Trp53−/− MEFs (D), Tsc2−/− Trp53+/+ 3T3 MEFs with stable reconstitution of TSC2 or empty vector (E), and Tsc2−/− 105K renal tumor cells with stable reconstitution of TSC2 or empty vector (F) were treated with vehicle, 20 nM rapamycin (Rap), or mizoribine (Miz: 2 μM (D); 3.5 μM (E); 5 μM (F)) for 18 hr in the presence or absence of serum, and mTORC1 signaling was assesed by immunoblot.
(G–I) Tsc2+/+ Trp53−/− and Tsc2−/− Trp53−/− MEFs (G), Tsc2−/− Trp53+/+ 3T3 MEFs with stable reconstitution of TSC2 or empty vector (H), and Tsc2−/− 105K renal tumor cells with stable reconstitution of TSC2 or empty vector (I) were treated for 72 hr with 20 nM rapamycin, and/or 2 μM mizoribine, with viable cells graphed as a percentage of vehicle-treated cells. n=6 biological replicates
(J) Cells from A were transfected with control or IMPDH1/2-targeting siRNAs, with viable cells measured over the following three days and graphed as a percentage of cells with control siRNAs (n=12 biological replicates). Knockdown efficiency is shown in the immunoblot below.
(K) Cells from A were treated with vehicle or mizoribine (3 μM, 72 hr) and stained with crystal violet, with a representative microscopic field shown. Scale bar = 0.2 mm
(L) Cells from A treated with vehicle, 3 μM mizoribine, 20 nM rapamycin, or 20 μM Q-VD-Oph for 72 hr. Cell death was quantified by Annexin V (Ann V)/propidium iodide (PI) staining and graphed as the percentage of the total cell population. n=4 biological replicates
(M,N) Immunoblots on cells from A treated with 2 μM mizoribine or 20 nM rapamycin for 48 hr (M), or transfected with siRNAs as in J for 72 hr (N).
Graphical data are represented as mean of indicated replicates, error bars represent ± SEM. *p<0.005 by two-tailed Student’s t test.
See also Figure S1.
Upon mizoribine treatment, TSC2-deficient cells undergo morphological changes and detachment consistent with cell death (Figure 1K). Nearly 70% of Tsc2−/− cells stained positive for Annexin V or both Annexin V and propidium iodide (PI) after 3 days of mizoribine treatment, compared to just 15% of Tsc2+/+ cells (Figure 1L). Rescuing effects with the pan-caspase inhibitor Q-VD-Oph suggest that apoptosis is primarily responsible for mizoribine-induced cell death. Importantly, not only did rapamycin fail to induce cell death in TSC2-deficient cells, it strongly protected these cells from mizoribine-induced death, demonstrating that mTORC1 activation is required for sensitization to mizoribine downstream of TSC2-loss (Figure 1L). Within 48 hr, mizoribine selectively induces caspase-3 activation in TSC2-deficient, but not wild-type cell lines, which is blocked by rapamycin co-treatment, while both lines display similar sensitivity to staurosporine (Figures 1M, S1I, S1J). TSC2 knockdown in HeLa cells similarly sensitizes to mizoribine-induced death, which is reduced by co-treatment with rapamycin (Figure S1K). Transient knockdown of IMPDH1 and 2 mimics mizoribine and selectively induces caspase-3 cleavage in Tsc2−/− cells (Figure 1N). These data indicate that elevated mTORC1 signaling sensitizes TSC2-deficient cells to apoptotic cell death upon IMPDH inhibition.
The IMPDH inhibitor mizoribine has anti-tumor efficacy in both genetic and xenograft tumor models of TSC
We next asked whether mizoribine can target tumors driven by TSC2-loss and elevated mTORC1 signaling. Tsc2+/− mice on the A/J strain background develop kidney tumors classified as cystadenomas with 100% penetrance by 4 months of age (Auricchio et al., 2012; Guo and Kwiatkowski, 2013). These lesions initially develop as hollow cysts and progressively fill with tumor cells, becoming adenomas that can progress to carcinomas. As rapamycin is used to treat TSC tumors and has been established as an effective anti-tumor agent in this mouse model, we directly compared mizoribine to rapamycin. Mice were treated with vehicle, mizoribine, or rapamycin for 1 month beginning at 6 months of age, when cystadenomas with extensive cellularity (or cyst filling) are well established (Figure 2A). Based on previous reports (Chen et al., 1990; Ushijima et al., 1991), mizoribine was initially administered at 200 mg/kg for the first 9 days. As some weight loss was observed, a 5-day break in treatment was taken, followed by continued treatment at a reduced dose of 100 mg/kg/day, under which mice regained weight back to the vehicle group (Figure S2A). Like rapamycin, mizoribine is used clinically as an immunosuppressant, and blood cell counts at the end of treatment revealed an expected reduction in lymphocyte and total white blood cell counts, similar to rapamycin-treated mice (Figure S2B). As a further confirmation of on-target effects of mizoribine, plasma concentrations of aminoimidazole carboxamide ribonucleotide (AICAR), an intermediate in the de novo purine synthesis pathway upstream of IMPDH, were elevated nearly 40-fold compared to vehicle or rapamycin-treated mice (Figure 2B). Mizoribine also caused AICAR accumulation in a dose dependent manner in the kidney, spleen, and liver of wild-type mice, with the greatest accumulation in the liver, where de novo nucleotide synthesis is most active (Figure S2C–S2E).
Figure 2. The IMPDH inhibitor mizoribine has anti-tumor efficacy in genetic and xenograft tumor models of TSC.
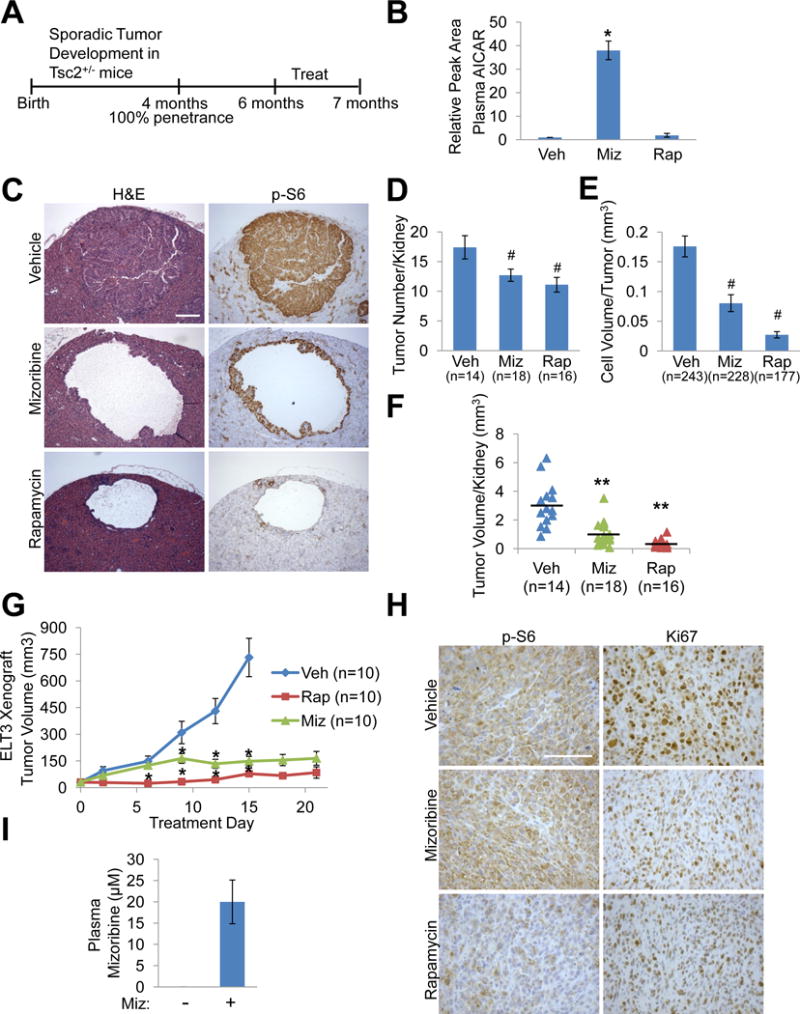
(A) Experimental design used in A–F. 6-month old Tsc2+/− mice were treated for 32 days with vehicle, mizoribine (days 1–9: 200 mg/kg/day; days 10–15: no treatment: days 16–32: 100 mg/kg/day), or rapamycin (1 mg/kg MWF).
(B) AICAR levels measured by LC-MS/MS in blood plasma collected on day 32, 3 hr after the final injection of vehicle (veh), mizoribine (miz), or rapamycin (rap), and graphed relative to vehicle. n=5 mice/group
(C) H&E and phospho-S6 (S235/236) IHC staining on serial kidney sections, with representative tumors from each treatment group shown. Scale bar = 0.2 mm
(D) Number of tumors (cysts, cystadenomas, and adenomas) per kidney (n=kidney number).
(E) Volume of tumor cell filling (cellularity) per tumor (n=tumor number).
(F) Total tumor volume per kidney, with each data point corresponding to one kidney and horizontal line indicating the mean (n=kidney number).
(G,H) Mice bearing Tsc2−/− ELT3 xenograft tumors were treated with vehicle, mizoribine (75 mg/kg/day), or rapamycin (1 mg/kg MWF) for 21 days. Tumor volume (n=tumor number) was measured every 3rd day (G). Representative IHC staining of tumors in each treatment group (H). Scale bar = 0.2 mm
(I) Mizoribine concentration in blood plasma collected 2.5 hr after the final injection of vehicle (-) or mizoribine in mice from G, measured by LC-MS/MS. n=3 mice/group
Graphical data are represented as mean ± SEM. #p<0.05, *p<0.009, **p<0.00005 by two-tailed Student’s t test.
See also Figure S2.
To determine the effects of mizoribine treatment on tumor volume, kidneys were serial sectioned and analyzed histologically (see methods). Like tumors in human TSC, tumors in the Tsc2+/− model arise due to sporadic loss of the remaining wild-type allele of Tsc2, resulting in aberrant activation of mTORC1 signaling (Auricchio et al., 2012; Guo and Kwiatkowski, 2013). Consistent with cell culture models (Figures 1D–1F), rapamycin but not mizoribine inhibited mTORC1 signaling in these kidney lesions, as scored by phospho-S6 staining (Figure 2C). Tumor number, size, and volume of cellularity were measured as described previously (Auricchio et al., 2012; Guo and Kwiatkowski, 2013), with 177–243 tumors analyzed per group. Mizoribine reduced the number of tumors per kidney by 27% and reduced tumor cellularity by 54%, resulting in a 68% reduction in total tumor volume per kidney, and per mouse (Figures 2D–2F and S2F). Rapamycin had similar effects on these parameters to those reported previously (Auricchio et al., 2012; Guo and Kwiatkowski, 2013). Rapamycin also reduced the average volume of individual cysts and the total cyst volume per kidney, which mizoribine did not influence (Figure S2G and S2H). Ultrasound monitoring of individual tumors before and after treatment indicated that the cellularity of established tumors was greatly reduced or eliminated after mizoribine, but not vehicle treatment (Figure S2I and S2J), demonstrating that mizoribine exerts anti-tumor activity on existing tumors. Therefore, while mizoribine has minimal effects on kidney cysts in this model, it has strong effects on adenomas and the tumor cells with high mTORC1 signaling that fill these cysts.
As a second widely used tumor model of TSC, mice bearing xenograft tumors established with the TSC2-deficient rat uterine leiomyoma ELT3 cell line (Hodges et al., 2002; Yu et al., 2009) were treated with vehicle, mizoribine (75 mg/kg/day), or rapamycin beginning 4 weeks after flank injection of ELT3 cells, when tumors first became palpable and measurable. Tumor volume measurements revealed that, after the first 6 days of treatment, mizoribine blocked tumor growth, an effect sustained for the duration of treatment and comparable to rapamycin (Figure 2G). Total body weight was not significantly affected in any treatment group (Figure S2K). Consistent with tumor volume measurements, both mizoribine and rapamycin reduced staining for the proliferation marker Ki67 (Figure 2H). In contrast to rapamycin, mizoribine did not affect phospho-S6 staining (Figure 2H). To assess plasma mizoribine concentrations, we developed an approach to measure mizorbine by LC-MS/MS, which we used to generate a standard curve (Figure S2L). Importantly, plasma concentrations of mizoribine following 21 days of treatment were measured at 20 μM (Figure 2I), well within the achievable pharmacological range in humans (Stypinski et al., 2007).
Mizoribine selectively affects the viability of cells with activated mTORC1 through the depletion of guanylate nucleotides
We next sought to determine the mechanism underlying the selective effects of mizoribine that are dependent on mTORC1 activation. Upon uptake into cells, mizoribine must be phosphorylated to produce the GMP analog mizoribine monophosphate (miz-MP) to inhibit IMPDH (Hager et al., 1995), a step proposed to be mediated by adenosine kinase (ADK) (Koyama and Tsuji, 1983) (Figure 3A). Following treatment, intracellular mizoribine levels were similar between Tsc2+/+ and Tsc2−/− cells (Figure 3B), but miz-MP levels were greatly reduced in Tsc2−/− cells, which correlated with reduced ADK expression (Figure 3B and 3C). Assaying this reaction in cell free extracts demonstrated that Tsc2−/− cells indeed have greatly decreased ability to generate the active miz-MP compound, a conversion confirmed by siRNA knockdowns to be dependent on ADK (Figures 3C and 3D). ADK knockdown also blocked the effects of mizoribine on cell proliferation, without affecting mTORC1 activity, demonstrating that ADK is required for the growth inhibitory effects of mizoribine (Figure 3C and 3E). Therefore, the selective effects of mizoribine are not due to differences in pharmacokinetics, as mizoribine selectively targets Tsc2−/− cells despite reduced levels of ADK and reduced production of miz-MP in these cells.
Figure 3. Mizoribine selectively affects the viability of cells with activated mTORC1 through the depletion of guanylate nucleotides.
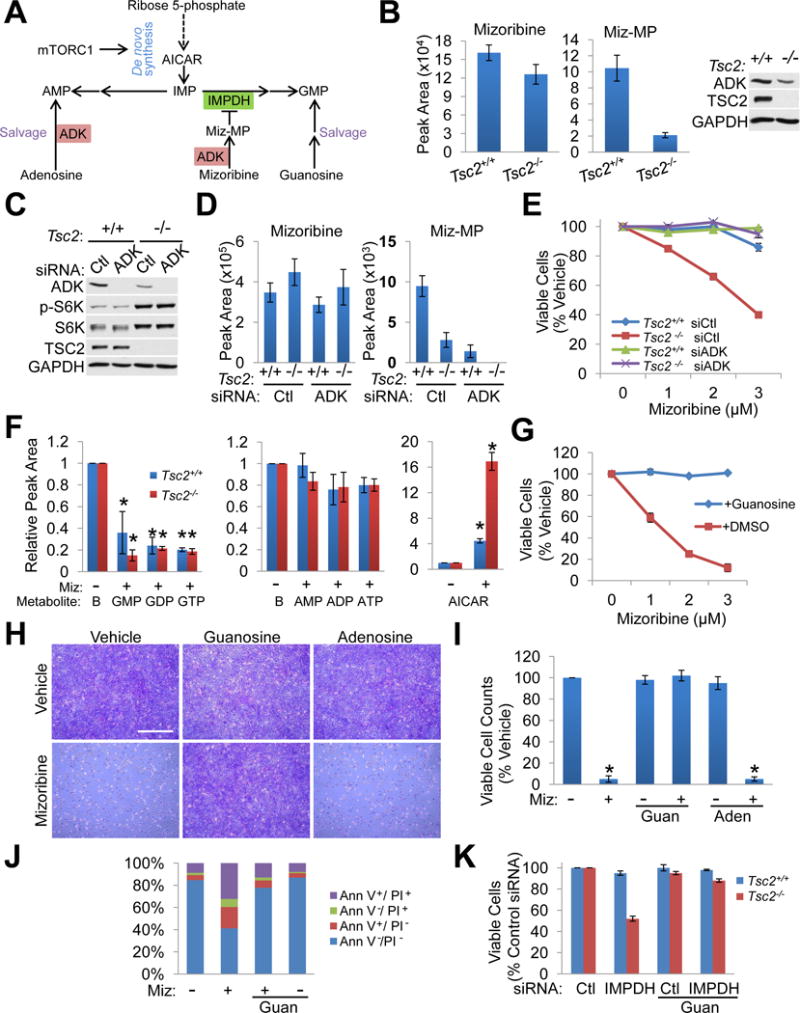
(A) Schematic of de novo purine synthesis and salvage pathways, and mizoribine conversion to mizoribine monophosphate (Miz-MP) by adenosine kinase (ADK).
(B) Intracellular mizoribine and Miz-MP levels measured by LC-MS/MS in Tsc2+/+ Trp53−/− and Tsc2−/− Trp53−/− MEFs treated with vehicle or mizoribine (2 μM, 18 hr). n=3 biological replicates. ADK levels were assessed by immunoblot (right).
(C) Tsc2+/+ Trp53−/− and Tsc2−/− Trp53−/− MEFs 48 hr post-transfection with control or ADK siRNAs were analyzed by immunoblotting.
(D) Conversion of mizoribine to Miz-MP in cell free extracts prepared from the cells in C. Mizoribine was added to extracts for 30 min, and mizoribine and Miz-MP levels were measured by LC-MS/MS. n=3 biological replicates
(E) Tsc2+/+ Trp53−/− and Tsc2−/− Trp53−/− MEFs were treated with mizoribine (48 hr). Cell viability is graphed as the percentage of vehicle-treated cells. n=6 biological replicates
(F) Relative abundance of the indicated metabolites, measured by LC-MS/MS and normalized to baseline, represented by left bars “B” (the abundance of each metabolite in vehicle-treated cells with values set to 1), from Tsc2+/+ Trp53−/− and Tsc2−/− Trp53−/− MEFs treated with vehicle or mizoribine (2 μM, 16 hr). n=3 biological replicates
(G) Viability of Tsc2−/− Trp53−/− MEFs treated for 72 hr with vehicle or mizoribine at the indicated concentrations with or without 50 μM guanosine and graphed as the percentage of vehicle-treated cells. n=6 biological replicates
(H,I) Tsc2−/− Trp53−/− MEFs treated with vehicle or mizoribine (3 μM, 72 hr) with or without 50 μM guanosine (guan) or 50 μM adenosine (aden), stained with crystal violet (H) or counted by trypan blue exclusion (I). Viable cell counts are graphed as the percentage of vehicle-treated cells (n=3 biological replicates). Scale bar = 1 mm
(J) Tsc2−/− Trp53−/− MEFs treated with vehicle or mizoribine (3 μM, 72 hr) and/or 50 μM guanosine. Cell death was quantified by Annexin V/PI staining and graphed as the percentage of the total cell population. n=3 biological replicates
(K) Viability of Tsc2+/+ Trp53−/− and Tsc2−/− Trp53−/− MEFs transfected with control (Ctl) or IMPDH1 and 2 siRNAs for 48 hr and cultured with or without 50 μM guanosine. Data are represented as the percentage of control siRNA-transfected cells. n=6 biological replicates
Graphical data are represented as mean of indicated replicates, error bars represent ± SEM.
*p<0.05 by two-tailed Student’s t test.
See also Figure S3.
Although TSC2-deficient cells are more sensitive to mizoribine in all 6 pairs of cell lines tested, the concentration range of mizoribine required for growth inhibition can vary from one cell pair to another, with the 621-101s being the least sensitive (Figure S1B). Unlike in MEFs, miz-MP was undetectable in these cells treated with 2 μM mizoribine, despite similar levels of total intracellular mizoribine (Figure S3A). The effective dose of mizoribine in 621–101 cells (300 μM) resulted in a 125-fold increase in intracellular mizoribine, but only a modest increase in intracellular miz-MP, which was comparable to that in Tsc2−/− MEFs treated with 2 μM mizoribine (Figure S3A). Consistent with this finding and the relative resistance of the 621-101 cells to mizoribine, these cells express very low levels of ADK, with the TSC2-deficient cells again having lower ADK levels compared to their wild-type controls (Figure S3B). These data further support that ADK levels influence sensitivity to mizoribine but do not underlie the selective effects on viability observed in TSC2-deficient cells.
IMPDH catalyzes the first committed step in guanylate nucleotide synthesis from IMP, a common precursor for both guanylate and adenylate nucleotides (Figure 3A). Steady state metabolomic profiling revealed that mizoribine reduces GMP, GDP, and GTP levels in MEFs (Figure 3F) and 621–101 cells (Figure S3C) without significant effects on adenylate nucleotides, confirming specific IMPDH inhibition. As observed in vivo (Figure 2B, S2C–S2E), mizoribine also caused an increase in AICAR, with greater AICAR accumulation in TSC2-deficient cells (Figure 3F, S3C), consistent with increased mTORC1-stimulated flux through the de novo purine synthesis pathway (Ben-Sahra et al., 2016). This mizoribine-induced increase in AICAR is not sufficient to activate AMPK, as scored by its phosphorylation and that of its substrate Raptor in either Tsc2−/− cells or the mouse liver (Figure S3D and S3E). Importantly, excess exogenous guanosine, which can be converted to GMP independent of IMPDH (Figure 3A), rescued mizoribine effects on Tsc2−/− cell growth and apoptosis, while adenosine had no effect (Figures 3G–3J, S3F, S3G). The selective effects of IMPDH knockdown on cell viability were also rescued by guanosine (Figure 3K). Collectively, these findings demonstrate that the selective effects of IMPDH inhibition on cells with activated mTORC1 result from guanylate nucleotide depletion.
IMPDH inhibition causes selective replication stress, DNA damage, and apoptosis in an mTORC1-dependent manner in TSC2-deficient cells and tumors
To further define the nature of the selective effects of mizoribine, cell cycle profiles were examined. Consistent with similar proliferation rates between TSC2-deficient and expressing cells (Figure S1D and S1F), cell cycle distributions in vehicle-treated cells were nearly identical (Figure 4A, 4B, S4A, S4B). However, mizoribine caused selective accumulation of TSC2-deficient cells in S-phase, an effect entirely rescued by guanosine. This effect is followed by the appearance of a subG0 cell population (Figure S4A), corresponding to the induction of apoptosis (Figure S1J). Thus, IMPDH inhibition selectively prevents TSC2-deficient cells, but not TSC2-expressing cells, from completing DNA replication.
Figure 4. IMPDH inhibition causes selective replication stress and DNA damage in TSC2-deficient cells due to elevated mTORC1 signaling and guanylate depletion.
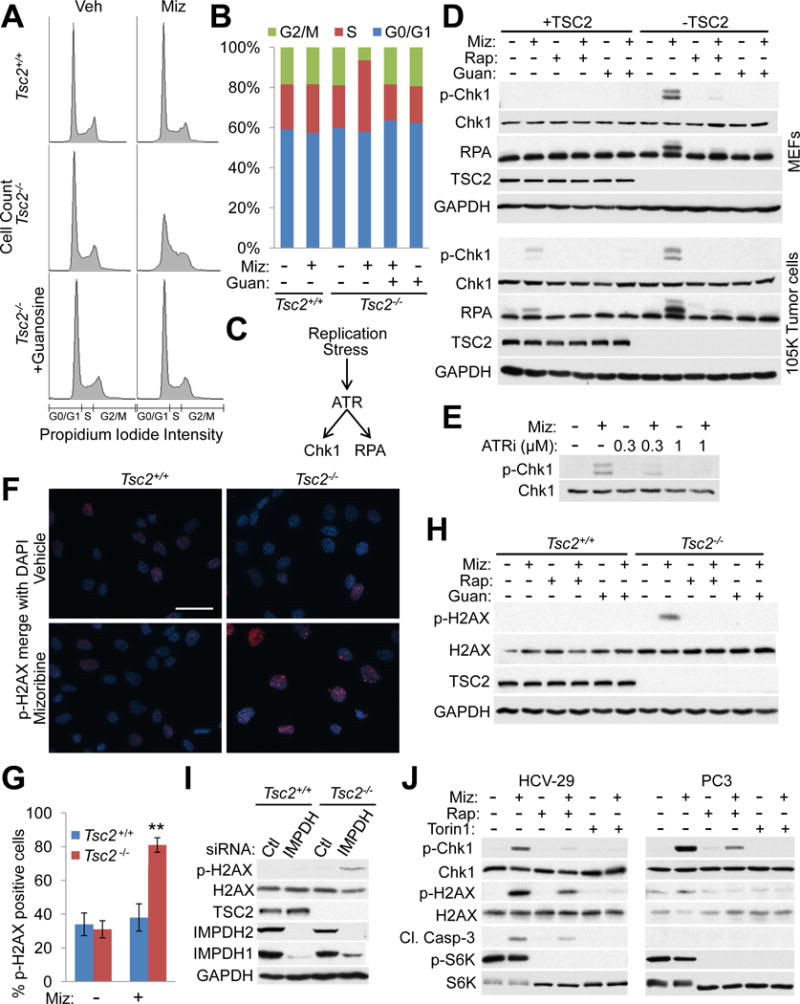
(A,B) Cell cycle distributions of Tsc2+/+ Trp53−/− and Tsc2−/− Trp53−/− MEFs treated with vehicle or mizoribine (2 μM, 24 hr) with or without 50 μM guanosine based on propidium iodide intensity (A) and graphed as percent of the total population (B). Data are represented as mean of biological replicates (n=4).
(C) Schematic of the intra-S phase checkpoint activated by replication stress.
(D) Immunoblots of the indicated proteins in Tsc2+/+ Trp53−/− and Tsc2−/− Trp53−/− MEFs (upper) and 105K tumor cells with stable TSC2 or vector addback (lower) treated for 24 hr with vehicle, mizoribine (2 μM (upper), 3.5 μM (lower)), 20 nM rapamycin, or 50 μM guanosine.
(E) Immunoblot of total and phosphorylated Chk1 inTsc2−/− Trp53−/− MEFs treated with mizoribine (2 μM, 24 hr) and/or the ATR inhibitor AZD6738.
(F,G) Tsc2+/+ Trp53−/− and Tsc2−/− Trp53−/− MEFs treated with vehicle or 2 μM mizoribine (48 hr) followed by immunofluorescence staining for phospho-H2AX (S139) (red). Representative images merged with DAPI (blue) are shown (F) and the percent of positive cells (n=94–110 cells/group) was quantified in (G). Data are presented as mean ± SEM. **p<0.00005 by two-tailed Student’s t test. Scale bar = 0.05 mm
(H) Immunoblots of p-H2AX in Tsc2+/+ Trp53−/− and Tsc2−/− Trp53−/− MEFs treated with vehicle, 2 μM mizoribine, 20 nM rapamycin, and/or 50 μM guanosine (48 hr).
(I) Immunoblots of p-H2AX from Tsc2+/+ Trp53−/− and Tsc2−/− Trp53−/− MEFs, 72 hr post-transfection with control (Ctl) or IMPDH1 and 2 siRNAs.
(J) HCV-29 (left) or PC3 (right) cells were treated for 72 hr with vehicle, mizoribine (500 μM), rapamycin (20 nM) or Torin1 (250 nM) and analyzed by immunoblotting for the indicated proteins.
See also Figure S4.
Nucleotide depletion can cause replication stress and activation of the intra S-phase checkpoint mediated by ATR, which phosphorylates Chk1 and RPA (Figure 4C) (Zeman and Cimprich, 2014). In all TSC2-deficient cells examined, mizoribine selectively induced phosphorylation of Chk1 at the ATR-specific site (S345), and of RPA as indicated by slower mobility (Olson et al., 2006), and these effects were blocked by co-treatment with rapamycin or guanosine (Figure 4D, S4C, S4D). Mizoribine-induced Chk1 phosphorylation was blocked by the ATR-selective inhibitor AZD6738 (Figure 4E) (Vendetti et al., 2015). Unresolved replication stress can lead to DNA damage (Zeman and Cimprich, 2014). Indeed, mizoribine selectively induced phosphorylation of histone H2AX (S139), a marker of DNA damage, in TSC2-deficient but not TSC2-expressing cells, and this was blocked by rapamycin or guanosine (Figure 4F–4H, S4E–S4I). IMPDH knockdown likewise induced phosphorylation of H2AX specifically in TSC2-deficient cells (Figure 4I). Importantly, the onset of DNA damage correlates temporally with induction of apoptosis (Figure S4F, S4G, S4I). Taken together, these data indicate that mizoribine selectively induces early replication stress in TSC2-deficient cells, an effect dependent on sustained mTORC1 signaling, and this unresolved stress leads to DNA damage and cell death. Mizoribine also induced replication stress, DNA damage, and apoptosis in TSC1-deficient bladder cancer cells (HCV-29), which was blocked by rapamycin or the mTOR kinase inhibitor torin1 (Figure 4J (left)), and in colorectal cancer cells with multiple mTORC1 pathway-activating mutations (HCT116), where torin1, but not rapamycin, blocked the effects of mizoribine (Figure S4J, S4K). In PTEN-deficient prostate cancer cells (PC3), mizoribine strongly induces Chk1 phosphorylation in an mTORC1-dependent manner, but does not cause DNA damage or apoptosis (Figure 4J (right)).
To determine whether mizoribine causes a similar response that is dependent on sustained mTORC1 signaling in a TSC tumor model, mice bearing ELT3 xenograft tumors were treated for 5 days with vehicle, mizoribine, rapamycin, or mizoribine plus rapamycin. In mice receiving mizoribine, the compound was readily detected in plasma (15–20 μM) and tumors, and it effectively depleted guanylate nucleotides in the tumors, with little effect on adenylate nucleotides (Figures 5A–5C, S5A). Mizoribine also caused AICAR accumulation in plasma and tumors, without affecting AMPK activity (Figure 5D, 5E, S5B). Interestingly, co-treatment with rapamycin attenuated the mizoribine-induced depletion of guanylates, without affecting plasma or tumor mizoribine levels (Figures 5A–5C), suggesting that the elevated mTORC1 signaling in these tumors contributes to guanylate consumption. Rapamycin also reduced AICAR accumulation in tumors, but not plasma, from mizoribine-treated mice, consistent with rapamycin reducing flux through de novo purine synthesis in these tumors (Figure 5E) (Ben-Sahra et al., 2016). Importantly, treatment with mizoribine, but not rapamycin, induced phosphorylation of Chk1 and H2AX in tumors, and caspase-3 activation in non-necrotic tumor regions, and these effects were blocked by co-treatment with rapamycin (Figures 5F, 5G, S5C). Not only did rapamycin fail to induce caspase-3 activation, it decreased the basal levels of cleaved caspase-3 in these tumors (Figure 5G, S5C). Mizoribine did not induce DNA damage or apoptosis in the normal tissues of these mice, including the kidney and liver (Figure S5C). Thus, mizoribine causes depletion of guanylate nucleotides, replication stress, DNA damage, and apoptosis in TSC2-deficient tumors, effects that are dependent on sustained mTORC1 signaling.
Figure 5. IMPDH inhibition induces replication stress, DNA damage, and apoptosis in TSC tumors, in an mTORC1-dependent manner.
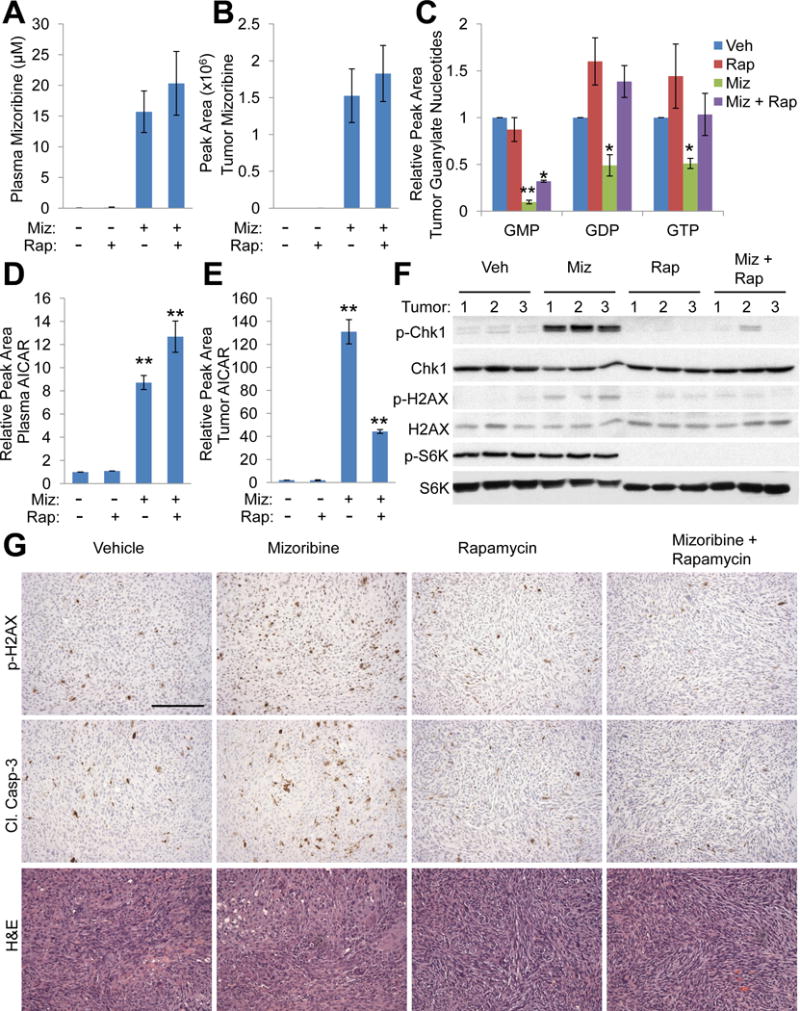
(A–G) Mice bearing ELT3 xenograft tumors were treated for 5 days with vehicle (Veh), mizoribine (Miz; 150 mg/kg/day), rapamycin (Rap; 1 mg/kg day 1, 3, 5), or mizoribine plus rapamycin. Blood plasma and tumors were collected 3 hr after the final treatment injection. n=3 mice/group
(A–E) Levels of mizoribine in blood plasma (A) or tumors (B), guanylate nucleotides in tumors (C), and AICAR in blood plasma (D) or tumors (E) were measured by LC-MS/MS and presented as mean ± SEM (n=3 mice/group). Data in C-E are graphed relative to vehicle-treated mice. *p<0.05, **p<0.005 by two-tailed Student’s t test.
(F,G) Analysis of indicated proteins by immunoblots (F) and IHC staining on non-necrotic regions (G) from tumors. Scale bar = 0.2 mm.
See also Figure S5.
Increased synthesis of rRNA downstream of mTORC1 sensitizes cells to nucleotide depletion and replication stress upon IMPDH inhibition
In proliferating cells transiting through S phase, ribonucleotides (NTPs) are converted to deoxyribonucleotides (dNTPs) by ribonucleotide reductase (RNR) to allow DNA replication. Unlike mizoribine, the RNR inhibitor hydroxyurea (HU) induced similar activation of the replication stress checkpoint in Tsc2+/+ and Tsc2−/− cells (Figure 6A), and did not selectively inhibit the viability of Tsc2−/− cells (Figure 6B). Furthermore, HU-induced Chk1 and RPA phosphorylation were not blocked by rapamycin (Figure 6C). Thus, TSC2 loss and mTORC1 activation do not increase sensitivity to dNTP depletion per se.
Figure 6. mTORC1-driven rRNA synthesis sensitizes cells to replication stress upon IMPDH inhibition.
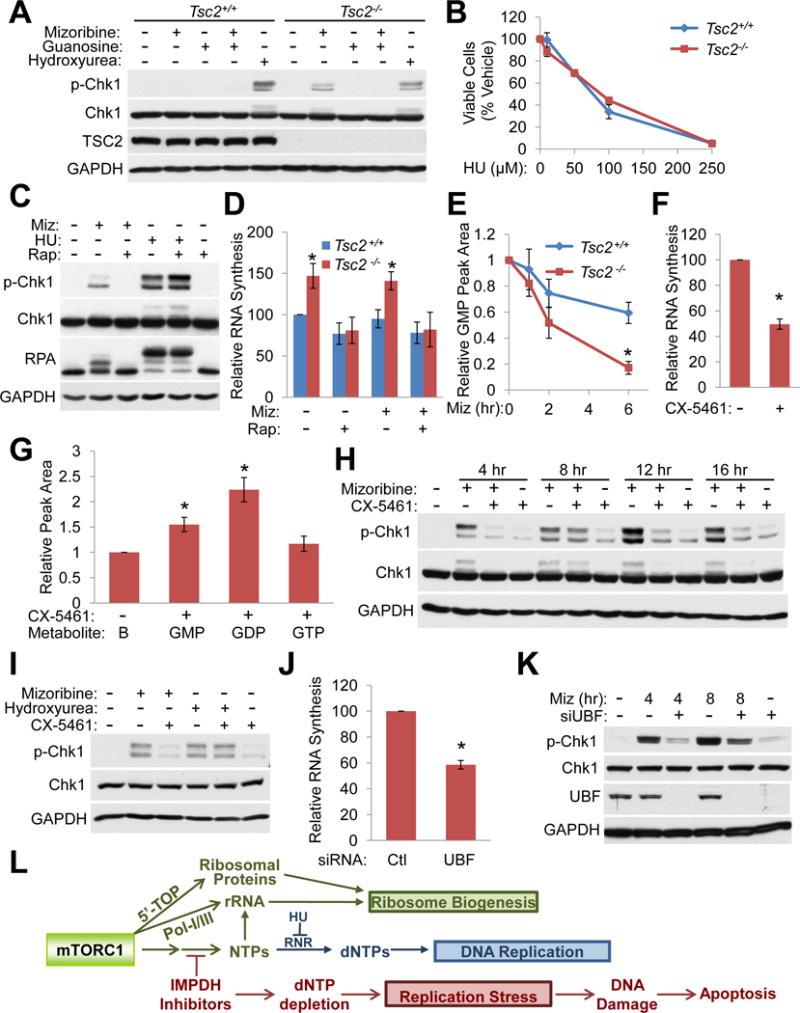
(A) Immunoblots from Tsc2+/+ Trp53−/− and Tsc2−/− Trp53−/− MEFs treated for 24 hr with 2 μM mizoribine and/or 50 μM guanosine, or for 3 hr with 3 mM hydroxyurea.
(B) Viability of Tsc2+/+ Trp53−/− and Tsc2−/− Trp53−/− MEFs treated for 72 hr with hydroxyurea (HU), graphed as percent of vehicle-treated cells. n=6 biological replicates
(C) Immunoblots from Tsc2−/− Trp53−/− MEFs treated for 4 hr with 2 μM mizoribine (Miz), 3 mM hydroxyurea (HU), or 20 nM rapamycin (Rap), as indicated.
(D) RNA synthesis was measured in Tsc2+/+ Trp53−/− and Tsc2−/− Trp53−/− MEFs treated for 5 hr with vehicle, 20 nM rapamycin, and/or 2 μM mizoribine. Cells were labeled for 1 hr with 2,8-3H-adenine. Total RNA was isolated and radioactive counts per minute were normalized to total RNA and graphed relative to vehicle-treated Tsc2+/+ cells. n=5 biological replicates
(E) Tsc2+/+ Trp53−/− and Tsc2−/− Trp53−/− MEFs were treated with 2 μM mizoribine for the indicated times, and cellular GMP, measured by LC-MS/MS, is graphed relative to untreated cells. n=3 biological replicates
(F) Tsc2−/− Trp53−/− MEFs were treated with the Pol I inhibitor CX-5461 (100 nM, 3.5 hr) before 1 hr labeling with 5,6-3H-uridine. Total RNA was isolated and radioactive counts per minute were normalized to total RNA and graphed relative to vehicle-treated cells. n=3 biological replicates
(G) Guanylate nucleotide levels were measured in Tsc2−/− Trp53−/− MEFs by LC-MS/MS and normalized to the abundance of each in vehicle-treated cells (represented by left bar “B”, with value set to 1) after CX-5461 treatment (100 nM, 6 hr). n=3 biological replicates
(H) Immunoblots from Tsc2−/− Trp53−/− MEFs treated with 2 μM mizoribine or 100 nM CX-5461.
(I) Immunoblots from Tsc2−/− Trp53−/− MEFs treated for 16 hr with 2 μM mizoribine, 3 mM hydroxyurea, or 100 nM CX-5461.
(J) Tsc2−/− Trp53−/− MEFs were transfected with control or siRNA targeting UBF for 48 hr before addition of 5,6-3H-uridine for 1 hr. Total RNA was isolated and radioactive counts per minute were normalized to total RNA and graphed relative to control siRNA. n=3 biological replicates
(K) Immunoblots from Tsc2−/− Trp53−/− MEFs transfected as in J followed by treatment with vehicle or 2 μM mizoribine for the indicated times.
(L) Model. mTORC1 promotes nucleotide synthesis to support rRNA synthesis as part of ribosome biogenesis. IMPDH inhibitors uncouple these processes, selectively reducing the guanine nucleotide pool available for DNA synthesis in cells with active mTORC1 signaling, resulting in replication stress, DNA damage, and apoptosis.
Graphical data are represented as mean ± SEM. *p<0.05 by two-tailed Student’s t test.
See also Figure S6.
mTORC1 stimulates ribosome biogenesis, in part, through increased Pol I-dependent synthesis of rRNAs (Iadevaia et al., 2014; Mayer and Grummt, 2006). The majority of nucleotides in growing cells reside in rRNA, with a disproportionate amount being guanylate (34.1%). Consistent with the role of mTORC1 in driving rRNA synthesis, TSC2-deficient cells exhibited an 80% increase in the rate of rRNA synthesis (Figure S6A), and a 50% increase in the rate of total RNA synthesis, which are entirely blocked by rapamycin yet unaffected by mizoribine (Figure 6D). Tsc2−/− cells also had ~30% more ribosomes, as measured by total rRNA and ribosomal protein per cell, which were not affected by mizoribine (Figure S6B, S6C). Mizoribine also did not affect ribosomal protein levels in ELT3 xenograft tumors (Figure S6D) or the rate of protein synthesis in wild-type or TSC2-deficient cells (Figure S6E).
We reasoned that increased rRNA synthesis downstream of mTORC1 might render nucleotide pools limiting for use in DNA replication. In support of this, a time course of mizoribine revealed that free GMP pools were depleted more rapidly in Tsc2−/− cells (Figure 6E), while free GDP and GTP were depleted at similar rates (Figure S6G), suggesting the remaining GMP is rapidly converted to GDP and GTP for use in nucleic acid synthesis. Rapamycin decreased RNA synthesis in the presence or absence of mizoribine (Figure 6D), and this correlated with a partial restoration of DNA synthesis in mizoribine-treated cells (Figure S6F). To specifically block rRNA synthesis, we used the RNA Pol I inhibitor CX-5461 (Drygin et al., 2011; Haddach et al., 2012). Pol I inhibition reduced RNA synthesis in Tsc2−/− cells by 50% and correspondingly increased guanylate nucleotide pools (Figures 6F, 6G). Importantly, Pol I inhibition decreased the mizoribine-mediated induction of replication stress in these cells, in a time and dose-dependent manner (Figure 6H, S6H, S6I), without effects on mTORC1 signaling (Figure S6H) or HU-induced replication stress (Figure 6I). Transient siRNA-mediated knockdown of the Pol I-specific transcription factor UBF likewise reduced RNA synthesis in TSC2-deficient cells (Figure 6J) and attenuated the replication stress induced by mizoribine (Figure 6K and S6J). As expected, long-term inhibition of Pol I with either CX-5461 or UBF knockdown, alone, induced cell death and thus was unable to rescue mizoribine effects on cell viability (Figure S6K).
These data indicate that an mTORC1-dependent increase in rRNA synthesis sensitizes TSC2-deficient cells to replication stress upon inhibition of guanylate nucleotide synthesis. We propose a model in which mTORC1 drives nucleotide synthesis to meet the increased nucleotide demand that accompanies its induction of ribosome biogenesis during anabolic growth and proliferation. IMPDH inhibition uncouples these processes by blocking nucleotide synthesis. In settings with sustained mTORC1 signaling, guanylate nucleotide pools available for DNA synthesis become depleted, leading to replication stress, DNA damage, and cell death (Figure 6L).
DISCUSSION
Here we find that mTORC1 promotes anabolic cell growth and proliferation by coordinately regulating ribosome biogenesis and nucleotide synthesis to maintain anabolic balance. This manifests as an increased dependence on nucleotide synthesis in cells with activated mTORC1, a metabolic vulnerability that can be targeted using the IMPDH inhibitor mizoribine. While IMPDH inhibitors have not been widely tested for their effects in cancer, mizoribine and MPA have been found to reduce the viability of certain cancer cells through guanylate nucleotide depletion (Barfeld et al., 2015; Liu et al., 2008; Sakaguchi et al., 1975a; Sakaguchi et al., 1975b). Here, we reveal a molecular mechanism that sensitizes cells to IMPDH inhibition. This study indicates that widely used clinical IMPDH inhibitors are good candidates for repurposing as anti-tumor agents for tumors with elevated mTORC1 signaling, such as those in TSC and LAM patients.
Our findings suggest that the primary reason that mTORC1 has evolved mechanisms to induce de novo nucleotide synthesis is to support its role in the production of new ribosomes, which by mass are over 50% RNA and are resident to the majority of nucleotides in a growing cell. mTORC1 drives the synthesis of both ribosomal proteins and rRNAs, the latter through both RNA Pol I and Pol III activation (Iadevaia et al., 2014; Mayer and Grummt, 2006), creating a great demand for more nucleotides. In settings with constitutive mTORC1 activation, the uncoupling of elevated rates of rRNA synthesis from increased nucleotide synthesis renders the existing nucleotide pools limiting, thereby causing DNA replication stress. A competition for nucleotides between the nucleic acid polymerases synthesizing rRNA and DNA is supported by our finding that transient inhibition of RNA Pol I-mediated rRNA synthesis spares nucleotides and protects cells from mizoribine-induced DNA replication stress.
For decades, cytotoxic therapeutics that target nucleotide metabolism, such as hydroxyurea, methotrexate, 6-mercaptopurine and others, have been components of chemotherapeutic cancer treatment regimens geared toward killing proliferating cells. Recent insights into how oncogenes and tumor suppressors, and their downstream effectors, exert direct control over nucleotide synthesis pathways have led to renewed interest in selectively targeting specific enzymes in nucleotide metabolism in specific genetic settings (Ben-Sahra et al., 2013; Ben-Sahra et al., 2016; Brown et al., 2017; Cunningham et al., 2014; Liu et al., 2008; Mathur et al., 2017; Robitaille et al., 2013; Ying et al., 2012). A key point in our findings is that the selective effects of IMPDH inhibitors are not dictated by differences in proliferation rates. While TSC-deficient cells have an mTORC1-dependent increase in cell size, the TSC2-expressing and -deficient cell lines examined proliferate at similar rates and exhibit similar cell cycle distributions within each pair. Furthermore, TSC2-deficient cells are not inherently more sensitive to replication stress, as blocking dNTP production with HU caused replication stress and cell death regardless of TSC2 status. Thus the selectivity of IMPDH inhibition is mechanistically distinct from DNA replication-inhibiting chemotherapeutic agents that target rapidly proliferating cells as their mode of action.
Rapalogs are the only therapeutics currently approved to treat TSC and LAM tumors, but evidence suggests that patients must remain on therapy indefinitely to prevent rapid tumor regrowth (Bissler et al., 2008; Franz et al., 2006). In a subset of patients, rapalog toxicity can limit treatment duration. Our preclinical data suggest that IMPDH inhibitors could potentially be repurposed to treat tumors with hyperactive mTORC1, such as in TSC, possibly leading to an effective alternative therapy to rapalogs that targets a unique metabolic feature of these tumors. It is important to note that our findings indicate that IMPDH inhibitors should not be used in combination with rapalogs, as their selective cytotoxic effects are dependent on sustained mTORC1 signaling. While the TSC tumor suppressors are only rarely mutated in sporadic cancers, a large network of the most common oncogenes and tumor suppressors underlying human malignancies converge on regulation of the TSC complex (Ilagan, 2016). Thus, the TSC complex is predicted to be dysfunctional in at least half of human cancers, across nearly all lineages, due to perturbations in upstream signaling pathways, leading to chronic activation of mTORC1. It will be important to determine in future studies which cancer settings with elevated mTORC1 signaling are similarly sensitive to IMPDH inhibition through the mechanism revealed here. In a limited survey of cancer cell lines, we found that mizoribine can induce replication stress in a manner that is blocked by mTOR inhibitors. IMPDH1 and 2 are transcriptional targets of the oncogene Myc, are upregulated in certain cancers, and have been suggested as potential targets in cells with Myc activation (Barfeld et al., 2015; Fellenberg et al., 2010; He et al., 2009; Liu et al., 2008; Mannava et al., 2008; Zou et al., 2015). It is interesting to note that, like mTORC1, Myc also drives both ribosome biogenesis and nucleotide synthesis (Liu et al., 2008; Stine et al., 2015), suggesting a similar mechanism of vulnerability.
Our study also identifies AICAR, an intermediate in the de novo purine synthesis pathway upstream of IMPDH, as an in vivo metabolic biomarker of IMPDH inhibition. AICAR levels are increased in blood plasma, tumors, and tissues of mizoribine-treated mice, suggesting an easily accessible biomarker that could aid in establishing dosing regimens in patients on IMPDH inhibitors, either as immunosuppressants or anti-tumor agents. AICAR is commonly used experimentally to activate AMPK (Corton et al., 1995), but mizoribine treatment did not activate AMPK in cells or tissues, suggesting that despite increases of up to 130-fold, as seen in our TSC tumor model, these levels are still lower than that achieved by exogenous addition of AICAR.
Our findings suggest an alternative approach to selectively target cells with aberrant activation of mTORC1. While rapalogs dampen the anabolic output of mTORC1 signaling, they do so in a relatively balanced manner, decreasing protein, lipid, and nucleotide synthesis along with the demand for these cellular building blocks. Thus, the anabolic balance achieved by the parallel activation of multiple biosynthetic processes downstream of mTORC1, is maintained by its direct inhibition. However, as illustrated in this study, inhibiting one anabolic branch of mTORC1 signaling in the face of sustained activation of the others creates an anabolic imbalance that induces cell death. Similarly, inhibiting mTORC1-driven lipid synthesis by knocking down the sterol regulatory element binding protein (SREBP) transcription factors can induce endoplasmic reticulum stress due to sustained protein synthesis and an inability to balance this with an expansion of cellular membrane lipids (Griffiths et al., 2013; Young et al., 2013). Thus, while IMPDH inhibitors are particularly interesting due to their widespread use and safety in humans, it is likely that other such targets to disrupt anabolic balance will emerge as we further define the points of metabolic control downstream of mTORC1.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Brendan Manning (bmanning@hsph.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Tsc2+/+; Trp53−/− and Tsc2−/−; Trp53−/− MEFs (Zhang et al., 2003), Tsc2−/− 3T3-immortalized MEFs stably expressing empty vector or TSC2 (Huang et al., 2008), Tsc2−/− 105Ks stably expressing empty vector or TSC2 (Filippakis et al., 2017), 621-101 cells stably expressing empty vector or TSC2 (Yu et al., 2004), HeLa cells stably expressing shRNAs targeting luciferase or TSC2 (Huang et al., 2008), Tsc2−/− ELT3 cells (Hodges et al., 2002), HCV-29, PC3, and HCT116 cells were grown in DMEM (VWR #45000-312) plus 10% heat-inactivated fetal bovine serum (ThermoFisher Scientific #10437-028). MCF10As stably expressing shRNAs targeting luciferase or TSC2 were cultured as previously described (Zhang et al., 2014).
Mouse studies
All animal procedures were approved by the Harvard Institutional Animal Care and Use Committee. Tsc2+/− mice on the A/J strain background were described previously (Auricchio et al., 2012; Guo and Kwiatkowski, 2013). Intraperitoneal injections of vehicle, mizoribine (200 mg/kg/day, Selleckchem #S1384), or rapamycin (1 mg/kg MWF, LC Laboratories #R-5000) were administered to both male (3–4 mice/treatment group, randomly assigned) and female (4–5 mice/treatment group, randomly assigned) mice beginning at 6 months of age. On the 9th day of treatment, mizoribine dose was reduced to 100 mg/kg/day in female mice and halted for 5 days in male mice due to weight loss. Mizoribine treatment resumed in male mice on Day 14 at 100mg/kg/day. Mice were treated until Day 32 and sacrificed for tissue harvesting and blood collection 3 hr after the final treatment injection.
For the xenograft tumor studies, 2.5 million ELT3 cells expressing luciferase (Yu et al., 2009) were subcutaneously injected in a 1:1 mixture with matrigel (BD #356237) into the flank of 6–7 week old female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice (Jackson Laboratory #005557). Treatment began 2–4 weeks later, when tumors first became palpable, with intraperitoneal injections of vehicle, mizoribine (75 (Figure 2) or 150 (Figure 5) mg/kg/day), or rapamycin (1 mg/kg M/W/F), with 10 (Figure 2) or 3 (Figure 5) mice per treatment group. Mice were assigned to treatment groups based on their tumor size, so that the average tumor size per group was the same among all groups at the start of treatment. Mice were sacrificed 2.5 hrs after the final treatment injection and tumors were harvested from the vehicle group on treatment day 15 (Figure 2) when tumors reached maximum allowable size, or day 5 (Figure 5), and from the mizoribine and rapamycin groups on day 21 (Figure 2) or 5 (Figure 5). Tumors were fixed and blood plasma was collected for analyses, as described below.
METHOD DETAILS
Chemical compounds
The following compounds were added into the cell culture medium at final concentrations indicated in the figure legends: rapamycin (EMD Millipore #53123-88-9) in DMSO, mizoribine (Selleckchem #S1384 and Sigma #M3047) in water, mycophenolic acid (Sigma #M3536) in methanol, ribavirin (Sigma #R9644) in water, azathioprine (Sigma #A4638) in DMSO, Q-VD-OPh (Sigma #SML0063) in DMSO, staurosporine (Tocris #1285) in DMSO, CX-5461 (Selleckchem #S2684) in DMF, hydroxyurea (Sigma #H8627) in water, AZD6738 (Selleckchem #S7693) in DMSO, guanosine (Sigma #G6752) in DMSO, adenosine (Sigma #A9251) in DMSO.
siRNA transfections
The following siRNAs (GE Dharmacon SMARTpools) were transfected at 12.5 nM using Lipofectamine RNAiMAX (ThermoFisher Scientific #13778150) according to manufacturer’s instructions: ADK (L-062728-00-0005), IMPDH2 (M-168757-00-0005), IMPDH1 (M-042235-00-0005), UBF (M-056732-01-0005), control (Non-targeting siRNA pool D-001810-10-50).
Cell viability assays
Cell viability was measured using the Cell Titer Glo Luminescent Cell Viability Assay (Promega #G7573) according to manufacturer’s instructions. Broad dose curves were generated for each IMPDH inhibitor and pair of cells, with only intermediate doses shown to exclude lower doses that had no effect and higher doses that all had the same, maximal effect. Where indicated, viable cell counts were determined by trypan blue (Sigma #T8154) exclusion using a hemocytometer. Annexin V/PI staining was performed using the Dead Cell Apoptosis Kit (ThermoFisher Scientific #V13245), according to manufacturer’s instructions. Staining was measured with a Becton Dickinson LSRFortessa flow cytometer, and analyzed with FlowJo Version 10.2 software.
Metabolite analyses by LC-MS/MS
Metabolites were extracted using 80% methanol and dried under nitrogen gas for targeted tandem mass spectrometry (LC-MS/MS) profiling via selected reaction monitoring (SRM) with polarity switching on a 5500 QTRAP mass spectrometer (AB/SCIEX) as previously described (Ben-Sahra et al., 2013; Yuan et al., 2012). Metabolites were extracted from 8μl plasma, 0.05–0.25g tissue, or 1 10cm cell culture dish (in triplicate) for LC-MS/MS analysis. Mizoribine was measured in negative ion mode using Q1/Q3 SRM transitions of 258.2/126 and mizoribine monophosphate in negative ion mode using 338/79. For standard curves, mizorbine was prepared at 10-fold dilutions from 1 mM to 1 nM in 50% methanol. Data were analyzed by calculating the Q3 peak areas using MultiQuant 2.1.1 software (AB/SCIEX).
Tumor volume measurements
For tumor volume measurements in Tsc2+/− mice, upon dissection, kidneys were fixed in 10% neutral buffered formalin (Sigma #HT501128) for 24 hr, then washed in ddH2O and stored in 70% ethanol. For analyses of kidney tumors, serial 1-mm sections, cut along the length of each kidney, were paraffin embedded and sectioned for H&E staining and immunostaining. Tumor volume measurements were taken from H&E stained slices from each 1-mm section across each kidney by a single blinded observer, as described previously (Auricchio et al., 2012; Guo and Kwiatkowski, 2013), with tumor volume for each cyst calculated as percent cellularity multiplied by cyst volume (1.333*π*r3). For xenograft experiments, tumor size was measured every three days using digital calipers.
Immunohistochemistry
IHC staining was performed on formalin-fixed, paraffin-embedded tissue sections. Sections were incubated in 3 washes of Histoclear (National Diagnostics, #HS-200) for 5 min each, followed by 2 washes in 100% ethanol and 2 washes in 95% ethanol for 10 min each, then 2 washes in ddH2O for 5 min each. Antigen retrieval was performed by boiling for 10 min in 10 mM sodium citrate, pH 6.0, followed by washing in ddH2O, then 3% hydrogen peroxide for 10 min, followed by ddH2O again. TBST plus 5% normal goat serum (CST #5425) was used as a blocking solution for 1 hr at room temperature. Primary antibodies, phospho-S6 S235/236 (CST #4858 1:400), Ki67 (Abcam #ab16667 1:100), p-H2AX S139 (CST #9178 1:100), Cleaved Caspase-3 (CST #9664 1:100), RPS10 (Abcam ab151550 1:50), and RPS11 (Abcam ab175213 1:50) were diluted in SignalStain Antibody Diluent (CST #8112) and incubated overnight at 4°C. Slides were washed three times for 5 min each in TBST and incubated in SignalStain Boost IHC Detection Reagent (CST #8114) for 30 min at room temperature, followed by three 5 min washes in TBST. Signal was detected using the SignalStain DAB Substrate Kit (CST #8059) according to manufacturer’s instructions. Slides were counterstained with hematoxylin (Sigma #GHS132) and washed twice for 5 min each in ddH2O, followed by 2 washes in 95% ethanol, 2 washes in 100% ethanol, then 2 washes in Histoclear, for 10 sec each. Coverslips were mounted with Permount Mounting solution (Fisher #SP15).
Blood Cell Counts
Blood was drawn by retroorbital insertion of a heparinized microcapillary tube (Fisher #22-362-566), collected in EDTA-coated microtainer tubes (BD #365973), centrifuged at 3000xg for 10min at room temperature, and plasma was removed. Blood cell counts were determined using a Mascot HemaVet 950FS Hematology Analyzer.
Ultrasound
Ultrasounds on kidneys before and after treatment were performed using a VisualSonics Vevo2100 Imaging Platform and analyzed using VisualSonics VevoLAB software.
Immunoblotting and antibodies
Cells were lysed in buffer containing 20 mM Tris pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 1% Triton X-100, 1 mM DTT, 50 mM NaF, protease inhibitor cocktail (Sigma #P8340) and phosphatase inhibitor cocktail #2 (Sigma #P5726) and #3 (Sigma #P0044) used at 1:100. Western blots were performed using the following antibodies at 1:1000 dilution unless otherwise indicated: phospho-p70 S6 Kinase T389 (CST #9234), p70 S6 Kinase (CST #2708), 4E-BP1 (CST #9644), TSC2 (CST #4308 and #3612), GAPDH (CST #5174), IMPDH1 (Abcam #ab137120), IMPDH2 (Abcam #ab131158), Cleaved Caspase-3 D175 (CST #9664), PARP (CST #9542), phospho-AMPKα T172 (CST #2535), AMPKα (CST #2532), phospho-Raptor S792 (CST #2083), Raptor (CST #2280), ADK (Santa Cruz #sc-514588, 1:500), phospho-Chk1 S345 (CST #2348), Chk1 (CST #2360), RPA32/RPA2 (Abcam #ab76420, 1:10000), phospho-Histone H2A.X S139 (CST #9718), Histone H2A.X (CST #7631), and UBF (Santa Cruz #sc9131).
Cell free mizoribine phosphorylation assay
Cell extract preparation and adenosine kinase reaction were carried out as described previously (Koyama and Kodama, 1982). Briefly, cells were lysed in buffer containing 20 mM sodium phosphate,pH 6.5 and 0.5% Triton X-100. Reaction mixtures containing 50 mM sodium phosphate, pH 6.5, 2.5 mM ATP, 0.25 mM MgCl2, cell lysate (150 μg total protein/reaction), and 10 μM mizoribine were incubated at 37°C for 30 min. Metabolites were extracted and analyzed as described above.
Cell cycle profiles
Single cell suspensions were washed in 1X PBS +0.1% BSA (Sigma #A7030) and fixed in 100% ethanol overnight at −20°C. Cells were then washed with cold PBS and incubated in PBS with 40 μg/mL propidium iodide (Sigma #P4170), 3.8 mM sodium citrate, and 0.5 μg/mL RNaseA (Worthington Biochemicals #LS005649) for 1 hr on ice. Staining was measured with a Becton Dickinson FACSCalibur flow cytometer and analyzed using FlowJo Version 8.8.6 software.
Immunofluorescence
Cells grown on glass coverslips were fixed in 4% formaldehyde (ThermoFisher Scientific #28908) for 15 min at room temperature, then washed in PBS. Antigen retrieval was performed by boiling for 10 min in 10 mM sodium citrate, pH 6.0, followed by washing in PBS. PBS with 5% normal goat serum (CST #5425) and 0.3% Triton X-100 was used as a blocking solution for 60 min at room temperature. Cells were incubated overnight at 4°C in primary antibody, phospho-Histone H2A.X S139 diluted 1:100. Fixed cells were then washed with PBS, incubated with Cy3-conjugated secondary antibody (Jackson ImmunoResearch #111-165-144) at 1:1000 for 1 hr at room temperature, counterstained with 0.5 μg/mL DAPI (ThermoFisher Scientific #62248), washed with PBS, and mounted with Fluoromount G (SouthernBiotech #0100-01). Representative images were taken with a Zeiss Axiotome fluorescence microscope with Apotome feature engaged and analyzed using AxioVision software.
RNA and rRNA synthesis and cellular ribosome content
Cells were labeled with 1 μCi/mL 5,6-3H-uridine (Perkin Elmer #NET367250UC) or 2,8-3H-adenine (Perkin Elmer #NET063001MC) for times indicated in the figure legends. Total RNA was extracted using the AllPrep RNA/DNA Mini Kit (Qiagen #80204) according to manufacturer’s instructions. Radiolabeled RNA was measured with a LS6500TD scintillation counter (Beckman-Coulter), and total RNA was quantified using a NanoDrop One/Onec UV-Vis Spectrophotometer (Thermo Scientific). Counts per minute (CPM) were normalized to total RNA. rRNA synthesis was measured as previously described (Ben-Sahra et al., 2013). Cells were labeled as above, and ribosomes were purified as described in Belin et al (Belin et al., 2010), followed by rRNA purification using the RNeasy RNA cleanup kit according to manufacturer’s instructions (Qiagen #74106), and then radiolabeled rRNA measured as above with CPM normalized to total rRNA. For quantification of rRNA and ribosomal protein per cell, RNA and protein were quantified in ribosome purifications as above, and by Bradford assay, respectively, and normalized to cells counted in parallel plates treated under the same conditions.
Quantification of protein synthesis
Protein synthesis was measured as described previously (Zhang et al., 2014). Cells were treated as described in the figure legend, then incubated in methionine-free medium for 30min prior to labeling with 167 μCi/ml 35S-methionine for 20 minutes, followed by cell lysis and total protein extraction as described above, and then SDS-PAGE.
QUANTIFICATION AND STATISTICAL ANALYSIS
Graphical data are represented as mean ± SEM. p values for pairwise comparisons were determined using an unpaired two-tailed Student’s t test. Statistical details for individual experiments can be found in their respective figure legends.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| phospho-p70 S6 Kinase T389 | Cell Signaling Technology | Cat# 9234 |
| p70 S6 Kinase | Cell Signaling Technology | Cat# 2708 |
| 4E-BP1 | Cell Signaling Technology | Cat# 9644 |
| TSC2 | Cell Signaling Technology | Cat# 4308 |
| TSC2 | Cell Signaling Technology | Cat# 3612 |
| GAPDH | Cell Signaling Technology | Cat# 5174 |
| IMPDH1 | Abcam | Cat# ab137120 |
| IMPDH2 | Abcam | Cat# ab131158 |
| Cleaved Caspase-3 D175 | Cell Signaling Technology | Cat# 9664 |
| PARP | Cell Signaling Technology | Cat# 9542 |
| phospho-AMPKα T172 | Cell Signaling Technology | Cat# 2535 |
| AMPKα | Cell Signaling Technology | Cat# 2532 |
| phospho-Raptor S792 | Cell Signaling Technology | Cat# 2083 |
| Raptor | Cell Signaling Technology | Cat# 2280 |
| ADK | Santa Cruz Biotechnology | Cat# sc-514588 |
| phospho-Chk1 S345 | Cell Signaling Technology | Cat# 2348 |
| Chk1 | Cell Signaling Technology | Cat# 2360 |
| RPA32/RPA2 | Abcam | Cat# ab76420 |
| phospho-Histone H2A.X S139 | Cell Signaling Technology | Cat# 9718 |
| Histone H2A.X | Cell Signaling Technology | Cat# 7631 |
| UBF | Santa Cruz Biotechnology | Cat# sc9131 |
| phospho-S6 S235/236 | Cell Signaling Technology | Cat# 4858 |
| Ki67 | Abcam | Cat# ab16667 |
| RPS10 | Abcam | Cat# ab151550 |
| RPS11 | Abcam | Cat# ab175213 |
| Cy3-conjugated anti-rabbit secondary antibody | Jackson ImmunoResearch | Cat# 111-165-144 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Rapamycin | EMD Millipore | Cat# 53123-88-9 |
| Rapamycin | LC Laboratories | Cat# R-5000 |
| Mizoribine | Selleckchem | Cat# S1384 |
| Mizoribine | Sigma | Cat# M3047 |
| Mycophenolic acid | Sigma | Cat# M3536 |
| Ribavirin | Sigma | Cat# R9644 |
| Azathioprine | Sigma | Cat# A4638 |
| Q-VD-OPh | Sigma | Cat# SML0063 |
| Staurosporine | T ocris | Cat# 1285 |
| CX-5461 | Selleckchem | Cat# S2684 |
| Hydroxyurea | Sigma | Cat# H8627 |
| AZD6738 | Selleckchem | Cat# S7693 |
| Guanosine | Sigma | Cat# G6752 |
| Adenosine | Sigma | Cat# A9251 |
| DAPI | ThermoFisher Scientific | Cat# 28908 |
| Trypan blue | Sigma | Cat# T8154 |
| Propidium iodide | Sigma | Cat# P4170 |
| Protease inhibitor cocktail | Sigma | Cat# P8340 |
| Phosphatase inhibitor cocktail #2 | Sigma | Cat# P5726 |
| Phosphatase inhibitor cocktail #3 | Sigma | Cat# P0044 |
| 5,6-3H-uridine | Perkin Elmer | Cat# NET367250UC |
| 2,8-3H-adenine | Perkin Elmer | Cat# NET063001MC |
| Critical Commercial Assays | ||
| Cell Titer Glo Luminescent Cell Viability Assay | Promega | Cat# G7573 |
| Dead Cell Apoptosis Kit | ThermoFisher Scientific | Cat# V13245 |
| AllPrep RNA/DNA Mini Kit | Qiagen | Cat# 80204 |
| SignalStain DAB Substrate Kit | Cell Signaling Technology | Cat# 8059 |
| Experimental Models: Cell Lines | ||
| Mouse: Tsc2+/+; Trp53−/− MEFs | Provided by DJ Kwiatkowski | Zhang et al., 2003 |
| Mouse: Tsc2−/−; Trp53−/− MEFs | Provided by DJ Kwiatkowski | Zhang et al., 2003 |
| Mouse: Tsc2−/− 3T3 MEFs +TSC2 | Generated in BD Manning lab | Huang et al., 2008 |
| Mouse: Tsc2−/− 3T3 MEFs +Vector | Generated in BD Manning lab | Huang et al., 2008 |
| Mouse: 105K +TSC2 | Generated in BD Manning lab | Filippakis et al., 2017 |
| Mouse: 105K +Vector | Generated in BD Manning lab | Filippakis et al., 2017 |
| Human: 621-101 +TSC2 | Provided by EP Henske | Yu et al., 2004 |
| Human: 621-101 +Vector | Provided by EP Henske | Yu et al., 2004 |
| Human: HeLa +shTSC2 | Generated in BD Manning lab | Huang et al., 2008 |
| Human: HeLa +shLuciferase | Generated in BD Manning lab | Huang et al., 2008 |
| Human: MCF10A +shTSC2 | Generated in BD Manning lab | Zhang et al., 2014 |
| Human: MCF10A +shLuciferase | Generated in BD Manning lab | Zhang et al., 2014 |
| Rat: ELT3 | Provided by CL Walker | Hodges et al., 2002 |
| Human: HCV-29 | Provided by DJ Kwiatkowski | RRID:CVCL_8228 |
| Human: PC3 | ATCC | Cat# CRL-1435, RRID:CVCL 0035 |
| Human: HCT116 | ATCC | Cat# CCL-247, RRID:CVCL 0291 |
| Experimental Models: Organisms/Strains | ||
| Mouse: A/J Tsc2+/− | Provided by DJ Kwiatkowski | Auricchio et al., 2012 |
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1W]l/SzJ | Jackson Laboratory | Cat# 005557 |
| Oligonucleotides | ||
| Non-targeting control siRNA | GE Dharmacon | Cat# D-001810-10-50 |
| ADK siRNA | GE Dharmacon | Cat# L-062728-00-0005 |
| IMPDH1 siRNA | GE Dharmacon | Cat# M-042235-00-0005 |
| IMPDH2 siRNA | GE Dharmacon | Cat# M-168757-00-0005 |
| UBF siRNA | GE Dharmacon | Cat#M-056732-01-0005 |
| Other | ||
| Matrigel | BD Biosciences | Cat# 356237 |
| Lipofectamine RNAiMAX | ThermoFisher Scientific | Cat# 13778150 |
| Normal goat serum | Cell Signaling Technology | Cat# 5425 |
| DMEM | VWR | Cat# 45000-312 |
| Fetal bovine serum | ThermoFisher Scientific | Cat# 10437-028 |
Supplementary Material
SIGNIFICANCE.
mTORC1 promotes anabolic cell growth and is aberrantly activated in the majority of sporadic cancers and genetic tumor syndromes, including tuberous sclerosis complex (TSC). However, mTORC1 inhibitors, such as rapamycin, have shown limited anti-tumor activity as single-agent therapies, as mTORC1 inhibition generally has cytostatic rather than cytotoxic effects in tumor cells. Here, we demonstrate that inhibiting nucleotide synthesis, an anabolic process stimulated by mTORC1, while maintaining mTORC1 in the active state induces selective cell death by creating a state of anabolic imbalance. Clinically used inhibitors of inosine monophosphate dehydrogenase, such as mizoribine, selectively target cell and tumor models of TSC. Thus, these well-tolerated compounds could be repurposed for the treatment of TSC and, perhaps, other tumors with elevated mTORC1 signaling.
HIGHLIGHTS.
IMPDH inhibition selectively kills TSC2-deficient cells in an mTORC1-dependent manner
The IMPDH inhibitor mizoribine has anti-tumor effects in in vivo models of TSC
IMPDH inhibition causes replication stress and DNA damage in TSC-deficient cells
mTORC1-driven rRNA synthesis sensitizes cells to inhibition of nucleotide synthesis
Acknowledgments
We thank Issam Ben-Sahra, Gerta Hoxhaj, James Hughes Hallet, Min Yuan and Jose Treviño-Villarreal for helpful discussions and technical assistance. This work was supported by postdoctoral fellowships from the American Cancer Society 127106-PF-14-254-01-TBE (AJV) and The LAM Foundation (HCL), The Engles Fund for TSC and LAM Research (EPH), NIH grants R01-HL118760 (EPH), P01-CA120964 (BDM), and R35-CA197459 (BDM), and a Rothberg Courage Award from the Tuberous Sclerosis Alliance (BDM). BDM is on the scientific advisory boards and is a shareholder of Navitor Pharmaceuticals and LAM Therapeutics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Conceptualization AJV, BDM, EPH, DJK, BEH, NP; Methodology AJV, BDM, HCL, EPH, DJK, JMA; Validation AJV, SKM, BDM; Investigation AJV, MT, AB, HCL, SKM, MCM, CB, JMA; Resources EPH, DJK, JMA; Writing/review AJV, BDM, HCL, EPH, DJK, BEH, NP; Visualization AJV, BDM; Supervision AJV, BDM; Funding Acquisition AJV, BDM, EPH, DJK, NP
References
- Auricchio N, Malinowska I, Shaw R, Manning BD, Kwiatkowski DJ. Therapeutic trial of metformin and bortezomib in a mouse model of tuberous sclerosis complex (TSC) PLoS One. 2012;7:e31900. doi: 10.1371/journal.pone.0031900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barfeld SJ, Fazli L, Persson M, Marjavaara L, Urbanucci A, Kaukoniemi KM, Rennie PS, Ceder Y, Chabes A, Visakorpi T, Mills IG. Myc-dependent purine biosynthesis affects nucleolar stress and therapy response in prostate cancer. Oncotarget. 2015;6:12587–12602. doi: 10.18632/oncotarget.3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belin S, Hacot S, Daudignon L, Therizols G, Pourpe S, Mertani HC, Rosa-Calatrava M, Diaz JJ. Purification of ribosomes from human cell lines. Curr Protoc Cell Biol. 2010 doi: 10.1002/0471143030.cb0340s49. Chapter 3, Unit 3 40. [DOI] [PubMed] [Google Scholar]
- Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013;339:1323–1328. doi: 10.1126/science.1228792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sahra I, Hoxhaj G, Ricoult SJ, Asara JM, Manning BD. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science. 2016;351:728–733. doi: 10.1126/science.aad0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sahra I, Manning BD. mTORC1 signaling and the metabolic control of cell growth. Curr Opin Cell Biol. 2017;45:72–82. doi: 10.1016/j.ceb.2017.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, Schmithorst VJ, Laor T, Brody AS, Bean J, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358:140–151. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KK, Spinelli JB, Asara JM, Toker A. Adaptive Reprogramming of De Novo Pyrimidine Synthesis Is a Metabolic Vulnerability in Triple-Negative Breast Cancer. Cancer Discov. 2017;7:391–399. doi: 10.1158/2159-8290.CD-16-0611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DY, Kikuchi H, Asamura M, Gamoh M, Wakui A. Evaluation of mizoribine as an immunosuppressant in subrenal capsule assay using immunocompetent mice. Jpn J Cancer Res. 1990;81:183–187. doi: 10.1111/j.1349-7006.1990.tb02546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- Cunningham JT, Moreno MV, Lodi A, Ronen SM, Ruggero D. Protein and nucleotide biosynthesis are coupled by a single rate-limiting enzyme, PRPS2, to drive cancer. Cell. 2014;157:1088–1103. doi: 10.1016/j.cell.2014.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibble CC, Manning BD. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol. 2013;15:555–564. doi: 10.1038/ncb2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drygin D, Lin A, Bliesath J, Ho CB, O’Brien SE, Proffitt C, Omori M, Haddach M, Schwaebe MK, Siddiqui-Jain A, et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011;71:1418–1430. doi: 10.1158/0008-5472.CAN-10-1728. [DOI] [PubMed] [Google Scholar]
- Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellenberg J, Kunz P, Sahr H, Depeweg D. Overexpression of inosine 5′-monophosphate dehydrogenase type II mediates chemoresistance to human osteosarcoma cells. PLoS One. 2010;5:e12179. doi: 10.1371/journal.pone.0012179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakis H, Alesi N, Ogorek B, Nijmeh J, Khabibullin D, Gutierrez C, Valvezan AJ, Cunningham J, Priolo C, Henske EP. Lysosomal regulation of cholesterol homeostasis in tuberous sclerosis complex is mediated via NPC1 and LDL-R. Oncotarget. 2017;8:38099–38112. doi: 10.18632/oncotarget.17485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz DN, Leonard J, Tudor C, Chuck G, Care M, Sethuraman G, Dinopoulos A, Thomas G, Crone KR. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59:490–498. doi: 10.1002/ana.20784. [DOI] [PubMed] [Google Scholar]
- Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140–156. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths B, Lewis CA, Bensaad K, Ros S, Zhang Q, Ferber EC, Konisti S, Peck B, Miess H, East P, et al. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab. 2013;1:3. doi: 10.1186/2049-3002-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Kwiatkowski DJ. Equivalent benefit of rapamycin and a potent mTOR ATP-competitive inhibitor, MLN0128 (INK128), in a mouse model of tuberous sclerosis. Mol Cancer Res. 2013;11:467–473. doi: 10.1158/1541-7786.MCR-12-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddach M, Schwaebe MK, Michaux J, Nagasawa J, O’Brien SE, Whitten JP, Pierre F, Kerdoncuff P, Darjania L, Stansfield R, et al. Discovery of CX-5461, the First Direct and Selective Inhibitor of RNA Polymerase I, for Cancer Therapeutics. ACS Med Chem Lett. 2012;3:602–606. doi: 10.1021/ml300110s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager PW, Collart FR, Huberman E, Mitchell BS. Recombinant human inosine monophosphate dehydrogenase type I and type II proteins. Purification and characterization of inhibitor binding. Biochem Pharmacol. 1995;49:1323–1329. doi: 10.1016/0006-2952(95)00026-v. [DOI] [PubMed] [Google Scholar]
- He Y, Mou Z, Li W, Liu B, Fu T, Zhao S, Xiang D, Wu Y. Identification of IMPDH2 as a tumor-associated antigen in colorectal cancer using immunoproteomics analysis. Int J Colorectal Dis. 2009;24:1271–1279. doi: 10.1007/s00384-009-0759-2. [DOI] [PubMed] [Google Scholar]
- Henske EP, Jozwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Primers. 2016;2:16035. doi: 10.1038/nrdp.2016.35. [DOI] [PubMed] [Google Scholar]
- Hodges LC, Houston KD, Hunter DS, Fuchs-Young R, Zhang Z, Wineker RC, Walker CL. Transdominant suppression of estrogen receptor signaling by progesterone receptor ligands in uterine leiomyoma cells. Mol Cell Endocrinol. 2002;196:11–20. doi: 10.1016/s0303-7207(02)00230-7. [DOI] [PubMed] [Google Scholar]
- Huang J, Dibble CC, Matsuzaki M, Manning BD. The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol Cell Biol. 2008;28 doi: 10.1128/MCB.00289-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadevaia V, Liu R, Proud CG. mTORC1 signaling controls multiple steps in ribosome biogenesis. Semin Cell Dev Biol. 2014;36:113–120. doi: 10.1016/j.semcdb.2014.08.004. [DOI] [PubMed] [Google Scholar]
- Ilagan E, Manning BD. Emerging Role of mTOR in the Response to Cancer Therapeutics. Trends in Cancer. 2016;2:241–251. doi: 10.1016/j.trecan.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H. Mizoribine and mycophenolate mofetil. Curr Med Chem. 1999;6:575–597. [PubMed] [Google Scholar]
- Kalluri HV, Hardinger KL. Current state of renal transplant immunosuppression: Present and future. World J Transplant. 2012;2:51–68. doi: 10.5500/wjt.v2.i4.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama H, Kodama H. Adenine phosphoribosyltransferase deficiency in cultured mouse mammary tumor FM3A cells resistant to 4-carbamoylimidazolium 5-olate. Cancer Res. 1982;42:4210–4214. [PubMed] [Google Scholar]
- Koyama H, Tsuji M. Genetic and biochemical studies on the activation and cytotoxic mechanism of bredinin, a potent inhibitor of purine biosynthesis in mammalian cells. Biochem Pharmacol. 1983;32:3547–3553. doi: 10.1016/0006-2952(83)90301-5. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski DJ, Manning BD. Molecular basis of giant cells in tuberous sclerosis complex. N Engl J Med. 2014;371:778–780. doi: 10.1056/NEJMcibr1406613. [DOI] [PubMed] [Google Scholar]
- Lane AN, Fan TW. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015;43:2466–2485. doi: 10.1093/nar/gkv047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YC, Li F, Handler J, Huang CR, Xiang Y, Neretti N, Sedivy JM, Zeller KI, Dang CV. Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS One. 2008;3:e2722. doi: 10.1371/journal.pone.0002722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- Maltzman JS, Koretzky GA. Azathioprine: old drug, new actions. J Clin Invest. 2003;111:1122–1124. doi: 10.1172/JCI18384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannava S, Grachtchouk V, Wheeler LJ, Im M, Zhuang D, Slavina EG, Mathews CK, Shewach DS, Nikiforov MA. Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle. 2008;7:2392–2400. doi: 10.4161/cc.6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur D, Stratikopoulos E, Ozturk S, Steinbach N, Pegno S, Schoenfeld S, Yong R, Murty VV, Asara JM, Cantley LC, Parsons R. PTEN Regulates Glutamine Flux to Pyrimidine Synthesis and Sensitivity to Dihydroorotate Dehydrogenase Inhibition. Cancer Discov. 2017;7:380–390. doi: 10.1158/2159-8290.CD-16-0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer C, Grummt I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene. 2006;25:6384–6391. doi: 10.1038/sj.onc.1209883. [DOI] [PubMed] [Google Scholar]
- Michels AA, Robitaille AM, Buczynski-Ruchonnet D, Hodroj W, Reina JH, Hall MN, Hernandez N. mTORC1 directly phosphorylates and regulates human MAF1. Mol Cell Biol. 2010;30:3749–3757. doi: 10.1128/MCB.00319-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson E, Nievera CJ, Klimovich V, Fanning E, Wu X. RPA2 is a direct downstream target for ATR to regulate the S-phase checkpoint. J Biol Chem. 2006;281:39517–39533. doi: 10.1074/jbc.M605121200. [DOI] [PubMed] [Google Scholar]
- Robitaille AM, Christen S, Shimobayashi M, Cornu M, Fava LL, Moes S, Prescianotto-Baschong C, Sauer U, Jenoe P, Hall MN. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science. 2013;339:1320–1323. doi: 10.1126/science.1228771. [DOI] [PubMed] [Google Scholar]
- Sakaguchi K, Tsujino M, Mizuno K, Hayano K, Ishida N. Effect of bredinin and its aglycone on L5178Y cells. J Antibiot (Tokyo) 1975a;28:798–803. doi: 10.7164/antibiotics.28.798. [DOI] [PubMed] [Google Scholar]
- Sakaguchi K, Tsujino M, Yoshizawa M, Mizuno K, Hayano K. Action of bredinin on mammalian cells. Cancer Res. 1975b;35:1643–1648. [PubMed] [Google Scholar]
- Shor B, Wu J, Shakey Q, Toral-Barza L, Shi C, Follettie M, Yu K. Requirement of the mTOR kinase for the regulation of Maf1 phosphorylation and control of RNA polymerase III-dependent transcription in cancer cells. J Biol Chem. 2010;285:15380–15392. doi: 10.1074/jbc.M109.071639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, Metabolism, and Cancer. Cancer Discov. 2015;5:1024–1039. doi: 10.1158/2159-8290.CD-15-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stypinski D, Obaidi M, Combs M, Weber M, Stewart AJ, Ishikawa H. Safety, tolerability and pharmacokinetics of higher-dose mizoribine in healthy male volunteers. Br J Clin Pharmacol. 2007;63:459–468. doi: 10.1111/j.1365-2125.2006.02779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushijima K, Nishida T, Oda T, Sugiyama T, Yakushiji M. A sequential immunosuppressive treatment with mizoribin (Bredinin) plus cyclosporin A on the subrenal capsule assay. Kurume Med J. 1991;38:195–198. doi: 10.2739/kurumemedj.38.195. [DOI] [PubMed] [Google Scholar]
- Vendetti FP, Lau A, Schamus S, Conrads TP, O’Connor MJ, Bakkenist CJ. The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of cisplatin to resolve ATM-deficient non-small cell lung cancer in vivo. Oncotarget. 2015;6:44289–44305. doi: 10.18632/oncotarget.6247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RM, Ackerman D, Quinn ZL, Mancuso A, Gruber M, Liu L, Giannoukos DN, Bobrovnikova-Marjon E, Diehl JA, Keith B, Simon MC. Dysregulated mTORC1 renders cells critically dependent on desaturated lipids for survival under tumor-like stress. Genes Dev. 2013;27:1115–1131. doi: 10.1101/gad.198630.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Astrinidis A, Howard S, Henske EP. Estradiol and tamoxifen stimulate LAM-associated angiomyolipoma cell growth and activate both genomic and nongenomic signaling pathways. Am J Physiol Lung Cell Mol Physiol. 2004;286:L694–700. doi: 10.1152/ajplung.00204.2003. [DOI] [PubMed] [Google Scholar]
- Yu JJ, Robb VA, Morrison TA, Ariazi EA, Karbowniczek M, Astrinidis A, Wang C, Hernandez-Cuebas L, Seeholzer LF, Nicolas E, et al. Estrogen promotes the survival and pulmonary metastasis of tuberin-null cells. Proc Natl Acad Sci U S A. 2009;106:2635–2640. doi: 10.1073/pnas.0810790106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Breitkopf SB, Yang X, Asara JM. A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc. 2012;7:872–881. doi: 10.1038/nprot.2012.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Cicchetti G, Onda H, Koon HB, Asrican K, Bajraszewski N, Vazquez F, Carpenter CL, Kwiatkowski DJ. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J Clin Invest. 2003;112:1223–1233. doi: 10.1172/JCI17222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Nicholatos J, Dreier JR, Ricoult SJ, Widenmaier SB, Hotamisligil GS, Kwiatkowski DJ, Manning BD. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature. 2014;513:440–443. doi: 10.1038/nature13492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Han Z, Zhou L, Cai C, Luo H, Huang Y, Liang Y, He H, Jiang F, Wang C, Zhong W. Elevated expression of IMPDH2 is associated with progression of kidney and bladder cancer. Med Oncol. 2015;32:373. doi: 10.1007/s12032-014-0373-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.