

ABSTRACT
Increased levels of fetal hemoglobin (HbF) are a hallmark of more than half of the children diagnosed with juvenile myelomonocytic leukemia (JMML). Elevated HbF levels in JMML are associated with DNA hypermethylation of distinct gene promoter regions in leukemic cells. Since the regulation of globin gene transcription is known to be under epigenetic control, we set out to study the relation of DNA methylation patterns at β-/γ-globin promoters, mRNA and protein expression of globins, and epigenetic modifications of genes encoding the globin-regulatory transcription factors BCL11A and KLF1 in nucleated erythropoietic precursor cells of patients with JMML. We describe several altered epigenetic components resulting in disordered globin synthesis in JMML. We identify a cis-regulatory upstream KLF1 enhancer sequence as highly sensitive to DNA methylation and frequently hypermethylated in JMML. The data indicate that the dysregulation of β-like globin genes is a genuine attribute of the leukemic cell clone in JMML and involves mechanisms not taking part in the normal fetal-to-adult hemoglobin switch.
KEYWORDS: Epigenetic regulation, fetal hemoglobin, juvenile myelomonocytic leukemia, KLF1
Introduction
Juvenile myelomonocytic leukemia (JMML) is an aggressive malignant haematopoietic disorder occurring in infants and young children.1 The leukemic stem/progenitor cell clone differentiates not only toward granulocytes and monocytes, but also the red cell lineage.2,3 A key characteristic observed in the majority of JMML patients is excessive production of fetal hemoglobin (HbF) even when corrected for patient age.4 The presence of elevated levels of HbF at diagnosis is a strong adverse prognostic factor linked with higher risk of treatment failure and shorter survival.4-6 Most studies on leukemic erythropoiesis in JMML date back several decades.7-12 With the caveat in mind that the diagnosis of JMML was not well defined at the time, the data suggested that the malignant clone maintained (or reverted to) a fetal type of erythropoiesis, as concluded from reverted red cell isoenzyme patterns, reduced expression of the I antigen, decreased hemoglobin A2 production, and shifted ratio of Gγ/Aγ globin chains.7-12
The erythropoietic transcription of human β-like globin genes, which are integrated in a broader locus on chromosome 11, switches from the ε-globin gene in embryonal and early fetal life to the γ-globin genes in most of fetal life and then on to β-globin late in fetal life and postnatally.13 The regulation of globin switching involves a set of powerful upstream enhancer elements (the “locus control region”), which are indispensable for high expression of the β-like globin genes. The elements represent active transcription factor-binding sites and form a complex structure that loops the chromosome to bring the control region near the appropriate globin gene, thereby delivering powerful coactivators to the site of transcription and insulating the locus from surrounding inactive chromatin.14,15 This process is supported by 2 major developmental-specific transcription factors, KLF116 and BCL11A.17 KLF1 is an activator of β-globin and primarily relevant during postnatal erythropoiesis.18 BCL11A interacts with the NuRD chromatin remodeling complex and other erythroid transcription factors and is a key repressor of γ-globin, probably through direct physical interaction.17 Importantly, the physiologic fetal-to-adult hemoglobin switch in humans is also associated with differential DNA methylation at intergenic cis-elements and globin gene promoters.19
Leukemic DNA hypermethylation of distinct genetic regions is a cardinal feature of JMML20-23 and defines a high-risk variety of JMML with poor prognosis.20,24 Aberrant DNA methylation and elevated levels of HbF were consistently shown to be highly concordant in JMML, in line with the adverse prognostic impact of both features.20-23 At least one previous case report demonstrated disordered globin synthesis in the absence of a β-globin gene mutation.25 All this led us to investigate how the globin-regulatory network in JMML erythroid cells is altered at the epigenetic level. We identify erythroid-specific epigenetic changes associated with the leukemic clone. We emphasize that these changes are different from mere reactivation of fetal physiology.
Materials and methods
Human cells
Human cells were collected after obtaining informed consent from participants or guardians and approval from institutional review committees. Spleen cells from 14 patients diagnosed with JMML were collected at study centers of the European Working Group of Myelodysplastic Syndromes in Childhood (EWOG-MDS). Splenectomies were performed for clinical indication. Cord blood was obtained from 6 healthy newborns, and peripheral blood (PB) was collected from 17 healthy volunteers.
Isolation of primary erythroblasts, in vitro-differentiated erythroblasts, granulocytes, and monocytes
Biocoll density gradient centrifugation (Biochrom, Berlin, Germany) was used to separate and cryopreserve mononuclear cells (MNC) and granulocytes from leukocyte suspensions. Glycophorin A-positive erythroid precursor (GPEP) cells were isolated from MNC by magnetic-activated cell sorting using CD235a MicroBeads (Miltenyi, Bergisch Gladbach, Germany) or by fluorescence-activated cell sorting after double-staining with anti-CD235a-fluorescein isothiocyanate (Biolegend, San Diego, CA) and diamidinophenylindole (DAPI). For human erythroid massive amplification (HEMA), progenitor cells were expanded from peripheral blood MNC for 10 d and differentiated for 4 d in culture media as described elsewhere.26 Monocytes (CD14+) cells were isolated from MNC using CD14 MicroBeads (Miltenyi).
Reverse transcriptase quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated using Quick-RNA MicroPrep (Zymo Research, Freiburg, Germany) and transcribed into cDNA using QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany). RT-qPCR analysis was performed with ABsolute qPCR SYBR Green Mix (Thermo Fisher Scientific, Waltham, MA) on a CFX384 system (Biorad, München, Germany) using GAPDH for internal normalization and HEMA GPEP cells from a healthy adult as calibrator. Primer sequences are provided in Table S1.
High-pressure liquid chromatography (HPLC)
Hemoglobin variants from filtered lysates of GPEP cells were separated using a PolyCAT A cation exchange column (PolyLC, Columbia, MD) and analyzed on an Elite LaChrom HPLC system (Hitachi, San Jose, CA) using gradient elution with a bis-tris buffer. Hemoglobins were detected by absorbance at 415 nm.
Quantitative high-resolution DNA methylation analysis
Genomic DNA was isolated using Gentra Puregene (Qiagen), bisulfite-converted using EZ DNA Methylation Kit (Zymo Research), and analyzed for HBG1/HGB2, BCL11A, and KLF1 methylation by EpiTYPER mass spectrometry (Agena, Hamburg, Germany) using protocols described previously,27 or for HBB methylation by bisulfite sequencing of subcloned alleles (pCR2.1-TOPO, Life Technologies). Primer sequences are provided in Table S1.
Dual luciferase reporter gene assay
The pCpG-free-promoter-Lucia plasmid (Invivogen, San Diego, CA) was used. In vitro methylation was performed using the M.SssI methyltransferase. Efficiency was monitored by methylation-sensitive cleavage with HpaII and methylation-insensitive cleavage with MspI (all enzymes from New England Biolabs, Ipswich, MA). The HEK293T human embryonal kidney cell line and the K562 erythroleukemia cell line were co-transfected with pCpG-free-promoter-Lucia or methylated/unmethylated pCpG-free-promoter-Lucia-KLF1 and a Firefly vector for normalization (pCpGL-CMV/EF1, courtesy of Michael Rehli, Regensburg, Germany). Twenty-four hours later, the cells were lysed and incubated with coelenterazine (Sigma-Aldrich, Steinheim, Germany) and D-luciferin (Biosynth, Staad, Switzerland). Reporter gene activity was measured on a luminometer at 480 nm (Lucia) and 560 nm (Firefly).
KLF1 target activation
Cells were double-stained with fluorescein isothiocyanate-labeled anti-human CD235a antibody and allophycocyanin-labeled anti-human CD44 antibody (both from Biolegend) and analyzed by flow cytometry.
Results
Globin expression analysis in JMML erythroid precursor cells
Epigenetic modifications of globin regulation cannot be studied in bulk red cells as erythrocytes do not contain DNA. We therefore isolated nucleated glycophorin A-positive erythroid precursor cells from MNC by immunomagnetic cell sorting with anti-CD235a beads (Fig. 1A). Cryopreserved mononuclear spleen cells of JMML patients (n = 14), peripheral blood of healthy adults (n = 17), and umbilical cord blood of healthy newborns (n = 6) were used. GPEP cell separation from cryopreserved samples containing excess dead cells was performed by fluorescence-activated cell sorting, thus permitting double selection for CD235a+/DAPI− cells. To obtain sufficient amounts of GPEP cells from peripheral blood, erythroid progenitors were first expanded by ex vivo differentiation in a 2-phase cell culture system (HEMA culture) and then purified immunomagnetically. The median purity of GPEP cells as assessed by CD235a+ flow cytometry was 92.2% (Table S2).
Figure 1.
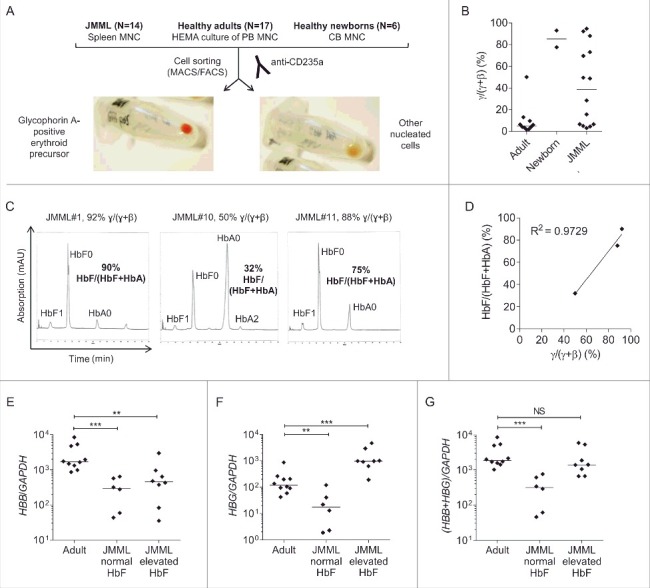
β-like globin expression in glycophorin A-positive erythroid precursor cells. (A) Isolation of GPEP cells and other nucleated cells from cryopreserved spleen cells of JMML patients, in vitro-differentiated blood cells of healthy adults and cord blood cells of healthy newborns. (B) β-like globin mRNA level [γ/(γ+β) mRNA quotient] measured by RT-qPCR in GPEP cells of adults, newborns, and JMML patients. (C) High pressure liquid chromatography to determine hemoglobin variants in GPEP cells of 3 patients with JMML. HbF0/HbA0/HbA2: unmodified, HbF1: glycosylated; for the calculation of %HbF as HbF/(HbF+HbA) the amount of unmodified and glycosylated HbF was considered. For each patient, the corresponding globin mRNA quotient is indicated above the chromatogram. (D) Correlation between globin mRNA quotient and %HbF in GPEP cells of 3 patients with JMML. (E) HBB transcript levels relative to the GAPDH reference gene in GPEP cells of adults, JMML patients classified as normal HbF, and JMML patients classified as elevated HbF. (F) HBG transcript levels relative to GAPDH in GPEP cells of adults, JMML patients classified as normal HbF, and JMML patients classified as elevated HbF. Due to high sequence homology, the assay did not discriminate between HBG1 and HBG2. (G) Total β-like globin transcription relative to GAPDH in GPEP cells of adults, JMML patients classified as normal HbF, and JMML patients classified as elevated HbF. Mann-Whitney test: NS, not significant; **P ≤ 0.01; ***P ≤ 0.001. MNC, mononuclear cells; HEMA, human erythroid massive amplification; CB, cord blood.
The expression level of β-like globins in GPEP cells was measured by RT-qPCR. As expected, GPEP cells from healthy adults predominantly expressed β-globin (HBB) mRNA [median γ/(γ+β) quotient 5.1%] whereas GPEP cells from healthy newborns expressed large amounts of γ-globin (HBG1/HBG2) mRNA and low levels of HBB mRNA [median γ/(γ+β) quotient 85.3%] (Fig. 1B, Table S3). The expression of β-like globin mRNA in JMML GPEP cells was highly variable between patients, with γ/(γ+β) quotients ranging from 3.1% to 95.0%. We next sought to determine how well the relative globin mRNA levels reflected the actual hemoglobin composition in red precursor cells. Although the limited number of purified GPEP cells posed a technical challenge, Hb-HPLC analysis succeeded in quantifying hemoglobin fractions of GPEP cells from 3 JMML patients (Fig. 1C). The percentage of HbF (%HbF) determined by Hb-HPLC was in good agreement with γ/(γ+β) mRNA quotient (Fig. 1D). This indicates that the relative expression of β-like globin mRNA translates directly into hemoglobin composition in JMML GPEP cells and that RT-qPCR of globin mRNA can be used to estimate %HbF if a direct determination by Hb-HPLC is not possible. Based on γ-globin mRNA quotient, 6 of 14 JMML patients were classified as patients with normal HbF and 8 were classified as JMML with elevated HbF (Fig. S1, Table S3). The %HbF values measured in bulk erythrocytes at time of diagnosis before first red blood cell transfusion were available from clinical records in 8 cases. The classification as “HbF normal” or “HbF elevated for age” was invariably consistent with the analysis of β-like globin mRNA levels in JMML GPEP cells (Table S3).
We observed that HBB transcript levels relative to GAPDH were lower in GPEP cells from JMML patients than healthy adults, whether HbF was elevated or not (Fig. 1E). HBG transcripts were increased in JMML patients with elevated HbF as expected (Fig. 1F). As a consequence, the overall transcription of β-like globins was reduced in GPEP cells from JMML patients with normal HbF compared with healthy adults (Fig. 1G). By contrast, increased HBG expression in JMML patients with elevated HbF resulted in total β-like globin transcription at a similar level as in healthy adults (Fig. 1G).
DNA methylation of globin gene promoters in JMML erythroid precursor cells
We next explored if altered β-like globin transcription was paralleled by aberrant DNA methylation of the β-like globin gene promoters in JMML patients. Quantitative mass spectrometry covering 6 of the 7 γ-globin promoter CpG dinucleotides (Fig. 2A) showed that the CpG methylation was dense in GPEP cells from healthy adults as well as JMML patients with normal HbF (Fig. 2B), in line with the low γ-globin expression observed in these groups. By contrast, the level of γ-globin promoter CpG methylation was low in GPEP cells from healthy newborns and JMML patients with increased HbF (Fig. 2B). Although a direct quantitative relationship between γ-globin promoter CpG methylation and γ-globin mRNA expression would not necessarily be expected because of the complex transcriptional regulation of hemoglobins, we observed a surprisingly robust correlation between γ-globin methylation and expression in JMML GPEP cells (Fig. 2C). We also measured the γ-globin promoter CpG methylation in non-GPEP cell populations (CD235a-negative mononuclear cells, CD14+ monocytes, and granulocytes) of patients and controls (Fig. 2D). Methylation was dense in all groups, consistent with lineage-specific silencing of hemoglobin transcription in non-GPEP cells.
Figure 2.
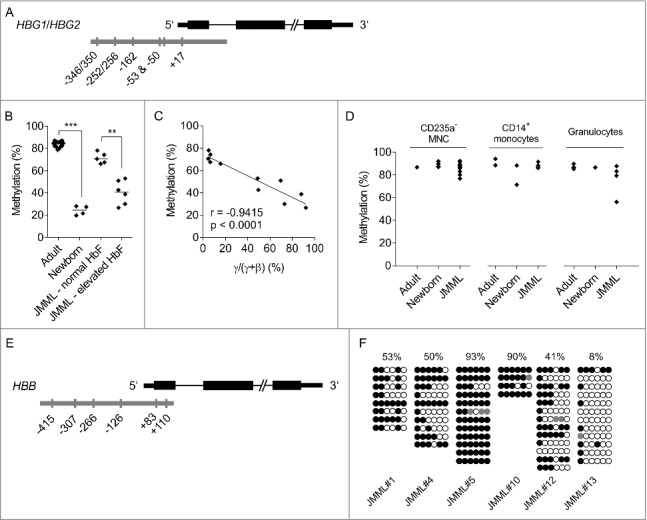
DNA methylation of the β-like globin gene promoters. (A) Target region (gray) in the γ-globin (HBG) promoter with CpG dinucleotide sites that were analyzed for methylation by mass spectrometry. Due to high sequence homology, the assay did not discriminate between HBG1 and HBG2. (B) γ-globin promoter CpG methylation (mean of 6 CpG sites) in GPEP cells of adults, newborns, JMML patients classified as normal HbF, and JMML patients classified as elevated HbF. (C) Correlation between globin expression [γ/(γ+β) mRNA quotient] and γ-globin promoter methylation in GPEP cells of patients with JMML. r, Pearson coefficient. (D) γ-globin promoter CpG methylation (mean of 6 CpG sites) in non-GPEP cell populations of adults, newborns, and patients with JMML. (E) Target region (gray) in the β-globin (HBB) promoter with CpG dinucleotide sites that were analyzed for methylation by bisulfite sequencing. (F) β-globin promoter CpG methylation in GPEP cells of 6 JMML patients assessed by bisulfite sequencing. Lines represent individual alleles; filled circles, methylated cytosines; open circles, unmethylated cytosines. The average level of methylation across CpG sites and alleles is indicated at the top for each patient. Mann-Whitney test: **p ≤ 0.01; ***p ≤ 0.001.
When attempting to analyze CpG methylation of the β-globin promoter we encountered technical limitations of mass spectrometry at this target region. We therefore resorted to the less quantitative method of bisulfite sequencing, with material availability allowing for a limited analysis of only 6 JMML cases (Fig. 2E). In contrast to healthy GPEP cells, which express β-globin and carry an unmethylated β-globin promoter,19 GPEP cells from children with JMML exhibited variable β-globin promoter hypermethylation (Fig. 2F), a finding that generally fits with diminished HBB expression levels as described above. However, the variability of HBB transcript levels (Fig. 1E) did not correlate well with variable promoter DNA methylation (Fig. 2F) in JMML GPEP cells.
Together, the results illustrate that aberrant hemoglobin expression in JMML is associated with epigenetic modification of the β-like globin promoters in GPEP cells. In addition, this is the first description of epigenetic differences between JMML cell fractions differentiating along various lineages.
The β-globin activator KLF1 is subject to DNA methylation changes in JMML erythroid precursor cells
We then asked if regulatory networks of β-like globin expression were affected by leukemic DNA hypermethylation in JMML GPEP cells. We focused on the 2 major regulators governing the fetal-to-adult hemoglobin switch, BCL11A and KLF1. KLF1 directly induces β-globin transcription in erythroid cells and acts as an activator of BCL11A, which in turn represses γ-globin mRNA expression (Fig. 3A).
Figure 3.
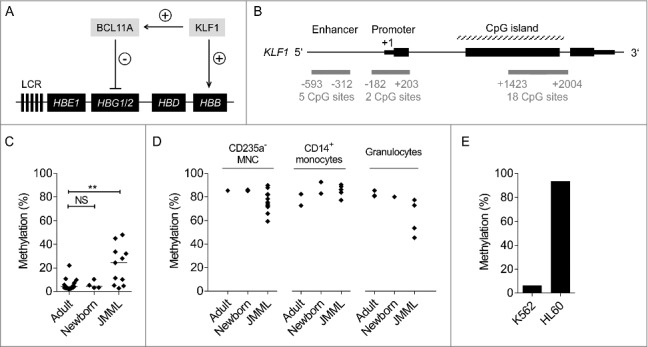
DNA methylation analysis of the KLF1 gene. (A) Regulation of β-like globin expression in erythropoietic cells by transcription factors BCL11A and KLF1. LCR, locus control region. (B) Target regions (gray) in the KLF1 gene locus that were analyzed for methylation by mass spectrometry. (C) CpG methylation of the KLF1 enhancer region (mean of 5 CpG sites) in GPEP cells of adults, newborns, and patients with JMML. (D) KLF1 enhancer methylation in non-GPEP cell populations of adults, newborns, and patients with JMML. (E) KLF1 enhancer methylation in the erythroleukemia cell line K562 and the non-erythroid myeloid leukemia cell line HL60. Mann-Whitney test: NS, not significant; **P ≤ 0.01.
We assessed CpG methylation by mass spectrometry at 4 BCL11A gene regions, 2 located in a promoter CpG island and 2 within intragenic CpG islands of BCL11A. All 4 regions were unmethylated in GPEP cells from JMML patients or healthy controls, as well as in non-erythroid cells from JMML patients or healthy controls (data not shown). We conclude that the BCL11A gene locus is not subject to changes in CpG island methylation, neither as a regulatory mechanism under physiologic conditions nor as a consequence of leukemic transformation in JMML.
Three KLF1 gene regions were investigated: the ∼0.5 kb upstream enhancer element, the ∼0.2 kb upstream promoter and the 5′ CpG island (Fig. 3B). The analysis showed that the KLF1 enhancer, but not the promoter and canonical CpG island were affected by CpG hypermethylation in GPEP cells from patients with JMML (Fig. 3C, Fig. S2). By contrast, all 3 regions were unmethylated in GPEP cells from healthy adults, compatible with active KLF1 function in this cell type. Of note, all 3 KLF1 regions were devoid of methylation in GPEP cells from healthy newborns although KLF1 transcription is downregulated in neonatal erythroblasts to enable physiologic HbF expression.18 By contrast, the KLF1 enhancer exhibited dense methylation in non-erythroid cells from healthy individuals or patients with JMML (Fig. 3D), suggesting that DNA methylation is used to repress transcription of the erythroid-specific factor KLF1 in cells not differentiating toward the red lineage. To corroborate the idea of lineage-specific epigenetic KLF1 control we compared the KLF1 enhancer DNA methylation in 2 acute myeloid leukemia (AML) cell lines: K562, derived from an erythroleukemia, and HL60, derived from an AML with myeloid maturation. Methylation was nearly absent in K562 whereas the region was densely methylated in HL60 (Fig. 3E).
Taken together, the data suggest that KLF1 enhancer DNA methylation is used under normal conditions to keep the gene silent in non-erythroid cells but does not participate in the fetal-to-adult hemoglobin switch in red cells. However, the KLF1 enhancer region acquires leukemia-associated hypermethylation specifically in the erythroid lineage in some patients with JMML.
DNA methylation of the KLF1 enhancer leads to transcriptional repression
To determine if the epigenetic changes observed alter the expression of KLF1 we used a dual luciferase reporter assay that allowed us to directly test how CpG methylation affects the function of the enhancer sequence. A 356-bp fragment of the KLF1 enhancer region was ligated into a reporter plasmid completely devoid of CpG dinucleotides and enhancer CpG sites were methylated enzymatically. The transcriptional activity of the reporter gene was assessed after transfecting unmethylated and methylated construct into erythroid K562 cells and non-erythroid HEK293T cells. We found that the unmethylated KLF1 enhancer construct was highly active in K562 cells, whereas methylation resulted in a dramatic loss of reporter gene transcription (Fig. 4A). By contrast, KLF1 enhancer activity was barely detectable in HEK293T cells irrespective of methylation (Fig. 4B), consistent with its specificity to the erythroid lineage.28 The experiment confirms the cis-regulatory nature of the KLF1 enhancer sequence and that epigenetic modification modulates its activity.
Figure 4.
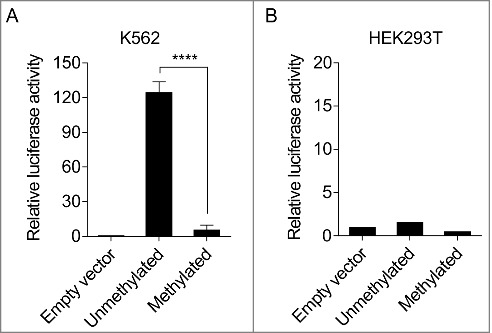
Hypermethylation inhibits the activity of the KLF1 enhancer. (A) Analysis of KLF1 enhancer activity by a luciferase reporter assay in K562 cells. Luciferase activity was normalized to the activity of the pCpGL-CMV/EF1 Firefly control vector. Normalized activity of the construct containing unmethylated KLF1 enhancer CpG sites and the construct containing methylated KLF1 enhancer CpG sites are shown relative to the normalized activity of the reporter construct not containing the KLF1 enhancer sequence (empty vector). Error bars represent standard deviation of 3 transfection experiments. T test: ****P ≤ 0.0001. (B) Same experiment in non-haematopoietic HEK293T cells, confirming specificity of KLF1 enhancer function to erythropoietic cells.
Variable expression of KLF1 in JMML erythroid precursor cells is reflected by expression changes of regulatory KLF1 target molecules
We next studied KLF1 mRNA expression in erythroid cells and its relation to downstream regulatory targets. RT-qPCR confirmed that the level of KLF1 mRNA expression was significantly lower in GPEP cells from healthy newborns than healthy adults (Fig. 5A), consistent with the role of KLF1 in hemoglobin switching. KLF1 expression in JMML GPEP cells was highly variable, with the median level being similar to that in healthy newborns (Fig. 5A). Consistent with the cis-regulatory effect of KLF1 enhancer methylation described above, there was a negative correlation between methylation and expression in JMML GPEP cells (Fig. 5B). As expected, the expression of KLF1 correlated positively with that of HBB (Fig. 5C). Unexpectedly, however, we failed to demonstrate a relationship between KLF1 and HBG expression in JMML GPEP cells (data not shown).
Figure 5.
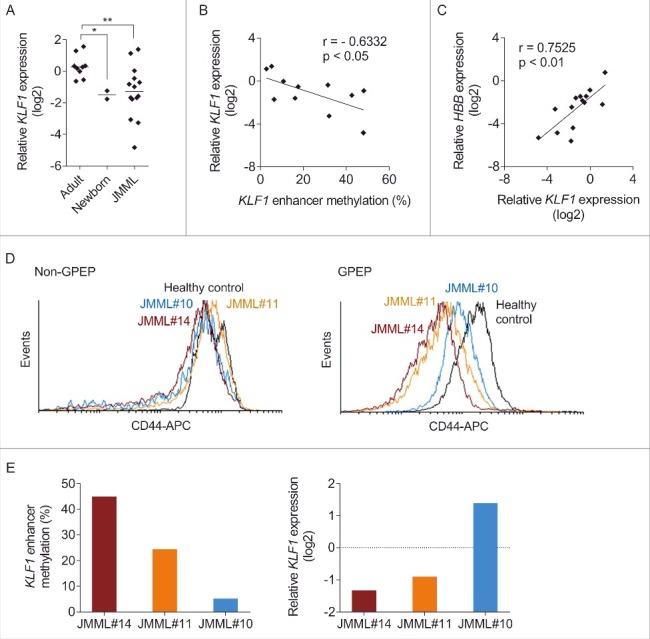
Expression analysis of KLF1 and its regulatory target CD44. (A) KLF1 transcript levels relative to the GAPDH reference gene measured by reverse transcriptase quantitative polymerase chain reaction in GPEP cells of adults, newborns, and patients with JMML. Cells of a healthy adult were used as calibrator (relative log2 expression = 0). Mann-Whitney test: *P ≤ 0.05; **P ≤ 0.01. (B) Correlation between KLF1 enhancer methylation and KLF1 mRNA expression in GPEP cells of patients with JMML. r, Pearson coefficient. (C) Correlation between KLF1 and HBB mRNA expression in GPEP cells of patients with JMML. r, Pearson coefficient. (D) Surface expression of CD44 on glycophorin A-negative mononuclear (non-GPEP) cells (left panel) or GPEP cells (right panel) of 3 JMML patients and a healthy control measured by flow cytometry. APC, allophycocyanin. (E) KLF1 enhancer methylation (left panel) and KLF1 mRNA expression relative to GAPDH (right panel) in GPEP cells of 3 JMML patients analyzed in.Fig. 2D Cells of a healthy control were used as calibrator (dashed line).
Assessing KLF1 protein expression in JMML GPEP cells proved difficult because of material constraints that led to insufficient specificity of the immunoblots. As an alternative we used flow cytometry to measure the surface expression of the CD44 protein, a direct regulatory target of KLF1.29 CD44 expression on non-erythroid cells was the same in JMML and control (Fig. 5D), in concordance with the lack of KLF1 function in non-erythroid cells.16 However, CD44 expression was reduced on GPEP cells from JMML patients compared with the control (Fig. 5D). As predicted, CD44 expression was lower on JMML GPEP cells with higher KLF1 enhancer methylation/lower KLF1 mRNA expression, and vice versa (Fig. 5E). Together, the data support that epigenetic alterations affect the function of the erythroid-specific transcription factor KLF1 in JMML and contribute, at least in part, to disordered β-like globin expression.
Discussion
JMML is a unique myeloproliferative disorder of childhood that has long been recognized as a model disease for oncogenic dysregulation of Ras signal transduction.30 In addition to genetic changes, the significance of leukemia-associated epigenetic modifications in JMML is now being increasingly appreciated. Non-random CpG island hypermethylation occurs in at least half of JMML cases and portends a poor prognosis.20,23,24 The concordance between abnormal hemoglobin composition and aberrant DNA methylation in high-risk JMML, together with the paradigmatic contribution of epigenetic mechanisms to globin gene regulation,19,31 prompted us to study the globin gene-regulatory network in JMML erythroid precursor cells with a focus on epigenetic alterations. We found that the canonical role of promoter CpG methylation for globin transcription was also reproducible in JMML cells. JMML GPEP cells with increased γ-globin transcripts exhibited decreased methylation of HBG promoter CpG dinucleotides as compared with those with low γ-globin expression. In JMML non-GPEP cell populations, the γ-globin promoter was silenced by dense methylation. We noted a surprisingly tight correlation of γ-globin transcript levels with HbF protein measured in leukemic red precursor cells or in full blood of JMML patients at time of diagnosis. The data indicate that increased HbF in JMML originates in the clonal leukemic erythropoiesis and is distinct from elevated levels of HbF observed under certain circumstances of stress erythropoiesis or in non-malignant haematopoietic disorders such as β-thalassemia or sickle cell disease.32-34
Increased levels of HbF are also noted in other malignant myeloid disorders, such as MDS or AML,9,35-37 but it must be emphasized that the levels are not nearly as high as those encountered in JMML.9,37 To our knowledge, it has not been determined experimentally in MDS or AML whether HbF production occurs in the neoplastic clone, as we show for JMML, or results from low-abundant normal erythroid precursor cells that express the γ-globin genes during adult erythroid development38 and expand under erythropoietic pressure. We prefer the latter possibility as it also helps explain the observation that, in contrast to JMML, the prognostic significance of increased HbF in MDS or AML is favorable.37,39
A remarkable finding is the specificity to the erythroid lineage of some leukemic epigenetic modifications, such as KLF1 enhancer hypermethylation or γ-globin hypomethylation. The JMML-initiating clone, deriving from an early haematopoietic multipotent stem/progenitor cell,40 differentiates not only to the myelomonocytic but also the erythroid lineage.2,3,12 Accordingly, gene mutations acquired during leukemogenesis are usually present in nucleated red cells of JMML patients, whether isolated by clonogenic progenitor cell culture3 or direct separation.20 Similarly, we described previously that epigenetic alterations identified in myelomonocytic cells of JMML patients could be traced back to the early progenitor compartment and were also reflected in the erythroid lineage.20 Hence, it appears that both global and lineage-specific epigenetic alterations occur in JMML. The observed variability of de novo KLF1 enhancer methylation in erythroid JMML cells may call into question that this is an early event during leukemogenesis. However, it is possible that the epimutation occurs early but becomes unstable in later stages of the disease.
With the identification of the KLF1 upstream enhancer element as highly sensitive to methylation and variably methylated in JMML cells, the present study is the first to describe the epigenetic control of a haematopoietic transcription factor in this leukemia. Importantly, erythroid-specific KLF1 enhancer methylation in JMML demonstrates that the leukemic dysregulation in JMML GPEP cells goes beyond reversion to an earlier or fetal developmental stage, as methylation of the KLF1 enhancer sequence does not occur during normal fetal-to-adult development of red cells. As expected, higher methylation of the KLF1 enhancer in JMML cells was associated with lower transcription of KLF1 and reduced expression of the KLF1 targets HBB and CD44. However, an unexpected limitation was that there was no obvious correlation of KLF1 methylation with HBG transcription or level of HbF. A possible explanation could be the influence of other γ-globin-regulating factors, such as LIN28B41 or LRF.42
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Marion Bähr for help with luciferase assays, and to Dr. Mirjam Kunze for providing access to umbilical cord blood. The study was supported by the German Research Foundation (CRC 992 “Medical epigenetics” C05 and SPP1463 “Epigenetic Dysregulation in Myeloid Neoplasia” FL345/4-1, FL345/4-2) (both to C.F.).
Authors contribution
S.F. devised the study, performed experiments, interpreted data, and wrote the manuscript. C.F.K., A.M., T.E. and O.M. performed experiments. C.P. interpreted data and contributed to manuscript writing. C.M.N. provided clinical samples, interpreted data and contributed to manuscript writing. C.F. devised the study, interpreted data and wrote the manuscript.
References
- 1.Locatelli F, Niemeyer CM. How I treat juvenile myelomonocytic leukemia. Blood. 2015;125(7):1083–90. https://doi.org/10.1182/blood-2014-08-550483. PMID:25564399. [DOI] [PubMed] [Google Scholar]
- 2.Busque L, Gilliland DG, Prchal JT, Sieff CA, Weinstein HJ, Sokol JM, Belickova M, Wayne AS, Zuckerman KS, Sokol L, et al.. Clonality in juvenile chronic myelogenous leukemia. Blood. 1995;85(1):21–30. PMID:7803795. [PubMed] [Google Scholar]
- 3.Flotho C, Valcamonica S, Mach-Pascual S, Schmahl G, Corral L, Ritterbach J, Hasle H, Arico M, Biondi A, Niemeyer CM. RAS mutations and clonality analysis in children with juvenile myelomonocytic leukemia (JMML). Leukemia. 1999;13(1):32–7. doi: 10.1038/sj.leu.2401240. PMID:10049057. [DOI] [PubMed] [Google Scholar]
- 4.Niemeyer CM, Aricò M, Basso G, Biondi A, Cantù-Rajnoldi A, Creutzig U, Haas O, Harbott J, Hasle H, Kerndrup G, et al.. Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. Blood. 1997;89(10):3534–43. PMID:9160658. [PubMed] [Google Scholar]
- 5.Passmore SJ, Hann IM, Stiller CA, Ramani P, Swansbury GJ, Gibbons B, Reeves BR, Chessells JM. Pediatric myelodysplasia: a study of 68 children and a new prognostic scoring system. Blood. 1995;85(7):1742–50. PMID:7703482. [PubMed] [Google Scholar]
- 6.Locatelli F, Nöllke P, Zecca M, Korthof E, Lanino E, Peters C, Pession A, Kabisch H, Uderzo C, Bonfim CS, et al.. Hematopoietic stem cell transplantation (HSCT) in children with juvenile myelomonocytic leukemia (JMML): results of the EWOG-MDS/EBMT trial. Blood. 2005;105(1):410–9. doi: 10.1182/blood-2004-05-1944. PMID:15353481. [DOI] [PubMed] [Google Scholar]
- 7.Weatherall DJ, Edwards JA, Donohoe WT. Haemoglobin and red cell enzyme changes in juvenile myeloid leukaemia. Br Med J. 1968;1(5593):679–81. doi: 10.1136/bmj.1.5593.679. PMID:4966603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maurer HS, Vida LN, Honig GR. Similarities of the erythrocytes in juvenile chronic myelogenous leukemia to fetal erythrocytes. Blood. 1972;39(6):778–84. PMID:4503341. [PubMed] [Google Scholar]
- 9.Weatherall DJ, Clegg JB, Wood WG, Callender ST, Sheridan BL, Pritchard J. Foetal erythropoiesis in human leukaemia. Nature. 1975;257(5528):710–2. doi: 10.1038/257710a0. PMID:810726. [DOI] [PubMed] [Google Scholar]
- 10.Dover GJ, Boyer SH, Zinkham WH, Kazazian HH, Pinney DJ, Sigler A. Changing erythrocyte populations in juvenile chronic myelocytic leukemia: evidence for disordered regulation. Blood. 1977;49(3):355–65. PMID:264791. [PubMed] [Google Scholar]
- 11.Gahr M, Schröter W. The pattern of reactivated fetal erythropoiesis in bone marrow disorders of childhood. Acta Paediatr Scand. 1982;71(6):1013–8. doi: 10.1111/j.1651-2227.1982.tb09565.x. PMID:6961727. [DOI] [PubMed] [Google Scholar]
- 12.Papayannopoulou T, Nakamoto B, Anagnou NP, Chui D, Dow L, Sanders J. Expression of embryonic globins by erythroid cells in juvenile chronic myelocytic leukemia. Blood. 1991;77(12):2569–76. PMID:2043763. [PubMed] [Google Scholar]
- 13.Bank A. Regulation of human fetal hemoglobin: new players, new complexities. Blood. 2006;107(2):435–43. doi: 10.1182/blood-2005-05-2113. PMID:16109777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stamatoyannopoulos G. Control of globin gene expression during development and erythroid differentiation. Exp Hematol. 2005;33(3):259–71. doi: 10.1016/j.exphem.2004.11.007. PMID:15730849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sankaran VG, Xu J, Orkin SH. Advances in the understanding of haemoglobin switching. Br J Haematol. 2010;149(2):181–94. doi: 10.1111/j.1365-2141.2010.08105.x. PMID:20201948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller IJ, Bieker JJ. A novel, erythroid cell-specific murine transcription factor that binds to the CACCC element and is related to the Kruppel family of nuclear proteins. Mol Cell Biol. 1993;13(5):2776–86. doi: 10.1128/MCB.13.5.2776. PMID:7682653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sankaran VG, Menne TF, Xu J, Akie TE, Lettre G, Van HB, Mikkola HK, Hirschhorn JN, Cantor AB, Orkin SH. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322(5909):1839–42. doi: 10.1126/science.1165409. PMID:19056937. [DOI] [PubMed] [Google Scholar]
- 18.Donze D, Townes TM, Bieker JJ. Role of erythroid Kruppel-like factor in human gamma- to beta-globin gene switching. J Biol Chem. 1995;270(4):1955–9. doi: 10.1074/jbc.270.4.1955. PMID:7829533. [DOI] [PubMed] [Google Scholar]
- 19.Mabaera R, Richardson CA, Johnson K, Hsu M, Fiering S, Lowrey CH. Developmental- and differentiation-specific patterns of human gamma- and beta-globin promoter DNA methylation. Blood. 2007;110(4):1343–52. doi: 10.1182/blood-2007-01-068635. PMID:17456718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olk-Batz C, Poetsch AR, Nöllke P, Claus R, Zucknick M, Sandrock I, Witte T, Strahm B, Hasle H, Zecca M, et al.. Aberrant DNA methylation characterizes juvenile myelomonocytic leukemia (JMML) with poor outcome. Blood. 2011;117(18):4871–80. doi: 10.1182/blood-2010-08-298968. PMID:21406719. [DOI] [PubMed] [Google Scholar]
- 21.Poetsch AR, Lipka DB, Witte T, Claus R, Nöllke P, Zucknick M, Olk-Batz C, Fluhr S, Dworzak M, de Moerloose B, et al.. RASA4 undergoes DNA hypermethylation in resistant juvenile myelomonocytic leukemia. Epigenetics. 2014;9(9):1252–60. doi: 10.4161/epi.29941. PMID:25147919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilhelm T, Lipka DB, Witte T, Wierzbinska JA, Fluhr S, Helf M, Mücke O, Claus R, Konermann C, Nöllke P, et al.. Epigenetic silencing of AKAP12 in juvenile myelomonocytic leukemia. Epigenetics. 2016;11(2):110–9. doi: 10.1080/15592294.2016.1145327. PMID:26891149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fluhr S, Boerries M, Busch H, Symeonidi A, Witte T, Lipka DB, Mücke O, Nöllke P, Krombholz CF, Niemeyer CM, et al.. CREBBP is a target of epigenetic, but not genetic, modification in juvenile myelomonocytic leukemia. Clin Epigenetics. 2016;8:50. doi: 10.1186/s13148-016-0216-3. PMID:27158276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sakaguchi H, Muramatsu H, Okuno Y, Makishima H, Xu Y, Furukawa-Hibi Y, Wang X, Narita A, Yoshida K, Shiraishi Y, et al.. Aberrant DNA methylation is associated with a poor outcome in juvenile myelomonocytic leukemia. PLoS One. 2015;10(12):e0145394. doi: 10.1371/journal.pone.0145394. PMID:26720758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hiwarkar P, Rao A. Revisiting Darwinism explains extinction of fetal erythroid progenitors in a leukaemogenic model of a paediatric myeloproliferative neoplasm. Br J Haematol. 2011;155(1):2. doi: 10.1111/j.1365-2141.2011.08731.x. PMID:21615380. [DOI] [PubMed] [Google Scholar]
- 26.Migliaccio G, Di Pietro R, di Giacomo V, Di Baldassarre A, Migliaccio AR, Maccioni L, Galanello R, Papayannopoulou T. In vitro mass production of human erythroid cells from the blood of normal donors and of thalassemic patients. Blood Cells Mol Dis. 2002;28(2):169–80. doi: 10.1006/bcmd.2002.0502. PMID:12064913. [DOI] [PubMed] [Google Scholar]
- 27.Claus R, Wilop S, Hielscher T, Sonnet M, Dahl E, Galm O, Jost E, Plass C. A systematic comparison of quantitative high-resolution DNA methylation analysis and methylation-specific PCR. Epigenetics. 2012;7(7):772–80. doi: 10.4161/epi.20299. PMID:22647397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiong Q, Zhang Z, Chang KH, Qu H, Wang H, Qi H, Li Y, Ruan X, Yang Y, Yang Y, et al.. Comprehensive characterization of erythroid-specific enhancers in the genomic regions of human Kruppel-like factors. BMC Genomics. 2013;14:587. doi: 10.1186/1471-2164-14-587. PMID:23985037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borg J, Papadopoulos P, Georgitsi M, Gutierrez L, Grech G, Fanis P, Phylactides M, Verkerk AJ, van der Spek PJ, Scerri CA, et al.. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat Genet. 2010;42(9):801–5. doi: 10.1038/ng.630. PMID:20676099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niemeyer CM. RAS diseases in children. Haematologica. 2014;99(11):1653–62. doi: 10.3324/haematol.2014.114595. PMID:25420281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ginder GD. Epigenetic regulation of fetal globin gene expression in adult erythroid cells. Transl Res. 2015;165(1):115–25. doi: 10.1016/j.trsl.2014.05.002. PMID:24880147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alter BP, Rappeport JM, Huisman TH, Schroeder WA, Nathan DG. Fetal erythropoiesis following bone marrow transplantation. Blood. 1976;48(6):843–53. PMID:793650. [PubMed] [Google Scholar]
- 33.Uda M, Galanello R, Sanna S, Lettre G, Sankaran VG, Chen W, Usala G, Busonero F, Maschio A, Albai G, et al.. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci U S A. 2008;105(5):1620–5. doi: 10.1073/pnas.0711566105. PMID:18245381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lettre G, Bauer DE. Fetal haemoglobin in sickle-cell disease: from genetic epidemiology to new therapeutic strategies. Lancet. 2016;387(10037):2554–64. doi: 10.1016/S0140-6736(15)01341-0. PMID:27353686. [DOI] [PubMed] [Google Scholar]
- 35.Bourantas KL, Georgiou I, Seferiadis K. Quantitation of HBF gamma-chain types by HPLC in patients with myelodysplastic syndrome. Haematologica. 1991;76(4):337–8. PMID:1724440. [PubMed] [Google Scholar]
- 36.Craig JE, Sampietro M, Oscier DG, Contreras M, Thein S. Myelodysplastic syndrome with karyotype abnormality is associated with elevated F-cell production. Br J Haematol. 1996;93(3):601–5. doi: 10.1046/j.1365-2141.1996.d01-1682.x. PMID:8652380. [DOI] [PubMed] [Google Scholar]
- 37.Lübbert M, Ihorst G, Sander PN, Bogatyreva L, Becker H, Wijermans PW, Suciu S, Bissé E, Claus R. Elevated fetal haemoglobin is a predictor of better outcome in MDS/AML patients receiving 5-aza-2′-deoxycytidine (decitabine). Br J Haematol. 2017;176(4):609–17. doi: 10.1111/bjh.14463. PMID:27905102. [DOI] [PubMed] [Google Scholar]
- 38.Papayannopoulou TH, Brice M, Stamatoyannopoulos G. Stimulation of fetal hemoglobin synthesis in bone marrow cultures from adult individuals. Proc Natl Acad Sci U S A. 1976;73(6):2033–7. doi: 10.1073/pnas.73.6.2033. PMID:1064874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheridan BL, Weatherall DJ, Clegg JB, Pritchard J, Wood WG, Callender ST, Durrant IJ, McWhirter WR, Ali M, Partridge JW, et al.. The patterns of fetal haemoglobin production in leukaemia. Br J Haematol. 1976;32(4):487–506. doi: 10.1111/j.1365-2141.1976.tb00952.x. PMID:816370. [DOI] [PubMed] [Google Scholar]
- 40.Lapidot T, Grunberger T, Vormoor J, Estrov Z, Kollet O, Bunin N, Zaizov R, Williams DE, Freedman MH. Identification of human juvenile chronic myelogenous leukemia stem cells capable of initiating the disease in primary and secondary SCID mice. Blood. 1996;88(7):2655–64. PMID:8839860. [PubMed] [Google Scholar]
- 41.Lee YT, de Vasconcellos JF, Yuan J, Byrnes C, Noh SJ, Meier ER, Kim KS, Rabel A, Kaushal M, Muljo SA, et al.. LIN28B-mediated expression of fetal hemoglobin and production of fetal-like erythrocytes from adult human erythroblasts ex vivo. Blood. 2013;122(6):1034–41. doi: 10.1182/blood-2012-12-472308. PMID:23798711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Masuda T, Wang X, Maeda M, Canver MC, Sher F, Funnell AP, Fisher C, Suciu M, Martyn GE, Norton LJ, et al.. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science. 2016;351(6270):285–9. doi: 10.1126/science.aad3312. PMID:26816381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.