

ABSTRACT
Aberrant DNA methylation patterns have been reported in inflamed tissues and may play a role in disease. We studied DNA methylation and gene expression profiles of purified intestinal epithelial cells from ulcerative colitis patients, comparing inflamed and non-inflamed areas of the colon. We identified 577 differentially methylated sites (false discovery rate <0.2) mapping to 210 genes. From gene expression data from the same epithelial cells, we identified 62 differentially expressed genes with increased expression in the presence of inflammation at prostate cancer susceptibility genes PRAC1 and PRAC2. Four genes showed inverse correlation between methylation and gene expression; ROR1, GXYLT2, FOXA2, and, notably, RARB, a gene previously identified as a tumor suppressor in colorectal adenocarcinoma as well as breast, lung and prostate cancer. We highlight targeted and specific patterns of DNA methylation and gene expression in epithelial cells from inflamed colon, while challenging the importance of epithelial cells in the pathogenesis of chronic inflammation.
KEYWORDS: DNA methylation, intestinal epithelial cell, inflammatory bowel disease, transcriptome, ulcerative colitis
Introduction
Ulcerative colitis (UC) is one of the major subtypes of inflammatory bowel disease (IBD). Its phenotype is characterized by chronic inflammation of the intestinal colonic mucosa.1 Individuals affected by long-standing UC experience multiple cycles of inflammation, damage to the colonic epithelium and repair.1,2 The factors that control the pathogenesis of ulcerative colitis are being elucidated but, to date, our knowledge of this process is incomplete. Current models of UC pathogenesis invoke exaggerated and prolonged dysregulated immune responses to normally innocuous intestinal microbial antigens that develop in genetically pre-disposed individuals.3–5 In the inflamed mucosa of UC patients, there is a greatly expanded acute and chronic immune infiltrate of the lamina propria, accompanied by an abundance of secreted factors from those cells, notably cytokines, such as TNF-α, IL-1, IL-6, and many others.6–9 The consequences of sometimes many years of exposure to those factors on the intestinal epithelial cells (IECs) of the colon are not known. In vitro studies of IECs have indicated that they are capable of immune functions, notably the secretion of cytokines and chemokines that could influence the immune infiltrate of the lamina propria.10–13 However, little is known about the specific role played by epithelial cells in the pathogenesis of UC from in vivo studies of patients.
In previous work, we showed that exposure of IECs to the pro-inflammatory cytokine IL-6 results in increased methylation of DNA, via stabilization of the DNA methylation enzyme DNMT1.14 IL-6-induced DNA methylation was accompanied by an altered phenotype of the IECs, including enhanced migration and ability to form foci in soft agar, 2 processes that are associated with neoplasia.14 However, whether the prolonged exposure of the colonic epithelium to inflammation in the disease setting of UC is also associated with altered DNA methylation of epithelial cells is not known. Variable DNA methylation patterns have been observed in colitis-associated cancer,15,16 and have been shown to contribute to aberrant epigenetic gene silencing in sporadic colorectal cancer.17,18 It is therefore plausible that aberrant DNA methylation might link chronic inflammation with carcinogenesis.
Prior studies have used genome-wide approaches to highlight distinct epigenetic patterns between affected diseased samples in IBD and non-affected controls.19–24 Interpretation of results from those studies, in which whole mucosal biopsies were used to extract DNA, must consider the cellular heterogeneity of the samples. Whole colonic biopsies consist of a mixture of different cell types, including epithelial cells, stromal cells (such as fibroblasts), immune cells (such as macrophages and lymphocytes), and endothelial cells. It is known that different cell types have different patterns of epigenetic and transcriptional regulation.25 Furthermore, the relative proportions of cell subtypes in the samples from which DNA and RNA are extracted can profoundly affect the overall DNA methylation and transcriptome pattern observed.26 One other important factor in the design of experiments to assess epigenetic profiles is the inter-individual variation that exists in epigenetic signatures,27 specifically in the presence of disease.28
To account for both of these considerations, we chose to isolate and purify intestinal epithelium from whole colonic biopsies obtained from human subjects with sub-total UC. We aimed to utilize pure epithelial cells to generate within-patient, genome-wide DNA methylation and gene expression maps of affected (i.e., inflamed) and matched unaffected (i.e., non-inflamed) areas of the large intestine. We aimed to use these intra-individual maps to reflect the potential epigenetic variation at the intestinal origin of UC pathogenesis. Moreover, we wish to utilize this data to gain insight into the molecular mechanism underlying the progression of IBD to colitis-associated cancer.
Results
We report DNA methylation analysis and transcriptome analysis comparing distal (inflamed) and proximal (non-inflamed) colonic regions in purified epithelial cells in human individuals with sub-total UC (n = 13 sample-pairs).
Establishment of colonic cell suspensions enriched in epithelial cells
Our method is a modification of 2 previously developed techniques29,30 that allows the detachment of whole epithelial crypts from mucosal biopsies of the colon. Flow cytometry with markers specific for IECs and bone marrow-derived cells were used to assess the cellular make-up of the suspensions resulting from the chelation procedure. It was found that more than 90% of the cell suspension comprised EpCAM positive cells, indicative of epithelial cells (Fig. 1). In this case, approximately 5% of the suspension cells in the inflamed samples were stained with the CD45 antibody, indicating bone marrow origin (Fig. 1D). The double negative cells in this analysis could be stromal cells, such as fibroblasts, or endothelial cells, which are neither epithelial nor of bone marrow origin. These data indicated a successful enrichment of epithelial cells in the suspension. We identified no significant difference between the proportion of CD45 positive cells between inflamed and non-inflamed regions of the colon. However, the absolute yield of DNA and RNA evaluated in each cell isolate in the distal and proximal region of diseased and non-diseased patients was also evaluated (Fig. S1). The mean yield of DNA in non-diseased specimens (7.83 ± 0.9 μg) was greater than that of diseased specimens (5.81 ± 0.6 μg). The total yield of RNA in each state also follows a similar trend (normal: 4.66 ± 0.64 μg; ulcerative colitis: 1.95 ± 0.31 μg), thus suggesting differences in epithelial cell yield between inflamed and non-inflamed states.
Figure 1.
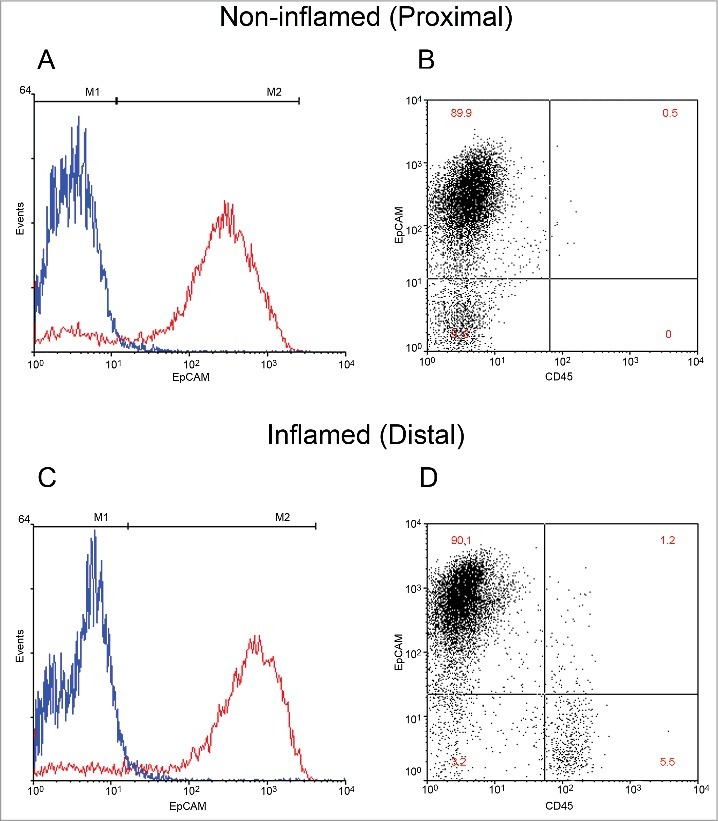
Classification of cellular proportions in non-inflamed and inflamed colonic regions. Intestinal epithelial cells (IECs) isolated from mucosal pinch biopsies are illustrated (A-D). IECs were labeled with fluorescent antibodies EpCAM and CD45 to distinguish cell populations. Representative histograms of EpCAM positive labeled cells (red) and its isotype control (blue) are illustrated in non-inflamed (A) and inflamed (C) regions. Quantification of the percentage of epithelial cells in the IEC isolate was then performed. Representative scatterplots of epithelial positive cells (upper left), CD45 positive cells (lower right) and double negative cells (lower left and upper right) are illustrated in non-inflamed (B) and inflamed (D) regions.
Genome-wide DNA methylation: Sequence data and coverage
DNA methylation was assayed in inflamed and non-inflamed samples using the HELP-tagging assay. Multiplexing of TruSeq HELP-tag library samples was done with 6 libraries per lane. DNA methylation was measured at ∼1.9 million CCGG sites, and ∼1.6 million sites remained when sites with fewer than 5 MspI reads were removed. DNA methylation levels were measured using a modified version of the angle methylation score; this ranged from 0 (no DNA methylation) to 100 (complete DNA methylation). The average number of reads for all samples varied from 13.5 to 24.6 million HpaII reads per sample with an average depth of coverage of CCGG sites between 11.6 and 19.4x (Table S1).
Genome wide patterns of DNA methylation
The majority of CCGG sites in the genome were methylated (DNA methylation score ≥70) (Fig. S2A). Consistent with previous reports31–34 a higher proportion of non-methylated CCGG sites (DNA methylation score ≤30) fell within the vicinity of the transcription start sites (TSS) of genes (±2 kb). The distribution in this region was bimodal, whereas other genomic locations such as the gene body and intergenic regions were predominantly methylated (Fig. S2A).
Gene regulation by epigenetic modification takes place at promoters and distally located regulatory elements.35 To characterize the DNA methylation patterns at promoter regions, the mammalian expression atlas generated by the FANTOM consortium36 was used to map CCGG sites that fall within the vicinity of site (TSS) ±2 kb. Results illustrated the relationship between CCGG rich and depleted regions and the relative DNA methylation state at these regions. As reported previously,34 the proportion of CCGG sites was higher at the TSS, becoming relatively depleted up and downstream of the TSS. However, DNA methylation decreased in close proximity to TSS (mean score = 21.0 at the TSS) and then increased both up- and down-stream from each TSS peak (mean score = 74.1 ±2 kb from TSS) (Fig. S2B). This characterized the typically unbalanced nature of DNA methylation in a normal state, with 70–80% of the genome being methylated, whereas non-methylated loci generally tended to cluster in groups around the TSS of protein-coding genes.37
Identification of differential methylation between inflamed and non-inflamed colonic regions in purified epithelial cell samples
During the initial analysis, a suspected mislabeling of paired samples was discovered. This was recognized during differential methylation analysis, as one pair was consistently methylated in the opposite direction of its grouped counterparts at each CCGG site (Fig. S3). This pattern was also present for the same sample in the transcriptome data set. Due to the manner in which samples are obtained, this consistent pattern observed in both DNA and RNA may therefore be attributable to upstream sample mishandling, perhaps during sample collection or cell isolation. For this reason, this pair of samples was excluded from any further analysis. Therefore, DNA methylation analysis comparing distal (inflamed) and proximal (non-inflamed) colonic regions in purified epithelial cell populations in individuals with sub-total UC (n = 12 pairs) is reported.
Using unsupervised hierarchal clustering, we identified no global differences in patterns of DNA methylation between inflamed and non-inflamed colonic regions, but numerous specific loci at which differential methylation occurred (n = 577). Of these differentially methylated sites (DMS), 371 (64%) showed higher methylation in inflamed regions compared with non-inflamed regions of the colon (Fig. 2A). A substantial number of genes contained multiple DMS mapping; of the 577 DMS, 324 mapped to 210 unique genes (Table S2). However, some variation may be attributable to the distinct epigenomic signatures observed in proximal and distal colonic regions; as identified previously.34 This is reflected by 41 intersecting DMS mapping to 28 genes identified in normal epithelia and in the presence of disease (Table S3).
Figure 2.
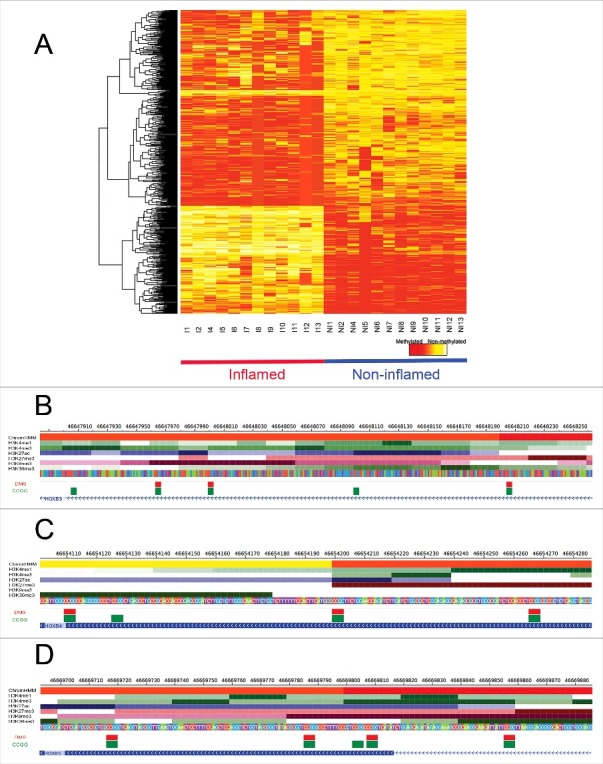
Site-specific differential methylation between non-inflamed and inflamed colonic regions. DNA methylation values of DMS using a color scale from red (high DNA methylation) to yellow (low DNA methylation). Columns represent samples (n = 12 pairs) and rows represent all differentially methylated CCGG sites (n = 577) in inflamed (red) and non-inflamed samples (blue) (A). DMS mapping to active TSS regions (red) and enhancer regions (yellow) of HOXB3 (B), HOXB4 (C), and HOXB5 (D), as well as the ChIP-seq profiles of the set of histone marks assayed in colonic mucosa is shown. Below the histone marks, DMS (red) and all CCGG sites (green) mapping to each gene segment are also illustrated.
Generally, genes containing multiple DMS had similar patterns of DNA methylation, as has been documented in previous studies.38,39 This was the case, for example, for HOXB3, HOXB4, HOXB5, HOXB6, HOXB7 HOXC4, FOXA2, TNS3, and OSR2, as these protein-coding genes showed similar patterns of differential methylation at multiple CCGG sites (Table S2). Among the DMS, 104 mapped to TSS peaks associated with 55 unique protein-coding genes (Table 2).
Table 2.
. List of significantly (Benjamini-Hochberg adjusted cutoff of 0.2) differentially methylated sites between inflamed and non-inflamed samples mapping to TSS peaks (±2 kb).
| DMS | Gene Symbol | Description | ΔMethylation | FDR |
|---|---|---|---|---|
| chr17-46683559 | HOXB7 | homeobox B7 | 84.6260971 | 2.45E-10 |
| chr17-46685296 | HOXB7 | homeobox B7 | 75.1675856 | 7.28E-06 |
| chr20-22563052 | FOXA2 | forkhead box A2 | -61.6184768 | 7.28E-06 |
| chr20-22561829 | FOXA2 | forkhead box A2 | -68.4510447 | 4.08E-05 |
| chr20-22562626 | FOXA2 | forkhead box A2 | -66.1910553 | 9.44E-05 |
| chr3-25469915 | RARB | retinoic acid receptor, beta | 67.3695157 | 0.000201385 |
| chr20-22562885 | FOXA2 | forkhead box A2 | -60.7635967 | 0.000287983 |
| chr12-54428544 | HOXC5 | homeobox C5 | -55.7262250 | 0.000536599 |
| chr2-234744505 | HJURP | holliday junction recognition protein | 75.5210131 | 0.000536599 |
| chr12-54423547 | HOXC6 | homeobox C5 | -65.0768474 | 0.00059452 |
| chr20-22563181 | FOXA2 | forkhead box A2 | -62.7972483 | 0.000957054 |
| chr20-22562585 | FOXA2 | forkhead box A2 | -61.3931252 | 0.001049545 |
| chr3-72939150 | GXYLT2 | glycosyltransferase 8 domain containing 4 | 65.3541771 | 0.001169168 |
| chr20-34147309 | FER1L4 | fer-1-like 4 (C. elegans) | 58.3370116 | 0.001579846 |
| chr12-54428644 | HOXC5 | homeobox C5 | -71.9161826 | 0.001873619 |
| chr2-95941784 | PROM2 | prominin 2 | 55.5342957 | 0.001873619 |
| chr12-54428892 | HOXC4 | homeobox C4 | -54.9707617 | 0.00190397 |
| chr17-46683018 | HOXB7 | homeobox B7 | 65.8624452 | 0.002011668 |
| chr4-6578275 | MAN2B2 | mannosidase, alpha, class 2B, member 2 | 50.7396690 | 0.002340171 |
| chr12-54423460 | HOXC6 | homeobox C6 | -60.5860321 | 0.003735155 |
| chr17-46671931 | HOXB6 | homeobox B6 | 55.5863964 | 0.004275325 |
| chr2-234744532 | HJURP | holliday junction recognition protein | 36.8117832 | 0.004425678 |
| chr2-232259527 | B3GNT7 | UDP-GlcNAc:betaGal beta-1,3-N-acetylglucosaminyltransferase 7 | -58.8405311 | 0.004575139 |
| chr17-46674011 | HOXB6 | homeobox B6 | 61.5163110 | 0.00460977 |
| chr12-54428601 | HOXC5 | homeobox C5 | -62.2630015 | 0.005523152 |
| chr16-89688808 | DPEP1 | dipeptidase 1 (renal) | -66.8219711 | 0.005523152 |
| chr20-22562467 | FOXA2 | forkhead box A2 | -63.0298815 | 0.010785706 |
| chr12-54428668 | HOXC5 | homeobox C5 | -60.969772 | 0.013669362 |
| chr20-34147353 | FER1L4 | fer-1-like 4 (C. elegans) | 52.260453 | 0.014286322 |
| chr10-86005070 | RGR | retinal G protein coupled receptor | 65.313282 | 0.014380736 |
| chr17-46654203 | HOXB4 | homeobox B4 | 49.255953 | 0.014380736 |
| chr15-41708401 | RTF1 | Rtf1, Paf1/RNA polymerase II complex component, homolog (S. cerevisiae) | 48.316353 | 0.015961603 |
| chr19-46274874 | DMPK | dystrophia myotonica-protein kinase | 59.4463585 | 0.017703077 |
| chr20-22563028 | FOXA2 | forkhead box A2 | -49.9576549 | 0.017722042 |
| chr11-126301918 | KIRREL3 | kin of IRRE like 3 (Drosophila) | -47.4455129 | 0.018426331 |
| chr17-46683607 | HOXB7 | homeobox B7 | 54.1100473 | 0.026995683 |
| chr20-61867695 | BIRC7 | baculoviral IAP repeat-containing 7 | 38.2431118 | 0.028259131 |
| chr4-184828474 | STOX2 | storkhead box 2 | -46.00453 | 0.03148721 |
| chr6-111983139 | FYN | FYN oncogene related to SRC, FGR, YES | 20.9468069 | 0.03487875 |
| chr12-5604367 | NTF3 | neurotrophin 3 | -49.2315213 | 0.036133471 |
| chr12-54424932 | HOXC5 | homeobox C5 | -58.4822544 | 0.036861493 |
| chrX-152710726 | TREX2 | three prime repair exonuclease 2; HAUS augmin-like complex, subunit 7 | 51.7780679 | 0.037115174 |
| chr2-20648535 | RHOB | ras homolog gene family, member B | 39.8321014 | 0.039985426 |
| chr17-46673605 | HOXB6 | homeobox B6 | 59.3137178 | 0.040288154 |
| chr17-46670094 | HOXB5 | homeobox B5 | 53.006026 | 0.041816188 |
| chr12-54425323 | HOXC5 | homeobox C5 | -57.0819331 | 0.05446436 |
| chr17-46672209 | HOXB6 | homeobox B6 | 59.4703343 | 0.055281525 |
| chr4-6578378 | MAN2B2 | mannosidase, alpha, class 2B, member 2 | 54.0144389 | 0.055439078 |
| chr17-46669859 | HOXB5 | homeobox B5 | 48.9420761 | 0.057845718 |
| chr20-22562795 | FOXA2 | forkhead box A2 | -55.262905 | 0.058748934 |
| chr17-46672379 | HOXB6 | homeobox B6 | 37.9220212 | 0.059719627 |
| chr17-46669811 | HOXB5 | homeobox B5 | 56.5792304 | 0.062007652 |
| chr16-774464 | CCDC78 | coiled-coil domain containing 78 | 50.2311308 | 0.067168718 |
| chr17-46651823 | HOXB4 | homeobox B4 | 54.0130147 | 0.070165337 |
| chr12-54408697 | HOXC6 | homeobox C6 | -44.2333111 | 0.072588767 |
| chr12-53496682 | SOAT2 | sterol O-acyltransferase 2 | -37.0650460 | 0.072799241 |
| chr8-67835854 | SNORD87 | small nucleolar RNA, C/D box 87 | 43.5438862 | 0.076412069 |
| chr4-13542884 | NKX3-2 | NK3 homeobox 2 | 57.5579881 | 0.088515052 |
| chr20-22562765 | FOXA2 | forkhead box A2 | -61.071834 | 0.088843008 |
| chr19-2716283 | DIRAS1 | DIRAS family, GTP-binding RAS-like 1 | 33.1088621 | 0.093550807 |
| chr2-1637565 | PXDN | peroxidasin homolog (Drosophila) | 51.3279908 | 0.102884331 |
| chr6-168417047 | KIF25 | kinesin family member 25 | 36.4742119 | 0.102884331 |
| chr1-9099850 | SLC2A5 | solute carrier family 2 (facilitated glucose/fructose transporter), member 5 | -55.1186093 | 0.104285117 |
| chr10-86005238 | RGR | retinal G protein coupled receptor | 42.1907947 | 0.104842784 |
| chr22-41075645 | MCHR1 | melanin-concentrating hormone receptor 1 | -40.1809805 | 0.108161774 |
| chr3-128199762 | GATA2 | GATA binding protein 2 | 48.3977178 | 0.110171566 |
| chr7-45146901 | SNORA5B | small nucleolar RNA, H/ACA box 5C | 36.5600523 | 0.110303096 |
| chr12-54447039 | HOXC4 | homeobox C4 | -50.1145258 | 0.111036454 |
| chr11-33277701 | HIPK3 | homeodomain interacting protein kinase 3 | 53.68370846 | 0.11209042 |
| chr5-92924000 | NR2F1 | nuclear receptor subfamily 2, group F, member 1 | 21.18993145 | 0.115121578 |
| chr20-61471911 | TCFL5 | transcription factor-like 5 (basic helix-loop-helix) | -44.30269566 | 0.116800473 |
| chr17-46669720 | HOXB5 | homeobox B5 | 22.19809798 | 0.118981641 |
| chr12-54423616 | HOXC6 | homeobox C6 | -53.3502713 | 0.12386958 |
| chr20-61471704 | TCFL5 | transcription factor-like 5 (basic helix-loop-helix) | -49.7622302 | 0.128802549 |
| chr12-54447386 | HOXC4 | homeobox C4 | -38.0307095 | 0.134526374 |
| chr17-46654113 | HOXB4 | homeobox B4 | 44.0713268 | 0.138722789 |
| chr6-101847583 | GRIK2 | glutamate receptor, ionotropic, kainate 2 | -54.4371579 | 0.14095978 |
| chr12-54428396 | HOXC5 | homeobox C5 | -47.9659770 | 0.142463163 |
| chr17-46651361 | HOXB4 | homeobox B4 | 59.0212241 | 0.148051962 |
| chr19-43858215 | CD177 | CD177 molecule | -52.8713803 | 0.151540502 |
| chr5-162931219 | MAT2B | methionine adenosyltransferase II | -49.2651751 | 0.153204861 |
| chr17-79992546 | DCXR | dicarbonyl/L-xylulose reductase | 52.1129033 | 0.15397301 |
| chr11-119180123 | MCAM | melanoma cell adhesion molecule | 48.8176834 | 0.164940401 |
| chr22-31032999 | SLC35E4 | solute carrier family 35, member E4 | 49.9240692 | 0.165230457 |
| chr17-46670995 | HOXB5 | homeobox B5 | 44.9380625 | 0.168400237 |
| chr13-107141057 | EFNB2 | ephrin-B2 | 44.95724607 | 0.172094399 |
| chr8-7306344 | SPAG11B | sperm associated antigen 11A; sperm associated antigen 11B | -29.7742479 | 0.172388783 |
| chr13-43597174 | DNAJC15 | DnaJ (Hsp40) homolog, subfamily C, member 15 | 49.5033384 | 0.176080002 |
| chr17-46669789 | HOXB5 | homeobox B5 | 57.8205074 | 0.178166434 |
| chr3-160474510 | PPM1L | protein phosphatase 1 (formerly 2C)-like | -27.9110318 | 0.178166434 |
| chr12-117580996 | FBXO21 | F-box protein 21 | 47.9564349 | 0.178915625 |
| chr20-22562989 | FOXA2 | forkhead box A2 | -48.453299 | 0.179191406 |
| chr20-22562221 | FOXA2 | forkhead box A2 | -43.4140179 | 0.179490127 |
| chr9-138553850 | LCN9 | lipocalin 9 | -36.5115718 | 0.185296539 |
| chr17-46671518 | HOXB5 | homeobox B5 | 49.4577331 | 0.186225094 |
| chr20-34148339 | FER1L4 | fer-1-like 4 (C. elegans) | 32.0504781 | 0.187717624 |
| chr2-95942146 | PROM2 | prominin 2 | 42.11374 | 0.19028988 |
| chr19-35781244 | MAG | myelin associated glycoprotein | -42.1237977 | 0.192763364 |
| chr17-46654269 | HOXB4 | homeobox B4 | 39.0674004 | 0.193710002 |
| chr1-204100320 | ETNK2 | ethanolamine kinase 2 | -36.2391408 | 0.19718686 |
| chr2-234744728 | HJURP | holliday junction recognition protein | 32.8961068 | 0.19718686 |
| chr20-22562511 | FOXA2 | forkhead box A2 | -55.6104561 | 0.19718686 |
| chr12-117580915 | FBXO21 | F-box protein 21 | 21.1321240 | 0.199050637 |
| chrX-152086803 | ZNF185 | zinc finger protein 185 (LIM domain) | 43.1109175 | 0.199122705 |
DMS: Differentially Methylated site; FDR: False Discovery Rate.
Gene ontology (GO) analysis was then applied to protein-coding genes with differential DNA methylation events using the R package Goseq. Following bias correction, 21 GO biologic process (GOBP) categories were significantly enriched [False discovery rate (FDR) <0.2; Table 3]. The enriched GO terms were associated with skeletal system morphogenesis (GO:0048705, BH adjusted P = 9e-03), embryological development and embryological morphogenesis (GO:0048704, BH adjusted P = 0.04). These included a considerable number of genes from the HOXB family (HOXB3, HOXB4, HOXB5, HOXB6, and HOXB7) (Fig. 2B-D), the HOXC family (HOXC4, HOXC5, and HOXC6), as well as genes FOXA2, RARB, TBX3, and SERP1. Other GO terms identified were associated with haematopoietic (GO:0048534, BH adjusted P = 0.06), tissue (GO:0009888, BH adjusted P = 0.09) and organ (GO:0048513, BH adjusted P = 0.05) development as well as development of the immune system (GO:0002520, BH adjusted P = 0.09) (Table 3). Genes that were associated with DMS involved in immune system development included RORA, GLI3, NKX3–2, HOXB3, NFATC1, LMO2, KIRREL3, RUNX1, LRP5, TAL1, PITX2, RHOH, GATA2, CACNB4, SATB1, HOXB4, CARD11, and HOXB7.
Table 3.
. Gene Ontology Biological processes (GOBP) categories of differentially methylated evens at protein-coding genes with FDR <0.2 from a gene set analysis (GSA) using GOseq.
| GOBPID |
Count |
P-Value |
FDR |
Term |
| GO:0048705 | 15 | 1.09E-06 | 0.009173675 | skeletal system morphogenesis |
| GO:0001501 | 22 | 1.46E-06 | 0.009173675 | skeletal system development |
| GO:0048568 | 19 | 1.28E-05 | 0.044713028 | embryonic organ development |
| GO:0048598 | 22 | 1.43E-05 | 0.044713028 | embryonic morphogenesis |
| GO:0060216 | 5 | 1.81E-05 | 0.044713028 | definitive hemopoieosis |
| GO:0060348 | 11 | 2.27E-05 | 0.044713028 | bone development |
| GO:0048562 | 15 | 2.49E-05 | 0.044713028 | embryonic organ morphogenesis |
| GO:0048706 | 10 | 2.87E-05 | 0.045099637 | embryonic skeletal system development |
| GO:0048513 | 61 | 3.31E-05 | 0.04625517 | organ development |
| GO:0048704 | 8 | 4.50E-05 | 0.056693684 | embryonic skeletal system morphogenesis |
| GO:0048534 | 18 | 5.75E-05 | 0.065834809 | haematopoietic or lymphoid organ development |
| GO:0002520 | 18 | 8.40E-05 | 0.088121986 | immune system development |
| GO:0061448 | 13 | 0.0001003 | 0.096219373 | connective tissue development |
| GO:0009888 | 38 | 0.0001070 | 0.096219373 | tissue development |
| GO:0009887 | 28 | 0.00012346 | 0.103615701 | organ morphogenesis |
| GO:0051216 | 11 | 0.000207756 | 0.163464742 | cartilage development |
| GO:0007389 | 17 | 0.000229614 | 0.170035874 | pattern specification process |
| GO:0043009 | 21 | 0.000256876 | 0.17171298 | chordate embryonic development |
| GO:0009790 | 30 | 0.000259159 | 0.17171298 | embryo development |
| GO:0009792 | 21 | 0.000274174 | 0.172578713 | embryo development ending in birth |
| GO:0003002 | 14 | 0.000299586 | 0.179594414 | regionalization |
GO analysis was then performed, removing the overlapping DMS mapping to genes previously identified in proximal and distal epithelia,34 thus identifying the enrichment of pathways, potentially more specific to inflammation. The enriched GO term that was identified was associated with bone morphogenesis (GO: 0060349, BH adjusted P = 0.09). These genes included FGFR3, GL13, BMPR1B, LRP5, AXI2, and RARB.
To further explore the epigenetic context of these DMS, we obtained publically available ChromHMM data specific to transcriptionally active and repressed histone marks (H3K4me3, H3K4me1, H3K36me3, H3K27me3, H3K9me3, and H3K27ac) in normal colonic mucosa (Fig. S4).40,41 These data were used to calculate enrichment of differential methylation at several genomic states including promoter, enhancer, and transcribed and repressed regions.
The majority of DMS and, indeed, the majority of all CCGGs mapped to regions in quiescent/low activity states (Fig. 3B), as was documented in previous studies (Fig. S4).41,42 We also tested for enrichment and depletion of DMS, conditional on the CCGG density, within each genomic state (Fig. 3B). Enrichment was identified upstream from active TSS (P = 2.6e-04, Fisher exact test), downstream from active TSS (P = 6e-03, Fisher exact test), at enhancer states (Active enhancer 1: P = 1e-03; Active enhancer 2: P = 4.7e-06; Weak enhancer: P = 4.8e-09; Bivalent enhancer: P = 2.3e-06; Fisher exact test), as well as at transcriptionally repressed polycomb regions (P = 7.8e-15, Fisher exact test) (Fig. 3B). Significant depletion was only identified at a quiescent/low activity state (P = 2.1e-14, Fisher exact test). DMS at the active TSS region of HOXB3, HOXB4, and HOXB5, as well as the ChIP-seq profiles of the chromatin marks assayed in colonic mucosa are shown in fig. 2B-D. These figures illustrate elevated levels of H3K27ac at these regions containing hypermethylated loci, a histone mark associated with active regulatory elements that differentiates active from inactive enhancers and promoters.
Figure 3.
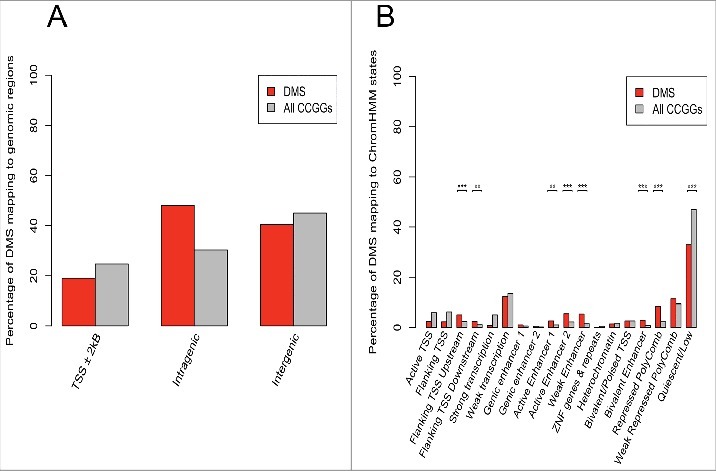
Annotation of CCGG sites to genomic regions and ChromHMM states. The percentage of CCGG sites (gray bars) and differentially methylated sites (DMS) (red bars) annotating to TSS, intragenic and intergenic regions is shown (A). A greater number of DMS mapped to intragenic regions as opposed to TSS (promoter) regions. The total percentage of probes is greater than 100% as several probes were classified as belonging to more than one class of genomic region. The percentage of CCGG sites (gray bars) and DMS (red bars) mapping to 18 active and repressed genomic states in colonic mucosa using the ChromHMM model is shown (B). Using the genomic states defined by the ChromHMM model, significant enrichment of DMS, conditioned on the CCGG density at each genomic state was tested. P-values were determined using a Fisher exact test (*P<0.05, ** P<0.005, *** P<0.0005). Results indicate significant enrichment at enhancers, TSS regions, as well as repressed polycomb regions.
Genome-wide differential expression analysis
mRNA expression was assayed in inflamed and non-inflamed samples using Affymetrix Human Transcriptome Array 2.0 (HTA 2.0) oligonucleotide arrays (see Materials and Methods). Genome-wide gene expression data were generated from purified epithelial cell populations from a subset of the same individuals that DNA methylation patterns were assessed. We report transcriptome analysis comparing inflamed and non-inflamed colonic regions from the same individuals affected by sub-total UC (n = 5 pairs).
We identified partial clustering between inflamed and non-inflamed samples (Fig. 4A) in a hierarchical clustering analysis. However, it is difficult to ascertain distinct global dissimilarity in gene expression patterns. Using the R package limma, we identified 73 transcripts that were differentially expressed between inflamed and non-inflamed IECs, corresponding to 62 unique known protein-coding genes (Fig. 4B, Table 4). A higher proportion of transcripts showed higher expression in inflamed regions (n = 46, 63%) compared with non-inflamed regions (n = 27, 37%). Differential expression at transcripts corresponding to cytokines, chemokines, and immune active soluble factors such as α and β defensins was relatively subtle however, and was not considered to be statistically significant. Relative expression levels of these genes are displayed in Table S4. Two related GOBP categories were significantly enriched (FDR <0.2) for differentially expressed genes. These biologic processes were ethanol oxidation (GO:0006069, BH adjusted P = 0.04) and ethanol metabolic processes (GO:0006067, BH adjusted P = 0.05). These genes included ADH1A, ADH1B, and ADH1C. Other genes that were differentially expressed included PRAC1, PRAC2 and HOXB13 (Fig. 4C-E). These 3 genes are located on chromosome 17q12.3 in relatively close proximity (∼4000 bp). It has previously been documented that these genes are highly expressed in the prostate (normal and cancerous) and distal parts of the colon and rectum in both human and mouse in a normal state.43–45
Figure 4.
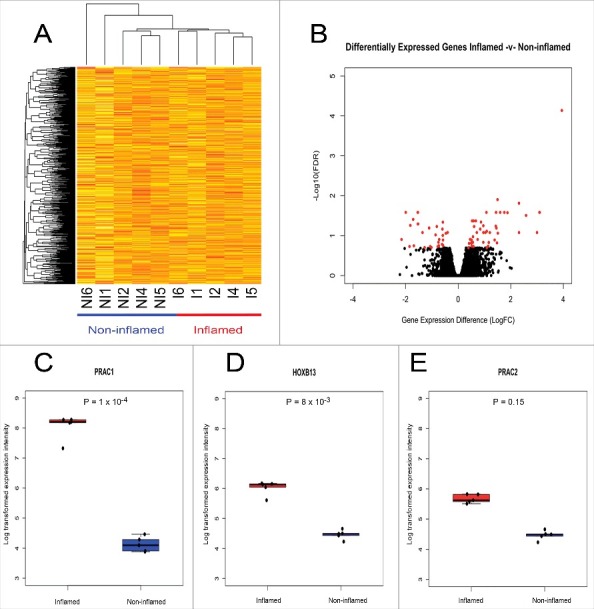
Genome-wide and site-specific gene expression patterns in non-inflamed and inflamed colonic regions. Unsupervised hierarchal clustering of global gene expression profiles (A). Each column represents individual samples [I: Inflamed; NI: Non-inflamed, (n = 5 pairs)] and each row represents individual transcripts. The relative gene expression differences are expressed by a color gradient intensity scale ranging from yellow (low expression) to red (high expression). Volcano plot of differential expression analysis (B): Log fold change (logFC) between inflamed and non-inflamed samples (X-axis), –Log10 for identified non-significant (black) and significant (red) (Benjamini-Hochberg adjusted P of < 0.2) transcripts (Y-axis). Boxplot with overlaying stripchart representing the relative gene expression differences between inflamed (red) and non-inflamed (blue) samples for genes PRAC1 (C), HOXB13 (D), and PRAC2 (E).
Table 4.
List of significantly (Benjamini-Hochberg adjusted P-value cutoff of 0.2) differentially expressed transcripts between inflamed and non-inflamed samples.
| Transcript ID |
Gene Symbol |
Description |
LogFC |
FDR |
| TC17001649.hg.1 | PRAC1 | prostate cancer susceptibility candidate | 3.8149 | 0.00012235 |
| TC15000405.hg.1 | GLDN | gliomedin | 1.2632 | 0.00830502 |
| TC15001837.hg.1 | ANPEP | alanyl (membrane) aminopeptidase | -2.2182 | 0.00830502 |
| TC17001651.hg.1 | HOXB13 | homeobox B13 | 1.4778 | 0.00830502 |
| TC09002921.hg.1 | ST6GALNAC6 | ST6 (α-N-acetyl-neuraminyl-2,3-β-galactosyl-1,3)-N-acetylgalactosaminide α-2,6-sialyltransferase 6 | 1.8220 | 0.01424986 |
| TC13000741.hg.1 | KCTD12 | potassium channel tetramerisation domain containing 12 | 2.3843 | 0.01424986 |
| TC21000989.hg.1 | B3GALT5 | UDP-Gal:βGlcNAc β 1,3 galactosyltransferase, polypeptide 5 | 1.5520 | 0.01514616 |
| TC08001294.hg.1 | CPA6 | carboxypeptidase A6 | 1.7881 | 0.01576238 |
| TC10002938.hg.1 | C10orf116 | chromosome 10 open reading frame 116 adipogenesis regulatory factor | -0.6264 | 0.01576238 |
| TC12001901.hg.1 | NT5DC3 | 5-nucleotidase domain containing 3 | 1.0920 | 0.02110773 |
| TC21000464.hg.1 | C21orf88 | chromosome 21 open reading frame 88 | 2.5530 | 0.02342417 |
| TC01002752.hg.1 | INSL5 | insulin-like 5 | 2.8999 | 0.02794573 |
| TC04001471.hg.1 | PITX2 | paired-like homeodomain 2 | -1.5796 | 0.02842635 |
| TC11000211.hg.1 | SPON1 | spondin 1, extracellular matrix protein | 1.4101 | 0.02842635 |
| TC04001410.hg.1 | ADH1B | alcohol dehydrogenase 1B (class I), β polypeptide | -0.8644 | 0.03071734 |
| TC04001409.hg.1 | ADH1A | alcohol dehydrogenase 1A (class I), α polypeptide | -0.9172 | 0.03323028 |
| TC07002589.hg.1 | LINC-PINT | long intergenic non-protein coding RNA,p53 induced transcript | 0.5743 | 0.03323028 |
| TC03003022.hg.1 | MYH15 | myosin, heavy chain 15 | 1.3573 | 0.03857138 |
| TC17002262.hg.1 | B4GALNT2 | β-1,4-N-acetyl-galactosaminyl transferase 2 (B4GALNT2) | -1.7076 | 0.03941676 |
| TC04001411.hg.1 | ADH1C | alcohol dehydrogenase 1C (class I), gamma polypeptide | -1.0960 | 0.03993123 |
| TC06001299.hg.1 | KIF13A | kinesin family member 13A | 0.5311 | 0.04292623 |
| TC05002796.hg.1 | FLJ00157 | Homo sapiens mRNA for FLJ00157 protein | -1.8563 | 0.04372244 |
| TC20000349.hg.1 | WFDC2 | WAP 4-disulfide core domain 2 | 1.0036 | 0.04455290 |
| TC0Y000341.hg.1 | *** | *** no description*** | 0.9024 | 0.05041901 |
| TC17000638.hg.1 | B4GALNT2 | β-1,4-N-acetyl-galactosaminyl transferase 2 | -1.5293 | 0.05129017 |
| TC01000723.hg.1 | ROR1 | receptor tyrosine kinase-like orphan receptor 1 | 0.5806 | 0.05660819 |
| TC0Y000275.hg.1 | *** | *** no description*** | 1.4056 | 0.05660819 |
| TC01003752.hg.1 | NUCKS1 | nuclear casein kinase and cyclin-dependent kinase substrate 1 | 0.6628 | 0.05878908 |
| TC03003359.hg.1 | GXYLT2 | glucoside xylosyltransferase 2 | -0.6263 | 0.06076798 |
| TC12001155.hg.1 | LPCAT3 | lysophosphatidylcholine acyltransferase 3 | -0.5652 | 0.07070229 |
| TC06003630.hg.1 | DAAM2 | disheveled associated activator of morphogenesis 2 | -0.8143 | 0.07543406 |
| TC05001095.hg.1 | PP7080 | uncharacterized LOC25845 | -1.4284 | 0.07636208 |
| TC04000168.hg.1 | GBA3 | glucosidase, β, acid 3 | -1.4168 | 0.07717089 |
| TC01004068.hg.1 | MIR3916 | microRNA 3916 | 0.5370 | 0.08218670 |
| TC06004132.hg.1 | MOCS1 | molybdenum cofactor synthesis 1 | -0.5321 | 0.08218670 |
| TC10000593.hg.1 | CDHR1 | cadherin-related family member 1 | 0.8484 | 0.08218670 |
| TC12000203.hg.1 | PTPRO | protein tyrosine phosphatase, receptor type, O | 0.8631 | 0.08218670 |
| TC03000131.hg.1 | RARB | retinoic acid receptor, β | -0.7255 | 0.08301883 |
| TC06000926.hg.1 | RFX6 | regulatory factor X, 6 | 0.5280 | 0.08301883 |
| TC15002698.hg.1 | *** | *** no description*** | -0.4019 | 0.08318291 |
| TC20001482.hg.1 | LINC00261 | long intergenic non-protein coding RNA 261 | 1.4472 | 0.08677308 |
| TC01002882.hg.1 | GCLM | glutamate-cysteine ligase, modifier subunit | -0.7496 | 0.09208306 |
| TC07000931.hg.1 | TMEM139 | transmembrane protein 139 | -0.6448 | 0.09320002 |
| TC02002479.hg.1 | GCG | glucagon | 2.1546 | 0.0972245 |
| TC05002797.hg.1 | SLC9A3 | solute carrier family 9, subfamily A (NHE3, cation proton antiporter 3), member 3 | -2.1590 | 0.09722454 |
| TC16000522.hg.1 | CA7 | carbonic anhydrase VII | 1.4832 | 0.09722453 |
| TC21000363.hg.1 | CLDN8 | claudin 8 | 2.8735 | 0.09722453 |
| TC06000173.hg.1 | HIST1H2AE | histone cluster 1, H2ae | -0.5886 | 0.09832936 |
| TC10001089.hg.1 | NEBL | nebulette | 0.5596 | 0.10402139 |
| TC14001475.hg.1 | LINC00341 | long intergenic non-protein coding RNA 341 | 0.5282 | 0.10402139 |
| TC01005141.hg.1 | *** | *** no description*** | 0.9118 | 0.10531854 |
| TC07003096.hg.1 | LHFPL3 | lipoma HMGIC fusion partner-like 3 | -1.6865 | 0.10531854 |
| TC12000329.hg.1 | ANO6 | anoctamin 6 | 1.6249 | 0.10531854 |
| TC21000729.hg.1 | C21orf88 | chromosome 21 open reading frame 88 | 0.9782 | 0.10531854 |
| TC04001299.hg.1 | CDKL2 | cyclin-dependent kinase-like 2 (CDC2-related kinase) | 0.6730 | 0.10721497 |
| TC16000969.hg.1 | CACNG3 | calcium channel, voltage-dependent, gamma subunit 3 | 0.5635 | 0.10608053 |
| TC16002090.hg.1 | CHST5 | carbohydrate (N-acetylglucosamine 6-O) sulfotransferase 5 | 1.6146 | 0.12788495 |
| TC20000726.hg.1 | APMAP | adipocyte plasma membrane associated protein | -0.5785 | 0.12831669 |
| TC05001096.hg.1 | SLC9A3 | solute carrier family 9, subfamily A (NHE3, cation proton antiporter 3), member 3 | -1.7126 | 0.12952132 |
| TC15000406.hg.1 | GLDN | gliomedin | 0.5266 | 0.12952132 |
| TC17001568.hg.1 | PYY | peptide YY | 1.1306 | 0.12987431 |
| TC17002257.hg.1 | PRAC2 | prostate cancer susceptibility candidate 2 | 0.5566 | 0.14674671 |
| TC17000317.hg.1 | PYY2 | peptide YY, 2 (pseudogene) | 0.5121 | 0.15335219 |
| TC04000895.hg.1 | *** | *** no description*** | -0.9907 | 0.15383132 |
| TC16002091.hg.1 | TMEM231 | transmembrane protein 231 | 0.4550 | 0.15516000 |
| TC04002356.hg.1 | *** | *** no description*** | -0.9819 | 0.17219695 |
| TC05003353.hg.1 | RANBP17 | RAN binding protein 17 | 0.5107 | 0.17219695 |
| TC16001964.hg.1 | TMEM231 | transmembrane protein 231 | 1.1312 | 0.18186527 |
| TC20000698.hg.1 | FOXA2 | forkhead box A2 | 0.5397 | 0.18186527 |
| TC02001391.hg.1 | B3GNT7 | UDP-GlcNAc:βGal β-1,3-N-acetylglucosaminyltransferase 7 | 1.1406 | 0.18308688 |
| TC07002442.hg.1 | CROT | carnitine O-octanoyltransferase | -0.7765 | 0.18626684 |
| TC09001528.hg.1 | RP11–388N2.1 | putative novel transcript | 0.5826 | 0.18626684 |
| TC03001525.hg.1 | PRICKLE2 | prickle homolog 2 (Drosophila) | 0.3886 | 0.19988562 |
Integrative analysis of DNA methylation and gene expression data of inflamed and non-inflamed colonic regions of intestinal epithelial cells
Differentially methylated genes were significantly enriched among genes that were differentially expressed (P = 3e-04, Fisher exact test). Gene ontology analysis also revealed that gene sets that were significantly differentially expressed or differentially methylated were not assigned to any GO categories associated with immune response or other disease or cancer relevant processes. Integrative analysis was performed using both the methylation data and transcriptome data generated from the same 10 pure epithelial cell samples (n = 5 pairs). Five genes were both differentially expressed and differentially methylated in inflamed and non-inflamed samples (PITX2, ROR1, GXYLT2, RARB, and FOXA2), several of which are related to Wnt signaling or to embryonic, cell or organism development. For 4 out of the 5 differentially expressed and differentially methylated genes (ROR1, GXYLT2, RARB, and FOXA2) DNA methylation and gene expression were significantly inversely correlated (Fig. 5), as expected. For example, we identified hypermethylation (ΔMethylation = 67.4, FDR = 2e-04) and downregulation (logFC = - 0.72, FDR = 0.08) at the active TSS of RARB (Fig. 5A,B). Hypomethylation at multiple CCGG sites (Fig. 5C,D, Table S5) and upregulation (logFC = 0.54, FDR = 0.18) at a bivalent enhancer region of FOXA2 was also identified.
Figure 5.
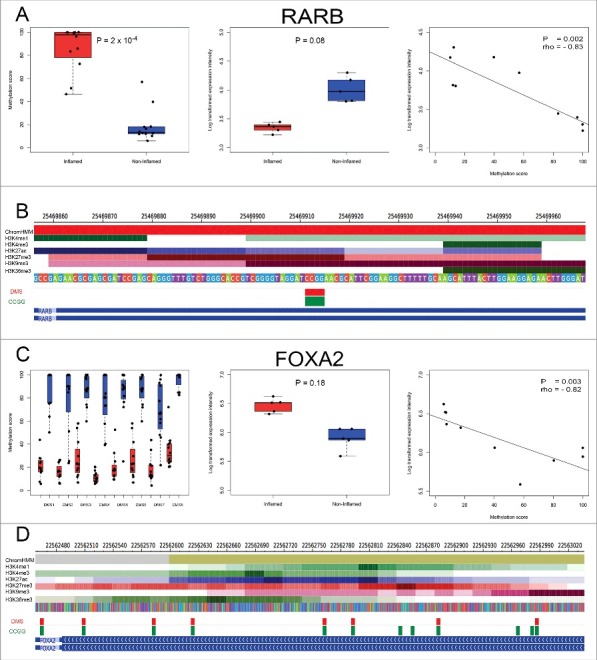
Integrative analysis of differential DNA methylation and gene expression patterns. DNA methylation patterns, gene expression patterns and spearman correlations for genes RARB (A) and FOXA2 (C) are illustrated. Left panel: bar plot and overlaying strip chart of DNA methylation levels between inflamed (red) and non-inflamed (blue) samples; middle panel: bar plot and overlaying strip chart of relative gene expression levels between inflamed (red) and non-inflamed (blue) samples; right panel: spearman correlation between DNA methylation levels (X-axis) and relative gene expression levels (Y-axis); P: P-value; rho: Spearman correlation value. DMS mapping to the active TSS (red) of RARB (B), bivalent enhancer (tan) and repressed polycomb regions (gray) of FOXA2 (D), as well as the ChIP-seq profiles of the set of 6 histone marks assayed in colonic mucosa is shown. Below the histone marks, DMS (red) and all CCGG sites (green) mapping to the given segment of RARB and FOXA2 are also illustrated.
Discussion
In this study, we aimed to generate an integrative epigenome data set, combining genome-wide DNA methylation data and transcriptome data. Our aim was for this data to illustrate the epigenetic variation induced by chronic inflammation of the colon. The objective was to gain insight into the molecular mechanisms of chronic colitis and potentially into its progression to colitis-associated cancer.
Using genome-wide DNA methylation analysis, 577 DMS mapping to 210 unique protein-coding genes were identified. Significant hypermethylation in the presence of inflammation at promoter regions of genes associated with embryonic development and regionalization was observed, most notably, members of the homeotic HOXB gene family. The enrichment of differential methylation at gene sets associated with haematopoietic, tissue, organ, and immune response development was also identified. However, one unanticipated limitation of this aspect of the study was the choice of comparing distal (inflamed) and proximal (non-inflamed) colonic regions from within the same patient. Our hypothesis was that variation in DNA methylation and gene expression patterns could be a product of inflammation. While this strategy allowed us to reduce the major confounding effect of DNA sequence variability on DNA methylation variation,46–48 some of the variability may also be attributable to the distinct epigenomic signatures observed in proximal and distal colonic regions, as we have recently reported.34 In fact, 41 of the 125 (32.8%) of the DMS identified in normal epithelia were also differentially methylated in inflamed epithelia (Table S3). However, although there were a high number of intersecting DMS between both studies, loci mapping to RARB were not differentially methylated in normal IECs.34 We can therefore identify genes with UC-associated DNA methylation changes that cannot be attributed to colonic location, loci that could potentially act as biomarkers to distinguish normal from inflamed colon epithelium.
Using genome-wide gene expression data generated from a subset of the same pure epithelial cell isolates, 73 differentially expressed transcripts, corresponding to 62 known unique protein-coding genes, were identified. GO analysis identified the enrichment of differentially expressed genes associated with ethanol oxidation (ADH1A, ADH1B, and ADH1C). Additionally, increased gene expression in the presence of inflammation at HOXB13 was identified, as well as prostate cancer susceptibility candidates PRAC1 and PRAC2. These genes have previously been identified to have higher expression levels in the distal colon in both humans and mouse in a normal state.43–45 However, very little is known about PRAC1 and PRAC2 in the context of UC. Interestingly, the majority of identified variably methylated or expressed gene sets were not directly or indirectly associated with immune response or other cancer-relevant processes. However, some of the variability may also be attributable to the distinct epigenomic signatures observed in proximal and distal colonic regions.34
IECs have several diverse functions, one of which involves acting as innate immune cells, controlling the interface between a potentially hostile colonic luminal environment and the host.49–51 Many studies, mostly using cultured intestinal epithelial cell lines, have demonstrated the secretion of cytokines and chemokines by those cells.10–13 Those results have supported the notion that intestinal epithelial cells promote chronic inflammation in IBD patients. In our DNA methylation and transcriptome analyses, we noted that several gene sets were enriched for processes associated with immune development. However, enrichment was greater for gene sets associated with skeletal system morphogenesis and development (Table 3). Our findings from purified epithelial cells are inconsistent with previous studies in UC that highlighted differential expression52–55 and differential methylation19,20,22 of key cytokines and inflammatory mediators (Table S4). However, those studies did not use purified epithelial cells, thus indicating that the cytokines/inflammatory mediators implicated in prior studies are likely to have been the route of confounding DNA methylation noise influenced by cell type heterogeneity; most probable representative of autoimmune cell phenotypes. Therefore, our results suggest that the role of colon epithelial cells in the pathogenesis of UC in vivo through cytokine and chemokine secretion may be less than previously estimated.
In a purified IECs isolate, 4 of the 5 genes that were both differentially expressed and differentially methylated had negative correlation between DNA methylation and expression levels (ROR1, GXYLT2, RARB, and FOXA2). RARB encodes the protein retinoic acid receptor β, a member of the thyroid-steroid hormone receptor family of nuclear transcription regulators. This receptor binds retinoic acid, which mediates cellular signaling in embryonic morphogenesis, cell growth, and differentiation. RARB has previously been identified as a tumor suppressor gene and found to be hypermethylated at promoter regions in several cancer phenotypes including breast cancer,56 lung cancer,57,58 and prostate cancer.59,60 However, it was first established as a tumor suppressor gene in colon cancer cell lines61 and, subsequently, in cancerous colonic mucosa.62 In this case, we demonstrated promoter hypermethylation and downregulation of RARB in an inflamed state. Although RARB has been previously highlighted as a tumor suppressor gene in colon cancer, this is the first report of RARB potentially being implicated in non-cancerous UC in purified IECs. Therefore, the interplay between RARB promoter methylation and downregulation may play an important role in the link between UC and colon cancer. Further research into epigenomic dysregulation of this gene in neoplastic IECs would be required to determine the potential of this gene as a clinical marker for colitis-associated cancer risk.
FOXA2 is a member of the forkhead box gene family. It acts as a transcriptional activator, essential for effective development of endoderm derived organs and tissue and thus considered a master regulator of that lineage.63 It has been suggested that FOXA2 may play a role in suppressing tumor development; for example, Zhu et al. identified FOXA2 as a tumor suppressor, demonstrating that it is downregulated in gastric cancer cell lines, and that inducing FOXA2 inhibits gastric cancer growth in vivo.64 We have identified hypomethylation at multiple CCGG sites and upregulation of FOXA2 in an inflamed state but any functional significance of this dysregulation in carcinogenesis remains speculative without subsequent mechanistic studies. Increased sample size and further technical validation are also required to support the biologic significance of these results.
This epigenomic study has reported distinct patterns of DNA methylation and gene expression in the inflamed epithelial cells of UC. These results have shown that differential methylation occurs at promoter regions of tumor suppressor genes, as well as a wide spectrum of both active and repressed coding regions. The relative paucity of variably methylated loci or differentially expressed transcripts corresponding to cytokines and inflammatory mediators also raises questions over the degree to which epithelial cell contribute to chronic colitis. The lack of differential methylation and differential expression at genes associated with colorectal cancer makes interpretation of biologic significance somewhat unclear. Differential methylation and expression may be a genome-wide effect that's not targeted to specific genes that are implicated already in colon cancer risk; effects may be subtle or act through previously unknown pathways. The clinical implications of these reported findings, and the interplay between genetic and epigenetic signatures in the pathway of colitis-associated carcinogenesis needs to be explored in further studies. However, this integrative epigenomic data set will enhance our understanding of UC pathophysiology, potentially bridging the gap between genetic predisposition to UC related disease and UC pathogenesis.
Materials and Methods
Patient recruitment and collection of biopsies
All patient recruitment and sample collection was performed under human subjects protocol approval from the Galway University Hospitals Research Ethics Committee. Patients enrolled in the study were attending University Hospital Galway undergoing colonoscopy for the evaluation of colitis symptoms. Biopsies were collected from 13 patients having subtotal UC where both the colonoscopic appearance and histological evaluation of biopsies confirmed inflammation of the distal colon and the absence of inflammation in the proximal colon. All patient information, including duration of colitis, medication, smoking history, and extent of colitis were recorded in our questionnaire and stored in an encrypted database on the day of recruitment. Clinical characteristics of the patients are included in Table 1. None of the patients was taking any medication known to alter the DNA methylome (folic acid, sulfasalazine, or valproic acid).
Table 1.
. Clinical characteristics of UC patients (n = 13). Current treatments include 5-aminosalicyclic acid, azathioprine, 6-mercaptopurine, methotrexate, infliximab, and adalimumab.
| Characteristic |
n |
| Age | |
| <40 | 6 |
| 40–50 | 2 |
| 50–60 | 3 |
| >60 | 2 |
| Sex | |
| Male | 6 |
| Female | 7 |
| Duration of Colitis (years) | |
| <5 | 3 |
| 5–10 | 3 |
| 10–15 | 1 |
| 15–20 | 4 |
| >20 | 2 |
| Severity (UCDAI scoring) | |
| <5 | 6 |
| 5–10 | 6 |
| >10 | 1 |
| Smoking History | |
| Non-smoker | 9 |
| Ex-smoker | 4 |
| Current smoker | 0 |
Isolation of epithelial cells from pinch biopsies
Previous techniques for isolating purified IECs relied on isolation of cells at room temperature or 37°C.65,66 Modifications to those techniques were developed that allow for epithelia to be obtained at 4°C to limit the detrimental effects to membrane integrity, cellular viability, and molecular degradation that can occur at higher temperatures.34,67
Colonoscopic pinch biopsies (n = 10) were taken from the proximal and distal areas of the colon from healthy patients and stored in ice-chilled PBS. Biopsies then underwent washing (3x) with 5 ml of ice-chilled PBS and centrifuged at 250 x g for 5 min at 4°C. After the third wash, the ice-chilled PBS was replaced with 25 ml of chelation buffer (1 mM EDTA, 1 mM EGTA, 0.5 M DTT, 55 mM D-Sorbitol, 44 mM Sucrose with distilled water at pH 7.3) and stored for 2 h at 4°C on a rocker. After chelation, the samples were then shaken by hand for 30 sec. The cell suspension consisting mostly of intact colon crypts was transferred to a new centrifuge tube and this step was repeated until no more visible cells were liberated. Finally, the cell suspension was centrifuged at 250 x g for 10 min at 4°C, the supernatant was discarded, and the pellet of cells was resuspended in 2 ml of 0.5% BSA in PBS. The sample was aliquoted (200 μl) for cell staining. The remainder of the cells was centrifuged at 250 x g at 4°C and the resulting cell pellet was used for DNA & RNA extraction. All reagents were purchased from Sigma-Aldrich.
Flow cytometry
Each sample was incubated with 5 μl of blocking IgG goat serum (Sigma-Aldrich) and stored for 20 min at 4°C. Samples were then double stained with an epithelial specific marker [FITC Anti-Human CD326 (EpCAM), Biolegend], an immune cell specific maker (APC Anti-Human CD45, Biolegend) or each marker's isotype control (FITC Mouse IgG2b, κ, APC Mouse IgG, κ, both Biolegend). Samples were then stored for a further 20 min in the dark at 4°C, centrifuged at 320 x g at 4°C, and washed twice with 200 μl of 0.5% BSA in PBS. Disaggregation of crypts into a single cell suspension was achieved through pipetting of the cell isolate. Subsequently, cells were examined by flow cytometry using FACSCanto and data analysis was performed using WinMDI (Version 2.8) software.
HELP-tagging assay library preparation
Extraction of genomic DNA from IECs was performed using the method derived from the Albert Einstein University online resource (http://wasp.einstein.yu.edu/index.php/Protocol). All steps outlined in the protocol were followed exactly. Please refer to Supplementary Materials and Methods for a detailed protocol description with no modifications.
Genomic DNA (500 ng) was digested in a 50 µl reaction (containing 5 µl NEB1, 1 µl HpaII, and water) overnight at 37°C. The digest (2 µl) was run on a 1% agarose gel. TE buffer (450 µl) was added to the rest of the digest as well as 500 µl of saturated phenol: chloroform (1:1) and mixed well. The sample was then spun in a micro centrifuge at top speed for 20 min. The aqueous phase of the sample was then transferred to a new tube and precipitated with 1 µl of Ethachinamate (supplied by Wako-chemicals) and 50 µl of 3 M Sodium Acetate. Isopropyl alcohol (800 µl) was added and the sample was incubated at -20°C for 2 h and then spun at top speed for 20 min. The supernatant was then removed and the pellet of DNA was washed with 70% ethanol. The sample was then air-dried and resuspended in 5 µl of TE buffer. Adapter EcoP15I side (TS_AE adaptor) ligation was performed in a 13 μl reaction containing 2x Quick ligase buffer, 0.5 μl of TS_AE adaptor (0.1 μM), digested DNA and 1 μl of Quick Ligase for 15 min at room temperature. All subsequent protocol steps up to polymerase chain reaction (PCR) amplification were performed using the HELP-tagging library preparation protocol developed by Suzuki et al.68 Please refer to Supplementary Materials and Methods for the exact protocol description.
The PCR product was extracted from a 3.5% low molecular weight agarose gel electrophoresis and purified by Mini-Elute gel Extraction Kit (Qiagen). Purified products were analyzed by Bioanalyzer to ensure integrity and purity followed by Illumina sequencing (end library size ∼160 bp). All enzymes used for the HELP-tagging assay were purchased from New England Biosciences unless otherwise stated. All adapters and primers were purchased from the WASP system at the Albert Einstein University. For a full list of adapters and primers used, refer to Table S6.
Processing of sequence data
Paired samples (n = 13) from UC patients met the pre-determined quality control standards and were analyzed. Sequencing was performed on an Illumina HiSeq 2000 at the Epigenomics Shared Facility of Albert Einstein University. For this assay, single-end 36–50 bp sequencing was required. The images generated by the Illumina sequencer were analyzed using Illumina Pipeline Software (version 1.4). Default read length of 36 bp was used for initial data pre-processing. Sequences for which adaptor sequence on the 3’end was found were then isolated. The adaptor sequence was replaced with a ploy(N) sequence of the same length and the Illumina ELAND pipeline was run on these sequences with the sequence length set to 27 bp. Data generated by the ELAND pipeline was used to count the number of aligned sequences overlapping each CCGG site in the hg19 build of the human genome. During the alignment process, a maximum of 2 mismatches in each sequence was accepted. For all non-unique alignments, sequences were assigned a partial count for each alignment location amounting to 1/n, where ‘n’ represented the total number of alignments. The number of sequences associated with each HpaII site was then divided by the total number of sequences (including partial counts) aligning to all HpaII sites in the same sample to normalize the data between experiments.68
To measure the level of DNA methylation at each CCGG site, the normalized accumulative proportion (NAP) count for the HpaII digested sample was compared with the reference NAP count for the MspI digest. The DNA methylation angle score was calculated using the arctangent of the ratio of HpaII NAP count and MspI NAP count as described previously.69 This allows normalization of HpaII counts in terms of variability of the MspI representation. DNA methylation levels reported here were calculated as one minus the DNA methylation angle score, for ease of interpretability, and range from 0 (no DNA methylation) to 100 (complete DNA methylation).34
DNA methylation analysis
CCGG sites with fewer than 5 MspI reads were excluded from all analyses to improve DNA methylation estimation accuracy. Quantile normalization was then performed using the preprocessCore package in R.70 The package limma71 was used to identify individual differentially methylated sites (DMS). A paired sample comparison was used, applying paired effects as a blocking factor in the design model, to compare inflamed and non-inflamed states within subjects. This linear model also incorporated covariates, including age, duration of colitis, severity of disease and total HpaII counts per sample. A false discovery rate analysis (Benjamini-Hochberg) was used to correct for multiple testing. A false discovery threshold of 0.2 was used to decide statistical significance. Differential methylation is expressed as delta methylation (ΔMethylation). ΔMethylation reflects the geometric mean methylation score of one sample group relative to another, a ΔMethylation value >0 reflects hypermethylation in the inflamed region (distal colon) and a ΔMethylation value <0 reflects hypermethylation in the non-inflamed region (proximal colon).
We tested for enrichment of gene ontology categories among genes for which at least one differentially methylated site was found close (±2 kb) to the transcription start site (TSS) as well as the rest of the gene body. The HELP-tagging assay profiles the DNA methylation status at CCGG sites. However, different genes may be associated with very different numbers of such sites, with genes associated with larger numbers of CCGG sites having a greater chance of being associated with at least one differentially methylated site. This can result in severe bias in gene set analysis.72 The R package Goseq was used to correct this bias. The Wallenius approximation was used to calculate the over and under representation of Gene Ontology (GO) categories among differentially methylated genes. A false discovery rate analysis (Benjamini-Hochberg) was used to correct for multiple testing. A false discovery threshold of 0.2 was used to decide statistical significance.
The UCSC table browser73 was used to obtain coordinates of genomic regions including gene bodies, intergenic and intragenic regions. The mammalian expression atlas and enhancer peaks were obtained from the FANTOM consortium.36,74 The epigenomics roadmap consortium was used to obtain coordinates for genomic regions for ChromHMM states in colonic mucosa.40,41 Enrichment of differential methylation was conditioned on the CCGG density at each state. Significant enrichment was measured using a fisher exact test. These states, as well as states for other cell lines and tissues can be downloaded from
Annotation of all CCGG sties and DMS to candidate genomic locations such as gene body, intergenic, intragenic, enhancers, TSS regions, and ChromHMM states was performed using customized python scripts.
Array hybridization: Human Transcriptome Array 2.0 (HTA 2.0)
Extraction of total RNA from IECs was performed using the method derived from the Albert Einstein University online resource (http://waspeinstein.yu.edu/index.php/Protocol). All steps outlined in the protocol were followed exactly. Please refer to supplementary materials and methods for a detailed protocol description with no modifications.
Total RNA sample processing and array hybridization took place at Core Unit Systems Medicine (SysMed) at the University of Wurzburg, Germany. The assessment of quality, integrity and quantity of total RNA, in vitro transcription for linear amplification, fragmentation, and biotin labeling was performed as outlined in the GeneChip WT Plus Reagent Kit user manual (Affymetrix). Preparation of buffers and staining for array hybridization was performed according to the GeneChip Hybridization Wash and Stain kit (Affymetrix) with no modifications. Samples were hybridized for 16 h at 45°C and 60 rpm to GeneChip Human Transcriptome Arrays 2.0; washing and staining was performed with a Fluidics Station FS450 using the fluidics script FS450_0001. Arrays were scanned with a GeneChip Scanner 3000 7G (Affymetrix) and processed by the Affymetrix GeneChip command console software (AGCC).
Transcriptome analysis
Raw signals of the arrays were processed using Affymetrix Power Tools,75 applying Robust Multi-array Average (RMA) for background correction, quantile normalization and median polish summarization of probe sets.76 Log transformed (log2) relative expression values for each probe were then annotated to each gene transcript. Identification of differentially methylated transcripts was performed using limma.71 A paired sample comparison was used, applying paired effects as a blocking factor in the design model, to compare inflamed and non-inflamed states within subjects. This model also incorporated covariates, including age, duration of colitis, and severity of disease. A false discovery rate analysis (Benjamini-Hochberg) was used to correct for multiple testing. A false discovery threshold of 0.2 was used to decide statistical significance.
Unsupervised hierarchical clustering of global DNA methylation and gene expression patterns was performed using complete linkage and Euclidean distance. Relationships between DNA methylation and gene expression data were examined using Spearman correlations.
Declarations
Ethical approval
All patient recruitment and sample collection was performed under human subjects protocol approval from the Galway University Hospitals Research Ethics Committee with written informed consent regarding participation. Consent for publication was also obtained under human subjects protocol approval from the Galway University Hospitals Research Ethics Committee.
Availability of supporting data
Methylome and transcriptome data sets generated and analyzed during the current study are available from the corresponding author on reasonable request. Coordinates for genomic regions for ChromHMM states in colonic mucosa can be downloaded from
The UCSC table browser73 was used to obtain coordinates of genomic regions. The Mammalian expression atlas and enhancer peaks were obtained from the FANTOM consortium.36,74
Supplementary Material
Funding Statement
This work was supported by the Irish Research Council and further funding was provided by AbbVie Ireland.
Disclosure of potential conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Coralie Mureau for her assistance during sample preparation. We also thank staff from Einstein's Center of Epigenomics including the Epigenomics Shared Facility and Computational Epigenomics group who provided technical support with next generation sequencing.
Author Contributions
AB performed the experiments and generated and analyzed the data. AB, AG, JG, CS and LE interpreted the data and drafted the manuscript. LE, CS and AG devised the study concept. AG, CS and LE obtained funding and supervised the study. All authors critically reviewed and approved the final version of the manuscript.
References
- 1.Conrad K, Roggenbuck D, Laass MW. Diagnosis and classification of ulcerative colitis. Autoimmun Rev 2014; 13:463–6; PMID:24424198; https://doi.org/10.1016/j.autrev.2014.01.028 [DOI] [PubMed] [Google Scholar]
- 2.Laass MW, Roggenbuck D, Conrad K. Diagnosis and classification of Crohn's disease. Autoimmun Rev 13:467–71; PMID:24424189; https://doi.org/10.1016/j.autrev.2014.01.029</bib> [DOI] [PubMed] [Google Scholar]
- 3.Li Y, Kundu P, Seow SW, de Matos CT, Aronsson L, Chin KC, Kärre K, Pettersson S, Greicius G. Gut microbiota accelerate tumor growth via c-jun and STAT3 phosphorylation in APCMin/+ mice. Carcinogenesis 2012; 33:1231–8; PMID:22461519; https://doi.org/10.1093/carcin/bgs137 [DOI] [PubMed] [Google Scholar]
- 4.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012; 491:119–24; PMID:23128233; https://doi.org/10.1038/nature11582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Araki A, Kanai T, Ishikura T, Makita S, Uraushihara K, Iiyama R, Totsuka T, Takeda K, Akira S, Watanabe M. MyD88-deficient mice develop severe intestinal inflammation in dextran sodium sulfate colitis. J Gastroenterol 2005; 40:16–23; PMID:15692785; https://doi.org/10.1007/s00535-004-1492-9 [DOI] [PubMed] [Google Scholar]
- 6.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 2008; 14:818–29; PMID:18539112; https://doi.org/10.1016/j.devcel.2008.05.009 [DOI] [PubMed] [Google Scholar]
- 7.Voronov E, Shouval DS, Krelin Y, Cagnano E, Benharroch D, Iwakura Y, Dinarello CA, Apte RN. IL-1 is required for tumor invasiveness and angiogenesis. Proc Natl Acad Sci U S A 2003; 100:2645–50; PMID:12598651; https://doi.org/10.1073/pnas.0437939100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang S, Liu Z, Wang L, Zhang X. NF-kappaB signaling pathway, inflammation and colorectal cancer. Cell Mol Immunol 2009; 6:327–34; PMID:19887045; https://doi.org/10.1038/cmi.2009.43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klampfer L. The role of signal transducers and activators of transcription in colon cancer. Front Biosci 2008; 13:2888–99; PMID:17981761; https://doi.org/10.2741/2893 [DOI] [PubMed] [Google Scholar]
- 10.Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of Colitis-associated cancer. Cancer Cell 2009; 15:103–13; PMID:19185845; https://doi.org/10.1016/j.ccr.2009.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dupaul-Chicoine J, Dagenais M, Saleh M. Crosstalk between the intestinal microbiota and the innate immune system in intestinal homeostasis and inflammatory bowel disease. Inflamm Bowel Dis [Internet] 2013. [cited 2015 Nov 4]; 19:2227–37. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23669404; PMID:23669404; https://doi.org/10.1097/MIB.0b013e31828dcac7 [DOI] [PubMed] [Google Scholar]
- 12.Coskun M. Intestinal epithelium in inflammatory bowel disease. Front Med 2014; 1:24; https://doi.org/10.3389/fmed.2014.00024</bib> [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henderson P, van Limbergen JE, Schwarze J, Wilson DC. Function of the intestinal epithelium and its dysregulation in inflammatory bowel disease. Inflamm Bowel Dis [Internet] 2011. [cited 2015 Nov 4]; 17:382–95. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20645321; PMID:20645321; https://doi.org/10.1002/ibd.21379 [DOI] [PubMed] [Google Scholar]
- 14.Foran E, Garrity-Park MM, Mureau C, Newell J, Smyrk TC, Limburg PJ, Egan LJ. Upregulation of DNA methyltransferase-mediated gene silencing, anchorage-independent growth, and migration of colon cancer cells by interleukin-6. Mol Cancer Res 2010; 8:471–81; PMID:20354000; https://doi.org/10.1158/1541-7786.MCR-09-0496 [DOI] [PubMed] [Google Scholar]
- 15.Garrity-Park MM, Loftus EV, Sandborn WJ, Bryant SC, Smyrk TC. Methylation status of genes in non-neoplastic mucosa from patients with ulcerative colitis-associated colorectal cancer. Am J Gastroenterol 2010; 105:1610–9; PMID:20160714; https://doi.org/10.1038/ajg.2010.22 [DOI] [PubMed] [Google Scholar]
- 16.Saito S, Kato J, Hiraoka S, Horii J, Suzuki H, Higashi R, Kaji E, Kondo Y, Yamamoto K. DNA methylation of colon mucosa in ulcerative colitis patients: Correlation with inflammatory status. Inflamm Bowel Dis 2011; 17:1955–65; PMID:21830274; https://doi.org/10.1002/ibd.21573 [DOI] [PubMed] [Google Scholar]
- 17.Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med 2000; 343:1350–4; PMID:11070098; https://doi.org/10.1056/NEJM200011093431901 [DOI] [PubMed] [Google Scholar]
- 18.Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, Markowitz S, Willson JK, Hamilton SR, Kinzler KW, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci U S A 1998; 95:6870–5; PMID:9618505; https://doi.org/10.1073/pnas.95.12.6870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cooke J, Zhang H, Greger L, Silva AL, Massey D, Dawson C, Metz A, Ibrahim A, Parkes M. Mucosal genome-wide methylation changes in inflammatory bowel disease. Inflamm Bowel Dis 2012; 18(11):2128–37; PMID:22419656; https://doi.org/10.1002/ibd.22942 [DOI] [PubMed] [Google Scholar]
- 20.Häsler R, Feng Z, Bäckdahl L, Spehlmann ME, Franke A, Teschendorff A, Rakyan VK, Down TA, Wilson GA, Feber A, et al. A functional methylome map of ulcerative colitis. Genome Res 2012; 22:2130–7; PMID:22826509; https://doi.org/10.1101/gr.138347.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olaru A V, Cheng Y, Agarwal R, Yang J, David S, Abraham JM, Yu W, Kwon JH, Lazarev M, Brant SR, et al. Unique patterns of CpG island methylation in inflammatory bowel disease-associated colorectal cancers. Inflamm Bowel Dis 2012; 18:641–8; PMID:21830278; https://doi.org/10.1002/ibd.21826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin Z, Hegarty JP, Cappel JA, Yu W, Chen X, Faber P, Wang Y, Kelly AA, Poritz LS, Peterson BZ, et al. Identification of disease-associated DNA methylation in intestinal tissues from patients with inflammatory bowel disease. Clin Genet 2011; 80:59–67; PMID:20950376; https://doi.org/10.1111/j.1399-0004.2010.01546.x [DOI] [PubMed] [Google Scholar]
- 23.Kim SW, Kim ES, Moon CM, Il Kim T, Kim WH, Cheon JH. Abnormal genetic and epigenetic changes in signal transducer and activator of transcription 4 in the pathogenesis of inflammatory bowel diseases. Dig Dis Sci 2012; 57:2600–7; PMID:22569826; https://doi.org/10.1007/s10620-012-2199-z [DOI] [PubMed] [Google Scholar]
- 24.Kraiczy J, Nayak K, Ross A, Raine T, Mak TN, Gasparetto M, Cario E, Rakyan V, Heuschkel R, Zilbauer M. Assessing DNA methylation in the developing human intestinal epithelium: potential link to inflammatory bowel disease. Mucosal Immunol [Internet] 2015. [cited 2015 Oct 8]; Available from: https://doi.org/10.1038/mi.2015.88; PMID:26376367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Won KJ, Zhang X, Wang T, Ding B, Raha D, Snyder M, Ren B, Wang W. Comparative annotation of functional regions in the human genome using epigenomic data. Nucleic Acids Res 2013; 41:4423–32; PMID:23482391; https://doi.org/10.1093/nar/gkt143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol 2014; 15:R31; PMID:24495553; https://doi.org/10.1186/gb-2014-15-2-r31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bock C, Walter J, Paulsen M, Lengauer T. Inter-individual variation of DNA methylation and its implications for large-scale epigenome mapping. Nucleic Acids Res 2008; 36:e55; PMID:18413340; https://doi.org/10.1093/nar/gkn122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rendeiro AF, Schmidl C, Strefford JC, Walewska R, Davis Z, Farlik M, Oscier D, Bock C. Chromatin accessibility maps of chronic lymphocytic leukaemia identify subtype-specific epigenome signatures and transcription regulatory networks. Nat Commun 2016; 7:11938; PMID:27346425; https://doi.org/10.1038/ncomms11938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flint N, Cove FL, Evans GS. A low-temperature method for the isolation of small-intestinal epithelium along the crypt-villus axis. Biochem J 1991; 280(Pt 2):331–4; PMID:1747105; https://doi.org/10.1042/bj2800331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whitehead RH, Demmler K, Rockman SP, Watson NK. Clonogenic growth of epithelial cells from normal colonic mucosa from both mice and humans. Gastroenterology 1999; 117:858–65; PMID:10500068; https://doi.org/10.1016/S0016-5085(99)70344-6 [DOI] [PubMed] [Google Scholar]
- 31.Illingworth RS, Bird AP. CpG islands–’a rough guide’. FEBS Lett 2009; 583:1713–20; PMID:19376112; https://doi.org/10.1016/j.febslet.2009.04.012 [DOI] [PubMed] [Google Scholar]
- 32.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 2007; 130:77–88; PMID:17632057; https://doi.org/10.1016/j.cell.2007.05.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim T-K, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007; 448:553–60; PMID:17603471; https://doi.org/10.1038/nature06008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barnicle A, Seoighe C, Golden A, Greally JM, Egan LJ. Differential DNA methylation patterns of homeobox genes in proximal and distal colon epithelial cells. Physiol Genomics [Internet] 2016; 48(4):257–73 [cited 2016 Feb 1];physiolgenomics.00046.2015. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26812987; PMID:26812987; https://doi.org/10.1152/physiolgenomics.00046.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blattler A, Yao L, Witt H, Guo Y, Nicolet CM, Berman BP, Farnham PJ. Global loss of DNA methylation uncovers intronic enhancers in genes showing expression changes. Genome Biol 2014; 15:469; PMID:25239471; https://doi.org/10.1186/s13059-014-0469-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Forrest ARR, Kawaji H, Rehli M, Baillie JK, de Hoon MJL, Lassmann T, Itoh M, Summers KM, Suzuki H, Daub CO, et al. A promoter-level mammalian expression atlas. Nature 2014; 507:462–70; PMID:24670764; https://doi.org/10.1038/nature13182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev 2002; 16:6–21; PMID:11782440; https://doi.org/10.1101/gad.947102 [DOI] [PubMed] [Google Scholar]
- 38.Lövkvist C, Dodd IB, Sneppen K, Haerter JO. DNA methylation in human epigenomes depends on local topology of CpG sites. Nucleic Acids Res 2016; 44:5123–32; PMID:26932361; https://doi.org/10.1093/nar/gkw124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaz AM, Wong CJ, Luo Y, Virgin JB, Kay Washington M, Willis JE, Leidner RS, Chak A, Grady WM. DNA methylation profiling in Barrett's esophagus and esophageal adenocarcinoma reveals unique methylation signatures and molecular subclasses. Epigenetics 2011; 6:1403–12; PMID:22139570; https://doi.org/10.4161/epi.6.12.18199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods 2012; 9:215–6; PMID:22373907; https://doi.org/10.1038/nmeth.1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Consortium RE, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. Integrative analysis of 111 reference human epigenomes. Nature 2015; 518:317–30; PMID:25693563; https://doi.org/10.1038/nature14248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lay FD, Triche TJ, Tsai YC, Su S-F, Martin SE, Daneshmand S, Skinner EC, Liang G, Chihara Y, Jones PA. Reprogramming of the human intestinal epigenome by surgical tissue transposition. Genome Res 2014; 24:545–53; PMID:24515120; https://doi.org/10.1101/gr.166439.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.LaPointe LC, Dunne R, Brown GS, Worthley DL, Molloy PL, Wattchow D, Young GP. Map of differential transcript expression in the normal human large intestine. Physiol Genomics 2008; 33:50–64; PMID:18056783; https://doi.org/10.1152/physiolgenomics.00185.2006 [DOI] [PubMed] [Google Scholar]
- 44.Liu XF, Olsson P, Wolfgang CD, Bera TK, Duray P, Lee B, Pastan I. PRAC: A novel small nuclear protein that is specifically expressed in human prostate and colon. Prostate 2001; 47:125–31; PMID:11340635; https://doi.org/10.1002/pros.1055 [DOI] [PubMed] [Google Scholar]
- 45.Sreenath T, Orosz A, Fujita K, Bieberich CJ. Androgen-independent expression of hoxb-13 in the mouse prostate. Prostate 1999; 41:203–7; PMID:10517879; https://doi.org/10.1002/(SICI)1097-0045(19991101)41:3%3c203::AID-PROS8%3e3.0.CO;2-J [DOI] [PubMed] [Google Scholar]
- 46.Gertz J, Varley KE, Reddy TE, Bowling KM, Pauli F, Parker SL, Kucera KS, Willard HF, Myers RM. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet 2011; 7:e1002228; PMID:21852959; https://doi.org/10.1371/journal.pgen.1002228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bell JT, Pai AA, Pickrell JK, Gaffney DJ, Pique-Regi R, Degner JF, Gilad Y, Pritchard JK. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol 2011; 12:R10; PMID:21251332; https://doi.org/10.1186/gb-2011-12-1-r10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grundberg E, Meduri E, Sandling JK, Hedman AK, Keildson S, Buil A, Busche S, Yuan W, Nisbet J, Sekowska M, et al. Global analysis of dna methylation variation in adipose tissue from twins reveals links to disease-associated variants in distal regulatory elements . Am J Hum Genet 2013; 93:876–90; PMID:24183450; https://doi.org/10.1016/j.ajhg.2013.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liévin-Le Moal V, Servin AL. The front line of enteric host defense against unwelcome intrusion of harmful microorganisms: Mucins, antimicrobial peptides, and Microbiota. Clin Microbiol Rev 2006; 19:315–37; PMID:16614252; https://doi.org/10.1128/CMR.19.2.315-337.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol 2014; 14:141–53; PMID:24566914; https://doi.org/10.1038/nri3608 [DOI] [PubMed] [Google Scholar]
- 51.Duerkop BA, Vaishnava S, Hooper L V. Immune responses to the Microbiota at the intestinal mucosal surface. Immunity 2009; 31:368–76; PMID:19766080; https://doi.org/10.1016/j.immuni.2009.08.009 [DOI] [PubMed] [Google Scholar]
- 52.Dieckgraefe BK, Stenson WF, Korzenik JR, Swanson PE, Harrington CA. Analysis of mucosal gene expression in inflammatory bowel disease by parallel oligonucleotide arrays. Physiol Genomics 2000; 4:1–11; PMID:11074008 [DOI] [PubMed] [Google Scholar]
- 53.Lawrance IC, Fiocchi C, Chakravarti S. Ulcerative colitis and Crohn's disease: distinctive gene expression profiles and novel susceptibility candidate genes. Hum Mol Genet 2001; 10:445–56; PMID:11181568; https://doi.org/10.1093/hmg/10.5.445 [DOI] [PubMed] [Google Scholar]
- 54.Planell N, Lozano JJ, Mora-Buch R, Masamunt MC, Jimeno M, Ordás I, Esteller M, Ricart E, Piqué JM, Panés J, et al. Transcriptional analysis of the intestinal mucosa of patients with ulcerative colitis in remission reveals lasting epithelial cell alterations. Gut [Internet] 2013. [cited 2017 Apr 7]; 62:967–76. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23135761; PMID:23135761; https://doi.org/10.1136/gutjnl-2012-303333 [DOI] [PubMed] [Google Scholar]
- 55.Granlund A van B, Flatberg A, Østvik AE, Drozdov I, Gustafsson BI, Kidd M, Beisvag V, Torp SH, Waldum HL, Martinsen TC, et al. Whole genome gene expression meta-analysis of inflammatory bowel disease colon mucosa demonstrates lack of major differences between Crohn's disease and ulcerative colitis. PLoS One [Internet] 2013. [cited 2017 Apr 7]; 8:e56818. Available from: http://dx.plos.org/10.1371/journal.pone.0056818; PMID:23468882; https://doi.org/10.1371/journal.pone.0056818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fackler MJ, McVeigh M, Evron E, Garrett E, Mehrotra J, Polyak K, Sukumar S, Argani P. DNA methylation of RASSF1A, HIN-1, RAR-beta, Cyclin D2 and Twist in in situ and invasive lobular breast carcinoma. Int J Cancer 2003; 107:970–5; PMID:14601057; https://doi.org/10.1002/ijc.11508 [DOI] [PubMed] [Google Scholar]
- 57.Kapitskaya KY, Azhikina TL, Ponomaryova AA, Cherdyntseva N V, Vlasov V V, Laktionov PP, Rykova EY. MIRA analysis of RARβ2 gene methylation in DNA circulating in the blood in lung cancer. Bull Exp Biol Med 2014; 157:516–9; PMID:25110096; https://doi.org/10.1007/s10517-014-2604-z [DOI] [PubMed] [Google Scholar]
- 58.Hua F, Fang N, Li X, Zhu S, Zhang W, Gu J. A meta-analysis of the relationship between RARβ gene promoter methylation and non-small cell lung cancer. PLoS One 2014; 9:e96163; PMID:24796328; https://doi.org/10.1371/journal.pone.0096163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gao T, He B, Pan Y, Li R, Xu Y, Chen L, Nie Z, Gu L, Wang S. The association of retinoic acid receptor beta2(RARβ2) methylation status and prostate cancer risk: a systematic review and meta-analysis. PLoS One 2013; 8:e62950; PMID:23675444; https://doi.org/10.1371/journal.pone.0062950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang D, Kryvenko ON, Mitrache N, Do KC, Jankowski M, Chitale DA, Trudeau S, Rundle A, Belinsky SA, Rybicki BA. Methylation of the RARB gene increases prostate cancer risk in black Americans. J Urol 2013; 190:317–24; PMID:23376149; https://doi.org/10.1016/j.juro.2013.01.083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Côté S, Sinnett D, Momparler RL. Demethylation by 5-aza-2’-deoxycytidine of specific 5-methylcytosine sites in the promoter region of the retinoic acid receptor beta gene in human colon carcinoma cells. Anticancer Drugs 1998; 9:743–50; PMID:9840718; https://doi.org/10.1097/00001813-199810000-00001 [DOI] [PubMed] [Google Scholar]
- 62.Youssef EM, Estecio MRH, Issa J-PJ. Methylation and regulation of expression of different retinoic acid receptor beta isoforms in human colon cancer. Cancer Biol Ther 2014; 3:82–6; https://doi.org/10.4161/cbt.3.1.591 [DOI] [PubMed] [Google Scholar]
- 63.Li Z, White P, Tuteja G, Rubins N, Sackett S, Kaestner KH. Foxa1 and Foxa2 regulate bile duct development in mice. J Clin Invest 2009; 119:1537–45; PMID:19436110; https://doi.org/10.1172/JCI38201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhu C-P, Wang J, Shi B, Hu P-F, Ning B-F, Zhang Q, Chen F, Chen W-S, Zhang X, Xie W-F. The transcription factor FOXA2 suppresses gastric tumorigenesis in vitro and in vivo. Dig Dis Sci 2015; 60:109–17; PMID:25129104; https://doi.org/10.1007/s10620-014-3290-4 [DOI] [PubMed] [Google Scholar]
- 65.Barnard JA, Beauchamp RD, Coffey RJ, Moses HL. Regulation of intestinal epithelial cell growth by transforming growth factor type beta. Proc Natl Acad Sci U S A 1989; 86:1578–82; PMID:2466294; https://doi.org/10.1073/pnas.86.5.1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Traber PG, Gumucio DL, Wang W. Isolation of intestinal epithelial cells for the study of differential gene expression along the crypt-villus axis. Am J Physiol 1991; 260:G895–903; PMID:2058677 [DOI] [PubMed] [Google Scholar]
- 67.Gallo-Payet N, Hugon JS. Insulin receptors in isolated adult mouse intestinal cells: Studies in vivo and in organ culture. Endocrinology 1984; 114:1885–901; PMID:6370669; https://doi.org/10.1210/endo-114-5-1885 [DOI] [PubMed] [Google Scholar]
- 68.Suzuki M, Jing Q, Lia D, Pascual M, Mclellan A, Greally JM. Optimized design and data analysis of tag-based cytosine methylation assays. Genome Biol 2010; 11(4):R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jing Q, McLellan A, Greally JM, Suzuki M. Automated computational analysis of genome-wide DNA methylation profiling data from HELP-tagging assays. Methods Mol Biol 2012; 815:79–87; PMID:22130985 [DOI] [PubMed] [Google Scholar]
- 70.Bolstad BM, Irizarry R, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003; 19:185–93; PMID:12538238; https://doi.org/10.1093/bioinformatics/19.2.185 [DOI] [PubMed] [Google Scholar]
- 71.Smyth G. limma: Linear Models for Microarray Data. In: Bioinformatics and computational biology solutions using R and Bioconductor. 2005:397–420 [Google Scholar]
- 72.Geeleher P, Hartnett L, Egan LJ, Golden A, Raja Ali RA, Seoighe C. Gene-set analysis is severely biased when applied to genome-wide methylation data. Bioinformatics 2013; 29:1851–7; PMID:23732277; https://doi.org/10.1093/bioinformatics/btt311 [DOI] [PubMed] [Google Scholar]
- 73.Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. The UCSC Table Browser data retrieval tool. Nucleic Acids Res 2004; 32:D493–6; PMID:14681465; https://doi.org/10.1093/nar/gkh103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, et al. An atlas of active enhancers across human cell types and tissues. Nature 2014; 507:455–61; PMID:24670763; https://doi.org/10.1038/nature12787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lockstone HE. Exon array data analysis using Affymetrix power tools and R statistical software. Brief Bioinform 2011; 12:634–44; PMID:21498550; https://doi.org/10.1093/bib/bbq086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 2003; 31:e15; PMID:12582260; https://doi.org/10.1093/nar/gng015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.