

ABSTRACT
Gestational diabetes mellitus (GDM) is associated with obesity in childhood. This suggests that consequences of in utero exposure to maternal hyperglycemia extend beyond the fetal development, possibly through epigenetic programming. The aims of this study were to assess whether placental DNA methylation (DNAm) marks were associated with maternal GDM status and to offspring body composition at 5 years old in a prospective birth cohort. DNAm levels were measured in the fetal side of the placenta in 66 samples (24 from GDM mothers) using bisDNA-pyrosequencing. Anthropometric and body composition (bioimpedance) were measured in children at 5 years of age. Mann-Whitney and Spearman tests were used to assess associations between GDM, placental DNAm levels at the lipoprotein lipase (LPL) locus and children's weight, height, body mass index (BMI), body fat, and lean masses at 5 years of age. Weight, height, and BMI z-scores were computed according to the World Health Organization growth chart. Analyses were adjusted for gestational age at birth, child sex, maternal age, and pre-pregnancy BMI. LPL DNAm levels were positively correlated with birth weight z-scores (r = 0.252, P = 0.04), and with mid-childhood weight z-scores (r = 0.314, P = 0.01) and fat mass (r = 0.275, P = 0.04), and negatively correlated with lean mass (r = −0.306, P = 0.02). We found a negative correlation between LPL DNAm and mRNA levels in placenta (r = −0.459; P < 0.001), which highlights the regulation of transcriptional activity by these epivariations. We demonstrated that alterations in fetal placental DNAm levels at the LPL gene locus are associated with the anthropometric profile in children at 5 years of age. These findings support the concept of fetal metabolic programming through epigenetic changes.
KEYWORDS: Development, DOHaD, epigenetics, hyperglycemia, in utero exposure, LPL, placenta, pregnancy
Abbreviations
- BIA
Bioelectrical impedance analysis
- BMI
Body mass index
- DNAm
DNA methylation
- DOHaD
Developmental Origins of Health and Disease
- E-21
Ecogene-21 birth cohort
- EWAS
Epigenome-wide association study
- FFA
Free fatty acid
- Gen3G
Genetics of Glucose regulation in Gestation and Growth
- GDM
Gestational diabetes mellitus
- HDL
High-density lipoprotein
- HOMA-IR
Homeostasis model assessment of insulin resistance
- IUGR
Intrauterine growth restriction
- LDL
Low-density lipoprotein
- LGA
Large for gestational age
- LPL
Lipoprotein lipase
- MVM
Microvillus membrane
- NGT
Normoglycemic
- OGTT
Oral glucose tolerance test
- TC
Total cholesterol
- TG
Total triglycerides
- VLDL
Very low-density lipoprotein
- WHO
World Health Organization
Introduction
There is a growing body of evidence suggesting that maternal metabolic disorders during pregnancy could contribute to alterations in childhood health. These maternal metabolic cues are more likely to have effects on offspring when they arise within critical periods of fetal development.1 Gestational diabetes mellitus (GDM) had been previously associated with childhood metabolic complications such as impaired glucose tolerance and insulin resistance,2 suggesting that the consequences of hyperglycemic prenatal insults may extend beyond the fetal development, possibly through epigenetic programming.
Epigenetic modifications are suspected to contribute to the intergenerational inheritance of obesity, diabetes, and cardiovascular complications. DNA methylation (DNAm) is well known for its implication in the transcriptional activity regulation of key processes, such as lipoprotein metabolism.3 DNAm is quite of interest considering that it can be of parental origin or programmed in the course of fetal development, while responding to maternal metabolic cues.4,5 Epigenetic modifications are highly suspected to contribute to the fetal programming of metabolic health since these modifications could be preserved through mitosis.6 This refers to the Developmental Origins of Health and Disease (DOHaD) concept, which supports that the environmental exposure during fetal development programs the susceptibility to disease later in life.7 Nevertheless, how epigenetic variations are implicated in the fetal metabolic programming is still poorly understood.
Previous studies, including ours, have demonstrated that GDM, through maternal-to-fetal hyperglycemia, is one of the intrauterine conditions affecting placental DNAm marks.8,9 Among other studies, we have reported associations between maternal blood glucose concentrations at 2nd trimester of pregnancy and fetal placental DNAm levels at the proximal promoter and intronic CpG island loci in the lipoprotein lipase (LPL) gene.9 Considering that GDM had been repeatedly associated with a long-term increase in obesity risk and diabetes in offspring, our results have suggested that maternal hyperglycemia, through epigenetic modifications at LPL gene loci, could contribute to the fetal metabolic programming of chronic metabolic disorders.10-12
LPL contributes to the metabolic homeostasis through triglycerides (TG) hydrolysis. Several studies have pointed out that the impairment of placental LPL in fetal development has significant impacts on fat accretion, growth, and development.13 Likewise, changes in placental LPL gene expression have been associated with an unfavorable intrauterine exposure, such as maternal obesity and GDM.14,15 Moreover, we have reported that decreased placental LPL gene expression changes could be induced, at least partially, by an increase in DNAm, following dysregulation.8,9 However, it is essential to understand how LPL epigenetic perturbations could translate into later life changes in lipid metabolism, fat accretion, and growth.
Hence, the aim of this study was to assess whether GDM-associated placental epivariations within the LPL proximal promoter locus are also associated with fat accretion and growth in childhood to better define the interplay between intrauterine GDM exposure, LPL epigenetic modifications, and childhood risk for metabolic disorders.
Results
Clinical characteristics of mothers and children are shown in Table 1. GDM and normoglycemic (NGT) mothers were similar in age and body mass index (BMI) at 1st trimester of pregnancy, gravidity, parity, smoking status in pregnancy and delivery mode. On average, they were aged 29 years and slightly overweight (BMI between 25.3 and 26.1), with normal fasting glucose levels. From the 66 recruited mothers, 24 had a GDM diagnosis [2 h post-75 g oral glucose tolerance test (OGTT) glucose ≥ 7.8 mmol/L] according to World Health Organization (WHO) criteria.16 GDM mothers were treated with a diet only (n = 13) or with a diet combined with daily insulin injections (n = 11).
Table 1.
Clinical characteristics of mothers and offspring from first trimester of pregnancy to 5 y of age.
| NGT | GDM | ||
|---|---|---|---|
| Participant clinical data | n = 42 | n = 24 | P value |
| Maternal characteristics | |||
| 1st trimester of pregnancy | |||
| Age (years) | 28.6 ± 3.7 | 29.0 ± 3.1 | 0.69 |
| BMI (kg/m2) | 25.3 ± 5.3 | 26.1 ± 4.1 | 0.54 |
| Fasting glucose (mmol/L) | 4.37 ± 0.38 | 4.49 ± 0.42 | 0.27 |
| 2nd trimester of pregnancy | |||
| Fasting glucose (mmol/L) | 4.29 ± 0.46 | 4.50 ± 0.52 | 0.18b |
| Fasting insulin (mU/L) | 10.42 ± 12.27 | 10.33 ± 5.30 | 0.97c |
| HOMA-IR | 2.11 ± 2.37 | 2.14 ± 1.33 | 0.96b |
| 2 h post-OGTT glucose concentration (mmol/L) | 6.25 ± 0.76 | 8.51 ± 0.73 | <0.001 |
| Weight gain 2nd – 1st of pregnancy (kg) | 6.00 ± 2.52 | 5.55 ± 2.13 | 0.44 |
| HDL (mmol/L) | 1.79 ± 0.45 | 1.73 ± 0.30 | 0.53 |
| LDL (mmol/L) | 3.6 ± 0.91 | 3.28 ± 0.77 | 0.10 |
| Cholesterol (mmol/L) | 6.28 ± 1.03 | 5.98 ± 0.76 | 0.19 |
| Triglycerides (mmol/L) | 1.85 ± 0.65 | 2.13 ± 0.73 | 0.13 |
| 3rd trimester of pregnancy | |||
| Fasting glucose (mmol/L) | 4.23 ± 0.80 | 4.24 ± 0.45 | 0.95d |
| Fasting insulin (mU/L) | 13.08 ± 7.80 | 9.02 ± 4.17 | 0.01e |
| HOMA-IR | 2.52 ± 1.89 | 1.71 ± 0.85 | 0.08e |
| Weight gain 3rd – 2nd of pregnancy (kg) | 6.00 ± 2.34 | 2.82 ± 2.26 | <0.001 |
| HDL (mmol/L) | 1.74 ± 0.44 | 1.73 ± 0.29 | 0.89d |
| LDL (mmol/L) | 3.97 ± 1.10 | 3.62 ± 1.00 | 0.20d |
| Cholesterol (mmol/L) | 6.93 ± 1.24 | 6.57 ± 1.09 | 0.24d |
| Triglycerides (mmol/L) | 2.74 ± 0.95 | 2.67 ± 0.69 | 0.73d |
| Newborns' characteristics | |||
| Gestational age (week) | 39.5 ± 1.0 | 39.5 ± 1.5 | 0.63 |
| Sex (Boys/Girls) | 19/23 | 12/12 | 0.78 |
| Birth weight (kg)l | 3.46 ± 0.35 | 3.33 ± 0.53 | 0.23 |
| Chest circumference (cm) | 33.52 ± 1.50 | 33.04 ± 2.05 | 0.43f |
| Head circumference (cm) | 34.41 ± 1.60 | 34.52 ± 1.44 | 0.77 g |
| Cord blood leptin (ng/mL) | 10.75 ± 8.22 | 6.79 ± 4.60 | 0.08h |
| Cord blood glucose (mmol/L) | 4.58 ± 0.87 | 4.59 ± 0.89 | 0.81i |
| Cord blood triglycerides (mmol/L) | 0.44 ± 0.15 | 0.51 ± 0.27 | 0.83j |
| C-Peptide (ng/mL) | 129.2 ± 81.9 | 174.6 ± 83.0 | 0.05k |
| Children' characteristics | |||
| Age (years) | 5.9 ± 0.7 | 5.9 ± 0.7 | 0.73 |
| Weight (kg)l | 20.39 ± 2.56 | 18.80 ± 2.23 | 0.03 |
| Height (m)l | 0.86 ± 0.05 | 0.88 ± 0.04 | 0.56 |
| BMI (kg/m2)l | 15.05 ± 1.35 | 14.55 ± 0.84 | 0.21 |
| Fat mass (%)m | 18.32 ± 2.50 | 17.62 ± 2.14 | 0.34 |
| Lean mass (%)m | 81.28 ± 3.64 | 82.40 ± 2.15 | 0.27 |
NGT, normal glucose tolerance; GDM, gestational diabetes mellitus.
n = 63 (40 NGT and 23 GDM);
n = 47 (29 NGT and 18 GDM);
n = 61 (41 NGT and 20 GDM);
n = 64 (42 NGT and 22 GDM);
n = 60 (41 NGT and 19 GDM);
n = 61 (40 NGT and 21 GDM);
n = 61 (40 NGT and 21 GDM);
n = 58 (35 NGT and 23 GDM);
n = 46 (29 NGT and 17 GDM);
n = 62 (38 NGT and 24 GDM);
n = 52 (29 NGT and 23 GDM);
Statistical analysis are performed with z-scores; n = 60 (38 NGT and 22 GDM); Continuous variables are mean ± s.d. Significant P values (≤ 0.05) are indicated in bold and trends (P < 0.1) are in italic.
Newborns' gestational age was similar and within normal range (37 to 42 weeks) between both groups. Birth weights and cord blood glucose and TG levels were also similar between groups. Cord blood C-peptide levels were higher in newborns exposed to GDM.
Correlation between maternal metabolic profile and placental DNA methylation at LPL locus
Fig. 1 shows the 3 epigenotyped regions in placenta biopsies on the fetal side and children's whole blood collected at 5 years old. It also shows a correlation map between analyzed CpGs in placenta.
Figure 1.
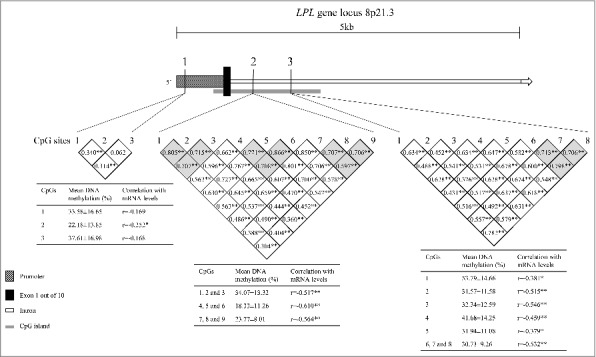
Schematic representation of the LPL gene and localization of the three epigenotyped regions. Region 1, containing 3 CpG sites, is located within the proximal promoter of the LPL gene. Regions 2 and 3, containing respectively 9 and 8 CpG sites, are located in the first intron of LPL gene, downstream the first exon. The grey square represents a CpG island. *p < 0.05; **p > 0.01.
We first tested associations between GDM exposure and placental DNAm levels. We found that LPL DNAm levels were lower in GDM-exposed placenta biopsies for CpG 1.1 (P = 0.004) and CpGs 2.1 to 2.3 (P = 0.05) (Table 2). The CpG 1.1 results remained statistically significant after Bonferroni correction (α = 0.05/12 tests; P ≤ 0.004). DNAm levels were 12% lower in GDM-exposed placenta as compared with non-exposed samples, at the CpG 1.1 locus. GDM exposure explained 7 to 14% of the DNA methylation variability at CpG 1.1 (P < 0.05). At 5 years of age, DNAm measured at CpG sites within R1 were no longer associated with GDM. All CpGs in R2 and R3 were hypomethylated (<10%) in children's blood cells.
Table 2.
Placental DNA methylation levels at the LPL loci in children exposed and not exposed to gestational diabetes mellitus during fetal development.
| CpG | NGT | GDMa | |||
|---|---|---|---|---|---|
| dinucleotides | n = 42 | n = 24 | P valueb | P valuec | P valued |
| CpG1.1 | 38.65 ± 17.71 | 25.67 ± 10.57 | 0.008 | 0.003 | 0.004 |
| CpG1.2 | 22.93 ± 13.41 | 21.51 ± 14.80 | 0.59 | 0.47 | 0.61 |
| CpG1.3 | 36.50 ± 15.99 | 39.41 ± 19.08 | 0.59 | 0.63 | 0.57 |
| Mean CpGs 2.1 to 2.3 | 36.87 ± 14.20 | 29.56 ± 10.65 | 0.05 | 0.04 | 0.04 |
| Mean CpGs 2.4 to 2.6 | 39.52 ± 11.06 | 36.38 ± 11.81 | 0.52 | 0.47 | 0.38 |
| Mean CpGs 2.7 to 2.9 | 23.85 ± 7.28 | 23.15 ± 9.11 | 0.98 | 0.95 | 0.97 |
| CpG 3.1 | 54.88 ± 13.86 | 52.22 ± 16.33 | 0.71 | 0.63 | 0.66 |
| CpG 3.2 | 32.01 ± 12.36 | 30.78 ± 10.60 | 0.82 | 0.69 | 0.59 |
| CpG 3.3 | 33.59 ± 14.08 | 30.46 ± 9.75 | 0.71 | 0.48 | 0.37 |
| CpG 3.4 | 44.09 ± 14.92 | 38.04 ± 12.52 | 0.11 | 0.06 | 0.06 |
| CpG 3.5 | 32.45 ± 11.71 | 30.69 ± 10.13 | 0.48 | 0.45 | 0.49 |
| Mean CpGs 3.6 to 3.8 | 31.99 ± 8.80 | 28.95 ± 9.91 | 0.32 | 0.31 | 0.14 |
NGT, normal glucose tolerance; GDM, gestational diabetes mellitus.
GDM diagnosed as 2 h post-75 g OGTT glucose ≥ 7.8 mmol/L.
Not adjusted P values;
P values adjusted for gestational age and sex;
P values adjusted for pre-pregnancy age and BMI, gestational age and sex of the children.
P values ≤ 0.05 are indicated in bold and P values <0.1 are in italic.
Associations between children's growth and maternal hyperglycemia
We further assessed whether GDM could impact the metabolic status of offspring. Interestingly, cord blood C-peptide levels were higher in GDM-exposed newborns (174.6 ± 83.0 ng/mL vs. 129.2 ± 81.9 ng/mL; P = 0.05) (Table 1), supporting an increase in insulin secretion. Moreover, cord blood leptin levels tended to be lower in the GDM-exposed offspring (6.79 ± 4.60 ng/mL vs. 10.75 ± 8.22 ng/mL; P = 0.08), suggesting that these newborns might have less adiposity, albeit the difference did not reach statistical significance. GDM-exposed children were also of lower weight (18.80 ± 2.32 kg vs. 20.39 ± 2.56 kg; P = 0.02) at age 5 (Table 1), and this result remained significant after adjustments for gestational age, sex of the children, maternal age, and pre-pregnancy BMI (Table 3), and also after consideration for maternal gestational weight gain, cord blood C-peptide levels, and birth weight (data not shown). Height, BMI, fat mass, and lean mass were not significantly different between the 2 groups. However, the weight and the BMI z-score of GDM-exposed children were significantly lower as compared with a zero z-score from the WHO growth chart reference for boys and girls (Fig. 2).
Table 3.
Growth in children exposed and not exposed to gestational diabetes mellitus during fetal development.
| NGT | GDMa | |||
|---|---|---|---|---|
| Children's body parameter | n = 42 | n = 24 | P valueb | P valuec |
| Birth weight z-scores | −0.01 ± 0.74 | −0.31 ± 1.00 | 0.17 | 0.10 |
| Weight z-scoresd | 0.05 ± 0.62 | −0.43 ± 0.87 | 0.03 | 0.03 |
| Height z-scores | 0.01 ± 0.03 | −0.01 ± 0.04 | 0.56 | 0.63 |
| BMI z-scorese | −0.17 ± 0.74 | −0.40 ± 0.76 | 0.21 | 0.15 |
| Fat mass (%)f | 17.08 ± 4.56 | 16.40 ± 4.61 | 0.24 | 0.24 |
| Lean mass (%)f | 82.53 ± 5.26 | 83.60 ± 4.55 | 0.22 | 0.25 |
NGT, normal glucose tolerance; GDM, gestational diabetes mellitus.
GDM diagnosed as 2 h post-75 g OGTT glucose ≥ 7.8 mmol/L.
P values adjusted for gestational age and sex (fat and lean masses).
P values adjusted for pre-gestational age and BMI and gestational age at birth and sex (fat and lean masses).
Weight and
BMI z-scores for GDM-exposed child were different from zero, according to a one-sample t-test.
n = 60 (38 NGT and 22 GDM).
P values ≤ 0.05 are indicated in bold and P values <0.1 are in italic.
Figure 2.
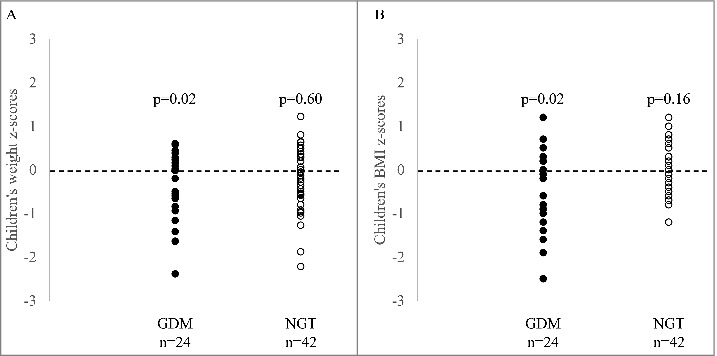
Comparison of anthropometric profiles in GDM and NGT offspring with WHO recommendations. A. Children's weight and; B. BMI z-scores were compared to WHO references using a one-sample T-test (with 0 as comparing group). Dotted line represent the WHO growth chart reference for boys and girls. The mean z-score is indicated by a bold grey line.
We also found that birth weight was correlated with children's weight and BMI, and these associations remained significant after adjustments for gestational age, age at mid-childhood measurements, and child sex (r = 0.364, P = 0.003 for weight; r = −0.287 and r = 0.251, P = 0.05 for BMI) (Fig. 3). Interestingly, placental weight was associated with both birth weight (r = 0.610, P < 0.001) and mid-childhood weight (r = 0.364, P = 0.004). Birth weight accounted for 15% of the variability of mid-childhood weight (P = 0.001), independently of the placental weight.
Figure 3.
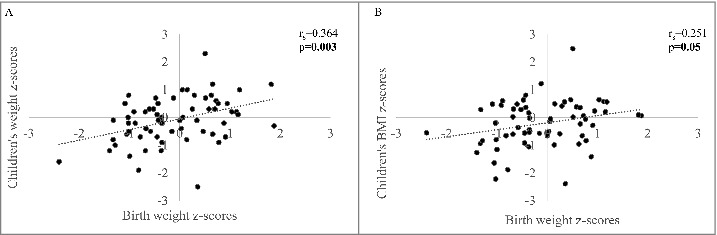
Associations between birth weight and body composition at age 5. Spearman correlation was evaluated between birth weight and children's A. weight and; B. BMI. p values are adjusted for children's age and sex. p values ≤0.05 are indicated in bold and p values <0.1 are in italic.
Associations between fetal placental DNA methylation levels at the LPL locus and children's anthropometric profile and body composition
We further investigated the relationship between placental DNAm levels at CpGs 1.1, 2.1–2.3, and 3.4 (all associated with GDM exposure at P < 0.1) and offspring anthropometric profile and body composition. First, we found that DNAm levels at CpG 3.4 were positively correlated with birth weight (r = 0.252, P = 0.04) (Fig. 4A). Placental DNAm levels at the same CpG were also correlated with mid-childhood weight (r = 0.314, P = 0.01) (Fig. 4B). The same CpG was also correlated with mid-childhood height (r = 0.299, P = 0.02) (Fig. 4C), lean mass (r = −0.306, P = 0.02) (Fig. 4D), fat mass (r = 0.275, P = 0.04) (Fig. 4E), and, possibly, BMI (r = 0.212, P = 0.09) (Fig. 4F). The correlation with mid-childhood weight remained significant after further adjustment for mid-childhood height (r = 0.328, P = 0.008). Interestingly, the correlation between placental DNAm levels and mid-childhood weight remained significant after adjustment for birth weight (r = 0.282, P = 0.02), albeit the associations with other anthropometric measures were lost. Correlations remained essentially unchanged after further adjustments for maternal 2 h post-OGTT, maternal GDM status, or for maternal fasting glucose at 3rd trimester (Table S1). Associations between DNAm levels and birth weight, as well as weight, height, and BMI at 5 years old seem to be limited to the GDM-exposed group, whereas those with body composition (fat mass and lean mass) were essentially similar in both GDM and NGT groups (Table S2).
Figure 4.
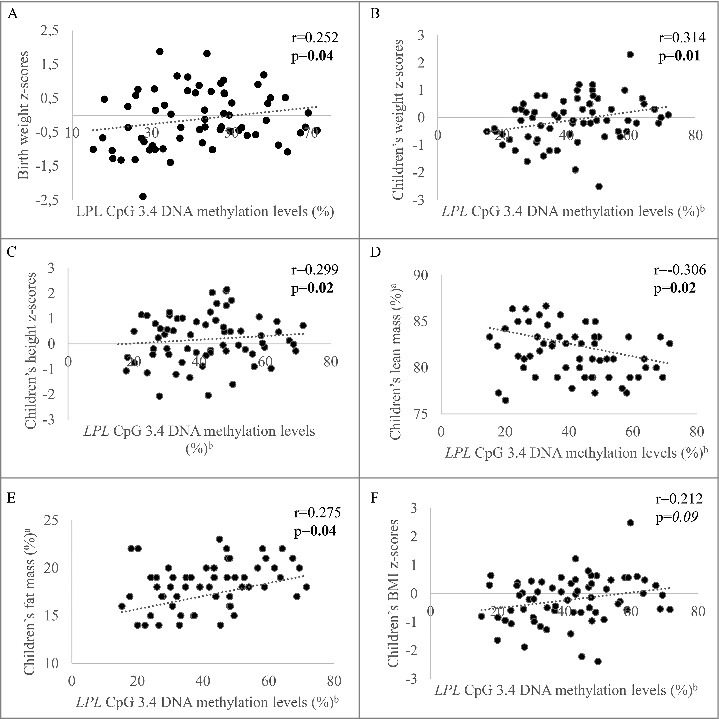
Associations between placental DNA methylation levels at LPL CpG dinucleotide 3.4 and children's body composition. Spearman correlation between DNA methylation levels and A. birth weight and children's; B. weight; C. BMI; D. height; E. lean mass and; F. fat mass. ap values adjusted for children's age and sex; bp values adjusted for gestational age and sex. p values ≤0.05 are indicated in bold and p values <0.1 are in italic.
Gene expression regulation and placental LPL DNAm levels
Potential impacts of reported epivariations on fetal placental LPL transcriptional activity were also tested. The results are reported in Fig. 1. We found a negative correlation for almost all CpG sites tested (Fig. 1), including at CpG 3.4 (r = −0.459, P < 0.001), suggesting that these epivariations could mediate LPL gene expression. Interestingly, the strongest correlations were observed in CpGs located within an intronic CpG island (R2 and 3), supporting that this locus might well be implicated in the regulation of LPL transcriptional activity. We also assessed the associations between placental LPL gene expression and mid-childhood anthropometric profile and body composition. Although the LPL mRNA levels were not significantly correlated with any children's variable, the direction of the association was as expected (Table S3).
Discussion
It is widely recognized that exposure to GDM is associated with an increased risk of adverse outcomes for mothers and offspring. Fetal exposure to untreated maternal GDM had been widely associated with large for gestational age (LGA) babies (birth weight >90th percentile for gestational age and sex) and macrosomia (birth weight >4 kg) through increased fetal growth.17,18 Although a better control of GDM in the 2nd and 3rd trimesters of pregnancy is known to decrease the risk of LGA and macrosomia,19 the long-term beneficial impacts of GDM treatment remains controversial.20-22 Mothers from our cohort were carefully followed up, and those with GDM were treated in accordance with established clinical practices. Accordingly, GDM mothers gained significantly less weight in the last trimester of pregnancy as compared with NGT mothers, suggesting that they were compliant to treatment. This might explain why we observed that GDM-exposed babies had a weight similar to that of non-exposed newborns. Nevertheless, we replicated results9,13,14 supporting that placental LPL gene DNAm is decreased in newborns exposed to GDM. Placental DNAm is known to show high inter-individual variability, making the analysis challenging.23 Although we showed that GDM exposure explains up to 16% of DNAm variability, the remaining percentage of DNAm variability might also be explained by several pre- and post-natal factors, as well as genetic factors.
We also demonstrated, for the first time, that GDM-associated placental LPL epigenetic variations are associated with birth weight, as well as anthropometric profile and body composition measured at 5 years old. Interestingly, children exposed to GDM had, on average, a lower body weight and BMI at age 5 than non-exposed children when compared with the WHO growth chart reference for boys and girls. These results suggest that GDM exposure, even with optimal follow-up and treatment, may impact fetal and childhood growth; whether this association is mediated by epigenetic mechanisms affecting the expression of the LPL gene still needs to be investigated.
Indeed, LPL plays a central role in lipid metabolism and transport. In peripheral vessels, it catalyzes the hydrolysis of TGs from circulating lipoproteins, including chylomicrons and very low-density lipoproteins (VLDL).24,25 This releases free fatty acids (FFAs) in circulation.24 The fetus is unable to synthetize enough FFAs, and thus relies on trans-placental transfer.26 This phenomena highlights the key role of LPL located in the microvillus membrane (MVM) of the placenta,27 where it initiates the transfer of TG-derived FFAs across the placenta to support fetal growth and development.13 In our study, we showed that lower DNAm levels at LPL CpG 3.4 (correlated with a higher LPL gene expression in placenta) were associated with lower birth weight and body composition at 5 years of age. GDM is characterized by maternal hyperglycemia (although well controlled after diagnosis late in 2nd trimester of pregnancy) and LPL is central to the trans-placental transport of FFAs to support fetal growth and fat mass accretion. Thus, a higher birth weight for GDM-exposed offspring would have been expected. However, similar associations (LPL mRNA levels were inversely correlated with birth weight) were reported in intrauterine growth restriction (IUGR) placentas,28,29 although these results are controversial.30,31 These studies suggested that LPL gene expression is regulated by the in utero environment, possibly in a IUGR severity-dependent manner.28 Nevertheless, the apparent discrepancy between the accepted role of LPL and the observed associations with lower weight and related phenotypes is not obvious to understand. We can speculate that GDM mothers were somehow “over-treated” given that their offspring were of slightly smaller weight at birth (in comparison with WHO growth reference charts), which would have impacted the placental LPL DNAm marks. However, the regulation of LPL activity is complex with transcriptional, posttranscriptional, and posttranslational mechanisms being involved.32 Indeed, it has been shown that increased LPL mRNA levels in IUGR placenta samples were not associated with an increased protein expression,31 although they were associated with a reduced LPL activity.33 Together, it might be the first evidence of a possible feedback regulation of LPL transcription and activity. Given the paucity of studies, these results highlight the need to confirm the transcriptional and functional impacts of LPL epivariations on protein levels and activity in placenta and their roles in growth and development.
Interestingly, the CpG at which DNAm levels are associated with GDM exposure, as we previously reported (cg16420199),9 and CpG 3.4 (located in close proximity to cg16420199), one of the GDM-associated CpG identified in this study, are both located within the proximal promoter region (within or very close exon 1) in a CpG island shore. Barrett et al. previously assessed whether placental DNAm levels at the LPL gene promoter locus were associated with the IUGR condition.31 Indeed, they investigated whether LPL gene expression could be modulated, at least partially, by IUGR-induced DNAm alterations. Although they found that the association between DNAm levels and mRNA was not statistically significant,31 the possible involvement of epigenetic mechanisms in LPL transcriptional activity regulation cannot be ruled out, since we reported correlations between LPL DNAm and mRNA levels in the current study. Moreover, the authors focused on CpG sites in the promoter area (not covered by the LPL CpG island) rather than those located within the LPL CpG island. DNAm within34 or flanking35 CpG islands are well known to regulate gene expression. Thus, more work is needed to characterize the transcriptional impact of the DNAm surrounding the LPL CpG island and its possible role in phenotypic variation. Also, there is evidence showing that alterations in DNAm in inter- and intra-genic regions, along with enhancers, are crucial to development, differentiation, and cell viability; such results need to be validated in LPL.35 However, ENCODE data reports transcription factors binding sites and enhancers characteristics including active histone marks (H3K4me1) near the epigenotyped regions. We will also need to detail how DNAm-induced perturbations in both the LPL promoter and CpG island impact growth, development, and disease risk.
This study has several strengths, including the prospective design and methodology used. First, we prospectively followed-up GDM and NGT mothers all through pregnancy, and we carefully phenotyped (anthropometric and metabolic data) newborns and children at 5 years of age, allowing us to better define the influence of placental maternally induced DNAm alterations on the anthropometric profile and body composition of offspring. However, these results will have to be replicated in a larger cohort. We also measured DNAm levels by bisulfite-pyrosequencing technology, which is highly reproducible and accurate.36,37 Nevertheless, this study has some shortcomings. One is the lack of genetics assessment. Although there are some genetic variants in the promoter of the LPL gene, none of them have been associated with GDM or childhood obesity. Moreover, the CpG sites in which we found significant associations with DNAm levels (GDM-induced or associated with children's variables) were fully unmethylated in blood collected in mid-childhood. Therefore, it was not possible to demonstrate that these epivariations are stable overtime or that they are associated with anthropometric measures and body composition at age 5. Since DNAm is tissue-specific,38-40 it would have probably been better to epigenotype tissues where LPL is highly active in lipid hydrolysis (adipose tissue, for instance). Accordingly, we have already demonstrated that LPL DNAm levels in adult blood samples are associated with gene expression and lipid profile.41 Understandably, adipose tissue is hardly accessible in healthy children's cohorts for obvious ethical reasons. In addition to the long-term stability of DNAm alterations, altered embryonic development and organogenesis in response to placental epigenetic changes might have occurred42 as well and could potentially explain the associations we have observed. Indeed, our results support that fetal development might have been perturbed by the exposure to maternal hyperglycemia, considering that we reported that GDM-exposed children were of lower birth weight compared with the WHO growth chart reference for boys and girls, and that birth weight reflects fetal growth. In addition, we already reported associations between DNAm levels at the LPL gene locus and lipid deregulation in adulthood.41 This metabolic dysfunction could be induced by a prenatal exposure to intrauterine insults during critical periods of epigenetic programming, which could increase the risk for cardiovascular complications later in life.21,22,43 Although our study added evidence supporting the implication of epigenetic modifications in the fetal metabolic programming of weight at birth and in mid-childhood, the impact on later life health and diseases, such as lipid metabolism impairments (according to the DOHaD hypothesis), needs to be studied. Nonetheless, it has been shown that metabolic dysfunctions later in life are predicted by birth weight and growth in childhood, for which placental LPL DNAm deregulation might be one of the underlying mechanisms.
Conclusion
In summary, we showed that epigenetic perturbations within the intergenic region of placental LPL gene were associated with birth weight, as well as with anthropometric profile and body composition at 5 years of age. Our results suggest that GDM-induced placental LPL epivariations impair fetal growth and fat accretion in childhood, which might result in metabolic dysfunctions later in life. Accordingly, these results provided supporting evidence of fetal metabolic programming of childhood obesity through epigenetic variations, underlining the harmful consequences of some in utero exposures.
Materials and methods
ECOGENE-21 (E-21) prospective Birth Cohort
For this study, we recruited 66 children from 5 to 6 years old between 2011 and 2016. These children were born from Caucasian women (initially recruited n = 241) from the Saguenay city area, Quebec, Canada. Women were all recruited at their 1st trimester of pregnancy from 2006 to 2010. Women were excluded if meeting any of these criteria: aged under 18 years old or over 40 years old, positive history of drug or alcohol consumption while pregnant, pre-gestational type 1 or 2 diabetes, polycystic ovary syndrome, hypercholesterolemia, morbid obesity [body mass index (BMI) >40], previous premature delivery (gestational age <37 weeks). This project, conducted according to the Declaration of Helsinki, was approved by the Chicoutimi Hospital Ethics Committee. All mothers gave their written consent before their inclusion in the study.
Maternal clinical data
Women were followed at each trimester of pregnancy (weeks 10–14, 24–28, and 36–39). Anthropometric parameters were measured by a research nurse at each visit, following our standardized procedures. 44 Blood samples were collected in the fasting state (12 hours). Glucose and insulin concentrations were measured on fresh serum samples at the Chicoutimi Hospital Clinical Laboratory. All glucose concentrations were measured on the Beckman analyzer (model Cx7; Beckman). Maternal insulin levels were assessed using a radioimmunoassay method (Advia Centaur, Simmens). The homeostatic model assessment for insulin resistance (HOMA-IR) was calculated as follows: (fasting glucose x insulin levels)/22.5.45 GDM was diagnosed for 24 out of the 66 mothers, according to WHO criteria (2 h post-75 g OGTT glucose ≥ 7.8 mmol/L) between 24–28 weeks of gestation.16
Total cholesterol (TC) and TG were measured with an enzymatic method (Beckman analyzer model Cx7, Beckman, Fullerton, CA). High-density lipoprotein-cholesterol (HDL) were isolated and measured from plasmatic very low-density lipoproteins (VLDL) and low-density lipoproteins (LDL) by precipitation in heparin and MnCl2. Furthermore, LDL levels were approximated using the Friedewald formula.46
Newborns' clinical data and samples
Cord blood samples and placental biopsies were collected at delivery. Cord blood glucose levels were measured as described for maternal samples. Cord C-peptide levels were assessed by ELISA and reflect insulin secretion in newborns. Newborns' clinical data were collected from medical files, including gestational age, sex, and anthropometric measurements. Gestational age was evaluated at the first visit and calculated from the last menstrual period; it was further corrected with data from ultrasound scans if necessary, as recommended.
Placenta biopsies were performed by well-trained medical staff within a few minutes after delivery (<15 min, recorded) and placenta expulsion. Two biopsies, 0.5 cm3 each, were sampled from the fetal side of the placenta within 2 cm of the insertion point of the umbilical cord. They consisted of chorionic plate with the chorionic villi. Samples were washed in PBS 1X to remove cord and maternal blood, then dissected to remove conjunctive tissue and kept in RNALater (Qiagen, Cat No. 76106) at −80°C until DNA and RNA extractions.
DNA and RNA were extracted from placenta biopsies using the DNA/RNA/Protein Mini Kit (Qiagen, Cat No. 80004). The RNA integrity number (RIN) was assessed with Agilent 6000 RNA Nano Chips (Agilent Technologies, Cat No. 5067–1511) and further analyzed when RIN >7.00. DNA purity was assessed using the ratio of absorbance at 260 nm to 280 nm with the Ultrospec 2000 UV/Visible Spectrophotometer (Pharmacia Biotech).
Children clinical data and samples
Children were followed-up at 5 years of age. Anthropometric measures were assessed by a research nurse according to our standardized procedures.44 Height was measured twice within the nearest 0.1 cm using a stadiometer (Standing Height, Holtain Ltd.). Body composition was estimated by bioelectrical impedance analysis (BIA) using the Tanita® SC-331s Total Body Composition Analyzer, which has been validated for children of 5 years old or more.47 Briefly, BIA measures the electrical impedance, which is the resistance of body tissues to an electric flow. This allows estimating the body weight as well as fat and lean masses (in kg and percentage). After verification in research records, 6 fat mass values lower than the 2nd percentile, as recommended by McCarthy and colleagues, were excluded from analyses.48
Whole blood samples were also collected from participating children and buffy coats conserved at −80°C. Leucocyte DNA was extracted manually from samples using the Gentra Puregene Blood kit (Qiagen, Cat No. 158389). DNA purity was assessed using the ratio of absorbance at 260 nm to 280 nm with Ultrospec 2000 UV/Visible Spectrophotometer (Pharmacia Biotech).
DNA methylation analysis in placenta and whole blood samples
Based on our previous results,8,9 we selected 3 loci in the proximal promoter of the LPL gene. DNA was beforehand treated by sodium-bisulfite (EpiTech Bisulfite Kit, Qiagen, Cat No. 59104), amplified by PCR (Pyromark PCR Kit, Qiagen, Cat No. 978705) and then pyrosequenced (Pyromark Gold Q24 Reagents, Qiagen, Cat No. 970802). Each PCR primer was first designed using the Pyromark Assay Design software (v2.0.1.15) (Qiagen). A total of 20 CpG dinucleotides consisting in 3, 9 and 8 CpGs were analyzed in the LPL proximal promoter first (R1), second (R2), and third (R3) region. The primer sequences are available in elsewhere.10
DNAm levels at the LPL locus were assessed at 20 CpG dinucleotides, distributed into 3 epigenotyped regions (Fig. 1). The 3 CpGs within R1 (1.1, 1.2, and 1.3) were not correlated with each other and were thus analyzed separately. Among R2, CpG 1 to 3 (i.e., 2.1, 2.2, and 2.3) were correlated (r≥ 0.6, P < 0.05), as well as CpG 4 to 5 (2.4, 2.5, and 2.6) and 7 to 9 (2.7, 2.8, and 2.9). Hence, they were included in statistical analyses as the mean DNAm levels for each group of CpGs. In R3, CpGs 6 to 8 (3.6, 3.7, and 3.8) were analyzed together since they were significantly correlated.
LPL mRNA levels in placenta
Placental RNA was converted in cDNA using a random primer hexamer from the High Capacity cDNA Archive Kit (Applied Biosystems, Cat No. 4368813). Amplifications were performed in duplicate in 20 μL containing equal amounts of cDNA and 10 μL of TaqMan® Universial PCR Master Mix (Applied Biosystems, Cat No. 4304437). Expression levels of LPL (LPL: Hs00173425_m1; Applied Biosystems) were measured by qRT-PCR with TaqMan technology using 7500 Real Time PCR System with Tyrosine 3-Monooxygenase/Tryptophan 5-Monooxygenase Activation Protein, Zeta (YWHAZ) as a reference gene (YWHAZ: Hs00237047_m1; Applied Biosystems). This gene has been shown to be stable in placenta in several studies.49-51
Statistical analyses
Mothers, newborns, and children clinical data, along with DNAm levels were tested for normality using a Shapiro-Wilk test. Log-transformation did not correct non-normality distribution for most of the variables. The Mann-Whitney test was thus applied to compare offspring anthropometric characteristics and DNAm levels between exposed and non-exposed to GDM groups. The comparison of birth weight and children z-scores variables with WHO recommendations was investigated using a one-sample t-test with 0 as the reference value. Epivariations associated with the intrauterine exposure to GDM were further tested for association with children clinical data (weight, height, BMI, fat and lean masses percentages) using Spearman's correlations (because of non-normality). We first performed statistical models with non-adjusted data. In the first-level adjusted model, we corrected for gestational age, child sex, and age at the mid-childhood visit; in the second-level adjusted models, we included maternal age and BMI at first prenatal visit. In some models, we further adjusted for maternal glycemia (represented as 2 h post-OGTT, or GDM status, or fasting glucose at 3rd trimester) and birth weight when relevant.52 To adjust for potential confounding factors, we computed unstandardized residuals. Residual scores of DNA methylation levels were obtained by using the unstandardized residuals computed by linear regressions that included gestational age and newborn's sex. Residual scores of children clinical data were obtained considering gestational age, newborn's sex, age of the children at follow-up, maternal pre-pregnancy BMI, maternal 2 h post-OGTT, maternal fasting glucose, HOMA-IR and HDL at 3rd trimester of pregnancy. Covariates were selected based on their association with mid-childhood outcomes (Table S3). The WHO age-sex z-score was used for children's weight, height and BMI instead of considering age and sex as confounding factors. We assessed associations between DNAm levels at significant LPL CpG sites and mRNA levels to investigate whether these DNAm marks were potentially functional (regulation of transcriptional activity) using Spearman's correlations. All analyses were performed on SPSS Statistics software v22 (IBM). Results were considered statistically significant when P < 0.05 (2-sided).
Declarations
Ethics approval and consent to participate
The Chicoutimi Hospital Ethics Committee approved the project. All women provided a written informed consent before their inclusion in the study, in accordance with the Declaration of Helsinki.
Consent for publication
Not applicable.
Availability of data and materials
Methylation data and analytical conditions can be provided upon request.
Disclosure of potential conflicts of interest
N.B. The authors have no conflict of interest or financial disclosure statements.
Supplementary Material
Funding Statement
This project was supported by ECOGENE-21, the Canadian Institutes of Health Research (CIHR team in community genetics (grant #CTP-82941)), Fonds de Recherche du Québec en Santé (FRQS), and Diabète Québec.
Acknowledgments
The authors acknowledge the contribution of Sébastien Claveau, MSc, ECOGENE-21 Laboratory; Stephanie-May Ruchat, Université de Sherbrooke for their dedicated work in this study. LB is a junior research scholar from the Fonds de la recherche du Québec en santé (FRQS). JPB is a senior research scholar from the FRQS. MFH was supported by a FRSQ junior scholar award and a Canadian Diabetes Association clinical scientist award; MFH is supported by an American Diabetes Association (ADA) Pathways Program Accelerator Early Investigator Award (1–15-ACE-26). LB, MFH, PP, and JPB are members of the FRQS-funded Center de recherche du CHUS (affiliated to the Center hospitalier universitaire de Sherbrooke). VGO received a research scholarship from the Faculté de médecine et des sciences de la santé de l'Université de Sherbrooke and a summer studenship award from Diabète Québec.
Author contributions
VGO performed the data collection and statistical analyses, and wrote the manuscript. AAH and SPG also contributed to the data collection and revised the manuscript. PP, JPB, DG, RG, DB and MFH participated in the conception of the study design and revised the manuscript. LB supervised all steps of the study and participated in manuscript writing and revision. Each author approved the manuscript.
References
- 1.Barker DJ. The origins of the developmental origins theory. J Intern Med 2007; 261:412–7; PMID:17444880; https://doi.org/10.1111/j.1365-2796.2007.01809.x [DOI] [PubMed] [Google Scholar]
- 2.Holder T, Giannini C, Santoro N, Pierpont B, Shaw M, Duran E, Caprio S, Weiss R. A low disposition index in adolescent offspring of mothers with gestational diabetes: a risk marker for the development of impaired glucose tolerance in youth. Diabetologia 2014; 57:2413–20; PMID:25168408; https://doi.org/10.1007/s00125-014-3345-2 [DOI] [PubMed] [Google Scholar]
- 3.Bird AP, Wolffe AP. Methylation-induced repression–belts, braces, and chromatin. Cell 1999; 99:451–4; PMID:10589672; https://doi.org/10.1016/S0092-8674(00)81532-9 [DOI] [PubMed] [Google Scholar]
- 4.Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, Slagboom PE, Heijmans BT. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet 2009; 18:4046–53; PMID:19656776; https://doi.org/10.1093/hmg/ddp353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heijmans BT, Tobi EW, Lumey LH, Slagboom PE. The epigenome: archive of the prenatal environment. Epigenetics 2009; 4:526–31; PMID:19923908; https://doi.org/10.4161/epi.4.8.10265 [DOI] [PubMed] [Google Scholar]
- 6.Wong CC, Caspi A, Williams B, Craig IW, Houts R, Ambler A, Moffitt TE, Mill J. A longitudinal study of epigenetic variation in twins. Epigenetics 2010; 5:516–26; PMID:20505345; https://doi.org/10.4161/epi.5.6.12226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hales CN, Barker DJ. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia 1992; 35:595–601; PMID:1644236; https://doi.org/10.1007/BF00400248 [DOI] [PubMed] [Google Scholar]
- 8.Ruchat SM, Houde AA, Voisin G, St-Pierre J, Perron P, Baillargeon JP, Gaudet D, Hivert MF, Brisson D, Bouchard L. Gestational diabetes mellitus epigenetically affects genes predominantly involved in metabolic diseases. Epigenetics 2013; 8:935–43; PMID:23975224; https://doi.org/10.4161/epi.25578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Houde AA, St-Pierre J, Hivert MF, Baillargeon JP, Perron P, Gaudet D, Brisson D, Bouchard L. Placental lipoprotein lipase DNA methylation levels are associated with gestational diabetes mellitus and maternal and cord blood lipid profiles. J Dev Orig Health Dis 2014; 5:132–41; PMID:24847699; https://doi.org/10.1017/S2040174414000038 [DOI] [PubMed] [Google Scholar]
- 10.Clausen TD, Mathiesen ER, Hansen T, Pedersen O, Jensen DM, Lauenborg J, Schmidt L, Damm P. Overweight and the metabolic syndrome in adult offspring of women with diet-treated gestational diabetes mellitus or type 1 diabetes. J Clin Endocrinol Metab 2009; 94:2464–70; PMID:19417040; https://doi.org/10.1210/jc.2009-0305 [DOI] [PubMed] [Google Scholar]
- 11.Hillier TA, Pedula KL, Schmidt MM, Mullen JA, Charles MA, Pettitt DJ. Childhood obesity and metabolic imprinting: the ongoing effects of maternal hyperglycemia. Diabetes Care 2007; 30:2287–92; PMID:17519427; https://doi.org/10.2337/dc06-2361 [DOI] [PubMed] [Google Scholar]
- 12.Deierlein AL, Siega-Riz AM, Chantala K, Herring AH. The association between maternal glucose concentration and child BMI at age 3 years. Diabetes Care 2011; 34:480–4; PMID:21216858; https://doi.org/10.2337/dc10-1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Magnusson-Olsson AL, Hamark B, Ericsson A, Wennergren M, Jansson T, Powell TL. Gestational and hormonal regulation of human placental lipoprotein lipase. J Lipid Res 2006; 47:2551–61; PMID:16926441; https://doi.org/10.1194/jlr.M600098-JLR200 [DOI] [PubMed] [Google Scholar]
- 14.Radaelli T, Lepercq J, Varastehpour A, Basu S, Catalano PM, Hauguel-De Mouzon S. Differential regulation of genes for fetoplacental lipid pathways in pregnancy with gestational and type 1 diabetes mellitus. Am J Obstet Gynecol 2009; 201:209 e1–10; PMID:19560108; https://doi.org/10.1016/j.ajog.2009.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dube E, Gravel A, Martin C, Desparois G, Moussa I, Ethier-Chiasson M, Forest JC, Giguere Y, Masse A, Lafond J. Modulation of fatty acid transport and metabolism by maternal obesity in the human full-term placenta. Biol Reprod 2012; 87:14:1–1; PMID:22553224; https://doi.org/10.1095/biolreprod.111.098095 [DOI] [PubMed] [Google Scholar]
- 16.Vandorsten JP, Dodson WC, Espeland MA, Grobman WA, Guise JM, Mercer BM, Minkoff HL, Poindexter B, Prosser LA, Sawaya GF, et al.. NIH consensus development conference: diagnosing gestational diabetes mellitus. NIH Consens State Sci Statements 2013; 29:1–31; PMID:23748438 [PubMed] [Google Scholar]
- 17.Stotland NE, Caughey AB, Breed EM, Escobar GJ. Risk factors and obstetric complications associated with macrosomia. Int J Gynaecol Obstet 2004; 87:220–6; PMID:15548393; https://doi.org/10.1016/j.ijgo.2004.08.010 [DOI] [PubMed] [Google Scholar]
- 18.Ostlund I, Hanson U, Bjorklund A, Hjertberg R, Eva N, Nordlander E, Swahn ML, Wager J. Maternal and fetal outcomes if gestational impaired glucose tolerance is not treated. Diabetes Care 2003; 26:2107–11; PMID:12832321; https://doi.org/10.2337/diacare.26.7.2107 [DOI] [PubMed] [Google Scholar]
- 19.Rice MM, Landon MB, Eunice Kennedy Shriver National Institute of Child H, Human Development Maternal-Fetal Medicine Units N . What we have learned about treating mild gestational diabetes mellitus. Semin Perinatol 2016; 40(5):298–302; PMID:27126295; https://doi.org/10.1053/j.semperi.2016.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gillman MW, Oakey H, Baghurst PA, Volkmer RE, Robinson JS, Crowther CA. Effect of treatment of gestational diabetes mellitus on obesity in the next generation. Diabetes Care 2010; 33:964–8; PMID:20150300; https://doi.org/10.2337/dc09-1810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malcolm JC, Lawson ML, Gaboury I, Lough G, Keely E. Glucose tolerance of offspring of mother with gestational diabetes mellitus in a low-risk population. Diabet Med 2006; 23:565–70; PMID:16681566; https://doi.org/10.1111/j.1464-5491.2006.01840.x [DOI] [PubMed] [Google Scholar]
- 22.Landon MB, Rice MM, Varner MW, Casey BM, Reddy UM, Wapner RJ, Rouse DJ, Biggio JR Jr, Thorp JM, Chien EK, et al.. Mild gestational diabetes mellitus and long-term child health. Diabetes Care 2015; 38:445–52; PMID:25414152; https://doi.org/10.2337/dc14-2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Houseman EA, Christensen BC, Yeh RF, Marsit CJ, Karagas MR, Wrensch M, Nelson HH, Wiemels J, Zheng S, Wiencke JK, et al.. Model-based clustering of DNA methylation array data: a recursive-partitioning algorithm for high-dimensional data arising as a mixture of beta distributions. BMC Bioinformatics 2008; 9:365; PMID:18782434; https://doi.org/10.1186/1471-2105-9-365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cryer A. Tissue lipoprotein lipase activity and its action in lipoprotein metabolism. Int J Biochem 1981; 13:525–41; PMID:7016622; https://doi.org/10.1016/0020-711X(81)90177-4 [DOI] [PubMed] [Google Scholar]
- 25.Wang CS, Hartsuck J, McConathy WJ. Structure and functional properties of lipoprotein lipase. Biochim Biophys Acta 1992; 1123:1–17; PMID:1730040; https://doi.org/10.1016/0005-2760(92)90165-R [DOI] [PubMed] [Google Scholar]
- 26.Baumann MU, Deborde S, Illsley NP. Placental glucose transfer and fetal growth. Endocrine 2002; 19:13–22; PMID:12583599; https://doi.org/10.1385/ENDO:19:1:13 [DOI] [PubMed] [Google Scholar]
- 27.Thomas CR, Lowy C. The interrelationships between circulating maternal esterified and non-esterified fatty acids in pregnant guinea pigs and their relative contributions to the fetal circulation. J Dev Physiol 1987; 9:203–14; PMID:3611637 [PubMed] [Google Scholar]
- 28.Tabano S, Alvino G, Antonazzo P, Grati FR, Miozzo M, Cetin I. Placental LPL gene expression is increased in severe intrauterine growth-restricted pregnancies. Pediatr Res 2006; 59:250–3; PMID:16439587; https://doi.org/10.1203/01.pdr.0000199441.62045.a1 [DOI] [PubMed] [Google Scholar]
- 29.Gauster M, Hiden U, Blaschitz A, Frank S, Lang U, Alvino G, Cetin I, Desoye G, Wadsack C. Dysregulation of placental endothelial lipase and lipoprotein lipase in intrauterine growth-restricted pregnancies. J Clin Endocrinol Metab 2007; 92:2256–63; PMID:17356047; https://doi.org/10.1210/jc.2006-2403 [DOI] [PubMed] [Google Scholar]
- 30.Laivuori H, Gallaher MJ, Collura L, Crombleholme WR, Markovic N, Rajakumar A, Hubel CA, Roberts JM, Powers RW. Relationships between maternal plasma leptin, placental leptin mRNA and protein in normal pregnancy, pre-eclampsia and intrauterine growth restriction without pre-eclampsia. Mol Hum Reprod 2006; 12:551–6; PMID:16870954; https://doi.org/10.1093/molehr/gal064 [DOI] [PubMed] [Google Scholar]
- 31.Barrett HL, Kubala MH, Scholz Romero K, Denny KJ, Woodruff TM, McIntyre HD, Callaway LK, Dekker Nitert M. Placental lipase expression in pregnancies complicated by preeclampsia: a case-control study. Reprod Biol Endocrinol 2015; 13:100; PMID:26336959; https://doi.org/10.1186/s12958-015-0098-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang H, Eckel RH. Lipoprotein lipase: from gene to obesity. Am J Physiol Endocrinol Metab 2009; 297:E271–88; PMID:19318514; https://doi.org/10.1152/ajpendo.90920.2008 [DOI] [PubMed] [Google Scholar]
- 33.Magnusson AL, Waterman IJ, Wennergren M, Jansson T, Powell TL. Triglyceride hydrolase activities and expression of fatty acid binding proteins in the human placenta in pregnancies complicated by intrauterine growth restriction and diabetes. J Clin Endocrinol Metab 2004; 89:4607–14; PMID:15356070; https://doi.org/10.1210/jc.2003-032234 [DOI] [PubMed] [Google Scholar]
- 34.Kulis M, Queiros AC, Beekman R, Martin-Subero JI. Intragenic DNA methylation in transcriptional regulation, normal differentiation and cancer. Biochim Biophys Acta 2013; 1829:1161–74; PMID:23938249; https://doi.org/10.1016/j.bbagrm.2013.08.001 [DOI] [PubMed] [Google Scholar]
- 35.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 2012; 13:484–92; PMID:22641018; https://doi.org/10.1038/nrg3230 [DOI] [PubMed] [Google Scholar]
- 36.Ge Y, Schimel JP, Holden PA. Analysis of run-to-run variation of bar-coded pyrosequencing for evaluating bacterial community shifts and individual taxa dynamics. PloS one 2014; 9:e99414; PMID:24911191; https://doi.org/10.1371/journal.pone.0099414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tost J, Gut IG. DNA methylation analysis by pyrosequencing. Nat Protocols 2007; 2:2265–75; PMID:17853883; https://doi.org/10.1038/nprot.2007.314 [DOI] [PubMed] [Google Scholar]
- 38.Houde AA, Legare C, Hould FS, Lebel S, Marceau P, Tchernof A, Vohl MC, Hivert MF, Bouchard L. Cross-tissue comparisons of leptin and adiponectin: DNA methylation profiles. Adipocyte 2014; 3:132–40; PMID:24719787; https://doi.org/10.4161/adip.28308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang X, Shao X, Gao L, Zhang S. Comparative DNA methylation analysis to decipher common and cell type-specific patterns among multiple cell types. Brief Funct Genomics 2016; 15(6):399–407; PMID:27107288; https://doi.org/10.1093/bfgp/elw013 [DOI] [PubMed] [Google Scholar]
- 40.Liu H, Liu X, Zhang S, Lv J, Li S, Shang S, Jia S, Wei Y, Wang F, Su J, et al.. Systematic identification and annotation of human methylation marks based on bisulfite sequencing methylomes reveals distinct roles of cell type-specific hypomethylation in the regulation of cell identity genes. Nucleic Acids Res 2016; 44:75–94; PMID:26635396; https://doi.org/10.1093/nar/gkv1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guay SP, Brisson D, Lamarche B, Marceau P, Vohl MC, Gaudet D, Bouchard L. DNA methylation variations at CETP and LPL gene promoter loci: new molecular biomarkers associated with blood lipid profile variability. Atherosclerosis 2013; 228:413–20; PMID:23623643; https://doi.org/10.1016/j.atherosclerosis.2013.03.033 [DOI] [PubMed] [Google Scholar]
- 42.Gabory A, Attig L, Junien C. Developmental programming and epigenetics. Am J Clin Nutr 2011; 94:1943S–52S; PMID:22049164; https://doi.org/10.3945/ajcn.110.000927 [DOI] [PubMed] [Google Scholar]
- 43.Silverman BL, Metzger BE, Cho NH, Loeb CA. Impaired glucose tolerance in adolescent offspring of diabetic mothers. Relationship to fetal hyperinsulinism. Diabetes Care 1995; 18:611–7; PMID:8585997; https://doi.org/10.2337/diacare.18.5.611 [DOI] [PubMed] [Google Scholar]
- 44.Lohman T. Anthropometric Standardization Reference Manual. Champaign, IL: 1988; Human Kinetics Books [Google Scholar]
- 45.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985; 28:412–9; PMID:3899825; https://doi.org/10.1007/BF00280883 [DOI] [PubMed] [Google Scholar]
- 46.Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem 1972; 18:499–502; PMID:4337382 [PubMed] [Google Scholar]
- 47.Barreira TV, Staiano AE, Katzmarzyk PT. Validity assessment of a portable bioimpedance scale to estimate body fat percentage in white and African-American children and adolescents. Pediatr Obes 2013; 8:e29–32; PMID:23239610; https://doi.org/10.1111/j.2047-6310.2012.00122.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCarthy HD, Cole TJ, Fry T, Jebb SA, Prentice AM. Body fat reference curves for children. Int J Obes (Lond) 2006; 30:598–602; PMID:16570089; https://doi.org/10.1038/sj.ijo.0803232 [DOI] [PubMed] [Google Scholar]
- 49.Meller M, Vadachkoria S, Luthy DA, Williams MA. Evaluation of housekeeping genes in placental comparative expression studies. Placenta 2005; 26:601–7; PMID:16085039; https://doi.org/10.1016/j.placenta.2004.09.009 [DOI] [PubMed] [Google Scholar]
- 50.Murthi P, Fitzpatrick E, Borg AJ, Donath S, Brennecke SP, Kalionis B. GAPDH, 18S rRNA and YWHAZ are suitable endogenous reference genes for relative gene expression studies in placental tissues from human idiopathic fetal growth restriction. Placenta 2008; 29:798–801; PMID:18684503; https://doi.org/10.1016/j.placenta.2008.06.007 [DOI] [PubMed] [Google Scholar]
- 51.Drewlo S, Levytska K, Kingdom J. Revisiting the housekeeping genes of human placental development and insufficiency syndromes. Placenta 2012; 33:952–4; PMID:23026742; https://doi.org/10.1016/j.placenta.2012.09.007 [DOI] [PubMed] [Google Scholar]
- 52.Fattah C, Farah N, Barry SC, O'Connor N, Stuart B, Turner MJ. Maternal weight and body composition in the first trimester of pregnancy. Acta Obstet Gynecol Scand 2010; 89:952–5; PMID:20380598; https://doi.org/10.3109/00016341003801706 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Methylation data and analytical conditions can be provided upon request.