

ABSTRACT
Understanding epigenetic processes holds immense promise for medical applications. Advances in Machine Learning (ML) are critical to realize this promise. Previous studies used epigenetic data sets associated with the germline transmission of epigenetic transgenerational inheritance of disease and novel ML approaches to predict genome-wide locations of critical epimutations. A combination of Active Learning (ACL) and Imbalanced Class Learning (ICL) was used to address past problems with ML to develop a more efficient feature selection process and address the imbalance problem in all genomic data sets. The power of this novel ML approach and our ability to predict epigenetic phenomena and associated disease is suggested. The current approach requires extensive computation of features over the genome. A promising new approach is to introduce Deep Learning (DL) for the generation and simultaneous computation of novel genomic features tuned to the classification task. This approach can be used with any genomic or biological data set applied to medicine. The application of molecular epigenetic data in advanced machine learning analysis to medicine is the focus of this review.
KEYWORDS: Active learning, deep learning, DNA methylation, epigenetics, epigenome, imbalanced-class learning, machine learning, molecular diagnostics
Abbreviations
- ML
Machine Learning
- ACL
Active Learning
- ICL
Imbalanced Class Learning
- DL
Deep Learning
- DMR
Differentially methylated DNA region
- AGQ+
Active Learner with Generalized Queries
- TAN
Tree-Augmented Naïve-Bayes
- MIP
Most Informative Positive
- CBAL
class Based Active Learning
- SSO
Subset Sample Optimization
- SNP
Single Nucleotide Polymorphism
- AIS
Artificial Immune Systems
- SVM
Support Vector Machines (Standard ML approach)
Introduction
Epigenetics is defined as “molecular factors around DNA that regulate genome activity independent of DNA sequence, and are mitotically stable”.1 In 1942, Conrad Waddington coined the term ‘epigenetics’ using studies of how environment influences development in conjunction with genotype, which leads to the development of the phenotype.1 Each cell type has a unique epigenome that allows a specific differentiation for the cell. Since a single genotype can be associated with many phenotypes, it is believed that for a single genome sequence infinite epigenomes may exist. One of the main epigenetic mechanisms is DNA methylation, which can influence gene expression without changing the DNA sequence. Additional epigenetic mechanisms include histone modifications, noncoding RNA (ncRNA), and chromatin structure.1 DNA methylation is one of the primary studied epigenetic mechanisms that has been shown to mediate generational inheritance through the male germ line.2 A number of studies show that epigenetic changes are essential for developmental processes (e.g., tissue formation, organ formation, sex determination). Epigenetic changes also lead to altered patterns of gene expression that can lead to adverse clinical outcomes, such as obesity, allergies, cancer, schizophrenia, or Alzheimer disease, to name a few. Recent epigenetic studies focus on how an environmental compound or exposure can promote an epigenetic disease state that can be transmitted through generations.1,3 Predicting regions of susceptibility to epigenetic changes that are associated with disease is crucial to understand epigenetics, biology, and disease.
A major goal of research in this area is to identify regions in the genome that are susceptible to epigenetic modification. This can include DNA methylation changes (e.g., CpG), histone modifications, ncRNA expression, or chromatin structural changes (e.g., nucleosome positioning). We have started to understand some of the underlying theory of epigenetics and the computational approaches necessary to identify regions that are associated for these changes. However, the extraction of biological data are time consuming and expensive due to the challenges of implementing experimental procedures that can produce epigenetic phenomena and several computational challenges to extract and analyze this data. Biological data sets have high dimensionality, but the cases of interest (e.g., disease states) are relatively rare. In epigenetic data sets, for example, DNA methylation data contain only a few differentially methylated DNA regions (DMRs) and many non-DMR sites, while both are described with numerous DNA sequence and genomic features. To address these challenges, an integrated approach that combines feature generation, feature selection, and machine learning on epigenetic data sets is needed. We envision 3 alternative approaches to this integration that involve combinations of Active Learning (ACL) to address the expense of generating epigenetic data, Imbalanced Class Learning (ICL) to address the relatively low occurrence of epimutations in the data, and Deep Learning (DL) to address the difficulty in manually defining relevant genomic features. Fig. 1 depicts these 3 alternative approaches: (i) ACL and ICL are used to learn efficiently from manually generated features; (ii) DL is used to automatically generate features for ACL/ICL and; (iii) a solely DL based approach incorporates these ML components into one paradigm that holds promise of applying recent dramatic successes in deep learning of sequential data to epigenetics. A list of the various types of Machine Learning (ML) approaches with their advantages and disadvantages are pointed out in Table 1.
Figure 1.

Machine Learning approaches to epigenetic data analysis: #1 ACL−ICL on manually generated features; #2 ACL-ICL on DL-generated features; #3 solely DL-based classification. Modified from.4
Table 1.
Machine learning approaches for biological data sets, along with their function, advantages, disadvantages, and recent examples.
| Machine Learning Approach | Function | Advantages | Disadvantages |
|---|---|---|---|
| Supervised Learning (e.g., support vector machine,84 random forest85) | Learn a model discriminating one class of biological phenomena from one or more other classes. | Precise model with predictive and interpretative properties. | Requires equally large number of examples from each class. |
| Unsupervised Learning (e.g., K-means,86 hierarchical clustering87) | Learn a model descriptive of the biological phenomena in the data. | Does not require class labels on data. | Sensitive to similarity measure; results difficult to interpret. |
| Semi-supervised Learning (e.g., transduction88) | Learn model from mixture of labeled and unlabeled data. | Utilize all available data; typically outperforms use just labeled data. | Sensitive to errors in propagating class labels from labeled to unlabeled data. |
| Feature Selection (e.g., PCA,89 LDA,90 wrapper91) | Reduce large number of features to fewer, more informative features. | Improves efficiency and accuracy of learning. | Sensitive to feature evaluation metric; may discard informative features. |
| Active Learning (e.g., uncertainty sampling,92 most informative instance93) | Identify most informative instances to label for accurate model learning. | Reduces number of examples needed to learn model; reduces burden on human expert and experiment cost. | May focus learner on outliers rather than prominent classes. |
| Imbalanced class Learning (e.g., minority over-sampling,94 boosting95) | Learn in the presence of large skew in the number of examples of each class. | Learn with relatively few examples of biological phenomenon of interest. | May underfit or overfit data depending on bias toward minority class. |
| Deep Learning (DeepBind,14 DeepMotif15) | Learns complex representations of concepts in the data. | General purpose and high accuracy. | Sensitive to parameter choices; long training times. |
The following sections first describe the main ML techniques recommended for prediction over epigenomic data: active learning, imbalanced class learning, and deep learning. Next, the application of these techniques to biological data sets, in general, and epigenetic data sets, in particular, are discussed along with the commensurate challenges. Recent results using active learning and imbalanced class learning for epigenetic feature selection and prediction are presented.4 Lastly, future applications of these ML techniques to molecular diagnostics and medicine are discussed.
Machine learning
Active learning
Biological and molecular data generally comes in raw form and needs to be annotated with class labels. This requires a domain expert and making the best use of the expert's knowledge and time. A new ML approach has arisen called Active Learning (ACL). ACL is designed to maximize the potential of the Oracle (the human expert) in labeling data by selecting only relevant instances and features. Instead of labeling all the instances, ACL methods can intelligently choose a small number of instances in a few iterations that quickly trains the learner while minimizing costs. ACL can produce better classifiers in less time and iterations.
In traditional ACL methods, it may not always be easy for the Oracle to label a query with many features, especially those with high precision values. Many of the queries may contain irrelevant features that have no effect on the final outcome (the label). A better approach is to remove some of the irrelevant features for a certain query such that it results in a shorter and more readable query, which is easier for the Oracle to label. Using such generalized queries will help achieve higher accuracy with fewer queries than traditional ACL methods. However, an overly general query may lead to an uncertain label, which may add noise to the learning process. Therefore, the goal of an ACL system should be to produce generalized queries with highly certain answers, from which it can learn a classifier quickly with fewer examples. Even when labeled data abounds, selecting a subset of the features on which to train the learner may result in a better classifier. This capability of ACL improves our ability to select feature subsets from both manually and automatically generated feature sets.
The most common and widely used form of ACL is uncertainty sampling,4 which chooses the most uncertain example as the next one for the Oracle to label. One problem with uncertainty sampling is that it may choose outliers, which are highly uncertain data points. Therefore, it does not always follow the underlying distribution of data points. The Active Learner with Generalized Queries (AGQ+)5 is an important ACL method that automatically generates meaningful new features, unlike previous approaches6 in which new features are manually adjusted. AGQ+ also constructs generalized queries with numeric attribute ranges that are automatically produced from raw numeric attribute data.
In recent work, we introduced an ACL method called GQAL, which is similar to AGQ+ but performs a local feature selection per query and achieves superior performance on classifying epigenetic data sets.7 GQAL uses pool-based uncertainty sampling for constructing a generalized query with don't-care features (irrelevant features in the most uncertain examples). GQAL uses the Tree-Augmented Naïve-Bayes (TAN) learner.8 TAN has been found to be superior to other learners in this setting,9 and it provides a probability of classification that is used by GQAL to find the instance whose classification probability is farthest from its distribution in the instance set (i.e., the most uncertain instance). GQAL generalizes the uncertain instance by identifying sets of features whose permuted values have no effect on the prediction for that instance. More detailed results of GQAL on epigenetic data are discussed later.
Imbalanced class learning
In many data sets, there are unequal numbers of instances in each class making an unequal distribution of samples. In this case, the classifier learns most of the target concepts of the majority class, but learns target concepts from the minority class poorly or not at all. Often, the interest is more on the minority class, such that getting rare instances from the minority class can be time consuming and costly. Such an unequal distribution between classes of a data set is known as the class imbalance problem. 10
In recent work, we introduced the TAN+AdaBoost ICL method that uses all the majority and minority class samples and uses boosting to ensure that each class is learned with equal priority. TAN+AdaBoost uses adaptive boosting (AdaBoost11), which learns a set, or ensemble, of classifiers using a base classifier (in this case TAN) repeatedly applied to the data set, but with incorrectly-classified examples receiving more weight to bias later classifiers toward correct classifications. While initial classifiers focus on the majority class, later classifiers focus on the minority class. The final classifier consists of the weighted majority vote of the individual classifiers. When applied to the 2 epigenetic data sets (sperm and somatic, described in Fig. 1), TAN+AdaBoost achieved the best overall performance compared with other imbalanced class learners using the combined average for AUC, F-measure, and G-mean, which are popular performance measures for ICL problems. 12
Deep learning
Deep Learning (DL) has recently demonstrated superior performance in several domains, most notably in image, speech, and natural language processing.4 Much of DL power resides in its ability to generate complex features while either learning to encode the input data in an unsupervised setting or learning to classify the input data in a supervised setting. The complex feature generation is accomplished using a multi-layer (deep) neural network with specialized nodes, e.g., convolutional input nodes to include neighboring information from around an input data point (e.g., motifs), and logistic or rectified linear units at the intermediate (hidden) and output layers, which reproduce (decode) or classify the input data. The weights on the interconnections between layers are trained using the standard backpropagation method. With typically 10 or more intermediate layers, where each layer's nodes compute a complex feature based on features from previous layers, the network generates complex features for representing the input data. It is this feature generation capability that is critical to an advanced DL-based approach to classification of genomic regions. As depicted by method #2 in Fig. 1, DL can be used to generate complex features from windows over the training DNA sequences. The sequence is then annotated with these features, and this newly annotated training data can be input to an ACL-ICL method to select the best features and data for learning. In addition to DL being used to generate new features, DL can also be used to minimize the number of relevant features; this would assist active learning. The generalized query based ACL technique becomes extremely computationally expensive when the number of features increases. As depicted by method #3 in Fig. 1, the DL network itself can be trained to identify genomic regions of interest, and then the learned network can be applied to the whole genome.
Machine learning in biological datasets
Machine Learning applied to biological data sets has a long history of success dating back to before 1990. A complete survey is beyond the scope of this article, but Table 1 summarizes the major ML techniques and specific approaches applied to biological data sets that are also applied to epigenetic data in particular, as described in the next section. Table 1 also describes the main advantages and disadvantages of each approach, as well as cites some recent examples from the literature. Supervised algorithms are used when there are labeled examples of 2 or more classes of interest (e.g., disease vs. healthy). Support vector machines and random forests of decision trees are among the most popular methods. Supervised algorithms have been used for the prediction of gene ontology and gene expression profiles across different environmental and experimental conditions. Unsupervised algorithms are used when the samples are not labeled. K-mean clustering and hierarchical clustering have been widely used in biological data sets. Chromatic data has been used with unsupervised learning algorithms for annotating the genomes to identify novel groups of functional elements. Semi-supervised algorithms fall between supervised and unsupervised, especially for cases when only a small portion of the samples is labeled. Semi-supervised algorithms have been used to identify functional relationships between genes and transcription factor binding sites. They are widely used for gene-finding approaches where the entire genome is the unlabeled set and only a collection of genes is annotated. Tentative labels are given after a first pass and the algorithm iterates to improve the learning model.13 Feature selection methods, such as principal component analysis, linear discriminant analysis, and wrapper methods, seek to reduce the dimensionality of data sets, identify informative features, and remove irrelevant features, to avoid overfitting the learned model.
Both ACL and ICL methods have applications in biological data sets. Retrieving good biological data can take months to years. Often, when experiments are done, researchers seek specific cases having a low incidence rate. So most biological data are naturally imbalanced. For example, among the 27,000 mouse genes, an experiment may observe only about 100 whose DNA methylation was changed within the experimental settings.14 Therefore, collecting data on such changes is a time consuming, multi-step process, and, naturally, results in a class imbalance problem.
Building a classifier based on such few instances requires the learner to choose instances and features that are most informative. By choosing few instances and features, if the learner can learn the target concept quickly, then a good classifier can be found without running more extensive experiments to obtain more rare instances. To address this issue, popular ML techniques, such as oversampling or undersampling, are used. However, these approaches have their own drawbacks. Oversampling the minority class leads to overfitting, whereas undersampling the majority class leads to underfitting. Instead, ML techniques like ACL certainly can help here. Therefore, both ACL and ICL methods have applications in biological data sets. Both types of methods have been widely used in other domains but, in biological data sets, only a few studies show the use of ACL, and even fewer studies show the use of the ICL methods in practice.
One ACL study15 used the Most Informative Positive (MIP) ACL method to find p53 mutants (mutated p53 is responsible for half of human cancers). In their ACL method, they train their classifier by only using positive instances that pass a certain score (which ranks all unlabeled instances) and include negative instances in the training set only if there are too few positive instances. Their approach looked for functionally active examples and, in their first in vivo experiment, the authors show that their MIP approach significantly increased discovery of novel positive mutants. A different study uses ACL techniques to annotate digital histopathology data. Their method, class Based Active Learning (CBAL), uses a mathematical model that calculates the cost of building a training set with a certain size and class ratio.16
Among the few studies addressing the imbalance class problem in biological data sets, subset sample optimization (SSO) uses an ensemble-based approach and different sets of classifiers in its optimal training set selection procedure and another set of classifiers for classification on the test set.16 They have used several medical data sets from the UCI ML Repository17 and used a genome-wide association study (GWAS, http://gwas.nih.gov/) data set that is based on single nucleotide polymorphism (SNP) of age-related macular degeneration. The Artificial Immune Systems (AIS)-based classification algorithm has performed well on highly skewed data sets as compared with other methods that use Support Vector Machine (SVM).18,19
Applications of DL to biological data sets have increased substantially in recent years.20 Much of this work has focused on biomedical imaging,21 but a significant number of studies have focused on genomic data.22 These ML tasks include protein structure prediction, protein classification, and gene expression regulation. Such applications are characterized by the computation of hundreds to thousands of predetermined features, such as motifs, which are input to a DL network. But this approach is essentially just replacing the ACL-ICL method #1 in Fig. 1 with a deep neural network. Interestingly, methods #2 and #3 avoid the predetermination of specific features, but instead allow the DL network to generate relevant features from lower-level sequence data. Some recent approaches have used DL networks to generate relevant motifs using convolution layers on windowed sequence data, such as in the DeepBind method.23 Other approaches have used a one-hot encoding input to a convolution layer, where each sequence window of size W is represented by a Wx4 array indicating which bases (A, G, C, T) are present in the sequence window, such as in the DeepMotif method.24 These methods have achieved classification performance competitive with top non-DL methods.
Epigenetics
Machine learning and epigenetics
The ability to identify regions of the genome susceptible to epimutations will greatly improve our ability to diagnose disease, and the recommended ML techniques have demonstrated this ability. Currently, diagnosis of disease is done through sign and symptoms followed by genomic testing and screening. This genomic testing can identify molecular biomarkers and can identify the risk of disease for the patients. However, personalized medicine is more about studying the genomic profile to predict and prevent the diseases a patient is predisposed to and recommend better care such patient through pharmaceuticals, lifestyle changes, and screening.
Advanced experimental and computational techniques have brought us closer to realizing this goal of personalized medicine. Recent advances in epigenomic technology have allowed research involving high-throughput data and ML-based bioinformatics to make significant contributions. To identify epigenetic changes and disease prediction, several approaches are useful. These approaches combine collection of genomic features, such as epigenetic marks and genetic alterations (SNPs, copy number variations, repeat elements, transcriptomes, and motifs]. Given its increased ability to collect data and identification of epigenetic-relevant features, ML continues to improve its accuracy at investigating the epigenome and identifying epimutation sites, as well as expanding the medical applications of epigenetic-based disease diagnosis.
There have been several studies using ML in epigenetics research4,25,26 (Table 2). Applications have included epigenome mapping,27-32 bioinformatics on complex data,33-35 biological investigations,25,36-40 disease detection,11,41-45 environmental exposure detection,4,26 and technology development.46-51 One of the initial studies looked into finding imprinted genes in human and mouse genomes. Imprinted genes are epigenetically modified genes52 that are also associated with various diseases. The genome-wide prediction of imprinted murine genes focused on comprehensive profiling of the mouse genes. The research group found thousands of relevant features for better prediction of the imprinted gene by mining the DNA sequence characteristics around 100 kb upstream and downstream of the imprinted genes.53 They used the Equbits Foresight (www.equbits.com) classifier and predicted 722 new sites. Their study looked into 23,788 annotated autosomal mouse genes and identified 600 mouse imprinted genes. The same group later mined the human genome for new imprinted sites.54 They again used the Equbits Foresight with SVM and 622 features and used their own sparse multinomial logistic regression (SMLR)55 classifier with 820 features to predict novel human imprinted genes. Another study looks into the correlation of different features to DNA methylation of CpG islands. They mined features from 190 CpG islands from human chromosome 21 and tested it on the rest of the CpG islands in the genome to find methylated CpG islands. They looked for correlation among features and found that different methylation profiles exist not only for different tissue types but also for different diseases.56 Wang et al.57 compared a standard ML approach (SVM) to a DL autoencoder approach called DeepMethyl using several tumor cell lines to assess CpG methylation and associated genomic topological features. Results show that the DL approach can improve over SVM in some cases. Although using lower resolution (50 kb windows), these observations show the value of using ML and DL to provide insight into epigenetics.
Table 2.
Machine Learning Applications in Epigenetics.
| Application | Observations | Literature |
|---|---|---|
| Epigenome mapping | Epigenetic site prediction | 27-32 |
| Bioinformatics of complex data | Mixed cell type analysis | 33-35,38 |
| Biological investigations | Predictions biological parameters (age, metabolism, neuroscience, evolution) | 25,36,37,39,40,52 |
| Disease detection | Disease diagnostics and prognosis | 9,11,41-45 |
| Exposure detection | Environmental exposure detection and impacts | 4,26 |
| Technology development | Improvement and advances in epigenetic analysis | 46-51 |
A previous study by the authors used a combined ACL−ICL method (Fig. 1, method #1) with previous epigenetic data sets of sperm promoter differential DMRs, termed epimutations from promoters.58 This involved a sequential approach of ACL followed by ICL on a gene promoter specific DMR set.1,59 The prediction for the genome-wide locations for potential DMRs identified 3,353 sites and the chromosomal locations (Fig. 2). One of the main advantages of using ACL- and ICL-based methods is that these approaches are classifier-independent; therefore, another classifier can be used for prediction purposes. Future studies will explore more advanced ML approaches and more complete genome-wide epigenetic data.4
Figure 2.
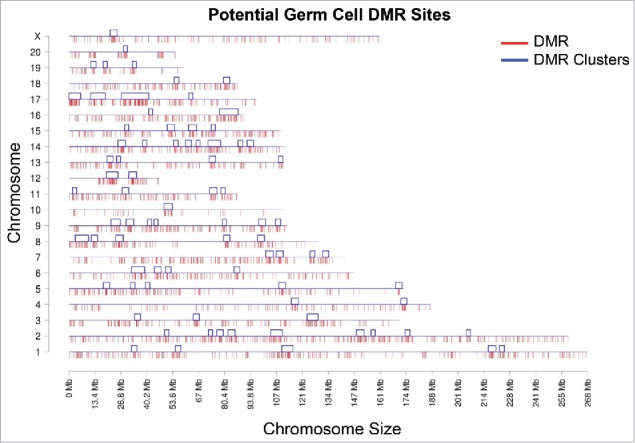
Genome-wide prediction of potential epimutation sites based on promoter only DMR training sets. Chromosomal plot of germ cell data set sperm shows the predicted 3+ sites and the clusters of DMR regions. Red lines below each chromosome line indicate predicted potential DMR sites (3,233) when sperm is used as the training set; blue boxes above each line indicate clusters.80 Y-axis shows each of the 21 chromosomes while X-axis shows the length of the chromosome with predicted potential DMR locations and the clusters. Clusters are regions that indicate over-representations of sites within a small sub-section of the genome.55 Modified from.4
Prediction of epigenetic states from relevant genomic features
Just as with any machine learning approach used for classification, the ML approach in epigenetics proceeds by training a classifier with relevant features, generating models, and then performing prediction on a set-aside test set. For the first phase of training, classifier-appropriate genomic features are needed, which are correlated with the label of the epigenetic data. Once the samples are properly labeled and features computed, the ML technique would build a predictive model.
Genomic features can include both DNA sequence and epigenetic components. Genetic features, such as repeat elements, CpG density, response elements, or specific sequences, are all DNA sequence-based features that impact the epigenome. In contrast, epigenetic features, such as DNA methylation or histone-mediated nucleosome positioning, and transitions between euchromatin and heterochromatin can impact gene expression and genetic features. More recently, epigenetic alterations have also been shown to influence genome stability and promote genetic sequence mutations.60,61 Therefore, the high degree of integration between genetics and epigenetics suggests that both features need to be considered in machine learning.
One of the main challenges of successful model building is how to use the high amount of available sample domain knowledge to guide the ML process. Having a good understanding and proper selection of genomic features is important for these kinds of tasks. Feature engineering or combining different features also needs to be considered. Appropriate pre-processing, data-cleaning, and careful selection of labeled data are important for building models with high accuracy. In the case of collecting genomic features, selection of a proper window size from which the features are collected is also important and benefits from the consideration of prior knowledge.
Since epigenetic data are expensive to acquire, alternative methods, such as prediction of potential epigenetic sites from DNA sequence, can act as a guide for future experimental epigenetic research or as a substitute for the data. The same is true for any genomic research. Mining of epigenetic profiles starts with extraction of interesting properties from DNA sequence data near base regions (location of epigenetic changes in the genome). After retrieving the training set, these locations are often annotated to find the name and orientation of the gene. FASTA files are created from up to 100 kb upstream and downstream of the target genes. After construction of FASTA files for extraction of genomic features, tools such as RepeatMasker4 are used to find SINE, LINE, ERVL, ERV, and other repeat elements to the upstream and downstream of the base locations. One of the common ways of extracting genomic features from sequences is through identification of repeat elements. Identifying repeat elements and consensus sites helps us detect interesting patterns from these sites. Other genomic features are GC content and CpG sites. Tools such as CpGislandSearcher4 can be used to find CpG islands in these regions. CpG islands work as catalysts as they overlap with promoter, enhancer, and other regulatory regions. Since over-representation of CpG islands can be due evolutionarily to reduced amount of DNA methylation, which then leads to less CpG to TpG mutation, lack of CpG islands can be a predictor of DNA methylation. In the previous study (shown in Fig. 2), one of the primary features was a low CpG density at epigenetic sites (Fig. 3). These are termed CpG deserts62 and will be a critical feature to consider.
Figure 3.
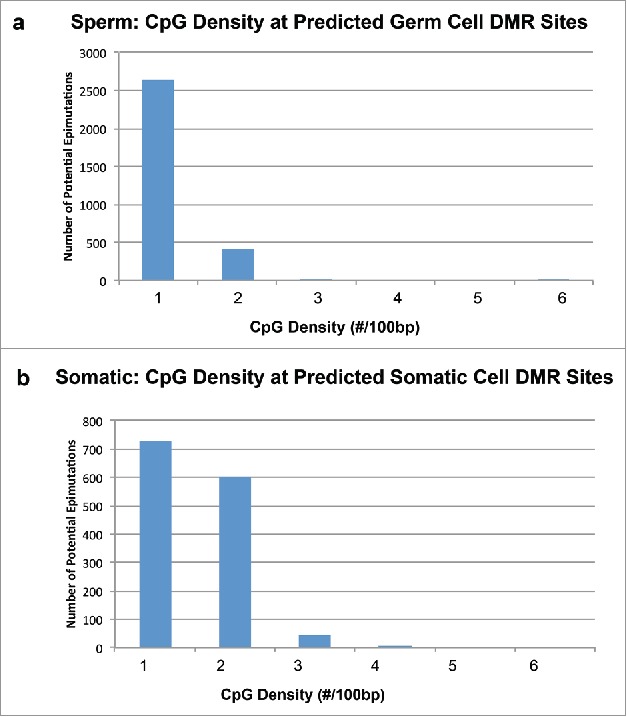
CpG density plot showing number of predicted DMR sites correlated with CpG density. (a) CpG density from the potential predicted germ cell DMR sites (3,234) when sperm is used as the training set to predict genome-wide. (b) CpG density from potential predicted somatic cell DMR sites (1,502) when somatic cell is used as training set to predict genome-wide CpGs. X-axis shows the number of CpGs per 100 bases on average, while Y-axis shows the number of sites. Modified from.4
Another important class of genomic feature is the DNA sequence motif.63,64 Common patterns among biologically relevant sites can be identified using motif finding tools. Motifs are short sequences that have biologically significant predicted roles. Motifs are identified with a probability matrix for each base position such that a certain combination of those sequences matches with every sub-sequence. Some motifs are also found to be unique to DNA methylation sites. Discovery tools like Oligo,65 LocalMotif,66,67 Prospector,68 and glam2,69 among other pattern discovery algorithms, have been used to find novel motifs, which are the best predictors of new DMR sites. These motifs are usually constructed by running these epigenetic sites from related experiments through some of the popular motif finding tools. For the murine imprinted gene project, the authors initially looked at 4 million genomic features, searching within a certain genomic distance. Most of these features were constructed by combining all combinations of features, ranking them based on which are more relevant, and then picking only the most relevant ones for final analysis.70,71
The above-mentioned DNA sequence characteristics (e.g., motifs, CpG islands), and many other features and techniques need to be used in the prediction of novel epigenetic sites. The amount of genomic features can be enormous, and finding relevant genomic features that help identify epigenetic sites is still a big challenge. Future research will need to develop more efficient and novel ML tools that combine computational approaches, including DL (for feature generation), ACL (to select the optimal feature set), and ICL (to improve accuracy when classes are imbalanced). Novel DL approaches tailored to predicting epigenetics alterations (or epimutations) are also promising, such as multi-dimensional convolution layers that are able to capture complex properties of the genome (e.g., CpG density) from the original sequence without formally defining such features. Although the current development and validation will involve epigenetics and genome-wide prediction of epimutations, these novel ML tools can be applied to other biological and non-biological data sets.
A previous study applied promoter DMR training sets in a preliminary ML approach on the rat genome and predicted 40,000 potential DMR sites genome-wide.4 Future research will need to advance the ML tool by using unbiased genome-wide data and advanced feature generation and selection. The preliminary rat ML tool was also applied to the human genome in a preliminary study and identified 20,000 potential human DMR sites susceptible to environmental reprogramming. Therefore, the previous studies support the need to develop more advanced ML tools for genomic and biological data.
Medical applications
Applications of machine learning and epigenetics to medicine
Machine learning was first applied to medicine with the use of electronic health records. An example is a comparison of approaches for heart failure cases.72 Recently, ML has been applied to pharmacology for improved therapy and pharmaceutical treatment design.73,74 Applications of ML in cardiovascular risk prediction,75 radiation oncology,76 and metabolic disease37 have been reported. ML has also been applied to clinical vision science77 and psychiatry.78 The application of ML to large molecular and clinical data sets will be critical in the future and have significant applications in medicine.79 The applications of machine learning and molecular epigenetics to medicine are outlined in Table 3.
Table 3.
Applications of machine learning and molecular epigenetics to medicine.
| Medical records and epidemiology studies |
| Molecular diagnostics for disease and disease susceptibility |
| Facilitating pharmacogenomics studies in therapy development and disease |
| Molecular diagnostics to facilitate treatment options for specific disease and medical conditions |
One of the first applications of epigenomics in medicine will be the development of molecular diagnostics for specific diseases or physiologic abnormalities. A number of disease conditions have been shown to be associated with epigenome modifications.1 Specific epimutations have been identified and correlated with specific physiologic abnormalities, such as in cancer, neurodegenerative disorders, fertility,80 obesity,81 ovarian disease,82 and gonadal function.83 Epigenetic programming and heterogeneity may play a role in standard therapies not being useful for many of these diseases. A combination of genetic and epigenetic approaches and diagnostic development is required for proper personalized medicine treatments. With the availability of massive amounts and novel types of data, there is more need to apply ML-based computational approaches to mine this data to extract meaningful insights. Using a trial-and-error approach to compare different classifiers and ML approaches is not very useful. To improve performance, there is a significant need for additional theoretical, experimental, and practical knowledge about ML techniques and specific research domain.
To realize the goal of personalized medicine, epigenetic modifications need to be identified. The prediction of genomic sites that are susceptible to epigenetic alterations will dramatically increase the potential to develop efficient molecular diagnostics for specific medical conditions. The application of ML to identify susceptible epimutation sites in the genome has been reported.4 Therefore, ML will not simply be used in medical records or population based epidemiology, but in the actual identification of molecular information to assist in the diagnostics and treatment of disease. We propose that the combination of Active Learning, Imbalanced Class Learning, and Deep Learning represents a promising and demonstrably successful direction toward realizing this goal.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Drs. Daniel Beck and Eric Nilsson for critical review of the manuscript and Ms. Heather Johnson for assistance in preparation of the manuscript.
Funding
This research was supported by NSF grant to LBH; NIH (ES012974–10) and Templeton (50183) grants to MKS.
Author contributions
LBH and MMH wrote and edited the manuscript. MKS conceived, wrote and edited the manuscript. All authors read and approved the final manuscript.
References
- 1.Skinner MK. Endocrine disruptor induction of epigenetic transgenerational inheritance of disease. Mol Cell Endocrinol 2014; 398(1-2):4-12; PMID:25088466; https://doi.org/ 10.1016/j.mce.2014.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manikkam M, M HM, Guerrero-Bosagna C, Nilsson E, Skinner M. Pesticide methoxychlor promotes the epigenetic transgenerational inheritance of adult onset disease through the female germline. PLoS One 2014; 9(7):e102091; PMID:25057798; https://doi.org/ 10.1371/journal.pone.0102091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waddington CH. Epigenetics and evolution. Symp Soc Exp Biol 1953; 7:186-99. [Google Scholar]
- 4.Haque MM, Holder LB, Skinner MK. Genome-wide locations of potential epimutations associated with environmentally induced epigenetic transgenerational inheritance of disease using a sequential machine learning prediction approach. PloS One 2015; 10(11):e0142274; PMID:26571271; https://doi.org/ 10.1371/journal.pone.0142274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dietterich TG, Lathrop RH, Lozano-Perez T. Solving the multiple instance problem with axis-parallel rectangles. Artif Intell 1997; 89:31-71; https://doi.org/ 10.1016/S0004-3702(96)00034-3 [DOI] [Google Scholar]
- 6.Settles B. Active Learning Literature Survey 2010. [Available from: http://www.cs.cmu.edu/∼bsettles/pub/settles.activelearning.pdf.
- 7.Du J, Ling CX. Active learning with generalized queries. Proceedings of the 9th IEEE International Conference on Data Mining, ICDM'2009 2009:120-8. [Google Scholar]
- 8.Smith JW, Everhart JE, Dickson WC, Knowler WC, Johannes RS. Using the ADAP learning algorithm to forecast the onset of diabetes mellitus. Proceedings of the Symposium on Computer Applications and Medical Care, IEEE Computer Society Press 1988:261-5. [Google Scholar]
- 9.Haque MM, Holder LB, Skinner MK, Cook DJ. Generalized query based active learning to identify differentially methylated regions in DNA. IEEE/ACM Trans Comput Biol Bioinform 2013; 10(3):632-44; PMID:24091397; https://doi.org/ 10.1109/TCBB.2013.38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hong-Bo S, Zhi-Hai W, Hou-Kuan H, Li-Ping J. Text classification based on the TAN model. TENCON'02 IEEE Region 10 Conference on Computers, Communications, Control and Power Engineering: IEEE; 2002. p. 43-6. [Google Scholar]
- 11.Jo T, Japkowicz N. Class imbalances versus small disjuncts. ACM SIGKDD Explorations Newsletter 2004; 6(1):40-9; https://doi.org/ 10.1145/1007730.1007737 [DOI] [Google Scholar]
- 12.Sun Y, Kamel MS, Wong AKC, Wang Y. Cost-sensitive boosting for classification of imbalanced data. Pattern Recogn 2007; 40(12):3358-78; https://doi.org/ 10.1016/j.patcog.2007.04.009 [DOI] [Google Scholar]
- 13.Libbrecht MW, Noble WS. Machine learning applications in genetics and genomics. Nat Rev Genet 2015; 16(6):321-32; PMID:25948244; https://doi.org/ 10.1038/nrg3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alipanahi B, Delong A, Weirauch M, Frey B. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat Biotechnol 2015; 33(8):831-8; PMID:26213851; https://doi.org/ 10.1038/nbt.3300 [DOI] [PubMed] [Google Scholar]
- 15.Lanchantin J, Singh R, Lin Z, Qi Y, editors. Deep Motif: Visualizing genomic sequence classifications. International Conference on Learning Representations; 2016. [Google Scholar]
- 16.Danziger SA, Baronio R, Ho L, Hall L, Salmon K, Hatfield GW, Kaiser P, Lathrop RH. Predicting positive p53 cancer rescue regions using Most Informative Positive (MIP) active learning. PLoS Comput Biol 2009; 5(9):e1000498; PMID:19756158; https://doi.org/ 10.1371/journal.pcbi.1000498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doyle S, Monaco J, Feldman M, Tomaszewski J, Madabhushi A. An active learning based classification strategy for the minority class problem: Application to histopathology annotation. BMC Bioinformatics 2011; 12:424; PMID:22034914; https://doi.org/ 10.1186/1471-2105-12-424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang P, Xu L, Zhou B, Zhang Z, Zomaya A. A particle swarm based hybrid system for imbalanced medical data sampling. BMC Genomics 2009; 10(Suppl 3):S34; PMID:19958499; https://doi.org/ 10.1186/1471-2164-10-S3-S34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang P, Zhang Z, Zhou BB, Zomaya AY. Sample subset optimization for classifying imbalanced biological data In Advances in Knowledge Discovery and Data Mining. Berlin Heidelberg: Springer; 2011. p. 333-44. [Google Scholar]
- 20.Freund Y, Schapire RE. A decision-theoretic generalization of on-line learning and an application to boosting. J Comput Syst Sci 1997; 55(1):119-39; https://doi.org/ 10.1006/jcss.1997.1504 [DOI] [Google Scholar]
- 21.Madabhushi A, Lee G. Image analysis and machine learning in digital pathology: Challenges and opportunities. Med Image Anal 2016; 33:170-5; PMID:27423409; https://doi.org/ 10.1016/j.media.2016.06.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Min S, Lee B, Yoon S. Deep learning in bioinformatics. Briefings Bioinform 2016. (epub) bbw068; PMID:27473064; https://doi.org/ 10.1093/bib/bbw068 [DOI] [PubMed] [Google Scholar]
- 23.Greenspan H, van Ginneken B, Summers R. Deep learning in medical imaging: Overview and future promise of an exciting new technique. IEEE T Med Imaging 2016; 35(5):1153-9; https://doi.org/ 10.1109/TMI.2016.2553401 [DOI] [Google Scholar]
- 24.Mamoshina P, Armando Vieira A, Putin E, Zhavoronkov A. Applications of deep learning in biomedicine. Mol Pharm 2016; 13(5):1445-54; PMID:27007977; https://doi.org/ 10.1021/acs.molpharmaceut.5b00982 [DOI] [PubMed] [Google Scholar]
- 25.Oh G, Wang SC, Pal M, Chen ZF, Khare T, Tochigi M, Ng C, Yang YA, Kwan A, Kaminsky ZA, et al. . DNA modification study of major depressive disorder: Beyond locus-by-locus comparisons. Biol Psychiatry 2015; 77(3):246-55; PMID:25108803; https://doi.org/ 10.1016/j.biopsych.2014.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ladd-Acosta C, Shu C, Lee BK, Gidaya N, Singer A, Schieve LA, Schendel DE, Jones N, Daniels JL, Windham GC, et al. . Presence of an epigenetic signature of prenatal cigarette smoke exposure in childhood. Environ Res 2016; 144(Pt A):139-48; PMID:26610292; https://doi.org/ 10.1016/j.envres.2015.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Angermueller C, Lee HJ, Reik W, Stegle O. DeepCpG: Accurate prediction of single-cell DNA methylation states using deep learning. Genome Biol 2017; 18(1):67; PMID:28395661; https://doi.org/ 10.1186/s13059-017-1233-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lutsik P, Slawski M, Gasparoni G, Vedeneev N, Hein M, Walter J. MeDeCom: Discovery and quantification of latent components of heterogeneous methylomes. Genome Biol 2017; 18(1):55; PMID:28340624; https://doi.org/ 10.1186/s13059-017-1182-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanchez R, Mackenzie SA. Genome-wide discriminatory information patterns of cytosine DNA methylation. Int J Mol Sci 2016; 17(6):938; PMID:27322251; https://doi.org/ 10.3390/ijms17060938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Capra JA. Extrapolating histone marks across developmental stages, tissues, and species: An enhancer prediction case study. BMC Genomics 2015; 16:104; PMID:25765133; https://doi.org/ 10.1186/s12864-015-1264-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cieslik M, Bekiranov S. Combinatorial epigenetic patterns as quantitative predictors of chromatin biology. BMC Genomics 2014; 15:76; PMID:24472558; https://doi.org/ 10.1186/1471-2164-15-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu X, Hoang S, Mayo MW, Bekiranov S. Application of machine learning methods to histone methylation ChIP-Seq data reveals H4R3me2 globally represses gene expression. BMC Bioinformatics 2010; 11:396. PMID:20653935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benton MC, Sutherland HG, Macartney-Coxson D, Haupt LM, Lea RA, Griffiths LR. Methylome-wide association study of whole blood DNA in the Norfolk Island isolate identifies robust loci associated with age. Aging (Albany NY) 2017; 9(3):753-68. PMID:28255110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shihab HA, Rogers MF, Campbell C, Gaunt TR. HIPred: An integrative approach to predicting haploinsufficient genes. Bioinformatics 2017; 33(12):1751–1757; PMID:28137713; https://doi.org/ 10.1093/bioinformatics/btx028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng Q, Ecker JR. Detection of allele-specific methylation through a generalized heterogeneous epigenome model. Bioinformatics 2012; 28(12):i163-71; PMID:22689757; https://doi.org/ 10.1093/bioinformatics/bts231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vidaki A, Ballard D, Aliferi A, Miller TH, Barron LP, Syndercombe Court D. DNA methylation-based forensic age prediction using artificial neural networks and next generation sequencing. Forensic Sci Int Genet 2017; 28:225-36; PMID:28254385; https://doi.org/ 10.1016/j.fsigen.2017.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tebani A, Afonso C, Marret S, Bekri S. Omics-based strategies in precision medicine: Toward a paradigm shift in inborn errors of metabolism investigations. Int J Mol Sci 2016; 17(9):1555; PMID:27649151; https://doi.org/ 10.3390/ijms17091555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kurum E, Benayoun BA, Malhotra A, George J, Ucar D. Computational inference of a genomic pluripotency signature in human and mouse stem cells. Biology Direct 2016; 11:47; PMID:27639379; https://doi.org/ 10.1186/s13062-016-0148-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He J, Sun MA, Wang Z, Wang Q, Li Q, Xie H. Characterization and machine learning prediction of allele-specific DNA methylation. Genomics 2015; 106(6):331-9; PMID:26407641; https://doi.org/ 10.1016/j.ygeno.2015.09.007 [DOI] [PubMed] [Google Scholar]
- 40.Chae H, Park J, Lee SW, Nephew KP, Kim S. Comparative analysis using K-mer and K-flank patterns provides evidence for CpG island sequence evolution in mammalian genomes. Nucleic Acids Res. 2013; 41(9):4783-91; PMID:23519616; https://doi.org/ 10.1093/nar/gkt144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silva TC, Colaprico A, Olsen C, D'Angelo F, Bontempi G, Ceccarelli M, Noushmehr H. TCGA Workflow: Analyze cancer genomics and epigenomics data using Bioconductor packages. F1000Res 2016; 5:1542; PMID:28232861; https://doi.org/ 10.12688/f1000research.8923.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Grembergen O, Bizet M, de Bony EJ, Calonne E, Putmans P, Brohee S, Olsen C, Guo M, Bontempi G, Sotiriou C, et al. . Portraying breast cancers with long noncoding RNAs. Sci Adv 2016; 2(9):e1600220; PMID:27617288; https://doi.org/ 10.1126/sciadv.1600220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li J, Ching T, Huang S, Garmire LX. Using epigenomics data to predict gene expression in lung cancer. BMC Bioinformatics 2015; 16 Suppl 5:S10; https://doi.org/ 10.1186/1471-2105-16-S5-S10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cai Z, Xu D, Zhang Q, Zhang J, Ngai SM, Shao J. Classification of lung cancer using ensemble-based feature selection and machine learning methods. Mol Biosyst 2015; 11(3):791-800; PMID:25512221; https://doi.org/ 10.1039/C4MB00659C [DOI] [PubMed] [Google Scholar]
- 45.Adorjan P, Distler J, Lipscher E, Model F, Muller J, Pelet C, Braun A, Florl AR, Gütig D, Grabs G, et al. . Tumour class prediction and discovery by microarray-based DNA methylation analysis. Nucleic Acids Res 2002; 30(5):e21; PMID:11861926; https://doi.org/ 10.1093/nar/30.5.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yalcin D, Hakguder ZM, Otu HH. Bioinformatics approaches to single-cell analysis in developmental biology. Mol Hum Reprod 2016; 22(3):182-92; PMID:26358759; https://doi.org/ 10.1093/molehr/gav050 [DOI] [PubMed] [Google Scholar]
- 47.Chen W, Feng P, Ding H, Lin H, Chou KC. iRNA-Methyl: Identifying N(6)-methyladenosine sites using pseudo nucleotide composition. Anal Biochem 2015; 490:26-33; PMID:26314792; https://doi.org/ 10.1016/j.ab.2015.08.021 [DOI] [PubMed] [Google Scholar]
- 48.Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning-based sequence model. Nat Methods 2015; 12(10):931-4; PMID:26301843; https://doi.org/ 10.1038/nmeth.3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pinello L, Lo Bosco G, Yuan GC. Applications of alignment-free methods in epigenomics. Brief Bioinform 2014; 15(3):419-30; PMID:24197932; https://doi.org/ 10.1093/bib/bbt078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schreiber J, Wescoe ZL, Abu-Shumays R, Vivian JT, Baatar B, Karplus K, Akeson M. Error rates for nanopore discrimination among cytosine, methylcytosine, and hydroxymethylcytosine along individual DNA strands. Proceedings of the National Academy of Sciences of the United States of America. 2013; 110(47):18910-5; PMID:24167260; https://doi.org/ 10.1073/pnas.1310615110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhuang J, Widschwendter M, Teschendorff AE. A comparison of feature selection and classification methods in DNA methylation studies using the Illumina Infinium platform. BMC Bioinformatics 2012; 13:59; PMID:22524302; https://doi.org/ 10.1186/1471-2105-13-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frank A, Asuncion A. UCI machine learning repository. URL http://archive ics uci edu/ml. 2010; 10. [Google Scholar]
- 53.Frank A, Asuncion A. UCI machine learning repository 2010. [Available from: http://archive.ics.uci.edu/ml.
- 54.Sotiropoulos G, Seitz AR, Series P. Perceptual learning in visual hyperacuity: A reweighting model. Vision Res 2011; 51(6):585-99; PMID:21316384; https://doi.org/ 10.1016/j.visres.2011.02.004 [DOI] [PubMed] [Google Scholar]
- 55.Luedi PP, Hartemink AJ, Jirtle RL. Genome-wide prediction of imprinted murine genes. Genome Res 2005; 15(6):875-84; PMID:15930497; https://doi.org/ 10.1101/gr.3303505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Luedi PP, Dietrich FS, Weidman JR, Bosko JM, Jirtle RL, Hartemink AJ. Computational and experimental identification of novel human imprinted genes. Genome Res 2007; 17(12):1723-30; PMID:18055845; https://doi.org/ 10.1101/gr.6584707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krishnapuram B, Carin L, Figueiredo MA, Hartemink AJ. Sparse multinomial logistic regression: Fast algorithms and generalization bounds. IEEE Trans Pattern Anal Mach Intell 2005; 27(6):957-68; PMID:15943426; https://doi.org/ 10.1109/TPAMI.2005.127 [DOI] [PubMed] [Google Scholar]
- 58.Wrzodek C, Buchel F, Hinselmann G, Eichner J, Mittag F, Zell A. Linking the epigenome to the genome: Correlation of different features to DNA methylation of CpG islands. PloS One 2012; 7(4):e35327; PMID:22558141; https://doi.org/ 10.1371/journal.pone.0035327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Y, Liu T, Xu D, Shi H, Zhang C, Mo Y-Y, Wang Z. Predicting DNA methylation state of CpG dinucleotide using genome topological features and deep networks. Sci Rep 2016; 6:19598; PMID:26237076 26797014; https://doi.org/ 10.1038/srep19598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Skinner MK, Guerrero-Bosagna C, Haque MM. Environmentally induced epigenetic transgenerational inheritance of sperm epimutations promote genetic mutations. Epigenetics 2015; 10(8):762-71; PMID:26237076; https://doi.org/ 10.1080/15592294.2015.1062207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McCarrey JR, Lehle JD, Raju SS, Wang Y, Nilsson EE, Skinner MK. Tertiary epimutations - A novel aspect of epigenetic transgenerational inheritance promoting genome instability. PloS One 2016; 11(12):e0168038; PMID:27992467; https://doi.org/ 10.1371/journal.pone.0168038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smit A, Hubley R, Green P. RepeatMasker 2015. [Available from: http://www.repeatmasker.org.
- 63.Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proceedings of the National Academy of Sciences of the United States of America. 2002; 99(6):3740-5; PMID:11891299; https://doi.org/ 10.1073/pnas.052410099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Takai D, Jones PA. The CpG island searcher: A new WWW resource. In Silico Biol 2003; 3(3):235-40. PMID:12954087. [PubMed] [Google Scholar]
- 65.Skinner MK, Guerrero-Bosagna C. Role of CpG deserts in the epigenetic transgenerational inheritance of differential DNA methylation regions. BMC Genomics 2014; 15(1):692; PMID:25142051; https://doi.org/ 10.1186/1471-2164-15-692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Das MK, Dai HK. A survey of DNA motif finding algorithms. BMC Bioinformatics 2007; 8 Suppl 7:S21; PMID:18047721; https://doi.org/ 10.1186/1471-2105-8-S7-S21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stormo GD. DNA binding sites: Representation and discovery. Bioinformatics 2000; 16(1):16-23; PMID:10812473; https://doi.org/ 10.1093/bioinformatics/16.1.16 [DOI] [PubMed] [Google Scholar]
- 68.Carro A, Rico D, Rueda OM, Diaz-Uriarte R, Pisano DG. waviCGH: A web application for the analysis and visualization of genomic copy number alterations. Nucleic Acids Res 2010; 38(Web Server issue):W182-7; PMID:20507915; https://doi.org/ 10.1093/nar/gkq441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu X, Brutlag DL, Liu JS. BioProspector: Discovering conserved DNA motifs in upstream regulatory regions of co-expressed genes. Pacific Symposium on Biocomputing Pacific Symposium on Biocomputing. 2001:127-38. PMID:11262934. [PubMed] [Google Scholar]
- 70.van Helden J, Rios AF, Collado-Vides J. Discovering regulatory elements in non-coding sequences by analysis of spaced dyads. Nucleic Acids Res 2000; 28(8):1808-18; PMID:10734201; https://doi.org/ 10.1093/nar/28.8.1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Narang V, Mittal A, Sung WK. Localized motif discovery in gene regulatory sequences. Bioinformatics 2010; 26(9):1152-9; PMID:20223835; https://doi.org/ 10.1093/bioinformatics/btq106 [DOI] [PubMed] [Google Scholar]
- 72.Blecker S, Katz SD, Horwitz LI, Kuperman G, Park H, Gold A, Sontag D. Comparison of approaches for heart failure case identification from electronic health record data. JAMA Cardiol 2016; 1(9):1014–1020; https://doi.org/ 10.1001/jamacardio.2016.3236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vanhaelen Q, Mamoshina P, Aliper AM, Artemov A, Lezhnina K, Ozerov I, Labat I, Zhavoronkov A. Design of efficient computational workflows for in silico drug repurposing. Drug Discovery Today 2017; 22(2):210-222; PMID:27693712. [DOI] [PubMed] [Google Scholar]
- 74.Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Gonçalves E, Barthorpe S, Lightfoot H, et al. . A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016; 166(3):740-54; PMID:27397505; https://doi.org/ 10.1016/j.cell.2016.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goldstein BA, Navar AM, Carter RE. Moving beyond regression techniques in cardiovascular risk prediction: Applying machine learning to address analytic challenges. Eur Heart J 2016. (epub) ehw302; https://doi.org/ 10.1093/eurheartj/ehw302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bibault JE, Giraud P, Burgun A. Big Data and machine learning in radiation oncology: State of the art and future prospects. Cancer Lett 2016; 382(1):110–117; PMID:27241666; https://doi.org/ 10.1016/j.canlet.2016.05.033 [DOI] [PubMed] [Google Scholar]
- 77.Caixinha M, Nunes S. Machine learning techniques in clinical vision sciences. Curr Eye Res 2017; 42(1):1-15. PMID:27362387 [DOI] [PubMed] [Google Scholar]
- 78.Iniesta R, Stahl D, McGuffin P. Machine learning, statistical learning and the future of biological research in psychiatry. Psychol Med 2016; 46(12):2455-65; PMID:27406289; https://doi.org/ 10.1017/S0033291716001367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mamoshina P, Vieira A, Putin E, Zhavoronkov A. Applications of deep learning in biomedicine. Mol Pharm 2016; 13(5):1445-54; PMID:27007977; https://doi.org/ 10.1021/acs.molpharmaceut.5b00982 [DOI] [PubMed] [Google Scholar]
- 80.Anway MD, Cupp AS, Uzumcu M, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 2005; 308(5727):1466-9; PMID:15933200; https://doi.org/ 10.1126/science.1108190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Skinner MK, Manikkam M, Tracey R, Nilsson E, Haque MM, Guerrero-Bosagna C. Ancestral dichlorodiphenyltrichloroethane (DDT) exposure promotes epigenetic transgenerational inheritance of obesity. BMC Medicine 2013; 11:228; PMID:24228800; https://doi.org/ 10.1186/1741-7015-11-228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nilsson E, Larsen G, Manikkam M, Guerrero-Bosagna C, Savenkova M, Skinner M. Environmentally induced epigenetic transgenerational inheritance of ovarian disease. PloS One 2012; 7(5):e36129; PMID:22570695; https://doi.org/ 10.1371/journal.pone.0036129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Guerrero-Bosagna C, Savenkova M, Haque MM, Sadler-Riggleman I, Skinner MK. Environmentally induced epigenetic transgenerational inheritance of altered sertoli cell transcriptome and epigenome: Molecular etiology of male infertility. PloS One 2013; 8(3):e59922; PMID:23555832; https://doi.org/ 10.1371/journal.pone.0059922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gillani Z, Akash MS, Rahaman MD, Chen M. CompareSVM: Supervised, Support Vector Machine (SVM) inference of gene regularity networks. BMC Bioinformatics 2014; 15:395; PMID:25433465; https://doi.org/ 10.1186/s12859-014-0395-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Huang BF, Boutros PC. The parameter sensitivity of random forests. BMC Bioinformatics 2016; 17(1):331; PMID:27586051; https://doi.org/ 10.1186/s12859-016-1228-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wu H, Li H, Jiang M, Chen C, Lv Q, Wu C. Identify high-quality protein structural models by enhanced K-means. Biomed Res Int 2017; 2017:7294519; PMID:28421198; https://doi.org/ 10.1155/2017/7294519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Reeb PD, Bramardi SJ, Steibel JP. Assessing dissimilarity measures for sample-based hierarchical clustering of RNA sequencing data using plasmode datasets. PLoS One 2015; 10(7):e0132310; PMID:26162080; https://doi.org/ 10.1371/journal.pone.0132310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Patel N, Wang JT. Semi-supervised prediction of gene regulatory networks using machine learning algorithms. J Biosci 2015; 40(4):731-40; PMID:26564975; https://doi.org/ 10.1007/s12038-015-9558-9 [DOI] [PubMed] [Google Scholar]
- 89.Ciucci S, Ge Y, Duran C, Palladini A, Jimenez-Jimenez V, Martinez-Sanchez LM, Wang Y, Sales S, Shevchenko A, Poser SW, et al. . Enlightening discriminative network functional modules behind Principal Component Analysis separation in differential-omic science studies. Sci Rep 2017; 7:43946; PMID:28287094; https://doi.org/ 10.1038/srep43946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Severson K, Monian B, Christopher Love J, Braatz RD. A method for learning a sparse classifier in the presence of missing data for high-dimensional biological datasets. Bioinformatics 2017. (epub ahead of print) btx224; PMID:28431087; https://doi.org/ 10.1093/bioinformatics/btx224 [DOI] [PubMed] [Google Scholar]
- 91.Franken H, Lehmann R, Häring H-U, Fritsche A, Stefan N, Zell A, editors. Wrapper- and ensemble-based feature subset selection methods for biomarker discovery in targeted metabolomics. Proceedings of the 6th IAPR International Conference on Pattern Recognition in Bioinformatics; 2011. [Google Scholar]
- 92.Padmanabhan RK, Somasundar VH, Griffith SD, Zhu J, Samoyedny D, Tan KS, Hu J, Liao X, Carin L, Yoon SS, et al. . An active learning approach for rapid characterization of endothelial cells in human tumors. PLoS One 2014; 9(3):e90495; PMID:24603893; https://doi.org/ 10.1371/journal.pone.0090495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cho H, Berger B, Peng J. Reconstructing causal biological networks through active learning. PLoS One 2016; 11(3):e0150611; PMID:26930205; https://doi.org/ 10.1371/journal.pone.0150611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lin W, Xu D. Imbalanced multi-label learning for identifying antimicrobial peptides and their functional types. Bioinformatics 2016; 32(24):3745-52; PMID:27565585; https://doi.org/ 10.1093/bioinformatics/btw560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kelchtermans P, Bittremieux W, De Grave K, Degroeve S, Ramon J, Laukens K, Valkenborg D, Barsnes H, Martens L. Machine learning applications in proteomics research: How the past can boost the future. Proteomics 2014; 14(4-5):353-66; PMID:24323524; https://doi.org/ 10.1002/pmic.201300289 [DOI] [PubMed] [Google Scholar]