

Abstract
Objective
Sarcomeric gene mutation carriers without overt left ventricular hypertrophy (G+/LVH−) can harbour subclinical changes in cardiovascular structure and function that precede the development of hypertrophic cardiomyopathy (HCM). We sought to investigate if circulating biomarkers of cardiovascular stress and collagen metabolism among G+/LVH− individuals, measured at rest and following exercise provocation, yield further insights into the underlying biology of HCM.
Methods
We studied 76 individuals with overt HCM, 50 G+/LVH− individuals and 41 genotype-negative related controls enrolled in a cross-sectional, multicentre observational study (HCMNet). Biomarkers of cardiac stress (N-terminal pro-B-type natriuretic peptide, NT-proBNP; high-sensitivity troponin I, hsTnI; soluble ST2) and fibrosis (carboxy-terminal propeptide of procollagen type I; C-terminal telopeptide of type I collagen; galectin-3; periostin) were measured.
Results
Individuals with overt HCM had elevated NT-proBNP and hsTnI compared with G+/LVH− subjects and controls at rest, along with an exaggerated increase in NT-proBNP and hsTnI in response to exercise. We found no detectable differences in resting or exercise-provoked biomarker profiles of cardiovascular stress and fibrosis among G+/LVH− individuals compared with healthy controls despite subtle echocardiographic differences in cardiac structure and function.
Conclusion
Dynamic exercise testing exaggerated resting differences in natriuretic peptides and troponin elevations among individuals with overt HCM. In contrast, we found no differences in biomarker profiles of cardiovascular stress and fibrosis among G+/LVH− individuals compared with controls even after maximal exercise provocation. Our findings highlight the need for continued investigation into early phenotypes of sarcomeric gene mutations and the evolution of HCM.
Keywords: hypertrophic cardiomyopathy, echocardiography, biomarker
Key questions.
What is already known about this subject?
Sarcomeric gene mutation carriers without overt left ventricular hypertrophy (G+/LVH−) can harbour subclinical changes in cardiovascular structure and function that precede the development of hypertrophic cardiomyopathy (HCM).
What does this study add?
Despite subtle subclinical changes in cardiovascular structure and function on echocardiography, we found no differences in biomarker profiles of cardiovascular stress and fibrosis among G+/LVH− individuals compared with controls even after maximal exercise provocation.
How might this impact on clinical practice?
The potential plasticity of early phenotypic manifestations in genotype-positive individuals may present future opportunities for prevention in the preclinical phase of the disease; however, our findings highlight the need for continued investigation into early phenotypes of sarcomeric gene mutations and the evolution of HCM.
Introduction
Hypertrophic cardiomyopathy (HCM) is frequently caused by sarcomeric gene mutations.1 Due to the autosomal dominant nature of inheritance, each immediate relative of an affected individual has a 50% chance of carrying the disease-causing mutation and potentially developing HCM. Therefore, current guidelines recommend longitudinal screening of first-degree family members of patients with this diagnosis.2 Most frequently serial clinical evaluation is performed; however, genetic testing provides the opportunity for more definitive identification of at-risk relatives, and its use has become more widespread. This has resulted in a growing population of genotype-positive individuals without overt left ventricular hypertrophy (LVH), and the unique opportunity to study preclinical phenotypes prior to the development of overt clinical manifestations of HCM.3 Prior studies suggest that adverse cardiovascular effects of pathogenic sarcomeric gene mutations may be at play long before the diagnostic clinical phenotype, LVH, is manifest. These findings include abnormal myocardial energetics, impaired left ventricular (LV) relaxation,4 altered intracellular calcium handling5 and transcriptional upregulation of fibrotic pathways.6 7 The potential plasticity of early phenotypic manifestations is supported by animal models and clinical data, suggesting that treatment of mutation carriers with diltiazem may improve cardiac remodelling if administered in the preclinical phase of the disease.5 8
A number of previous imaging studies focused on genotype-positive/LVH-negative (G+/LVH−) individuals have demonstrated abnormalities in cardiac structure and function.4 9–11 In contrast, there are few studies of circulating biomarkers of cardiovascular stress and collagen metabolism in G+/LVH− individuals with conflicting results to date.12 13 Therefore in this study, we sought to examine specific biomarkers reflecting cardiac stress (N-terminal pro-B-type natriuretic peptide, NT-proBNP; high-sensitivity troponin I, hsTnI; soluble ST2) and fibrosis (carboxy-terminal propeptide of procollagen type I, PICP; C-terminal telopeptide of type I collagen, CITP; galectin-3; periostin) in a well-characterised genotyped population, to attempt to examine potential pathways underlying subclinical disease in G+/LVH− individuals.
Lastly, we explored the potential for exercise stress to unmask and provoke latent phenotypic traits. Cardiopulmonary responses to exercise are predictors of outcome among patients with HCM.14 Exercise can also enhance detection of cardiovascular dysfunction, including abnormal LV strain and twisting reserve in HCM.15 Among general cardiology patients referred for clinical exercise testing, transient stress-induced ischaemia can be detected via ultrasensitive troponin assays.16 Exercise has not been examined as a means to uncover subclinical disease in G+/LVH− individuals. Examining such provoked phenotypes is of particular interest to further explore mechanisms underlying the formation of myocardial fibrosis in HCM, a key feature of clinically overt HCM.
Increased replacement and interstitial fibrosis are histopathological hallmarks of disease, and the majority of adults with HCM have demonstrable late gadolinium enhancement on cardiac MRI (CMR).17 Although the processes driving scar formation have not been rigorously characterised, it is generally considered to be a secondary consequence of ischaemic and haemodynamic stressors associated with HCM. However, both animal models and studies on preclinical human populations have suggested that profibrotic pathways are activated early in the pathogenesis of HCM, prior to the development of LVH.12 A potential hypothesis is that the presence of a sarcomeric gene mutation may make the myocyte more susceptible to injury. Although this susceptibility may not be detectable at rest, it may be unmasked by stress. We therefore sought to examine provoked phenotypes, as reflected by circulating biomarker levels measured after physiological stress, to investigate whether exercise testing might unmask subclinical phenotypes and a predisposition to myocardial injury among sarcomeric gene mutation carriers.
Methods
Study sample
Participants with overt HCM (G+/LVH+), mutation carriers without LVH (G+/LVH−) and healthy genotype-negative members of families with known mutations were enrolled in a cross-sectional, multicentre observational study (HCMNet) based at 11 HCM specialty centres in the USA (see online supplementary table 1). Genotype-positive individuals >5 years of age were included if sarcomeric gene mutations were deemed pathogenic or likely pathogenic based on segregation, conservation and absence from control populations according to standard criteria, and reassessed at the time of data analysis.18 All genetic testing was performed at Clinical Laboratory Improvement Amendments (CLIA_-approved facilities. Individuals with overt HCM were defined as sarcomeric gene mutation carriers with LV wall thickness ≥12 mm in adults >18 years or z-score of ≥3 in subjects under 18 years of age as previously described.12 Participants were deemed G+/LVH− if they were mutation carriers with a normal LV wall thickness (defined as maximal thickness under the limits above). Healthy genotype-negative relatives with normal LV wall thickness by echocardiogram comprised the control population. Participants with known hypertension, cardiovascular disease other than HCM, prior cardiac surgery or alcohol septal ablation, or comorbid conditions potentially altering collagen turnover (cirrhosis, liver, pulmonary, renal fibrosis, inflammatory states) were excluded from the study. Individuals >40 years of age were excluded from G+/LVH− and control groups. All participants provided informed consent.
openhrt-2017-000615supp001.pdf (107KB, pdf)
Study procedures
Participants underwent comprehensive medical and family history, physical exam, and symptom-limited treadmill exercise tolerance testing using a standard Bruce protocol.
Transthoracic echocardiography was performed, including two-dimensional, spectral, colour and tissue Doppler interrogation. Left atrial (LA), LV dimensions and standard parameters of diastolic function, including spectral Doppler transmitral early (E) and late (A) velocities and E wave deceleration time, were measured according to published guidelines.19 Tissue Doppler velocities of the lateral, septal, anterior and inferior mitral annulus were measured, and global e' velocity calculated as the average of these four values.11 Studies were analysed at a central core laboratory blinded to clinical and genotype status. Echocardiographic measurements were converted to z-scores (number of SD from the normal mean value relative to age or body surface area) due to the wide age range in our study cohort.20
CMR with gadolinium enhancement was performed in available subjects without contraindications. ECG-gated images were obtained during breath-hold using 1.5T or 3T systems using cine steady-state free precession imaging for LV function and mass.21 Late gadolinium enhancement was determined as previously described, and quantified as a percentage of the total LV mass.11 All CMR analyses were performed using QMass MR (V.7.4, Medis, Leiden, The Netherlands) by a core laboratory blinded to clinical and genotype status.
Biomarker assessment
Venous blood samples were obtained at rest, immediately after peak exercise and 4 hours post exercise, and serum and K3-EDTA plasma samples processed and frozen at −80°C for further analysis by the biomarker core laboratories (Brigham and Women’s Hospital, Boston, Massachusetts, USA, for NT-proBNP, hsTnI, soluble ST2 and galectin-3; University of Navarra, Pamplona, Spain, for PICP, CITP, periostin and bone alkaline phosphatase (BAP)). Biomarkers were assayed using commercially available reagents by personnel blinded to clinical status and genotype. For assay details and characteristics, please see online supplementary methods.
Statistical analysis
Of 193 individuals enrolled in HCMNet, 15 were excluded due to genetic variants deemed to be of uncertain significance at the time of data analysis, and 11 individuals were excluded due to missing baseline NT-proBNP measures. Baseline characteristics were summarised by the three study groups (G+/LVH+, G+/LVH− and controls) and compared using analysis of variance (ANOVA), and pairwise comparisons assessed using two-sample t-tests. Bonferroni-corrected p values of less than 0.017 were considered statistically significant. Biomarkers were log-transformed due to skewed distributions, and values below the detectable threshold were set to the lower detection limit (n=11 for NT-proBNP). The ratio of PICP/CITP was examined as an integrated measure of collagen type I synthesis and degradation.22 The ratio of PICP to BAP was examined to account for bone collagen turnover.23 NT-proBNP, soluble ST2 (sST2), hsTnI and galectin-3 concentrations before and after exercise testing were assessed, and change in biomarker concentrations compared between groups. Differences between groups were assessed using generalised estimating equations (GEEs) adjusted for age, sex and familial correlation using an exchangeable correlation structure.
In exploratory analyses, we compared biomarkers before and after exercise between MYH7 and MYBPC3 mutation carriers, as well as between thick filament (MYBPC3, MYH7, MYL2, MYL3) and thin filament mutation carriers (ACTC, TTNT2, TNNI3, TPM1). Correlations between biomarkers and measures of cardiac structure and function on echocardiography and CMR and exercise testing were assessed using GEE after adjusting for age, sex and family correlation. For correlation analyses, we accounted for six biomarkers tested, and deemed a p value of 0.01 as significant. All analyses were conducted using the Statistical Analysis System V.9.3.
Results
A total of 167 genotyped individuals were enrolled in this study, including 76 with overt HCM (G+/LVH+), 50 genotype-positive, LVH-negative individuals (G+/LVH−) and 41 genotype-negative controls. Individuals with overt HCM were older than G+/LVH− individuals (27±14 vs 20±11 years, p<0.05; table 1). The majority of individuals with overt HCM were asymptomatic, with 83% New York Heart Association class I symptoms and 33% taking beta-blockers. Genotyping revealed the majority of mutations were present in MYH7 and MYBPC3 in both G+/LVH+ and G+/LVH− individuals.
Table 1.
Baseline characteristics by study group (n=167)
| Overt HCM (n=76) | Preclinical HCM (n=50) | Control (n=41) | p for ANOVA | |
| Clinical characteristics | ||||
| Age, years | 27 (14)*† | 20 (11) | 17 (8) | 0.0002 |
| Women, n (%) | 25 (33)*† | 28 (56) | 24 (56) | 0.007 |
| Systolic blood pressure, mm Hg | 115 (15) | 113 (13) | 113 (13) | 0.71 |
| Diastolic blood pressure, mm Hg | 67 (9) | 68 (9) | 66 (9) | 0.55 |
| Height, cm | 168 (18)*† | 162 (14) | 159 (18) | 0.012 |
| Weight, kg | 72 (24)*† | 62 (18) | 58 (21) | 0.002 |
| Body surface area, kg/m2 | 1.83 (0.42)*† | 1.67 (0.32) | 1.59 (0.39) | 0.003 |
| NYHA class, n (%) | – | |||
| Class I | 63 (83) | 50 (100) | 41 (100) | |
| Class II | 9 (12) | 0 (0) | 0 (0) | |
| Class III | 2 (3) | 0 (0) | 0 (0) | |
| Class IV | 2 (3) | 0 (0) | 0 (0) | |
| Medication use, n (%) | – | |||
| Beta-blocker | 25 (33) | 0 (0) | 0 (0) | |
| Calcium channel blocker | 9 (12) | 1 (2) | 0 (0) | |
| ACE inhibitor/ARB | 4 (5) | 0 (0) | 0 (0) | |
| Genetic background, n (%) | 0.51 | |||
| MYH7 | 26 (34) | 20 (40) | ||
| MYBPC3 | 39 (51) | 26 (52) | ||
| TNNT2 | 7 (9) | 1 (2) | ||
| Imaging characteristics | ||||
| Left atrial diameter, cm | 3.7 (0.9)*† | 3.0 (0.5)* | 2.8 (0.5) | <0.0001 |
| LV end-diastolic dimension, z-score | −2.6 (1.6)*† | −1.8 (1.2)* | −1.2 (1.3) | <0.0001 |
| LV end-systolic dimension, z-score | −2.5 (2.0)*† | −1.6 (1.2)* | −0.7 (1.4) | <0.0001 |
| LV wall thickness, z-score | 10.0 (6.7)*† | 1.4 (1.3) | 1.1 (1.3) | <0.0001 |
| LV ejection fraction, % | 67 (10)* | 67 (8)* | 63 (8) | 0.048 |
| E/A ratio | 1.6 (0.5)*† | 2.0 (0.6) | 2.0 (0.5) | <0.0001 |
| Deceleration time, ms | 214 (67)*† | 184 (43) | 182 (44) | 0.003 |
| Mitral valve global e', cm/s | 11.3 (3.6)*† | 15.2 (1.9)* | 16.2 (2.3) | <0.0001 |
| Mitral valve global s', cm/s | 9.0 (2.0)† | 10.3 (1.9) | 9.6 (1.8) | 0.003 |
| CMR LGE present, n (%) | 18 (46) | – | – | – |
| CMR LGE, % LV mass | 5.9 (7.6) | NA | NA | – |
| Exercise testing | ||||
| Duration, min | 11.6 (3.0) | 12.2 (2.2) | 11.9 (3.0) | 0.48 |
| Peak MET, mL O2/kg/min | 13.1 (3.8) | 14.3 (2.6) | 14.3 (3.2) | 0.08 |
Values are mean (SD) unless otherwise indicated.
Cardiac MRI performed on a subset of n=41 overt HCM, n=32 preclinical HCM and n=23 controls, of whom LGE was assessed in n=39, 29 and 23 individuals.
*Indicates p<0.05 versus control.
†Indicates p<0.05 versus preclinical HCM.
ANOVA, analysis of variance; ARB, angiotensin receptor blocker; CMR, cardiac MRI; HCM, hypertrophic cardiomyopathy; LGE, late gadolinium enhancement; LV, left ventricular; MET, metabolic equivalent; NA, not applicable; NYHA, New York Heart Association; MYH7, myosin heavy chain 7; MYBPC3, myosin binding protein C; TNNT2, troponin T2.
On echocardiography, those with overt HCM had larger LA dimensions, smaller LV dimensions, greater LV wall thickness and lower mitral annular tissue Doppler velocities compared with the G+/LVH− and genotype-negative groups (table 1). In contrast to overt HCM, differences in LA size, LV dimensions and tissue Doppler were less pronounced between G+/LVH− individuals and genotype-negative controls. Among a subset of individuals (n=96) who underwent CMR, late gadolinium enhancement was ascertained in 39 G+/LVH+, 29 G+/LVH− and 23 controls. Among this group, 44% of the G+/LVH+ group and none in the G+/LVH− group had evidence of late gadolinium enhancement.
Baseline biomarker characterisation of overt and preclinical HCM
Individuals with overt HCM had higher baseline NT-proBNP (median 137 pg/mL (25th–75th percentile 60, 569)) compared with G+/LVH− and controls (45 pg/mL (20, 72) and 32 pg/mL (15, 51), respectively, adjusted p for each pairwise comparison <0.0001; table 2). Similarly, those with overt HCM had higher baseline hsTnI concentrations (6.3 (3.2, 11.7)) compared with G+/LVH− and control groups (1.9 (1.3, 5.0), p=0.008; and 1.4 (0.9, 2.4) ng/mL, p=0.03, respectively). There were no significant differences in NT-proBNP or hsTnI between G+/LVH− and controls. Further, there were no significant between-group differences detected for soluble ST2, or biomarkers of fibrosis, including galectin-3, PICP, CITP, PICP/CITP ratio, BAP, PICP/BAP ratio and periostin (p for ANOVA >0.05 for all; table 2).
Table 2.
Baseline and postexercise cardiovascular biomarkers by study group
| Overt HCM (n=76) | Preclinical HCM (n=50) | Control (n=41) | p for ANOVA | |
| Baseline biomarkers, median (25th–75th percentile) | ||||
| NT-proBNP, pg/mL | 137 (60, 569)*† | 45 (20, 72) | 32 (15, 51) | 0.0005 |
| High-sensitivity troponin, ng/mL | 6.3 (3.2, 11.7)*† | 1.9 (1.3, 5.0) | 1.4 (0.9, 2.4) | 0.03 |
| Soluble ST2, ng/mL | 29.2 (21.5, 37.3) | 26.5 (20.0, 35.5) | 22.8 (18.5, 31.3) | 0.83 |
| Galectin-3, pg/mL | 11.1 (9.7, 13.4) | 11.4 (10.2, 12.8) | 11.5 (9.7, 12.9) | 0.97 |
| PICP, μg/L | 95.4 (67.6, 141.2) | 102.5 (84.5, 144.8) | 114.9 (73.3, 184.9) | 0.94 |
| CITP, μg/L | 4.4 (3.1, 10.0) | 6.1 (3.5, 11.5) | 5.2 (3.3, 11.4) | 0.32 |
| PICP/CITP ratio | 18.7 (12.6, 32.0) | 15.8 (10.3, 26.1) | 17.6 (13.8, 26.5) | 0.09 |
| BAP, U/L | 1.5 (1.1, 2.4) | 1.4 (1.1, 2.6) | 1.3 (1.0, 1.9) | 0.72 |
| PICP/BAP ratio | 57.1 (38.8, 114.6) | 68.9 (37.9, 107.8) | 87.7 (50.9, 129.0) | 0.81 |
| Periostin | 70.5 (56, 93.5) | 73.5 (61, 95) | 73 (60, 92) | 0.99 |
| Immediate or 4-hour postexercise biomarkers, median (25th–75th percentile) | ||||
| NT-proBNP, pg/mL | 199 (84, 775)*† | 54 (27, 93) | 41 (23, 77) | 0.0004 |
| Change in NT-proBNP, pg/mL | 37 (14, 133)*† | 9 (5, 18) | 8 (3, 25) | 0.0004 |
| % Change in NT-proBNP (%) | 27 (15, 43) | 27 (13, 40) | 38 (13, 70) | 0.08 |
| 4-Hour hsTnI, ng/mL | 8.9 (5.4, 18.0)* | 2.6 (1.4, 7.2) | 2.0 (1.3, 3.8) | 0.03 |
| Change in hsTnI, ng/mL | 1.5 (−0.4, 4.5) | 0.3 (−0.2, 1.3) | 0.4 (−0.2, 1.0) | 0.43 |
| % Change in hsTnI (%) | 28 (−4, 67) | 17 (−11, 58) | 28 (−10, 76) | 0.25 |
| 4-Hour soluble ST2, ng/mL | 36.1 (26.1, 42.8) | 34.5 (25.7, 45.6) | 31.5 (24.3, 37.8) | 0.66 |
| Change in sST2, ng/mL | 4.5 (0.5, 9.8) | 7.9 (2.4, 10.9) | 6.3 (2.0, 11.0) | 0.68 |
| % Change in sST2 (%) | 17 (1, 36) | 28 (10, 42) | 19 (13, 41) | 0.99 |
*Indicates p<0.05 versus control.
†Indicates p<0.05 versus preclinical HCM in generalised estimating equations regression analyses accounting for age, sex and familial correlation.
ANOVA, analysis of variance; BAP, bone alkaline phosphatase; CITP, C-terminal telopeptide of type I collagen; HCM, hypertrophic cardiomyopathy; hsTnI, high-sensitivity troponin I; NT-proBNP, N-terminal pro-B-type natriuretic peptide; PICP, propeptide of procollagen type I; sST2, soluble ST2.
In exploratory analyses, there were no biomarker differences when stratified based on underlying genotype (MYH7 (n=46) vs MYBPC3 (n=65) gene mutations) (see online supplementary table 2). Similarly, there were no significant differences in biomarkers between individuals with thick filament (MYBPC3, MYH7, MYL2, MYL3) compared with thin filament mutations (ACTC, TTNT2, TNNI3, TPM1; see online supplementary table 3).
Exercise effects on biomarkers of cardiovascular stress
Exercise provocation enhanced differences in NT-proBNP between the G+/LVH+ group compared with G+/LVH− and controls. Specifically, NT-proBNP increased by 37 pg/mL (27%) in the G+/LVH+ group after exercise, compared with a 9 pg/mL increase (20%) in the G+/LVH− group, and 8 pg/mL increase (25%) in the control groups (p<0.0001 for both comparisons; table 2). Similarly, hsTnI increased by 1.5 ng/mL (24%) 4 hours after exercise in the G+/LVH+ group, compared with 0.3 ng/mL (16%) in the G+/LVH− and 0.4 ng/mL (29%) in the control groups, although between-group differences were statistically not significant (p for ANOVA 0.22). Lastly, soluble ST2 concentrations increased in all three groups 4 hours after exercise with no significant between-group differences (p for ANOVA 0.97).
Biomarkers correlate with cardiac structure and function in genotype-positive individuals
Among genotype-positive individuals, NT-proBNP was significantly correlated with measures of cardiac structure and function (figure 1). Specifica lly, NT-proBNP was correlated with greater LA diameter, greater LV wall thickness, lower mitral e' tissue Doppler velocity and greater late gadolinium enhancement by CMR (p<0.001 for all analyses adjusted for age, sex and familial correlation; table 3).
Figure 1.
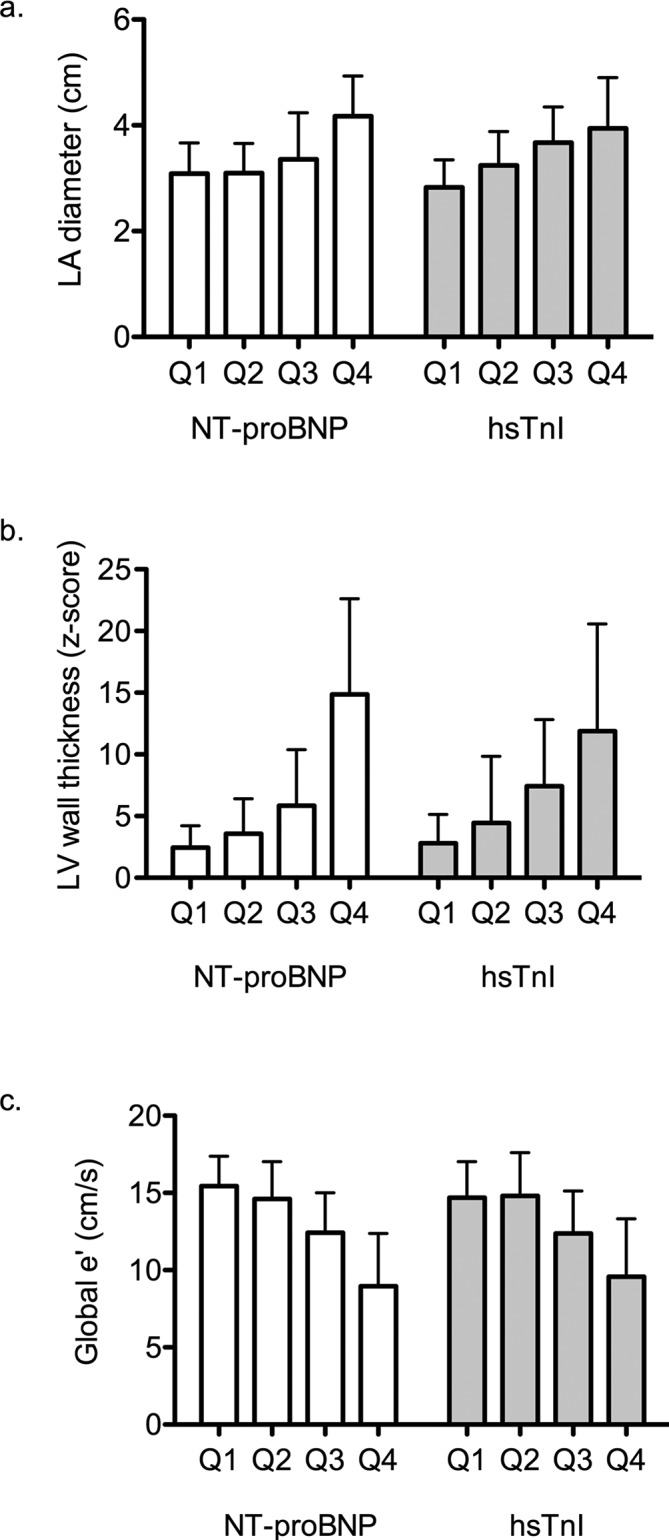
Association of biomarker quartiles and echocardiographic traits. Panel A shows left atrial diameter across NT-proBNP and hsTnI quartiles; panel B shows left ventricular wall thickness; and panel C shows tissue Doppler velocity for global mitral annular velocity. p for trend <0.0001 for all traits shown. hsTnI, high-sensitivity troponin I; LA, left atrial; LV, left ventricular; NT-proBNP, N-terminal pro-B-type natriuretic peptide.
Table 3.
Correlations between biomarkers and imaging characteristics among genotype-positive individuals
| NT-proBNP | hsTnI | Soluble ST2 | Galectin-3 | Periostin | ||||||
| Beta (SE) | p Value | Beta (SE) | p Value | Beta (SE) | p Value | Beta (SE) | p Value | Beta (SE) | p Value | |
| LA diameter | 0.67(0.12) | <0.0001 | 0.64 (0.11) | <0.0001 | 0.14 (0.12) | 0.22 | 0.22 (0.13) | 0.09 | 0.11 (0.12) | 0.35 |
| LVEDD z-score | −0.14 (0.06) | 0.018 | −0.01 (0.06) | 0.91 | 0.01 (0.05) | 0.79 | 0.03 (0.07) | 0.61 | 0.04 (0.06) | 0.58 |
| LVESD z-score | −0.12 (0.06) | 0.033 | 0.02 (0.05) | 0.72 | 0.02 (0.05) | 0.68 | 0.04 (0.06) | 0.49 | 0.02 (0.05) | 0.67 |
| LVWT z-score | 0.11 (0.01) | <0.0001 | 0.06 (0.01) | <0.0001 | −0.01 (0.01) | 0.59 | 0.01 (0.02) | 0.63 | 0.02 (0.01) | 0.15 |
| LVEF, % | 0.01 (0.01) | 0.20 | −0.002 (0.01) | 0.84 | −0.002 (0.01) | 0.79 | −0.02 (0.01) | 0.09 | 0.001 (0.01) | 0.96 |
| E/A ratio | −0.19 (0.14) | 0.19 | −0.34 (0.15) | 0.03 | −0.10 (0.13) | 0.45 | −0.49 (0.14) | 0.001 | 0.03 (0.16) | 0.84 |
| Deceleration time | 0.0004 (0.002) | 0.81 | 0.004 (0.002) | 0.02 | 0.002 (0.001) | 0.06 | 0.002 (0.002) | 0.23 | −0.002 (0.002) | 0.21 |
| Global e' z-score | −0.21(0.02) | <0.0001 | −0.16 (0.02) | <0.0001 | 0.01 (0.03) | 0.65 | −0.05 (0.03) | 0.15 | −0.03 (0.03) | 0.21 |
| Global s' z-score | −0.20 (0.05) | 0.0002 | −0.13 (0.04) | 0.003 | 0.08 (0.03) | 0.02 | −0.04 (0.05) | 0.42 | −0.07 (0.04) | 0.10 |
| CMR LGE, % | 0.07 (0.01) | <0.0001 | 0.02 (0.01) | 0.02 | 0.02 (0.01) | 0.03 | 0.01 (0.01) | 0.24 | 0.07 (0.01) | <0.001 |
| Exercise duration | −0.13 (0.03) | 0.0001 | −0.04 (0.03) | 0.15 | 0.01 (0.03) | 0.66 | −0.03 (0.03) | 0.31 | −0.03 (0.03) | 0.40 |
| Peak MET | −0.12 (0.02) | <0.0001 | −0.05 (0.03) | 0.07 | 0.02 (0.02) | 0.46 | −0.05 (0.03) | 0.12 | −0.03 (0.03) | 0.19 |
Beta coefficient represents the change in imaging characteristic per 1 SD increase in log-transformed biomarker, after adjustment for age, sex and familial correlation.
CMR, cardiac MRI; hsTnI, high-sensitivity troponin I; LA, left atrial; LGE, late gadolinium enhancement; LVEDD, left ventricular end-diastolic dimension; LVEF, left ventricular ejection fraction; LVESD, left ventricular end-systolic dimension; LVWT, left ventricular wall thickness; MET, metabolic equivalent; NT-proBNP, N-terminal pro-B-type natriuretic peptide.
Similarly, hsTnI was correlated with larger LA diameter, greater LV wall thickness and lower mitral e' velocity (p<0.0001 for all). We did not find significant associations of soluble ST2 and cardiac structure or function. Lastly, galectin-3 concentrations were associated with lower E/A ratio (p=0.001), but no other associations with cardiac measures were observed (table 3).
Discussion
Among a young population of sarcomeric gene mutation carriers with overt HCM, mutation carriers without LVH and healthy genotype-negative members of families with known mutations, we found that both NT-proBNP and hsTnI were elevated in those with overt HCM. This difference was further enhanced by dynamic exercise testing, with an exaggerated increase in natriuretic peptides in response to exercise among individuals with overt HCM. Interestingly, despite detectable echocardiographic differences in cardiac structure and function among mutation carriers and genotype-negative controls, we found no significant difference in resting or exercise-provoked biomarker profiles of cardiovascular stress and fibrosis among preclinical mutation carriers compared with healthy controls.
The growing use of genetic testing has allowed identification of an intriguing population of individuals who carry disease-causing sarcomeric gene mutations at a stage when LVH or diagnostic clinical manifestations of HCM are absent.24 Although the penetrance of sarcomeric gene mutations may be variable or incomplete, this preclinical population is at high risk for developing HCM and therefore provides a unique opportunity to examine early phenotypes that may herald adverse cardiovascular remodelling that ultimately leads to clinical disease. Previous studies have demonstrated subclinical abnormalities in diastolic function, myocardial deformation and other structural abnormalities among mutation carriers.10 25 26 In addition to differences detected on imaging modalities, early transcriptional changes and altered myocardial energetics may also be at play.6 7 Notably, few studies have examined circulating biomarkers in this preclinical population.
Two smaller previous studies have shown no difference in resting NT-proBNP concentrations among mutation carriers compared with unaffected relatives.11 13 We now extend these results to include biomarker profiles obtained during exercise testing as a dynamic assessment of cardiovascular response to stress, and demonstrate that even with provocation, NT-proBNP does not differ among mutation carriers and unaffected relatives. In contrast, baseline differences in NT-proBNP among individuals with overt HCM are further unmasked with exercise.
Second, we present a broader range of biomarkers of cardiovascular stress and fibrosis among preclinical mutation carriers compared with prior studies. Prior studies have demonstrated mixed results with regard to markers of collagen metabolism among patients with HCM.27–29 In a single-centre study, we previously observed a higher concentration of PICP among mutation carriers.12 This was not replicated in the current multicentre cohort, and may be due to age differences in the control group. It may be that older samples with more advanced disease may display different biomarker profiles, including markers of neurohormonal activation, myocyte injury and cardiac fibrosis. Of note, PICP concentrations were quite high in our control group. This may be in part due to the young age of mutation-negative controls (mean age 17 years), raising the possibility of bone turnover and growth during adolescence as potential confounder of PICP concentrations that is not fully accounted for by assessing BAP.30 In addition, physical exercise is a known source of bone turnover, and it has been shown in adolescent males (aged 15–17 years) that PICP increases with chronic aerobic/endurance training,31 which may have influenced our study results. Lastly, given that apparently healthy controls were still selected from families with HCM participants, there may be other potential confounding factors at play, including possible unrecognised genetic and familial determinants of circulating PICP.32
Galectin-3 is a potential mediator of cardiac fibrosis in experimental studies,33 and also reflects the degree of cardiac fibrosis in non-ischaemic cardiomyopathy as assessed by late gadolinium enhancement on CMR.34 Circulating galectin-3 can be elevated years prior to clinically apparent heart failure35; thus, we sought to investigate whether galectin-3 may be elevated early in the course of HCM. We did not find a difference in galectin-3 among patients with HCM compared with genotype-negative related controls. These findings contrast with a prior case–control study showing that galectin-3 was higher in 40 patients with HCM compared with 35 unrelated healthy controls.36 This discrepancy may be due to differences in age, as our sample was over 20 years younger on average, compared with the prior case–control study. It is also important to note that galectin-3 levels are in part genetically determined,37 which may blunt potential differences among related individuals such as in our study. Among genotype-positive individuals, galectin-3 was negatively correlated with E/A ratio, although we found no association with other measures of diastolic function or late gadolinium enhancement by CMR.
Lastly, we examined soluble ST2, an interleukin-1 receptor family member that is upregulated in cardiomyocytes in response to stress.38 Although patients with HCM had numerically higher soluble ST2 concentrations, there was no statistical difference among groups.
In contrast to the paucity of studies on biomarkers in mutation carriers, numerous previous studies have examined cardiovascular biomarkers among patients with overt HCM. Our findings are congruent with others showing elevated natriuretic peptide and troponin concentrations in HCM, which in turn predict functional capacity and clinical prognosis among this patient population.39–42 Among genotype-positive individuals, we also found that NT-proBNP and hsTnI were correlated with cardiac remodelling, including LA size, LV wall thickness, diastolic function as assessed by tissue Doppler mitral annular e' velocity, and regional fibrosis based on late gadolinium enhancement, similar to prior studies.39 43 44
Several additional limitations deserve mention. The utility of circulating biomarkers among mutation carriers without LVH remains unclear, particularly since we ascertained biomarkers at a single timepoint among otherwise relatively young, healthy individuals. Future studies should focus on serial measurements to examine whether biomarkers may be useful in detecting dynamic subclinical cardiovascular remodelling as a potential screening strategy. We acknowledge that, despite a multicentre effort, our sample size is modest, and may preclude detection of smaller effect sizes. Based on prior studies on subjects with coronary artery disease,16 we ascertained biomarkers immediately after peak exercise, and again 4 hours after maximal stress testing. It is possible that provoked changes with exercise might be better captured during recovery or later following exercise. Furthermore, it is possible that sustained, submaximal exercise, rather than maximal, symptom-limited exercise may be a more appropriate physiological stress to detect abnormalities in cardiac biomarker release.45
In sum, we showed that dynamic exercise testing exaggerated resting differences in natriuretic peptides and troponin elevations among individuals with overt HCM compared with controls. It is important to note that while we observed changes in NT-proBNP and hsTnI with exercise in overt HCM, the clinical implications of this finding remain unknown. The benefits of aerobic exercise in the general population are well-known, and whether this can be extended to patients with overt HCM is an active area of investigation.46 Despite the presence of altered cardiac structure and function by imaging, we found no differences in biomarker profiles of cardiovascular stress and fibrosis among preclinical mutation carriers without LVH compared with controls even after maximal exercise provocation. Our findings highlight that much remains to be elucidated with regard to the early phenotypes of sarcomeric gene mutations and the evolution of HCM. Continued investigation to identify pathways involved early in the disease processes, as well as serum biomarkers that reliably reflect their alteration, will yield valuable insights with regard to characterising disease development and targeting early preventive strategies to at-risk individuals.
Footnotes
Contributors: All authors have contributed to this investigation as follows: study concept and proposal (JEH, EJO, CYH), data acquisition (SMD, SDC, MWR, JAT, MVS, CEC, JLJ, AM, MT, LM, ALC, LAS, PJ, BL, AG, JD, CYH), data analysis (JEH, LS, EJO, CYH), drafting of manuscript (JEH, LS, CYH) and critical revisions of the manuscript (all authors).
Funding: This work was supported by the National Heart, Lung, and Blood Institute at the National Institutes of Health (K23-HL116780 to JEH, 1P20HL101408 to CYH).
Competing interests: PJ has received research support from Abbott Laboratories, Amgen, AstraZeneca LP, Beckman Coulter, Daiichi Sankyo, GlaxoSmithKline, Merck & Co, Roche Diagnostics Corporation, Takeda Global Research and Development Center, and Waters Technologies Corporation.
Ethics approval: The study protocol was approved by the appropriate individual institutional review boards.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: There are no additional data available for this paper.
References
- 1.Seidman JG, Seidman C. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell 2001;104:557–67. [DOI] [PubMed] [Google Scholar]
- 2.Gersh BJ, Maron BJ, Bonow RO, et al. . 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association task force on practice guidelines. Circulation 2011;124:e783–831. 10.1161/CIR.0b013e318223e2bd [DOI] [PubMed] [Google Scholar]
- 3.Ho CY. Genetics and clinical destiny: improving care in hypertrophic cardiomyopathy. Circulation 2010;122:2430–40. 10.1161/CIRCULATIONAHA.110.978924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ho CY, Sweitzer NK, McDonough B, et al. . Assessment of diastolic function with Doppler tissue imaging to predict genotype in preclinical hypertrophic cardiomyopathy. Circulation 2002;105:2992–7. [DOI] [PubMed] [Google Scholar]
- 5.Semsarian C, Ahmad I, Giewat M, et al. . The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J Clin Invest 2002;109:1013–20. 10.1172/JCI200214677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim JB, Porreca GJ, Song L, et al. . Polony multiplex analysis of gene expression (PMAGE) in mouse hypertrophic cardiomyopathy. Science 2007;316:1481–4. 10.1126/science.1137325 [DOI] [PubMed] [Google Scholar]
- 7.Crilley JG, Boehm EA, Blair E, et al. . Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J Am Coll Cardiol 2003;41:1776–82. 10.1016/S0735-1097(02)03009-7 [DOI] [PubMed] [Google Scholar]
- 8.Ho CY, Lakdawala NK, Cirino AL, et al. . Diltiazem treatment for pre-clinical hypertrophic cardiomyopathy sarcomere mutation carriers: a pilot randomized trial to modify disease expression. JACC Heart Fail 2015;3:180–8. 10.1016/j.jchf.2014.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ho CY, Carlsen C, Thune JJ, et al. . Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2009;2:314–21. 10.1161/CIRCGENETICS.109.862128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gruner C, Chan RH, Crean A, et al. . Significance of left ventricular apical-basal muscle bundle identified by cardiovascular magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Eur Heart J 2014;35:2706–13. 10.1093/eurheartj/ehu154 [DOI] [PubMed] [Google Scholar]
- 11.Ho CY, Abbasi SA, Neilan TG, et al. . T1 measurements identify extracellular volume expansion in hypertrophic cardiomyopathy sarcomere mutation carriers with and without left ventricular hypertrophy. Circ Cardiovasc Imaging 2013;6:415–22. 10.1161/CIRCIMAGING.112.000333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ho CY, López B, Coelho-Filho OR, et al. . Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N Engl J Med 2010;363:552–63. 10.1056/NEJMoa1002659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silva D, Madeira H, Almeida A, et al. . Tissue Doppler imaging and plasma N-terminal probrain natriuretic peptide for the identification of hypertrophic cardiomyopathy mutation carriers. Am J Cardiol 2013;112:996–1004. 10.1016/j.amjcard.2013.05.039 [DOI] [PubMed] [Google Scholar]
- 14.Finocchiaro G, Haddad F, Knowles JW, et al. . Cardiopulmonary responses and prognosis in hypertrophic cardiomyopathy: a potential role for comprehensive noninvasive hemodynamic assessment. JACC Heart Fail 2015;3:408–18. 10.1016/j.jchf.2014.11.011 [DOI] [PubMed] [Google Scholar]
- 15.Soullier C, Obert P, Doucende G, et al. . Exercise response in hypertrophic cardiomyopathy: blunted left ventricular deformational and twisting reserve with altered systolic-diastolic coupling. Circ Cardiovasc Imaging 2012;5:324–32. 10.1161/CIRCIMAGING.111.968859 [DOI] [PubMed] [Google Scholar]
- 16.Sabatine MS, Morrow DA, de Lemos JA, et al. . Detection of acute changes in circulating troponin in the setting of transient stress test-induced myocardial ischaemia using an ultrasensitive assay: results from TIMI 35. Eur Heart J 2009;30:162–9. 10.1093/eurheartj/ehn504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rubinshtein R, Glockner JF, Ommen SR, et al. . Characteristics and clinical significance of late gadolinium enhancement by contrast-enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010;3:51–8. 10.1161/CIRCHEARTFAILURE.109.854026 [DOI] [PubMed] [Google Scholar]
- 18.Alfares AA, Kelly MA, McDermott G, et al. . Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med 2015;17:880–8. 10.1038/gim.2014.205 [DOI] [PubMed] [Google Scholar]
- 19.Lang RM, Badano LP, Mor-Avi V, et al. . Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr 2015;28:1–39. 10.1016/j.echo.2014.10.003 [DOI] [PubMed] [Google Scholar]
- 20.Sluysmans T, Colan SD. Theoretical and empirical derivation of cardiovascular allometric relationships in children. J Appl Physiol 2005;99:445–57. 10.1152/japplphysiol.01144.2004 [DOI] [PubMed] [Google Scholar]
- 21.Chavhan GB, Babyn PS, Jankharia BG, et al. . Steady-state MR imaging sequences: physics, classification, and clinical applications. Radiographics 2008;28:1147–60. 10.1148/rg.284075031 [DOI] [PubMed] [Google Scholar]
- 22.López B, González A, Díez J. Circulating biomarkers of collagen metabolism in cardiac diseases. Circulation 2010;121:1645–54. 10.1161/CIRCULATIONAHA.109.912774 [DOI] [PubMed] [Google Scholar]
- 23.Trivedi P, Risteli J, Risteli L, et al. . Serum concentrations of the type I and III procollagen propeptides as biochemical markers of growth velocity in healthy infants and children and in children with growth disorders. Pediatr Res 1991;30:276–80. 10.1203/00006450-199109000-00016 [DOI] [PubMed] [Google Scholar]
- 24.Maron BJ, Ho CY, Cy H. Hypertrophic cardiomyopathy without hypertrophy: an emerging pre-clinical subgroup composed of genetically affected family members. JACC Cardiovasc Imaging 2009;2:65–8. 10.1016/j.jcmg.2008.09.008 [DOI] [PubMed] [Google Scholar]
- 25.Nagueh SF, McFalls J, Meyer D, et al. . Tissue Doppler imaging predicts the development of hypertrophic cardiomyopathy in subjects with subclinical disease. Circulation 2003;108:395–8. 10.1161/01.CIR.0000084500.72232.8D [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rüssel IK, Brouwer WP, Germans T, et al. . Increased left ventricular torsion in hypertrophic cardiomyopathy mutation carriers with normal wall thickness. J Cardiovasc Magn Reson 2011;133 10.1186/1532-429X-13-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ellims AH, Taylor AJ, Mariani JA, et al. . Evaluating the utility of circulating biomarkers of collagen synthesis in hypertrophic cardiomyopathy. Circ Heart Fail 2014;7:271–8. 10.1161/CIRCHEARTFAILURE.113.000665 [DOI] [PubMed] [Google Scholar]
- 28.Roldán V, Marín F, Gimeno JR, et al. . Matrix metalloproteinases and tissue remodeling in hypertrophic cardiomyopathy. Am Heart J 2008;156:85–91. 10.1016/j.ahj.2008.01.035 [DOI] [PubMed] [Google Scholar]
- 29.Lombardi R, Betocchi S, Losi MA, et al. . Myocardial collagen turnover in hypertrophic cardiomyopathy. Circulation 2003;108:1455–60. 10.1161/01.CIR.0000090687.97972.10 [DOI] [PubMed] [Google Scholar]
- 30.Crofton PM, Evans N, Taylor MR, et al. . Procollagen type I amino-terminal propeptide: pediatric reference data and relationship with procollagen type I carboxyl-terminal propeptide. Clin Chem 2004;50:2173–6. 10.1373/clinchem.2004.039958 [DOI] [PubMed] [Google Scholar]
- 31.Eliakim A, Raisz LG, Brasel JA, et al. . Evidence for increased bone formation following a brief endurance-type training intervention in adolescent males. J Bone Miner Res 1997;12:1708–13. 10.1359/jbmr.1997.12.10.1708 [DOI] [PubMed] [Google Scholar]
- 32.Sorva A, Tähtelä R, Risteli J, et al. . Familial high serum concentrations of the carboxyl-terminal propeptide of type I procollagen. Clin Chem 1994;40:1591–3. [PubMed] [Google Scholar]
- 33.Sharma UC, Pokharel S, van Brakel TJ, et al. . Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation 2004;110:3121–8. 10.1161/01.CIR.0000147181.65298.4D [DOI] [PubMed] [Google Scholar]
- 34.Vergaro G, Del Franco A, Giannoni A, et al. . Galectin-3 and myocardial fibrosis in nonischemic dilated cardiomyopathy. Int J Cardiol 2015;184:96–100. 10.1016/j.ijcard.2015.02.008 [DOI] [PubMed] [Google Scholar]
- 35.Ho JE, Liu C, Lyass A, et al. . Galectin-3, a marker of cardiac fibrosis, predicts incident heart failure in the community. J Am Coll Cardiol 2012;60:1249–56. 10.1016/j.jacc.2012.04.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yakar Tülüce S, Tülüce K, Çil Z, et al. . Galectin-3 levels in patients with hypertrophic cardiomyopathy and its relationship with left ventricular mass index and function. Anatol J Cardiol 2016;16 10.5152/AnatolJCardiol.2015.6191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Boer RA, Verweij N, van Veldhuisen DJ, et al. . A genome-wide association study of circulating galectin-3. PLoS One 2012;7:e47385 10.1371/journal.pone.0047385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weinberg EO, Shimpo M, De Keulenaer GW, et al. . Expression and regulation of ST2, an interleukin-1 receptor family member, in cardiomyocytes and myocardial infarction. Circulation 2002;106:2961–6. 10.1161/01.CIR.0000038705.69871.D9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moreno V, Hernández-Romero D, Vilchez JA, et al. . Serum levels of high-sensitivity troponin T: a novel marker for cardiac remodeling in hypertrophic cardiomyopathy. J Card Fail 2010;16:950–6. 10.1016/j.cardfail.2010.07.245 [DOI] [PubMed] [Google Scholar]
- 40.Saura D, Marín F, Climent V, et al. . Left atrial remodelling in hypertrophic cardiomyopathy: relation with exercise capacity and biochemical markers of tissue strain and remodelling. Int J Clin Pract 2009;63:1465–71. 10.1111/j.1742-1241.2009.02127.x [DOI] [PubMed] [Google Scholar]
- 41.Kubo T, Kitaoka H, Yamanaka S, et al. . Significance of high-sensitivity cardiac troponin T in hypertrophic cardiomyopathy. J Am Coll Cardiol 2013;62:1252–9. 10.1016/j.jacc.2013.03.055 [DOI] [PubMed] [Google Scholar]
- 42.Cambronero F, Marín F, Roldán V, et al. . Biomarkers of pathophysiology in hypertrophic cardiomyopathy: implications for clinical management and prognosis. Eur Heart J 2009;30:139–51. 10.1093/eurheartj/ehn538 [DOI] [PubMed] [Google Scholar]
- 43.Kawasaki T, Sakai C, Harimoto K, et al. . Usefulness of high-sensitivity cardiac troponin T and brain natriuretic peptide as biomarkers of myocardial fibrosis in patients with hypertrophic cardiomyopathy. Am J Cardiol 2013;112:867–72. 10.1016/j.amjcard.2013.04.060 [DOI] [PubMed] [Google Scholar]
- 44.Kubo T, Kitaoka H, Okawa M, et al. . Serum cardiac troponin I is related to increased left ventricular wall thickness, left ventricular dysfunction, and male gender in hypertrophic cardiomyopathy. Clin Cardiol 2010;33:E1–E7. 10.1002/clc.20622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shave R, Baggish A, George K, et al. . Exercise-induced cardiac troponin elevation: evidence, mechanisms, and implications. J Am Coll Cardiol 2010;56:169–76. 10.1016/j.jacc.2010.03.037 [DOI] [PubMed] [Google Scholar]
- 46.Day SM. Exercise in hypertrophic cardiomyopathy. J Cardiovasc Transl Res 2009;2:407–14. 10.1007/s12265-009-9134-5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
openhrt-2017-000615supp001.pdf (107KB, pdf)